
Analysis of the Ammonia Production Rates by Nitrogenase


Abstract
1. Introduction
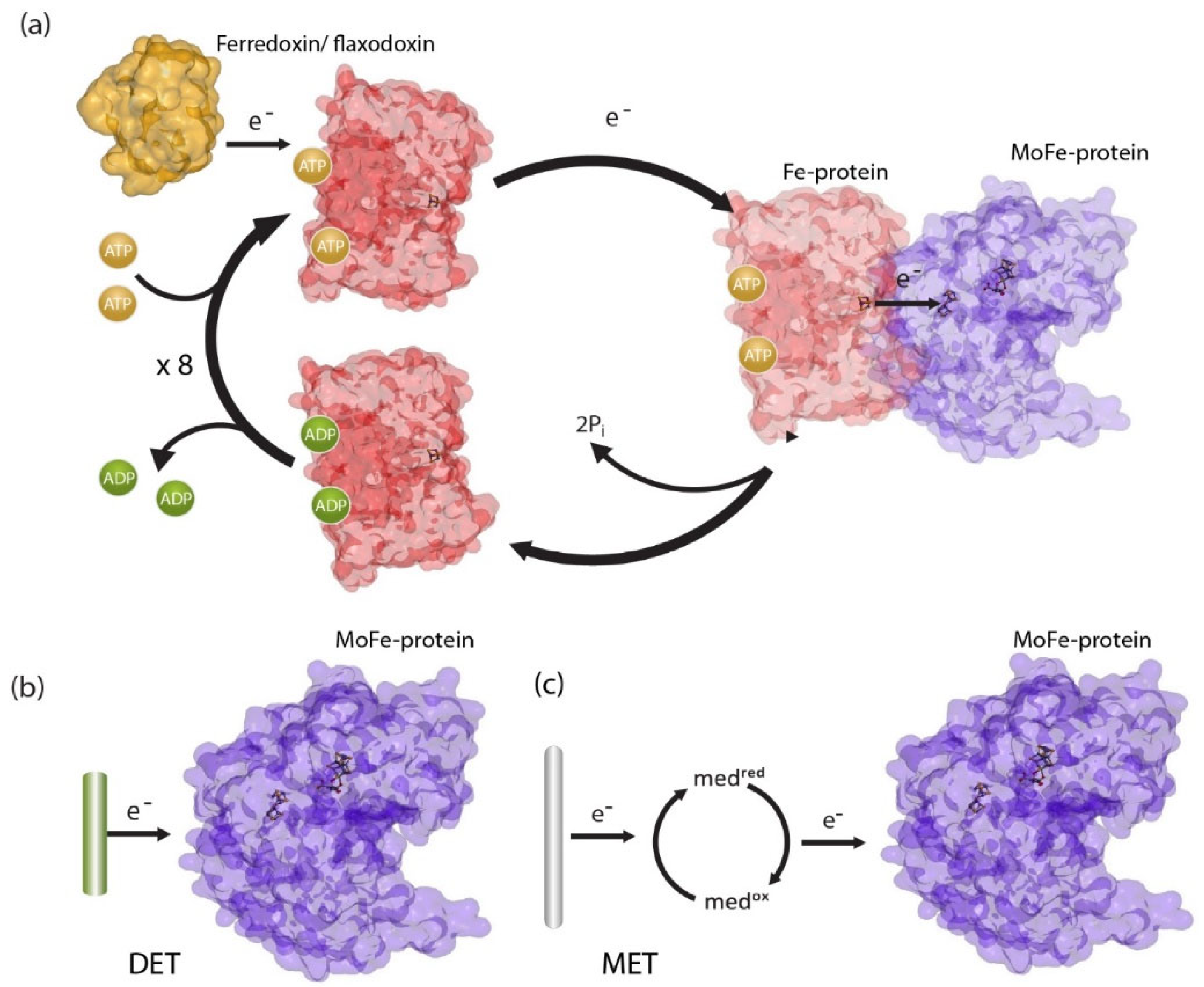
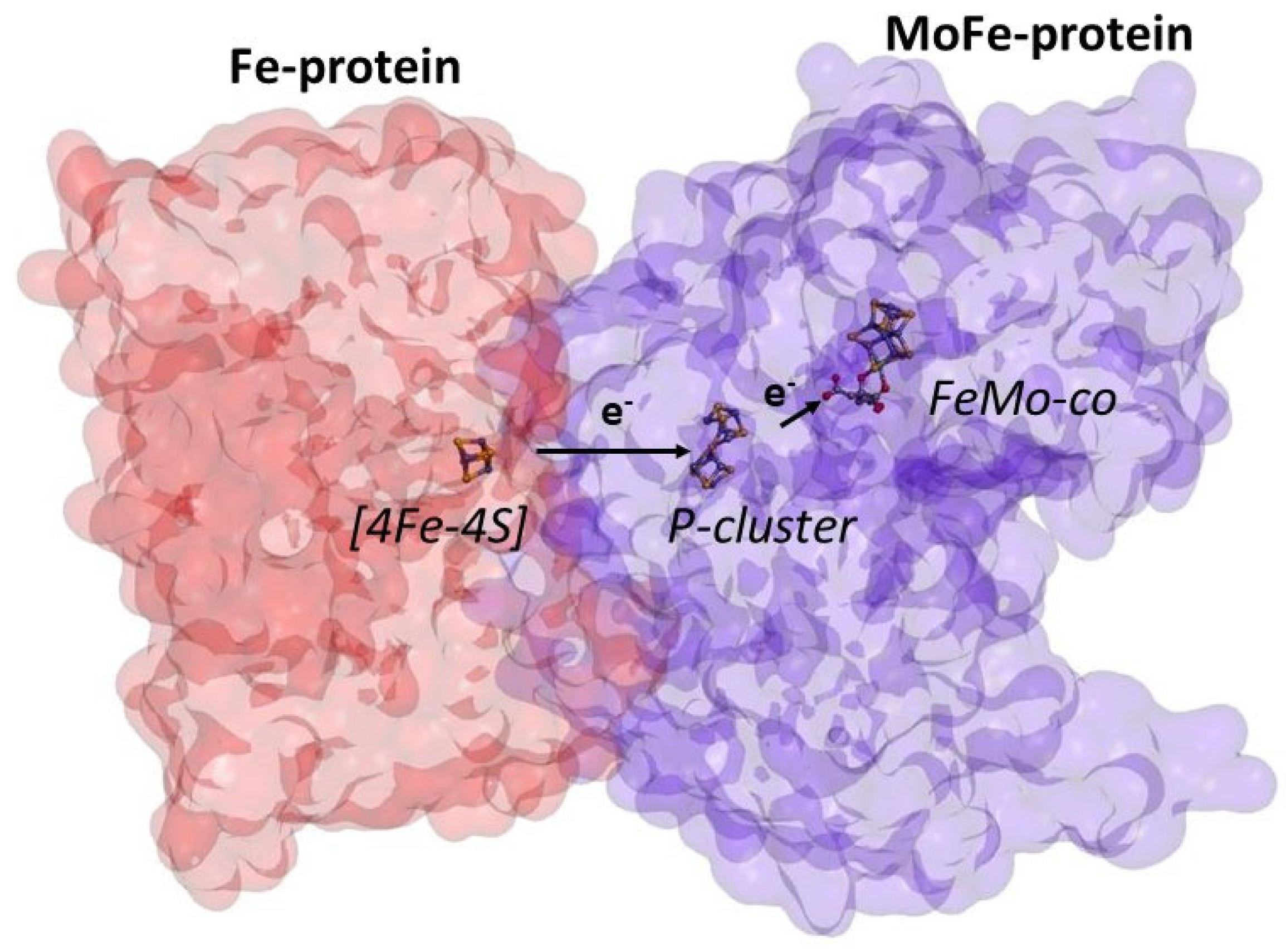
2. ATP-Free Ammonia Production
3. The Role of ATP in Nitrogenase Catalysis
4. Calculating the Theoretical Maximal Rate of Ammonia Production by Nitrogenase in Comparison to the Haber–Bosch Process
5. Avenues to Stabilise Nitrogenase
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Mather, T.A.; Pyle, D.M.; Allen, A.G. Volcanic Source for Fixed Nitrogen in the Early Earth’s Atmosphere. Geology 2004, 32, 905–908. [Google Scholar] [CrossRef]
- Huebert, B.; Vitousek, P.; Sutton, J.; Elias, T.; Heath, J.; Coeppicus, S.; Howell, S.; Blomquist, B. Volcano Fixes Nitrogen into Plant-Available Forms. Biogeochemistry 1999, 47, 111–118. [Google Scholar] [CrossRef]
- Tuck, A.F. Production of Nitrogen Oxides by Lightning Discharges. Q. J. R. Meteorol. Soc. 1976, 102, 749–755. [Google Scholar] [CrossRef]
- Burris, R.H.; Wilson, P. Biological Nitrogen Fixation. Annu. Rev. Biochem. 1945, 14, 685–708. [Google Scholar] [CrossRef]
- Burns, R.C.; Bulen, W.A. ATP-Dependent Hydrogen Evolution by Cell-Free Preparations of Azotobacter Vinelandii. Biochim. Biophys. Acta 1965, 105, 437–445. [Google Scholar] [CrossRef]
- Crookes, W. Address of the President before the British Association for the Advancement of Science. Science 1898, 8, 561–575. [Google Scholar] [CrossRef]
- Yanes, J. The Lifesavers of Science. Available online: https://www.bbvaopenmind.com/en/science/leading-figures/the-lifesavers-of-science/ (accessed on 20 June 2022).
- Smith, C.; Hill, A.K.; Torrente-Murciano, L. Current and Future Role of Haber-Bosch Ammonia in a Carbon-Free Energy Landscape. Energy Environ. Sci. 2020, 13, 331–344. [Google Scholar] [CrossRef]
