
Organic Chemistry and Synthesis Rely More and More upon Catalysts


Abstract
1. Introduction
2. Invention Is Quicker if Physical Organic Chemistry Principles Are Integrated in Preparative Chemistry
3. Thermochemical Data Establish Fundamental Concepts of Reactivity
4. Mnemonic Devices Are Very Useful to Learn Chemistry, but They Must Be Applied Critically
| X = | Me | Et | n-Pr | OH | SH | NH2 | F | Cl | Br | I |
| ΔrH°(8): | −2.0 | −1.6 | −1.8 | −4.2 | −4.0 | −3.2 | −1.8 | −3.1 | −3.0 | −2.4 kcal mol−1 |
5. Models Must Be Based on Measurements and Thermochemical Data
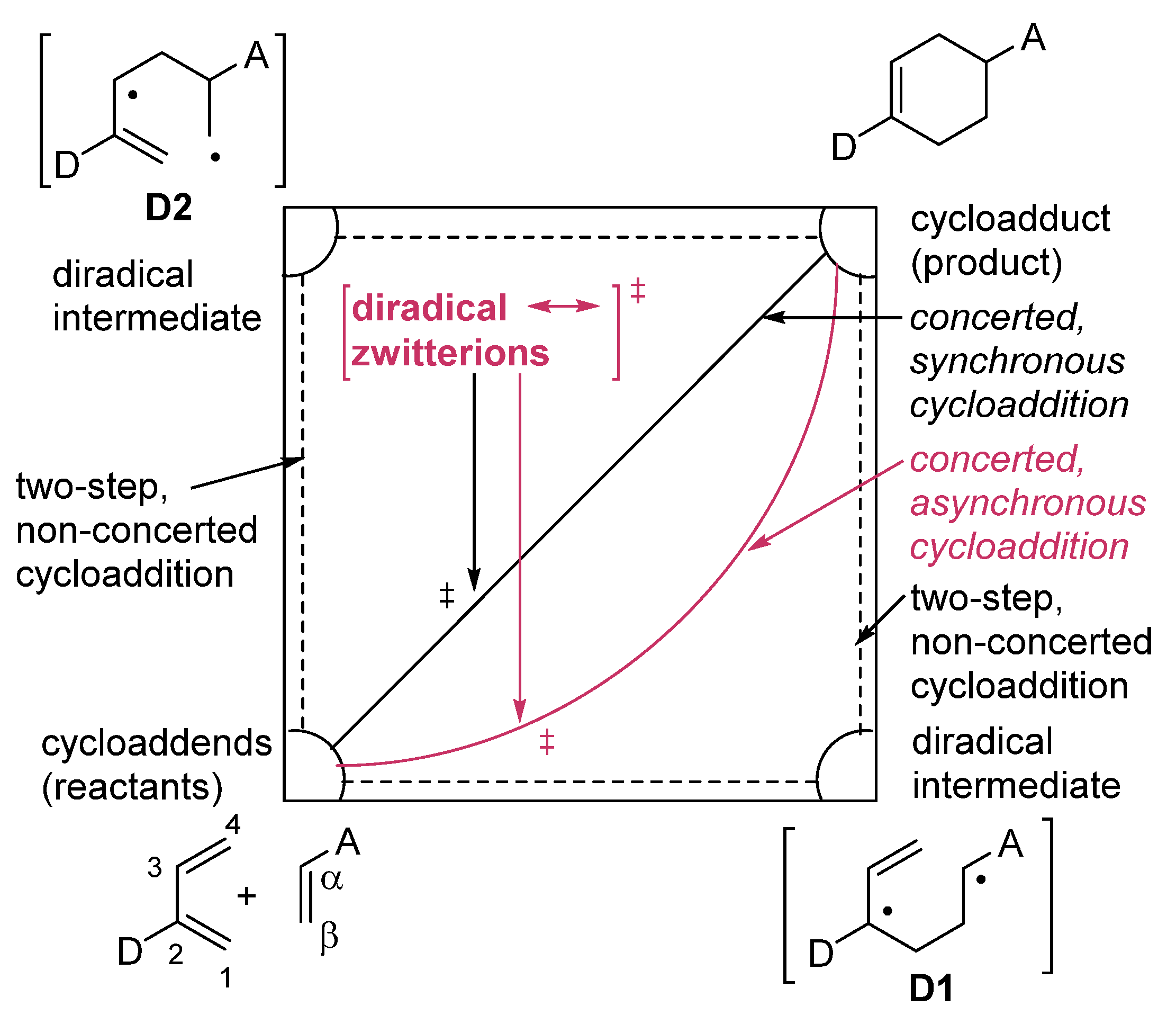
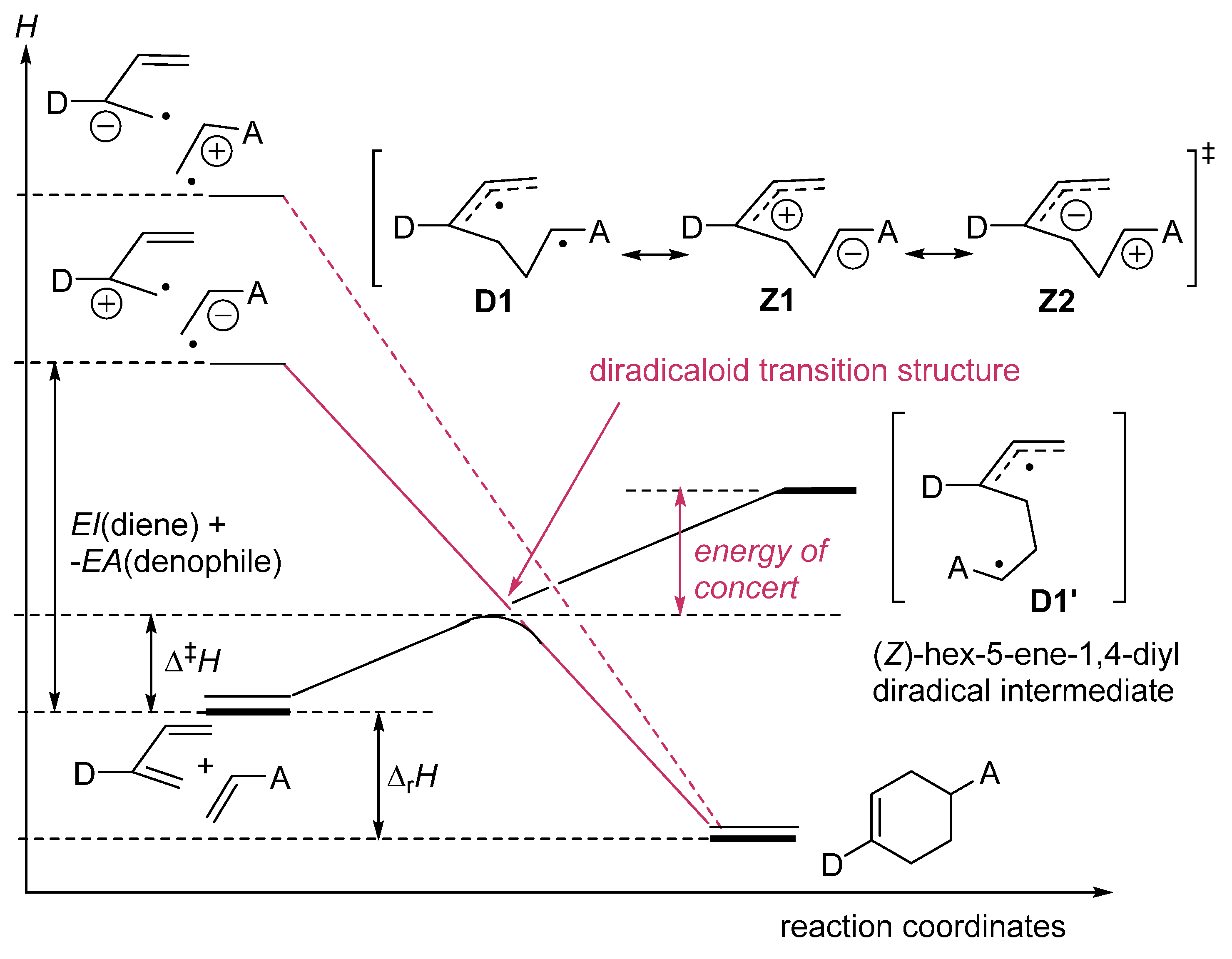
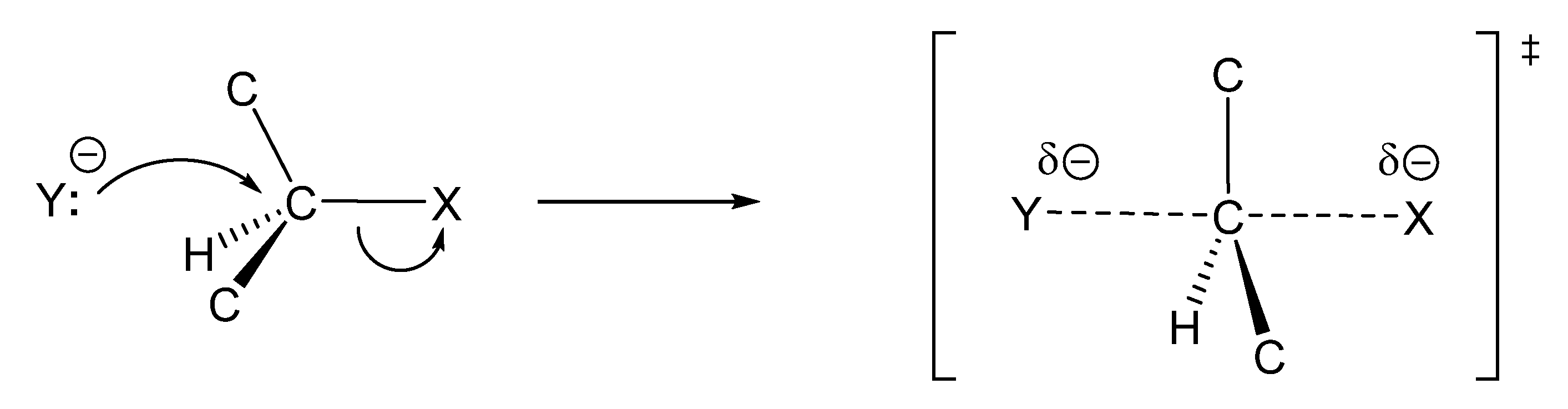
 zwitterion species. Thus, without having to calculate molecular orbitals, by using just thermochemical data (homolytical bond dissociation enthalpies: DH°(R•/H•) or DH°(R•/X•)), the substituent effect on the relative stability of radicals, cations and anions, ionization energies (IE(R•) [68], and electron affinities of radicals (–EA(R•)) [69] can be used to predict relative rates and chemo-, regio-, and stereoselectivities of concerted, one-step pericyclic reactions. The diradicaloid model, advocated by Dewar for Diels–Alder reactions [70,71], is simple because it is based on thermochemical data (measurements) and it can be applied to catalyzed pericyclic reactions.
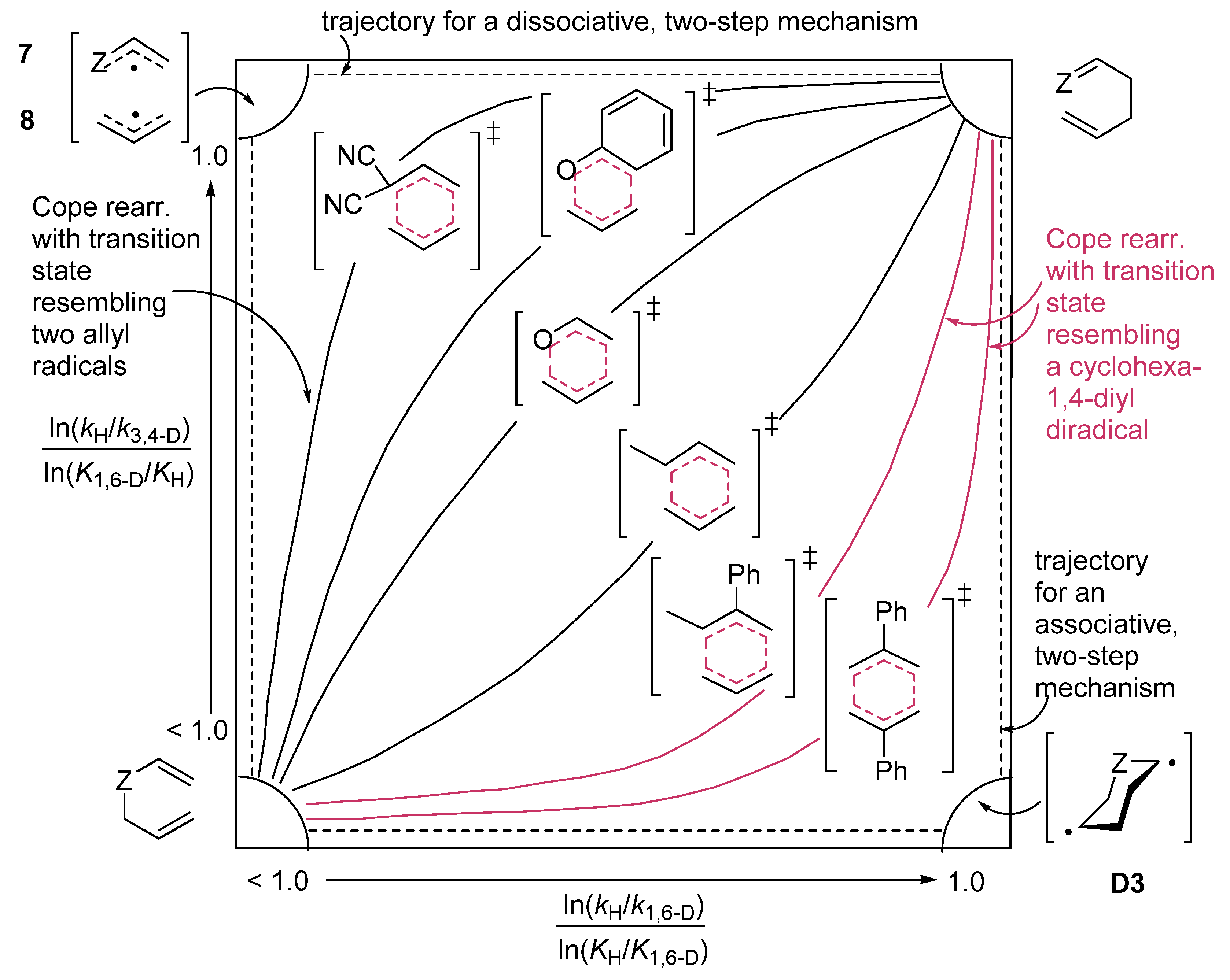
zwitterion species. Thus, without having to calculate molecular orbitals, by using just thermochemical data (homolytical bond dissociation enthalpies: DH°(R•/H•) or DH°(R•/X•)), the substituent effect on the relative stability of radicals, cations and anions, ionization energies (IE(R•) [68], and electron affinities of radicals (–EA(R•)) [69] can be used to predict relative rates and chemo-, regio-, and stereoselectivities of concerted, one-step pericyclic reactions. The diradicaloid model, advocated by Dewar for Diels–Alder reactions [70,71], is simple because it is based on thermochemical data (measurements) and it can be applied to catalyzed pericyclic reactions.6. Diradicaloids Are Transition Structures of Concerted Cycloadditions
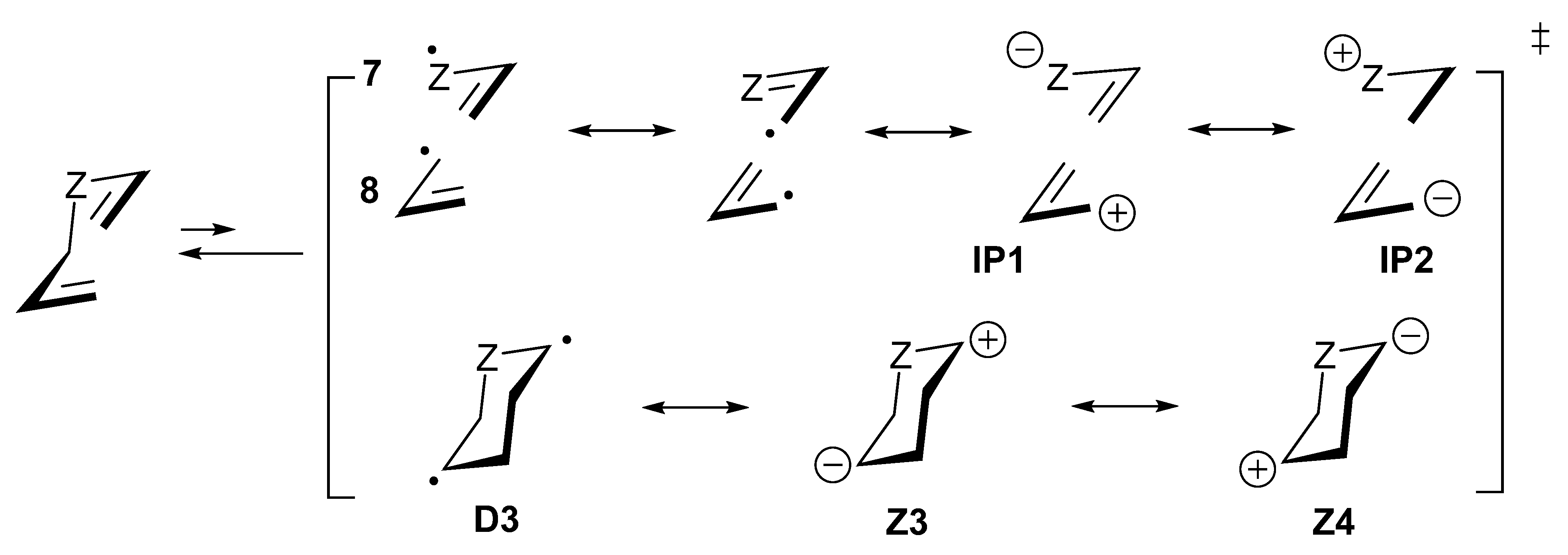
Z1 Z2.7. Diradicaloid Transition Structures of Concerted Sigmatropic Rearrangements
8. Diradicaloid Transition Structures in Concerted Ene-Reactions and Related Processes
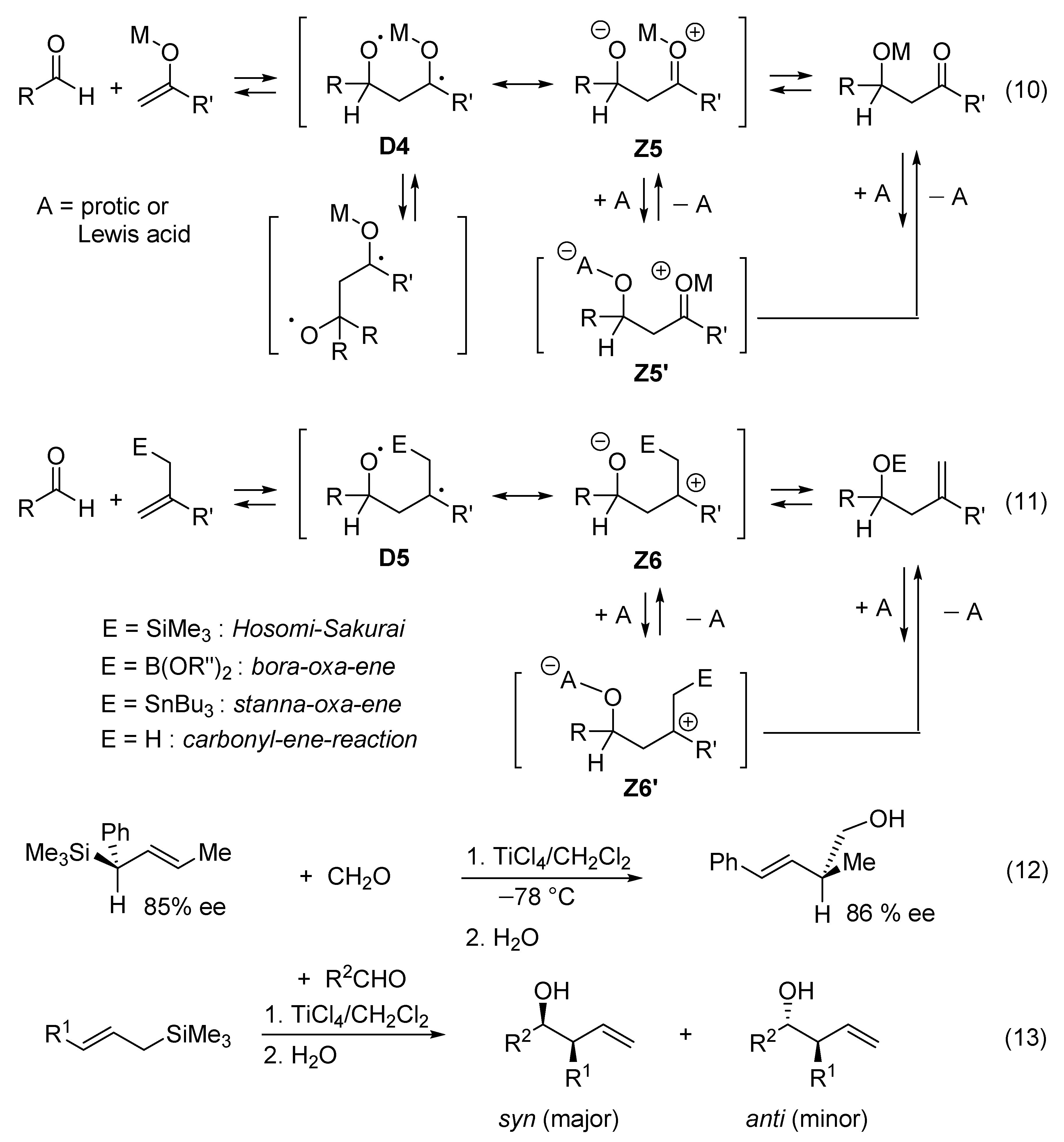
1,4-zwitterion) of type D4 Z5 if it follows a concerted one-step mechanism (Figure 6, reaction (10)). The role of the acid catalyst A is to stabilize the zwitterionic limiting structure Z5 by equilibrating with zwitterion Z5’. As a consequence, the reaction may follow a multi-step mechanism in which Z5’ is one reaction intermediate that can adopt quasi-cyclic or acyclic conformations. Similar diradicaloid models (D5 Z6) can be considered for metalla-carbonyl-ene reactions promoted by a Lewis acid (Figure 6, reaction (11)). Very electrophilic ketones such as hexafluoroacetone react with allylsilanes without catalyst [80]. The Hosomi–Sakurai reaction promoted by TiCl4 (Figure 6, reaction (11)) is not only regiospecific, but also stereospecifically anti with respect to the silyl group, which leads to the formation of a (E)-homoallylic alcohol [80,81,82]. ((E)-crotyl)trimethylsilane and ((E)-cinnamyl)trimethylsilane react with aldehydes in the presence of TiCl4 to give homoallyl alcohols with over 93% syn-selectivity (Figure 6, reactions (12) and (13)). Lower syn-selectivity is observed for the reactions of (Z)-crotylsilanes [83,84]. The stereoselectivity is generally accepted to be determined by chair-like six-membered (“closed”) transition states [85,86,87,88,89,90].9. Valence Bond Theory Can Be Used to Explain Structural and Medium Effects on the Rates of Displacement Reactions
10. Hydrocarbation of Unsaturated Compounds by R-H Reagents Do Not Produce Waste
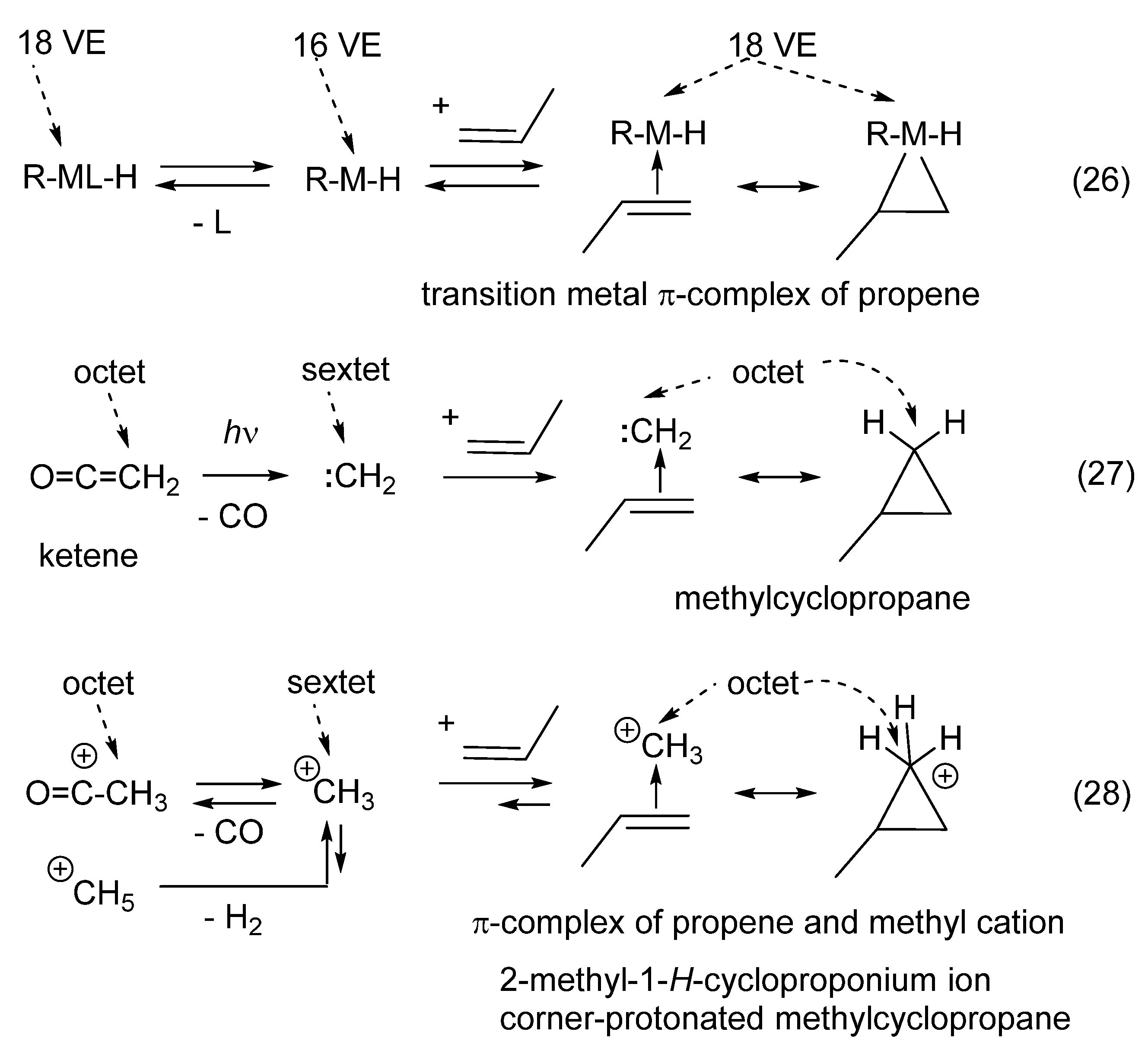
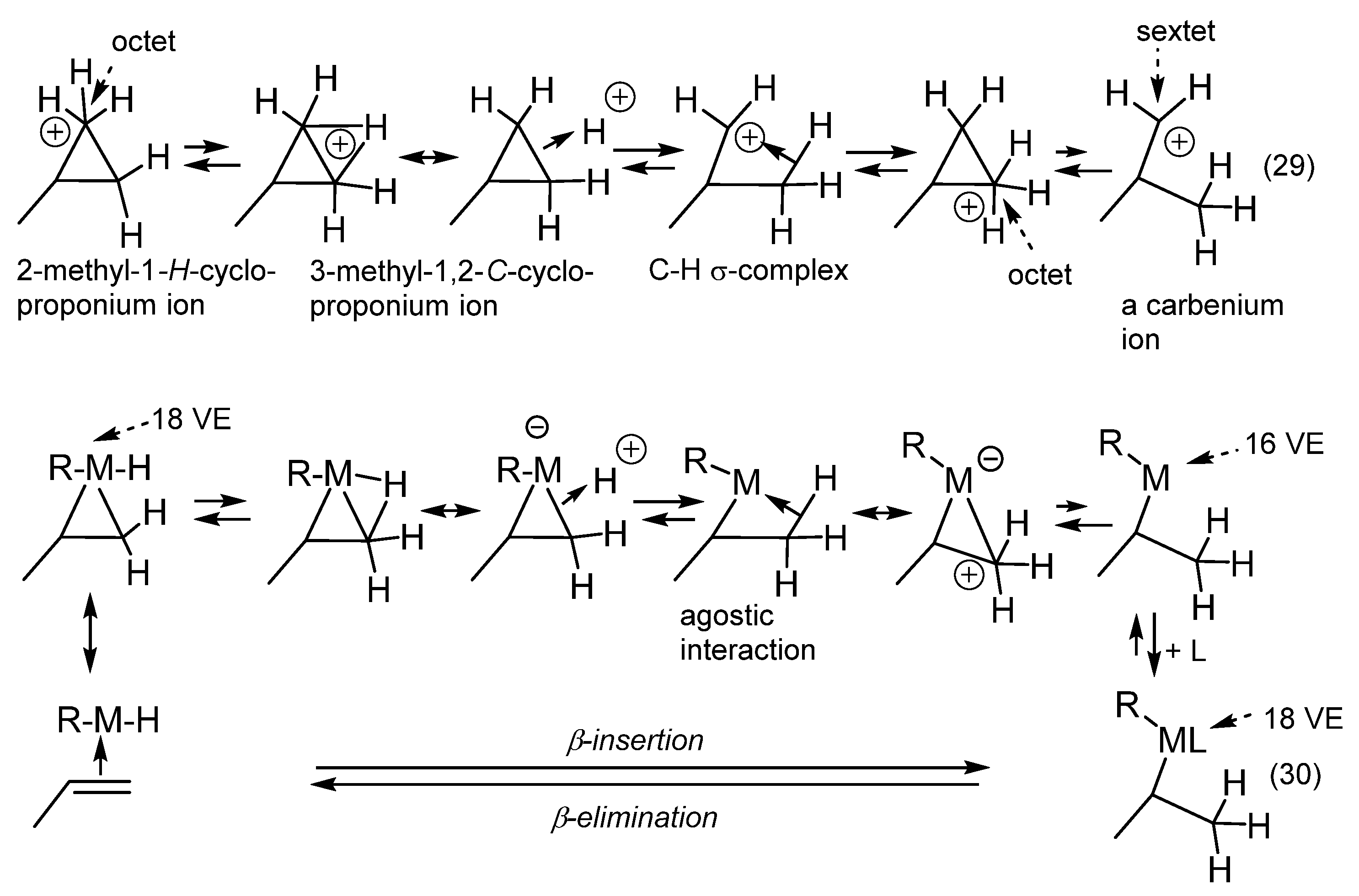
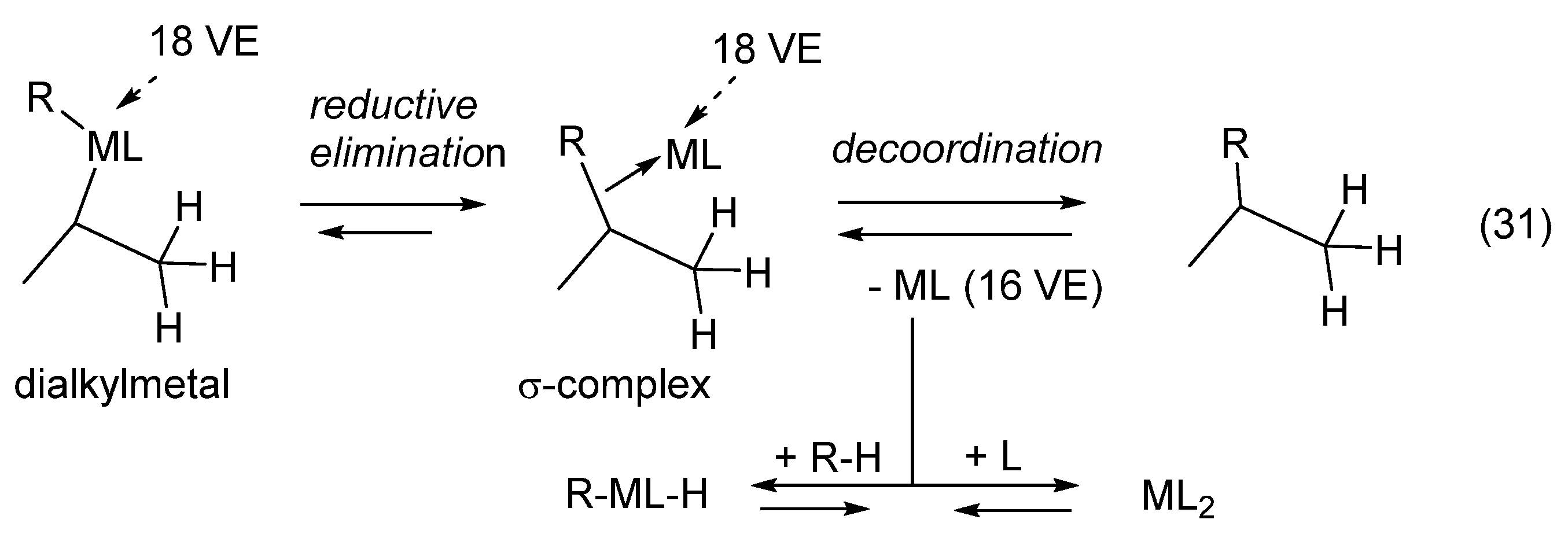
11. Transition Metal-Catalyzed Hydrocarbations
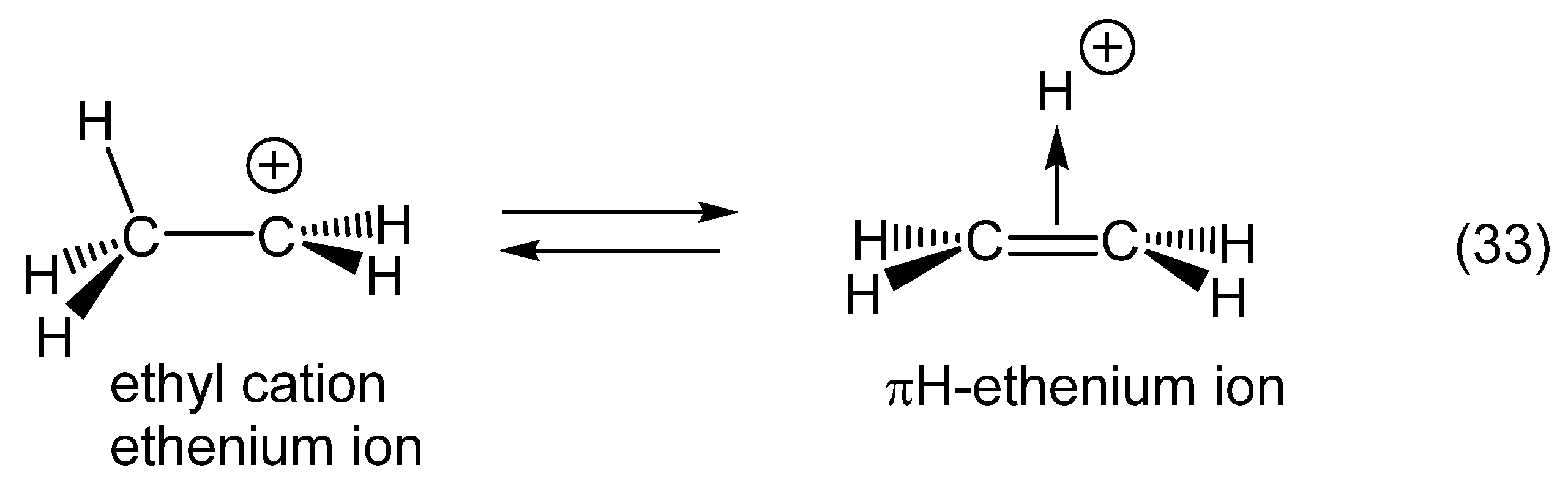
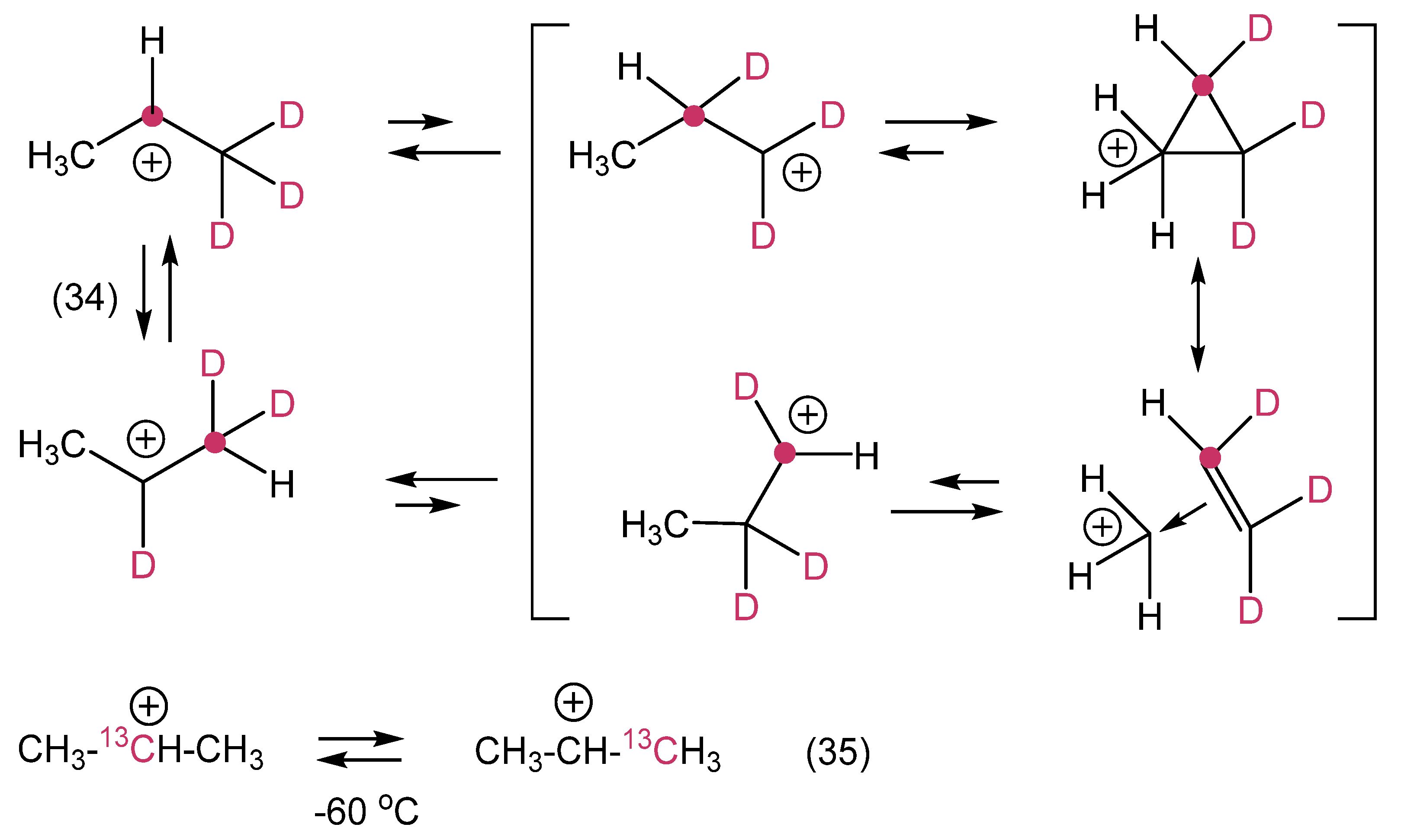
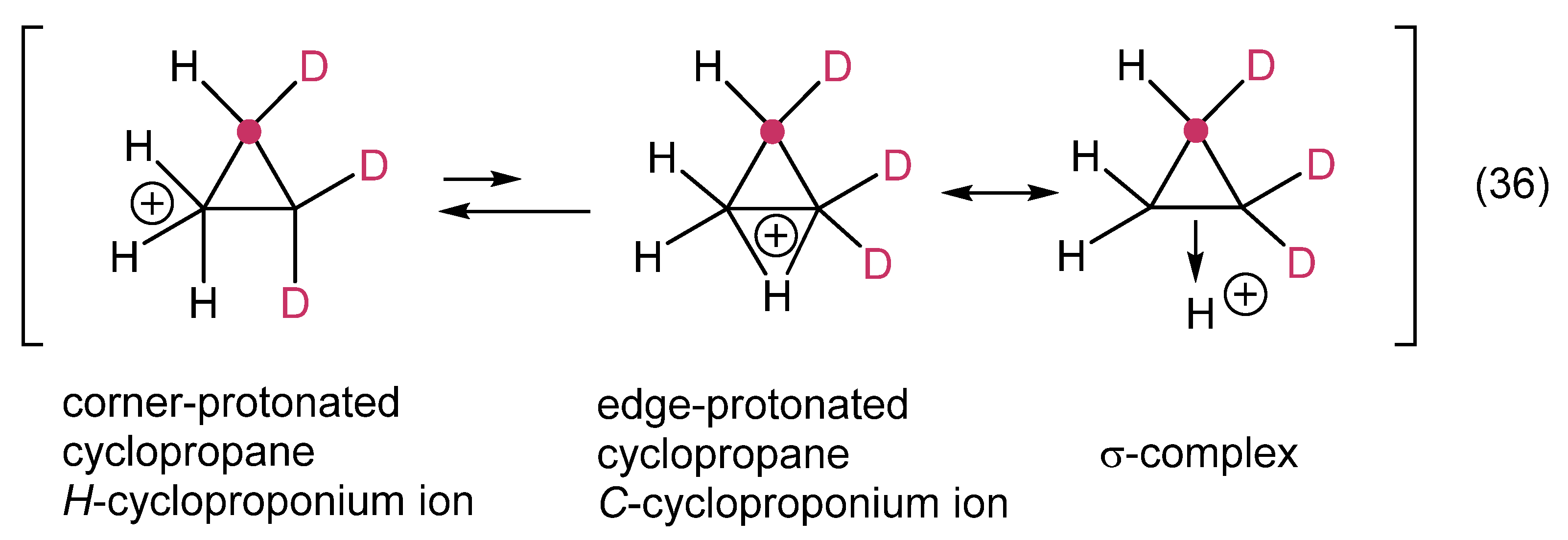
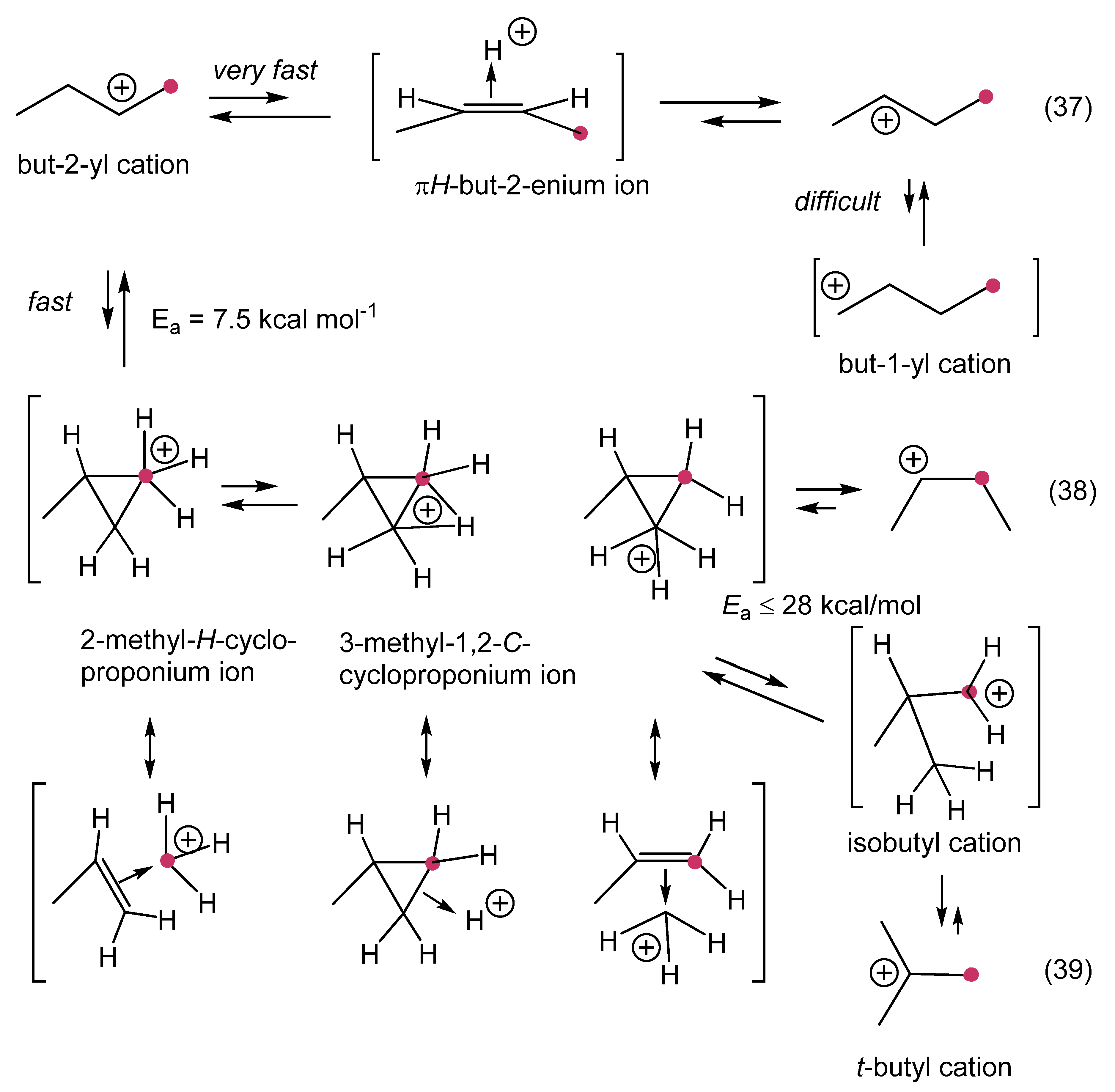
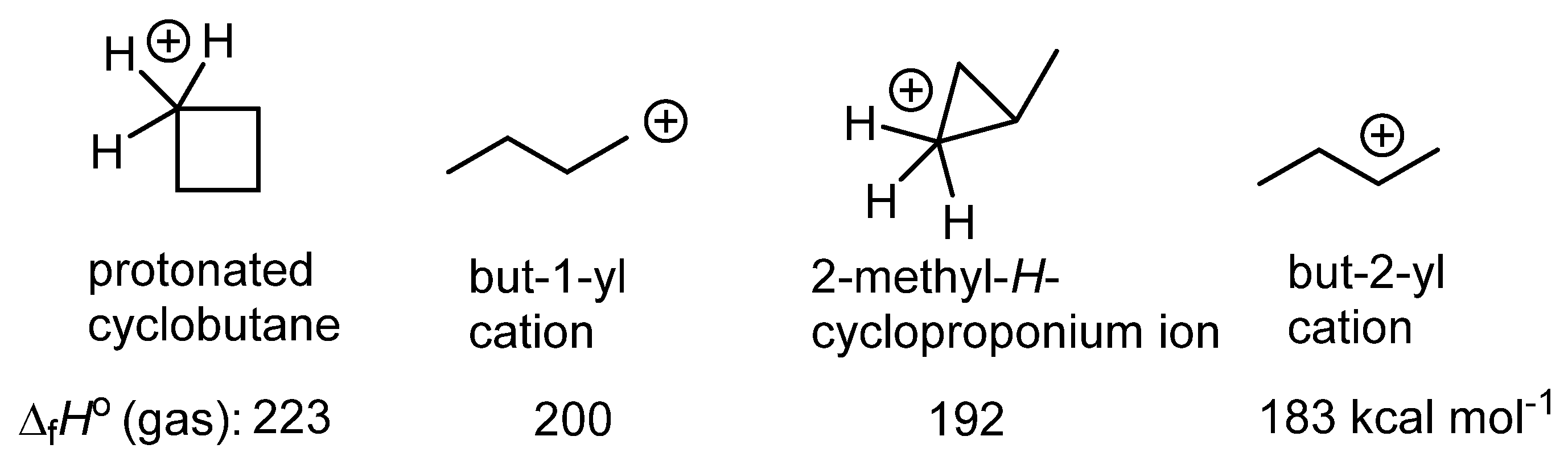
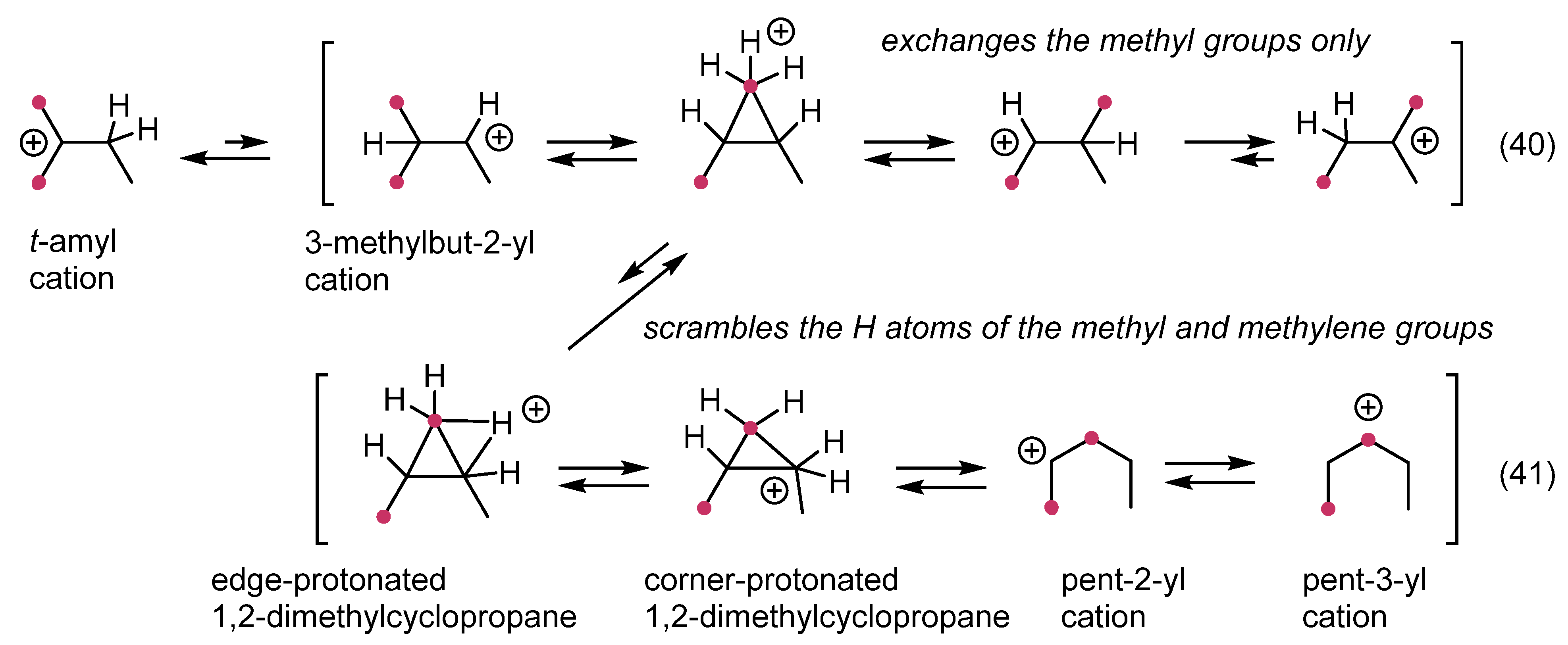
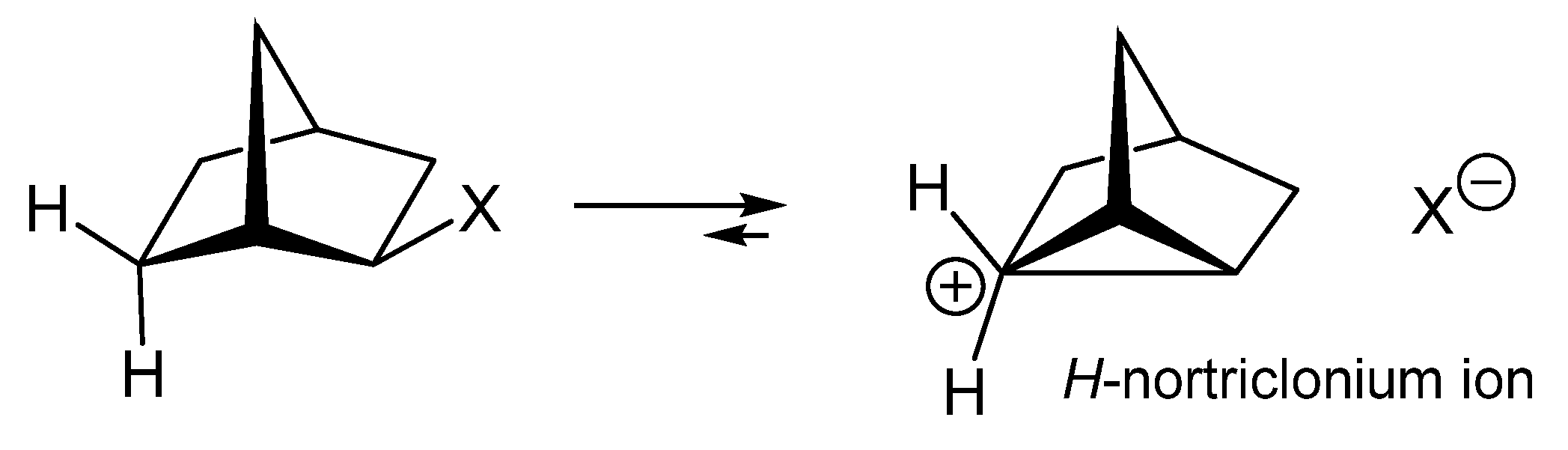
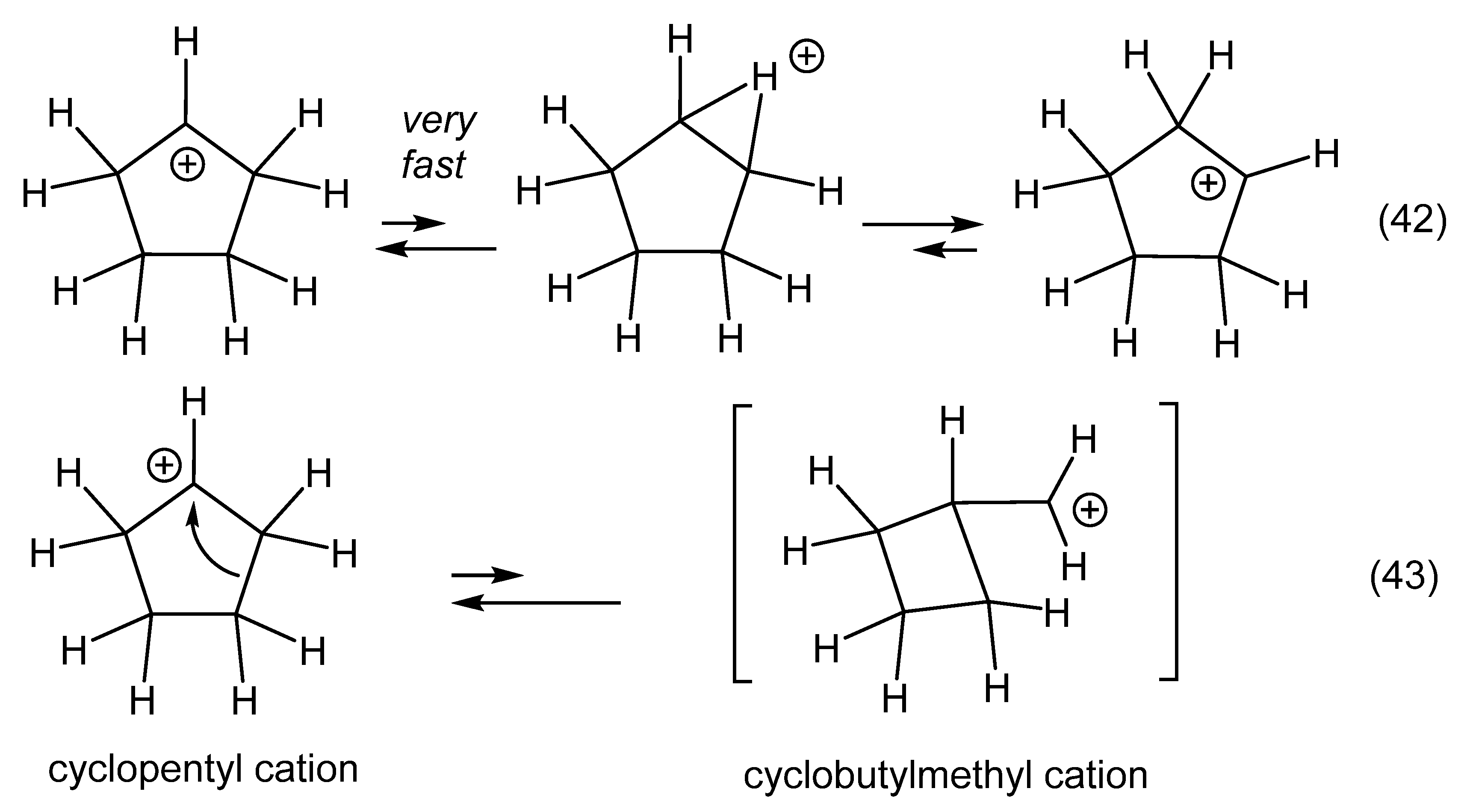
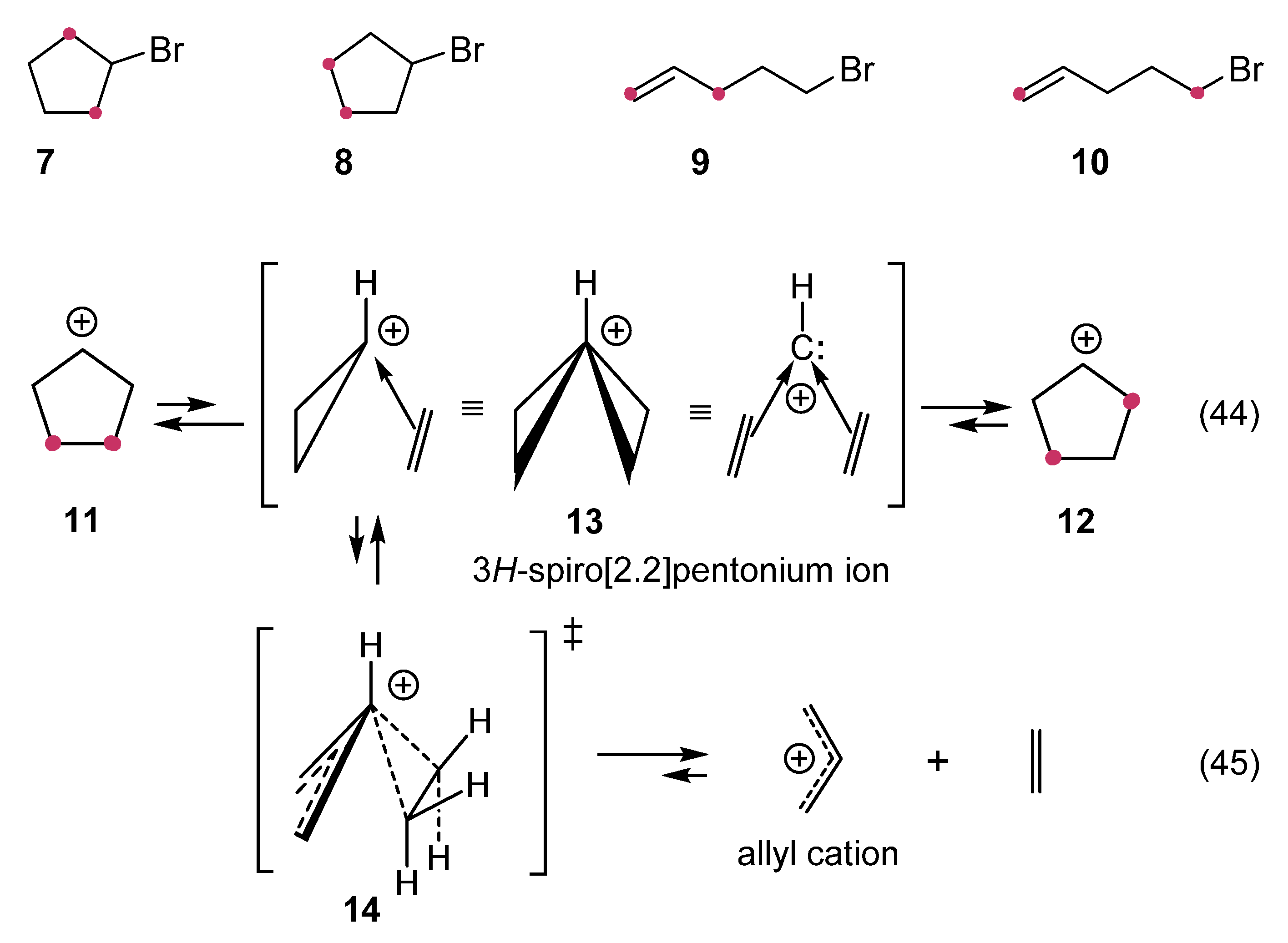
12. Review of the Fundamental Reactions of Simple Carbocations
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perkin, W.H. LXXIV. On mauveine and allied colouring matters. J. Chem. Soc. Trans. 1879, 35, 717–732. [Google Scholar] [CrossRef]
- Travis, A.S. Perkin’s Mauve: Ancestor of the Organic Chemical Industry. Technol. Cult. 1990, 31, 51–82. [Google Scholar] [CrossRef]
- Hubner, K. History–150 Years of mauveine. Chem. Unserer Zeit 2006, 40, 274–275. [Google Scholar] [CrossRef]
- Liebig, J. Ueber die Zersetzung des Alkohols durch Chlor. Ann. Der Pharm. 1832, 1, 31–32. [Google Scholar] [CrossRef][Green Version]
- Butler, T.C. The introduction of chloral hydrate into medical practice. Bull. Hist. Med. 1970, 44, 168–172. [Google Scholar]
- Jones, A.W. Early drug discovery and the rise of pharmaceutical chemistry. Drug Test. Anal. 2011, 3, 337–344. [Google Scholar] [CrossRef]
- Ng, R. Drugs: From Discovery to Approval, 3rd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 1–560. ISBN 1118907272/9781118907276. [Google Scholar]
- Inglis, L. Milk of Paradise: A History of Opium; Pan MacMillan: London, UK, 2019; pp. 1–440. ISBN 9781760782276. [Google Scholar]
- Lev, E. Traditional healing with animals (zootherapy): Medieval to present-day Levantine practice. J. Ethnopharmacol. 2003, 85, 107–118. [Google Scholar] [CrossRef]