- Chen, J.G.; Crooks, R.M.; Seefeldt, L.C.; Bren, K.L.; Bullock, R.M.; Darensbourg, M.Y.; Holland, P.L.; Hoffman, B.; Janik, M.J.; Jones, A.K.; et al. Beyond Fossil Fuel–Driven Nitrogen Transformations. Science 2018, 360, eaar6611. [Google Scholar] [CrossRef]
- Rutledge, H.L.; Tezcan, F.A. Electron Transfer in Nitrogenase. Chem. Rev. 2020, 120, 5158–5193. [Google Scholar] [CrossRef]
- Einsle, O.; Rees, D.C. Structural Enzymology of Nitrogenase Enzymes. Chem. Rev. 2020, 120, 4969–5004. [Google Scholar] [CrossRef]
- Lanzilotta, W.N.; Seefeldt, L.C. Electron Transfer from the Nitrogenase Iron Protein to the [8Fe-(7/8)S] Clusters of the Molybdenum-Iron Protein. Biochemistry 1996, 35, 16770–16776. [Google Scholar] [CrossRef]
- Brown, K.A.; Harris, D.F.; Wilker, M.B.; Rasmussen, A.; Khadka, N.; Hamby, H.; Keable, S.; Dukovic, G.; Peters, J.W.; Seefeldt, L.C.; et al. Light-Driven Dinitrogen Reduction Catalyzed by a CdS:Nitrogenase MoFe Protein Biohybrid. Science 2016, 352, 448–450. [Google Scholar] [CrossRef]
- Hodgetts, R.Y.; Kiryutin, A.S.; Nichols, P.; Du, H.L.; Bakker, J.M.; MacFarlane, D.R.; Simonov, A.N. Refining Universal Procedures for Ammonium Quantification via Rapid 1H NMR Analysis for Dinitrogen Reduction Studies. ACS Energy Lett. 2020, 5, 736–741. [Google Scholar] [CrossRef]
- Andersen, S.Z.; Čolić, V.; Yang, S.; Schwalbe, J.A.; Nielander, A.C.; McEnaney, J.M.; Enemark-Rasmussen, K.; Baker, J.G.; Singh, A.R.; Rohr, B.A.; et al. A Rigorous Electrochemical Ammonia Synthesis Protocol with Quantitative Isotope Measurements. Nature 2019, 570, 504–508. [Google Scholar] [CrossRef]
- Yang, Z.-Y.; Ledbetter, R.; Shaw, S.; Pence, N.; Tokmina-Lukaszewska, M.; Eilers, B.; Guo, Q.; Pokhrel, N.; Cash, V.L.; Dean, D.R.; et al. Evidence That the Pi Release Event Is the Rate-Limiting Step in the Nitrogenase Catalytic Cycle. Biochemistry 2016, 55, 3625–3635. [Google Scholar] [CrossRef]