- Bade, R.; Chan, H.-F.; Reynisson, J. Characteristics of known drug space, natural products, their derivatives and synthetic drugs. Eur. J. Med. Chem. 2010, 45, 5646–5652. [Google Scholar] [CrossRef]
- Li, J.J. History of drug discovery. In Drug Discovery; Li, J.J., Corey, E.J., Eds.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2013; pp. 1–42. [Google Scholar]
- Vogel, P. Sustainable Development: The Roles of Carbon and Bio-Carbon; an Introduction to Molecular Sciences; World Scientific Publishing: Hackensack, NJ, USA; London, UK; Singapore, 2022; pp. 1–585. ISBN 978-9811240485/9811240485. [Google Scholar]
- Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry, 2nd ed.; Oxford University Press: Oxford, UK, 2012; pp. 1–1234. ISBN 978-0199270293/0199270295. [Google Scholar]
- Smith, M.B. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th ed.; John Wiley & Sons. Inc.: Hoboken, NJ, USA, 2013; pp. 1–2047. ISBN 0470462590/1118854683/9788126556588/9780470462591/9781118854686/9788126556588. [Google Scholar]
- Bruice, P.Y. Organic Chemistry, 8th ed.; Pearson Education: Upper Saddle River, NJ, USA, 2016; pp. 1–1344. ISBN 978-0134042282/013404228X. [Google Scholar]
- Hartwig, J.F. Organotransition Metal Chemistry. From Bonding to Catalysis; University Science Books: Melville, NY, USA, 2010; pp. 1–1160. ISBN 978-1891389535. [Google Scholar]
- Vogel, P.; Houk, K.N. Organic Chemistry: Theory, Reactiviy and Mechanisms in Modern Synthesis; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2019; pp. 1–1352. ISBN 9783527345328/3527345329. [Google Scholar]
- Anslyn, E.V.; Dougherty, D.A. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, USA, 2006; pp. 1–1095. ISBN 1891389319/9781891389313. [Google Scholar]
- Wang, Z.; Wille, U.; Juaristi, E. (Eds.) Encyclopedia of Physical Organic Chemistry; John Wiley & Sons, Inc.: New York, NY, USA, 2017; Volume 6, pp. 1–4464. ISBN 978-1-118-47045-9. [Google Scholar]
- Reymond, J.-L. The chemical space project. Acc. Chem Res. 2015, 48, 727–730. [Google Scholar] [CrossRef]
- Awale, M.; Sirockin, F.; Stiefl, N.; Reymond, J.-L. Medicinal chemistry database GDBMedChem. Mol. Inform. 2019, 38, 1900031. [Google Scholar] [CrossRef]
- Boström, J.; Brown, D.G. Stuck in a rut with old chemistry. Drug Discov. Today 2016, 21, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; Boström, J. Analysis of past and present synthetic methodologies on medicinal chemistry: Where have all the new reactions gone? J. Med. Chem. 2016, 59, 4443–4458. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Hao, X.; Jing, L.; Wu, G.; Kang, D.; Liu, X.; Zhan, P. Recent applications of click chemistry in drug discovery. Expert Opin. Drug Discov. 2019, 14, 779–889. [Google Scholar] [CrossRef] [PubMed]
- Takayama, Y.; Kusamori, K.; Nishikawa, M. Click chemistry as a tool for cell engineering and drug delivery. Molecules 2019, 24, 172. [Google Scholar] [CrossRef] [PubMed]
- Dreher, S.D. Catalysis in medicinal chemistry. Reaction Chem. Eng. 2019, 4, 1530–1535. [Google Scholar] [CrossRef]
- Siddiqui, S.; Sharma, S.; Sharma, B.; Siddiqui, A.A. Role of serendipity in drug discovery. J. Pharm. Res. 2010, 9, 49–55. [Google Scholar] [CrossRef][Green Version]
- Pedreira, J.G.B.; Franco, L.S.; Barreiro, E.J. Chemical intuition in drug design and discovery. Curr. Top. Med. Chem. 2019, 19, 1679–1693. [Google Scholar] [CrossRef]
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, Last Update to Data 2022. Available online: https://webbook.nist.gov/chemistry/ (accessed on 15 May 2022).
- Hendrickson, J.B. Systematic synthesis design. 4. Numerical codification of construction reactions. J. Am. Chem. Soc. 1975, 97, 5784–5800. [Google Scholar] [CrossRef]
- Newhouse, T.; Baran, P.S.; Hoffmann, R.W. The economies of synthesis. Chem. Soc. Rev. 2009, 38, 3010–3021. [Google Scholar] [CrossRef]
- Gaich, T.; Baran, P.S. Aiming for the ideal synthesis. J. Org. Chem. 2010, 75, 4657–4673. [Google Scholar] [CrossRef]
- Morawetz, H.; Otaki, P.S. Kinetics and equilibria of amide formation in aqueous media. J. Am. Chem. Soc. 1963, 85, 463–468. [Google Scholar] [CrossRef]
- de Azambuja, F.; Parac-Vogt, T.N. Water-tolerant and atom economical amide bond formation by metal-substituted polyoxometalate catalysts. ACS Catal. 2019, 9, 10245–10252. [Google Scholar] [CrossRef]
- Perrot, P. A to Z of Thermodynamics; Oxford University Press: Oxford, UK; New York, NY, USA; Tokyo, Japan, 1998; pp. 1–336. ISBN 0198565569/9780198565567. [Google Scholar]
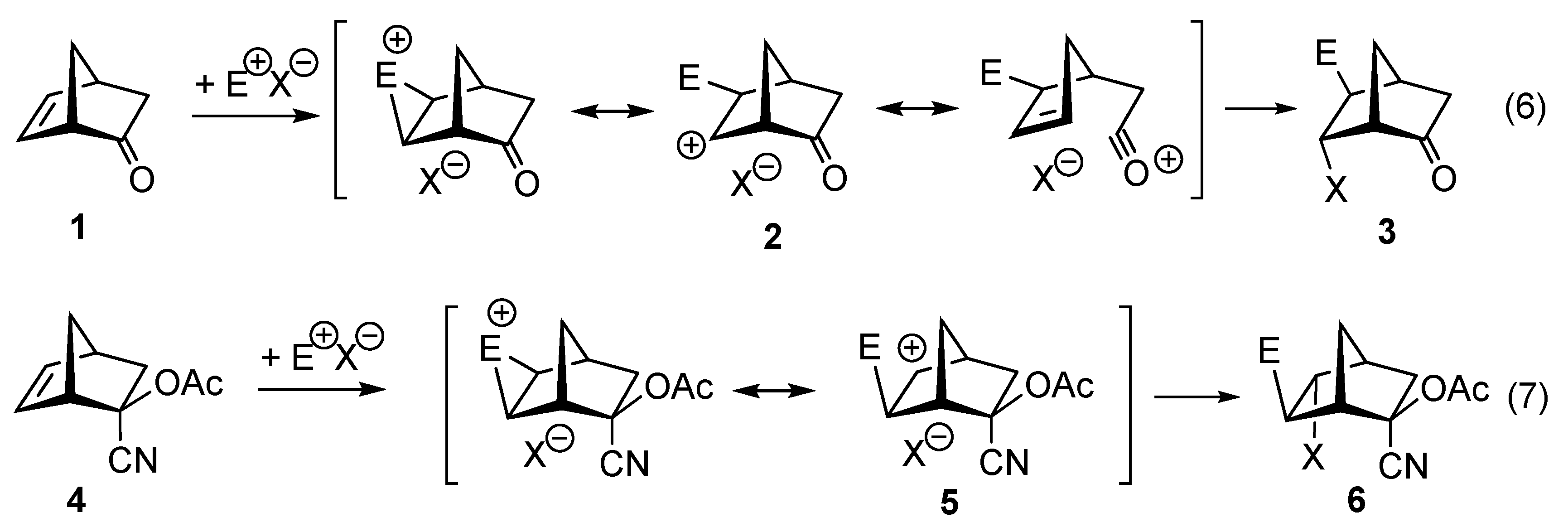
- Kernen, P.; Vogel, P. The homoconjugated electron-releasing carbonyl group of 1-methyl-7-oxabicyclo[2.2.1]hept-5-en-2-one. Regioselective syntheses of 5-chloro- and 6-chloro-1-methyl-7-oxabicyclo[2.2.1]hept-5-en-2-one. Helv. Chim. Acta 1993, 76, 2338–2343. [Google Scholar] [CrossRef]
- Vogel, P. The electron-releasing carbonyl group. Part 1: Discovery and theoretical studies. Chim. Oggi 1997, 15, 18–25. [Google Scholar]
- Vogel, P. The electron-releasing carbonyl group. Part 2. Spectroscopic detection and synthetic applications. Chim. Oggi 1997, 15, 37–43. [Google Scholar] [CrossRef]
- Carrupt, P.-A.; Vogel, P. Regioselective additions of electrophiles to olefins remotely perturbed–The carbonyl groups as a homoconjugated electron-donating substituent. Tetrahedron Lett. 1982, 23, 2563–2566. [Google Scholar] [CrossRef]
- Carrupt, P.-A.; Vogel, P. The carbonyl group as homoconjugated electron-releasing substituent. Regioselective electrophilic additions at bicyclo[2.2.1]hept-5-en-2-one, bicyclo[2.2.2]oct-5-en-2-one, and derivatives. Helv. Chim. Acta 1989, 72, 1008–1028. [Google Scholar] [CrossRef]
- Ruggiu, A.A.; Lysek, R.; Moreno-Clavijo, E.; Moreno-Vargas, A.J.; Robina, I.; Vogel, P. The regioselectivity of the addition of benzeneselenyl chloride to 7-azanorborn-5-ene-2-yl derivatives is controlled by the 2-substituent: New entry into 3- and 4-hydroxy-5-substituted prolines. Tetrahedron 2010, 66, 7309–7315. [Google Scholar] [CrossRef]
- Carrupt, P.-A.; Vogel, P. The carbonyl group as homoconjugated electron-donating substituent–Ab initio STO3G MO calculations. Tetrahedron Lett. 1984, 25, 2879–2882. [Google Scholar] [CrossRef]
- Sordo, J.A.; Varela-Alvarez, A.; Giani, S.; Vogel, P. Quantum calculations on the acid catalyzed rearrangements of norborn-5-en-2-one, 7-oxanorborn-5-en-2-one and 7-azanorborn-5-en-2-one. Appl. Catal. A General 2008, 336, 72–78. [Google Scholar] [CrossRef]
- Le Drian, C.; Vogel, P. Acid-catalyzed rearrangement of 5,6-exo-epoxy-7-oxabicyclo[2.2.1]hept-2-yl derivatives–Migratory apitudes of acyl vs. alkyl groups in Wagner-Meerwein transpositions. Helv. Chim. Acta 1987, 70, 1703–1720. [Google Scholar] [CrossRef]
- Berner, D.; Dahn, H.; Vogel., P. Unambiguous proof for alcoxycarbonyl group migration in Wagner-Meerwein rearrangements. Helv. Chim. Acta 1980, 63, 2538–2553. [Google Scholar] [CrossRef]
- Scuseria, G.E.; Miller, M.D.; Jensen, F.; Geertsen, J. The dipole moment of carbon monoxide. J. Chem. Phys. 1991, 94, 6660–6663. [Google Scholar] [CrossRef]
- De Proft, F.; Martin, J.M.L.; Geerlings, P. On the performance of density functional methods for describing atomic populations, dipole moments and infrared intensities. Chem. Phys. Lett. 1996, 250, 393–401. [Google Scholar] [CrossRef]
- Abboud, J.L.M.; Alkorta, I.; Davalos, J.Z.; Müller, P.; Quintanilla, E. Thermodynamic stabilities of carbocations. Adv. Phys. Org. Chem. 2002, 37, 57–135. [Google Scholar] [CrossRef]
- Aue, D.H. Carbocations. Wiley Interdiscip. Rev.-Comput. Mol. Sci. 2011, 1, 487–508. [Google Scholar] [CrossRef]
- Klump, D.A. Carbocations. In Organic Reaction Mechanisms Series; Knipe, A.C., Ed.; Wiley Online Library, John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2020; Chapter 6; pp. 337–367. [Google Scholar] [CrossRef]
- Wodrich, M.D.; Wannere, C.S.; Mo, Y.; Jarowski, P.D.; Houk, K.N.; Schleyer, P.V.R. The concept of protobranching and its many paradigm shifting implications for energy evaluations. Chem. Eur. J. 2007, 13, 7731–7744. [Google Scholar] [CrossRef]
- Aizman, A.; Contreras, R.; Galvan, M.; Cedillo, A.; Santos, J.C.; Chamorro, E. The Markovnikov regioselectivity rule in the light of site activation models. J. Phys. Chem. A 2002, 106, 7844–7849. [Google Scholar] [CrossRef]
- Hughes, P. Was Markovnikov’s rule an inspired guess? J. Chem. Educ. 2006, 83, 1152–1154. [Google Scholar] [CrossRef]
- Kerber, R.C. Markovnikov’s rule. J. Chem. Educ. 2007, 84, 1109. [Google Scholar] [CrossRef]
- Ilich, P.P. Markovnikov’s rule-Replies. J. Chem. Educ. 2007, 84, 1109. [Google Scholar] [CrossRef][Green Version]
- Zavitsas, A.A.; Rogers, D.W.; Matsunaga, N. Heats of formation of organic compounds by simple calculation. J. Org. Chem. 2010, 75, 6502–6515. [Google Scholar] [CrossRef] [PubMed]