- Hickey, D.P.; Lim, K.; Cai, R.; Patterson, A.R.; Yuan, M.; Sahin, S.; Abdellaoui, S.; Minteer, S.D. Pyrene Hydrogel for Promoting Direct Bioelectrochemistry: ATP-Independent Electroenzymatic Reduction of N2. Chem. Sci. 2018, 9, 5172–5177. [Google Scholar] [CrossRef]
- Hickey, D.P.; Cai, R.; Yang, Z.Y.; Grunau, K.; Einsle, O.; Seefeldt, L.C.; Minteer, S.D. Establishing a Thermodynamic Landscape for the Active Site of Mo-Dependent Nitrogenase. J. Am. Chem. Soc. 2019, 141, 17150–17157. [Google Scholar] [CrossRef]
- Lee, Y.S.; Ruff, A.; Cai, R.; Lim, K.; Schuhmann, W.; Minteer, S.D. Electroenzymatic Nitrogen Fixation Using an Organic Redox Polymer-Immobilized MoFe Protein System. Angew. Chemie Int. Ed. 2020, 59, 16511–16516. [Google Scholar] [CrossRef]
- Lee, Y.S.; Yuan, M.; Cai, R.; Lim, K.; Minteer, S.D. Nitrogenase Bioelectrocatalysis: ATP-Independent Ammonia Production Using a Redox Polymer/MoFe Protein System. ACS Catal. 2020, 10, 6854–6861. [Google Scholar] [CrossRef]
- Meirovich, M.M.; Bachar, O.; Yehezkeli, O. Artificial, Photoinduced Activation of Nitrogenase Using Directed and Mediated Electron Transfer Processes. Catalysts 2020, 10, 979. [Google Scholar] [CrossRef]
- Brown, K.A.; Ruzicka, J.; Kallas, H.; Chica, B.; Mulder, D.W.; Peters, J.W.; Seefeldt, L.C.; Dukovic, G.; King, P.W. Excitation-Rate Determines Product Stoichiometry in Photochemical Ammonia Production by CdS Quantum Dot-Nitrogenase MoFe Protein Complexes. ACS Catal. 2020, 10, 11147–11152. [Google Scholar] [CrossRef]
- Tezcan, F.A.; Kaiser, J.T.; Mustafi, D.; Walton, M.Y.; Howard, J.B.; Rees, D.C. Nitrogenase Complexes: Multiple Docking Sites for a Nucleotide Switch Protein. Science 2005, 309, 1377–1380. [Google Scholar] [CrossRef]
- Lanzilotta, W.N.; Ryle, M.J.; Seefeldt, L.C. Nucleotide Hydrolysis and Protein Conformational Changes in Azotobacter Vinelandii Nitrogenase Iron Protein: Defining the Function of Aspartate 1291. Biochemistry 1995, 34, 10713–10723. [Google Scholar] [CrossRef]