- Dimroth, O. Beziehungen zwischen Affinität und Reaktiongeschwindichkeit. Angew. Chem. 1933, 52, 571–576. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Further considerations on the thermodynamics of chemical equilibria and reaction rates. Trans. Faraday Soc. 1936, 32, 1333–1359. [Google Scholar] [CrossRef]
- Bell, R.P. The kinetics of proton transfer reactions. Trans. Faraday Soc. 1938, 34, 0229–0236. [Google Scholar] [CrossRef]
- Levy, D.E. Arrow-Pushing in Organic Chemistry, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 1–400. [Google Scholar] [CrossRef]
- Woodward, R.B.; Hoffmann, R. Conservation of orbital symmetry. Angew. Chem. Int. Ed. 1969, 8, 781–853. [Google Scholar] [CrossRef]
- Longuet-Higgins, H.C.; Abrahamson, E.W. The electronic mechanism of electrocyclic reactions. J. Am. Chem. Soc. 1965, 87, 2045–2046. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Houk, K.N. Analyzing reaction rates with the distortion/interaction-activation strain model. Angew. Chem. Int. Ed. 2017, 56, 10070–10088. [Google Scholar] [CrossRef]
- Dewar, M.J.S. Aromaticity and pericyclic reactions. Angew. Chem. Int. Ed. 1971, 10, 761–776. [Google Scholar] [CrossRef]
- Zimmerman, H.E. Möbius-Hückel concept in organic chemistry-Application to organic molecules and reactions. Acc. Chem. Res. 1971, 4, 272–280. [Google Scholar] [CrossRef]
- Evans, M.G. The activation energies of reactions involving conjugated systems. Trans. Faraday Soc. 1939, 35, 0824–0834. [Google Scholar] [CrossRef]
- Houk, K.N. Frontier molecular orbital theory and organic reactions. Nature 1977, 266, 662. [Google Scholar] [CrossRef]
- Lias, S.G.; Bartmess, J.E.; Liebman, J.F.; Holmes, J.L.; Levin, R.D.; Mallard, W.G. Gas-Phase Ion and Neutral Thermochemistry. J. Phys. Chem. Reference Data 1988, 17 (Suppl. 1), 861. [Google Scholar] [CrossRef]
- Rienstra-Kiracofe, J.C.; Tschumper, G.S.; Schaefer, H.F.I.; Nandi, S.; Ellison, G.B. Atomic and molecular electron affinities: Photoelectron experiments and theoretical computations. Chem. Rev. 2002, 102, 231–282. [Google Scholar] [CrossRef]
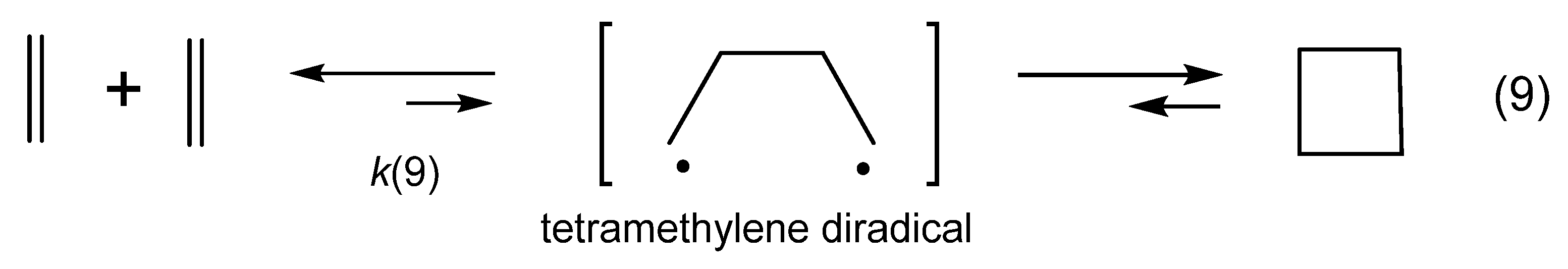
- Dewar, M.J.S.; Pyron, R.S. Nature of transition state in some Diels-Alder reactions. J. Am. Chem. Soc. 1970, 92, 3098–3103. [Google Scholar] [CrossRef]
- Dewar, J.J.S.; Pierini, A.B. Mechanism of the Diels-Alder reaction. Studies of the addition of maleic anhydride to furan and methylfurans. J. Am. Chem. Soc. 1984, 106, 203–208. [Google Scholar] [CrossRef]
- Pedersen, S.; Herek, J.L.; Zewail, A.H. The validity of the diradical hypothesis-Direct femtosecond studies of the transition-state structures. Science 1994, 266, 1359–1364. [Google Scholar] [CrossRef]
- Woodward, R.B.; Katz, T.J. The mechanism of the Diels-Alder reaction. Tetrahedron 1959, 5, 70–89. [Google Scholar] [CrossRef]
- Horn, B.A.; Herek, J.L.; Zewail, A.H. Retro-Diels-Alder femtosecond reaction dynamics. J. Am. Chem. Soc. 1996, 118, 8755–8756. [Google Scholar] [CrossRef]
- Littmann, E.R. The mechanism of the diene synthesis. J. Am. Chem. Soc. 1936, 58, 1316–1317. [Google Scholar] [CrossRef]
- Houk, K.N.; Gonzalez, J.; Li, Y. Pericyclic reaction transition-states-Passions and punctilios, 1935–1995. Acc. Chem. Res. 1995, 28, 81–90. [Google Scholar] [CrossRef]
- Houk, K.N. The frontier molecular orbital theory of cycloaddition reactions. Acc. Chem. Res. 1975, 8, 361–369. [Google Scholar] [CrossRef]
- Chen, P.-P.; Ma, P.; He, X.; Svatunek, D.; Liu, F.; Houk, K.N. Computational exploration of ambiphilic reactivity of azides and Sustmann’s paradigmatic parabola. J. Org. Chem. 2021, 86, 5792–5804. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, J.J.; Conrad, N.D. Variable transition-state structure in the Cope rearrangement as deduced from secondary deuterium kinetic isotope effects. J. Am. Chem. Soc. 1978, 100, 6269–6270. [Google Scholar] [CrossRef]
- Hosomi, A.; Sakurai, H. Syntheses of γ,δ-unsaturated alcohols from allylsilanes and carbonyl compounds in presence of Ti tetrachloride. Tetrahedron Lett. 1976, 17, 1295–1298. [Google Scholar] [CrossRef]
- Hosomi, A. Characteristics in the reactions of allylsilanes and their applications to versatile synthetic equivalents. Acc. Chem. Res. 1988, 21, 200–206. [Google Scholar] [CrossRef]
- Biamonte, M.A. Sakurai allylation. In Name Reactions for Homologation, Part I; Li, J.J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 539–575. ISBN 0470487011/9780470487013. [Google Scholar]
- Hayashi, T.; Kabeta, K.; Hamachi, I.; Kumada, M. Erythro-selectivity in addition of γ-substituted allylsilanes to aldehydes in the presence of Ti tetrachloride. Tetrahedron Lett. 1983, 24, 2865–2868. [Google Scholar] [CrossRef]
- Deleris, G.; Dunoguès, J.; Calas, R. Synthèse d’alcools γ,δ-éthyléniques ou propargyliques par voie organosilicique. Tetrahedron Lett. 1976, 28, 2449–2450. [Google Scholar] [CrossRef]
- Hoffmann, R.W.; Zeiss, H.J. Stereoselective synthesis of alcohols. 8. Diastereoselective synthesis of β-methylhomoallyl alcohols via crotylboronates. J. Org. Chem. 1981, 46, 1309–1314. [Google Scholar] [CrossRef]
- Collum, D.B.; McDonald, J.H.; Still, W.C. Synthesis of the polyether antibiotic Monensin. 2. Preparation of intermediates. J. Am. Chem. Soc. 1980, 102, 2118–2120. [Google Scholar] [CrossRef]
- Sato, F.; Iida, K.; Iijima, S.; Moriya, H.; Sato, M. Threo-selective synthesis of β-methylhomoallyl alcohols via but-2-enyltitanium compounds. J. Chem. Soc. Chem. Commun. 1981, 1140–1141. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Maruyama, K. Crotylzirconium derivatives as a new reagent for the threo-selective synthesis of β-methylhomoallyl alcohols. Tetrahedron Lett. 1981, 22, 2895–2898. [Google Scholar] [CrossRef]
- Hiyama, T.; Kimura, K.; Nozaki, H. Chromium(II) mediated threo-selective synthesis of homoallyl alcohols. Tetrahedron Lett. 1981, 22, 1037–1040. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Yatagai, H.; Naruta, Y.; Maruyama, K. Erythro-selective addition of crotyltrialkyltins to aldehydes regardless of the geometry of the crotyl unit-Stereoselection independent of the stereochemistry of precursors. J. Am. Chem. Soc. 1980, 102, 7107–7109. [Google Scholar] [CrossRef]
- Shaik, S.S.; Pross, A. SN2 Reactivity of CH3X derivatives-A valence bond approach. J. Am. Chem. Soc. 1982, 104, 2708–2719. [Google Scholar] [CrossRef]
- Pross, A. Relationship between rates and equilibria and the mechanistic significance of the Brønsted parameter-A qualitative valence-bond approach. J. Org. Chem. 1984, 49, 1811–1818. [Google Scholar] [CrossRef]
- Shaik, S.S.; Schlegel, H.B.; Wolfe, S. Theoretical Aspects of Physical Organic Chemistry. The SN2 Mechanism; John Wiley & Sons Inc.: New York, NY, USA, 1992; pp. 1–285. ISBN 0471840416. [Google Scholar]
- Shaik, S.S.; Ioffe, A.; Reddy, A.C.; Pross, A. Is the avoided crossing state a good approximation for the transition state of a chemical reaction-An analysis of Menschutkin and ionic SN2 reactions. J. Am. Chem. Soc. 1994, 116, 262–273. [Google Scholar] [CrossRef]
- Pross, A. Theoretical & Physical Principles of Organic Reactivity; John Wiley & Sons, Inc.: New York, NY, USA, 1995; pp. 1–312. ISBN 0471555991/9780471555995. [Google Scholar]
- Anglada, J.M.; Besalú, E.; Bofill, J.M.; Crehuet, R. Prediction of approximate transition states by Bell-Evans-Polanyi principle: I. J. Comput. Chem. 1999, 20, 1112–1129. [Google Scholar] [CrossRef]
- Bofill, J.M.; Anglada, J.M.; Besalú, E.; Crehuet, R. Quantum chemical reactivity: Beyond the study of small molecules. In Fundamentals of Molecular Similarity; Springer: Boston, MA, USA, 2001; pp. 125–141. [Google Scholar] [CrossRef]
- Wester, R.B.A.E.; Davis, A.V.; Neumark, D.M. Time-resolved study of the symmetric SN2-reaction I-+CH3I. J. Chem. Phys. 2003, 119, 10032–10039. [Google Scholar] [CrossRef]
- Smith, M.B. Aliphatic Substitution, Nucleophilic and Organometallic. In March’s Advanced Organic Chemistry, Reactions, Mechanisms, and Structure; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Chapter 10; pp. 373–568. [Google Scholar] [CrossRef]
- Crampton, M.R. Electrophilic aromatic substitution. In Organic Reaction Mechanisms Series; Knipe, A.C., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2014; Chapter 6; pp. 257–283. [Google Scholar] [CrossRef]
- Rowlands, G.J. Radicals in organic synthesis: Part 2. Tetrahedron 2010, 66, 1593–1636. [Google Scholar] [CrossRef]
- Tebben, L.; Studer, A. Nitroxides: Applications in synthesis and in polymer chemistry. Angew. Chem. Int. Ed. 2011, 50, 5034–5068. [Google Scholar] [CrossRef] [PubMed]
- Quiclet-Sire, B.; Zard, S.Z. Fun with radicals: Some new perspectives for organic synthesis. Pure Appl. Chem. 2011, 83, 519–551. [Google Scholar] [CrossRef]
- Lapointe, G.; Kapat, A.; Weidner, K.; Renaud, P. Radical azidation reactions and their application in the synthesis of alkaloids. Pure Appl. Chem. 2012, 84, 1633–1641. [Google Scholar] [CrossRef]