- Watt, G.D.; Knotts, R.R.; Wang, Z.C. Redox Reactions of and Nucleotide Binding to the Iron Protein of Azotobacter Vinelandii. Biochemistry 1986, 25, 8156–8162. [Google Scholar] [CrossRef]
- Duval, S.; Danyal, K.; Shaw, S.; Lytle, A.K.; Dean, D.R.; Hoffman, B.M.; Antony, E.; Seefeldt, L.C. Electron Transfer Precedes ATP Hydrolysis during Nitrogenase Catalysis. Proc. Natl. Acad. Sci. USA 2013, 110, 16414–16419. [Google Scholar] [CrossRef]
- Rutledge, H.L.; Cook, B.D.; Nguyen, H.P.M.; Herzik, M.A.; Tezcan, A. CryoEM Structures of the Nitrogenase Complex during Catalytic Turnover. bioRxiv 2022, 1–8. [Google Scholar] [CrossRef]
- Gray, H.B.; Ellis, W.R. Electron Transfer. In Bioinorganic Chemistry; Bertini, I., Gray, H.B., Lippard, S.J., Valentine, J.S., Eds.; University Science Books: Sausalito, CA, USA, 1994; pp. 315–363. [Google Scholar]
- Chica, B.; Ruzicka, J.; Kallas, H.; Mulder, D.W.; Katherine, A.; Peters, J.W.; Seefeldt, L.C.; Dukovic, G.; King, P.W.; Chica, B.; et al. Defining Intermediates of Nitrogenase MoFe Protein during N Reduction under Photochemical Electron Delivery from CdS Quantum Dots Defining Intermediates of Nitrogenase MoFe Protein During N2 Reduction under Photochemical Electron Delivery from CdS Quantum dots. J. Am. Chem. Soc. 2020, 142, 14324–14330. [Google Scholar] [CrossRef]
- Badalyan, A.; Yang, Z.Y.; Seefeldt, L.C. A Voltammetric Study of Nitrogenase Catalysis Using Electron Transfer Mediators. ACS Catal. 2019, 9, 1366–1372. [Google Scholar] [CrossRef]
- Humphreys, J.; Lan, R.; Tao, S. Development and Recent Progress on Ammonia Synthesis Catalysts for Haber–Bosch Process. Adv. Energy Sustain. Res. 2021, 2, 2000043. [Google Scholar] [CrossRef]
- Newton, W.E.; Dilworth, M.J. Assays of Nitrogenase Reaction Products. Methods Mol. Biol. 2011, 766, 105–127. [Google Scholar] [CrossRef]
- Milton, R.D.; Cai, R.; Abdellaoui, S.; Leech, D.; De Lacey, A.L.; Pita, M.; Minteer, S.D. Bioelectrochemical Haber–Bosch Process: An Ammonia-Producing H2/N2 Fuel Cell. Angew. Chem. Int. Ed. 2017, 56, 2680–2683. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Minteer, S.D. Nitrogenase Bioelectrocatalysis: From Understanding Electron-Transfer Mechanisms to Energy Applications. ACS Energy Lett. 2018, 3, 2736–2742. [Google Scholar] [CrossRef]
- Puetz, J.; Wurm, F.M. Recombinant Proteins for Industrial versus Pharmaceutical Purposes: A Review of Process and Pricing. Processes 2019, 7, 476. [Google Scholar] [CrossRef]
- Chen, H.; Prater, M.B.; Cai, R.; Dong, F.; Chen, H.; Minteer, S.D. Bioelectrocatalytic Conversion from N2 to Chiral Amino Acids in a H2/α-Keto Acid Enzymatic Fuel Cell. J. Am. Chem. Soc. 2020, 142, 4028–4036. [Google Scholar] [CrossRef]
- Mus, F.; Khokhani, D.; MacIntyre, A.M.; Rugoli, E.; Dixon, R.; Ané, J.-M.; Peters, J.W. Genetic Determinants of Ammonium Excretion in NifL Mutants of Azotobacter Vinelandii. Appl. Environ. Microbiol. 2022, 88, e01876-21. [Google Scholar] [CrossRef]
- Leddy, J.; Paschkewitz, T.M. Ammonia Production Using Bioelectrocatalytical Devices. U.S. Patent No. US2014/0011252 AI, 9 January 2016. [Google Scholar]
- Knoche, K.L.; Aoyama, E.; Hasan, K.; Minteer, S.D. Role of Nitrogenase and Ferredoxin in the Mechanism of Bioelectrocatalytic Nitrogen Fixation by the Cyanobacteria Anabaena Variabilis SA-1 Mutant Immobilized on Indium Tin Oxide (ITO) Electrodes. Electrochim. Acta 2017, 232, 396–403. [Google Scholar] [CrossRef]
- Dong, F.; Lee, Y.S.; Gaffney, E.M.; Liou, W.; Minteer, S.D. Engineering Cyanobacterium with Transmembrane Electron Transfer Ability for Bioelectrochemical Nitrogen Fixation. ACS Catal. 2021, 11, 13169–13179. [Google Scholar] [CrossRef]