- Sebren, L.J.; Devery, J.J.; Stephenson, C.R. Catalytic radical domino reactions in organic synthesis. ACS Catal. 2014, 4, 703–716. [Google Scholar] [CrossRef]
- Denes, F.; Pichowicz, M.; Povie, G.; Renaud, P. Thiyl radicals in organic synthesis. Chem. Rev. 2014, 114, 2587–2693. [Google Scholar] [CrossRef]
- Smith, M.B. Addition to carbon-carbon multiple bonds. In March’s Advanced Organic Chemistry, Reactions, Mechanisms, and Structure; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Chapter 15; pp. 859–902. [Google Scholar] [CrossRef]
- Trost, B.M. The atom economy—A search for synthetic efficiency. Science 1991, 254, 1471–1477. [Google Scholar] [CrossRef]
- Shin, I.; Montgomery, T.P.; Krische, M.J. Catalytic C-C bond formation and the Hendricksonian ideal: Atom- and redox-economy, stereo- and site-selectivity. Aldrichimica Acta 2015, 48, 15. [Google Scholar]
- Smith, M.B. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 8th ed.; John Wiley & Sons. Inc.: Hoboken, NJ, USA, 2022; Chapters 15 & 16; pp. 891–1272. ISBN 978-1119371809/1119371805. [Google Scholar]
- Arundale, E.; Mikeska, L.A. The olefin aldehyde condensation-The Prins reaction. Chem. Rev. 1952, 51, 505–555. [Google Scholar] [CrossRef]
- Overman, L.E.; Velthuisen, E.J. Scope and facial selectivity of the Prins-pinacol synthesis of attached rings. J. Org. Chem. 2006, 71, 1581–1587. [Google Scholar] [CrossRef]
- Li, C.J. The development of catalytic nucleophilic additions of terminal alkynes in water. Acc. Chem. Res. 2010, 43, 581–590. [Google Scholar] [CrossRef]
- Engle, K.M.; Mei, T.S.; Wasa, M.; Yu, J.Q. Weak coordination as a powerful means for developing broadly useful C-H functionalization reactions. Acc. Chem. Res. 2012, 45, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.A.; Tsai, A.S.; Bergman, R.G.; Ellman, J.A. Rhodium-catalyzed chelation-assisted C-H bond functionalization reactions. Acc. Chem. Res. 2012, 45, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Rogge, T.; Kaplaneris, N.; Chatani, N.; Kim, J.; Chang, S.; Punji, B.; Schafer, L.L.; Musaev, D.G.; Wencel-Delord, J.; Roberts, C.A.; et al. C-H activation. Nat. Rev. Methods Primers 2021, 1, 43. [Google Scholar] [CrossRef]
- Jang, H.Y.; Huddleston, R.R.; Krische, M.J. Reductive generation of enolates from enones using elemental hydrogen: Catalytic C-C bond formation under hydrogenative conditions. J. Am. Chem. Soc. 2002, 124, 15156–15157. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.Y.; Krische, M.J. Catalytic hydrogen-mediated cross-coupling of enones and carbonyl compounds: Aldol condensation by hydrogenation. Eur. J. Org. Chem. 2004, 2004, 3953–3958. [Google Scholar] [CrossRef]
- Ngai, M.Y.; Kong, J.R.; Krische, M.J. Hydrogen-mediated C-C bond formation: A broad new concept in catalytic C-C coupling. J. Org. Chem. 2007, 72, 1063–1072. [Google Scholar] [CrossRef]
- Nguyen, K.D.; Park, B.Y.; Luong, T.; Sato, H.; Garza, V.J.; Krische, M.J. Metal-catalyzed reductive coupling of olefin-derived nucleophiles: Reinventing carbonyl addition. Science 2016, 354, 300. [Google Scholar] [CrossRef]
- Holmes, M.; Schwartz, L.A.; Krische, M.L. Intermolecular metal-catalyzed reductive coupling of dienes, allenes, and enynes with carbonyl compounds and imines. Chem. Rev. 2018, 118, 6026–6052. [Google Scholar] [CrossRef]
- Ngai, M.Y.; Rucas, E.; Krische, M.J. Ruthenium-catalyzed C-C bond formation via transfer hydrogenation: Branch-selective reductive coupling of allenes to paraformaldehyde and higher aldehydes. Org. Lett. 2008, 10, 2705–2708. [Google Scholar] [CrossRef][Green Version]
- Shibahara, F.; Bower, J.F.; Krische, M.J. Ruthenium-catalyzed C-C bond forming transfer hydrogenation: Carbonyl allylation from the alcohol or aldehyde oxidation level employing acyclic 1,3-dienes as surrogates to preformed allyl metal reagents. J. Am. Chem. Soc. 2008, 130, 6338–6339. [Google Scholar] [CrossRef]
- Sam, B.; Breit, B.; Krische, M.J. Paraformaldehyde and methanol as C1 feedstocks in metal-catalyzed C-C couplings of -unsaturated reactants: Beyond hydroformylation. Angew. Chem. Int. Ed. 2015, 54, 3267–3274. [Google Scholar] [CrossRef] [PubMed]