- Dong, F.; Lee, Y.S.; Gaffney, E.M.; Grattieri, M.; Haddadin, H.; Minteer, S.D.; Chen, H. An Engineered, Non-Diazotrophic Cyanobacterium and Its Application in Bioelectrochemical Nitrogen Fixation. Cell Rep. Phys. Sci. 2021, 2, 100444. [Google Scholar] [CrossRef]
- Liu, C.; Sakimoto, K.K.; Colón, B.C.; Silver, P.A.; Nocera, D.G. Ambient Nitrogen Reduction Cycle Using a Hybrid Inorganic–Biological System. Proc. Natl. Acad. Sci. USA 2017, 114, 6450–6455. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Nitrogenase System | Method of Reduction | Method of Quantification | NH3 Production Rate a | Ref. |
|---|---|---|---|---|
| MoFe-protein + Fe-protein + ATP | Chemical | Spectrophotometric | 119 | [13] |
| MoFe-protein + CdS nanorod | Light driven | Spectrophotometric | 75 | [13] |
| MoFe-protein in pyrene hydrogel | DET a | Spectrophotometric | 0.5 | [17] |
| MoFe-protein in pyrene hydrogel | DET b | Spectrophotometric + 15N2 | 0.13 | [18] |
| MoFe-protein in an organic redox polymer | MET c | Spectrophotometric + 15N2 | 3.3 | [19] |
| MoFe-protein in a redox polymer | MET b | Spectrophotometric + 15N2 | 2.3 | [20] |
| MoFe-protein + CdS | Light driven | Spectrophotometric | 0.3 | [21] |
| MoFe-protein + CdS quantum dots | Light driven | 15N2 | 3.5 | [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rapson, T.D.; Wood, C.C. Analysis of the Ammonia Production Rates by Nitrogenase. Catalysts 2022, 12, 844. https://doi.org/10.3390/catal12080844
Rapson TD, Wood CC. Analysis of the Ammonia Production Rates by Nitrogenase. Catalysts. 2022; 12(8):844. https://doi.org/10.3390/catal12080844
Chicago/Turabian StyleRapson, Trevor D., and Craig C. Wood. 2022. "Analysis of the Ammonia Production Rates by Nitrogenase" Catalysts 12, no. 8: 844. https://doi.org/10.3390/catal12080844
APA StyleRapson, T. D., & Wood, C. C. (2022). Analysis of the Ammonia Production Rates by Nitrogenase. Catalysts, 12(8), 844. https://doi.org/10.3390/catal12080844