- Geary, L.M.; Woo, S.K.; Leung, J.C.; Krische, M.J. Diastereo- and enantioselective Ir-catalyzed carbonyl propargylation from the alcohol or aldehyde oxidation level: 1,3-Enynes as allenylmetal equivalents. Angew. Chem. Int. Ed. 2012, 51, 2972–2976. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.D.; Herkommer, D.; Krische, M.J. Ruthenium-BINAP catalyzed alcohol C-H tert-prenylation via 1,3-enyne transfer hydrogenation: Beyond stoichiometric carbanions in enantioselective carbonyl propargylation. J. Am. Chem. Soc. 2016, 138, 5238–5241. [Google Scholar] [CrossRef] [PubMed]
- Speith, J.G. The Chemistry and Technology of Petroleum; CRC Press, Francis & Taylor Group: Boca Raton, FL, USA, 2006; pp. 1–984. ISBN 1420008382/9781420008388. [Google Scholar]
- Corma, A.; Martínez, A. Chemistry, catalysts, and processes for isoparaffin–olefin alkylation: Actual situation and future trends. Rev. Sci. Eng. 1993, 35, 483–570. [Google Scholar] [CrossRef]
- Li, X.; Nagoaka, K.; Simon, L.J.; Olindo, R.; Lercher, J.A. Mechanism of butane skeletal isomerization on sulfated zirconia. J. Catal. 2005, 232, 456–466. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, J.; Wang, G.; Li, C.; Ang, C. Nature of active sites and deactivation mechanism for n-butane isomerization over alumina-promoted sulfated zirconia. J. Catal. 2016, 338, 124–134. [Google Scholar] [CrossRef]
- Olah, G.A.; Farooq, O.; Husain, A.; Ding, N.; Trivedi, N.J.; Olah, J.A. Superacid FSO3H/HF catalyzed butane isomerization. Catal. Lett. 1991, 10, 239–247. [Google Scholar] [CrossRef]
- Kennedy, J.P. Living cationic polymerization of olefins. How did the discovery come about. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 2285–2293. [Google Scholar] [CrossRef]
- Puskas, J.E.; Peng, H.H. Kinetic simulation of living carbocationic polymerizations. I. Simulation of living isobutylene polymerization. Polym. React. Eng. 1999, 7, 553–576. [Google Scholar] [CrossRef]
- Rudin, A.; Choi, P. The Elements of Polymer Science and Engineering, 3rd ed.; Academic Press: New York, NY, USA; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1–563. ISBN 978-0-12-382178-2. [Google Scholar]
- Vasilenko, I.V.; Yeong, H.Y.; Delgado, M.; Ouardad, S.; Peruch, F.; Voit, B.; Ganachaud, F.; Kostjuk, S.V. A catalyst platform for unique cationic (co)polymerization in aqueous emulsion. Angew. Chem. Int. Ed. 2015, 54, 12728–12732. [Google Scholar] [CrossRef]
- Ouardad, S.; Wirotius, A.L.; Kostjuk, S.; Ganachaud, F.; Peruch, F. Carbocationic polymerization of isoprene using cumyl initiators: Progress in understanding side reactions. RSC Adv. 2015, 5, 59218–59225. [Google Scholar] [CrossRef]
- Goriainow, W.; Butlerow, A. Ueber die Polyolene und die Umwandlung von Aethylen in Aethylalkohol. Liebigs Ann. Chem. 1873, 169, 146–149. [Google Scholar] [CrossRef]
- Kostjuk, S.V. Recent progress in the Lewis acid co-initiated cationic polymerization of isobutylene with 1,3-dienes. RSC Advances 2015, 5, 13125–13144. [Google Scholar] [CrossRef]
- Rajasekhar, T.; Singh, G.; Kapur, G.S.; Ramakumar, S.S.V. Recent advances in catalytic chain transfer polymerization of isobutylene: A review. RSC Adv. 2020, 10, 18180–18191. [Google Scholar] [CrossRef]
- Mattay, J. Photo-induced electron-transfer in organic synthesis. Synthesis 1989, 1989, 233–256. [Google Scholar] [CrossRef]
- Garcia, H.; Roth, H.D. Generation and reactions of organic radical-cations in zeolites. Chem. Rev. 2002, 102, 3977–4007. [Google Scholar] [CrossRef] [PubMed]
- Broggi, J.; Rollet, M.; Clément, J.-L.; Canard, G.; Terme, T.; Gigmes, D.; Vanelle, P. Polymerization initiated by organic electron donors. Angew. Chem. Int. Ed. 2016, 55, 5994–5999. [Google Scholar] [CrossRef]
- Elschenbroich, C. Organometallics, 3rd Completely Revised and Extended Edition; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. 1–817. ISBN 978-3-527-29390-2. [Google Scholar]
- Ishii, Y.; Tsutsui, M. Organotransition-Metal Chemistry; Springer: Boston, MA, USA, 1975; pp. 1–398. [Google Scholar] [CrossRef]
- Crabtree, R.H. An organometallic future in green and energy chemistry? Organometallics 2011, 30, 17–19. [Google Scholar] [CrossRef]
- Mathey, F. Transition Metal Organometallic Chemistry; SpringerBriefs in Molecular Sciences; Springer: Singapore, 2013; pp. 1–100. ISBN 978-981-4451-09-3. [Google Scholar]
- Frenking, G. Chemical bonding in transition metals. In The Chemical Bond: Chemical Bonding Across the Periodic Table; Frenking, G., Sason, S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014; pp. 175–218. [Google Scholar] [CrossRef]
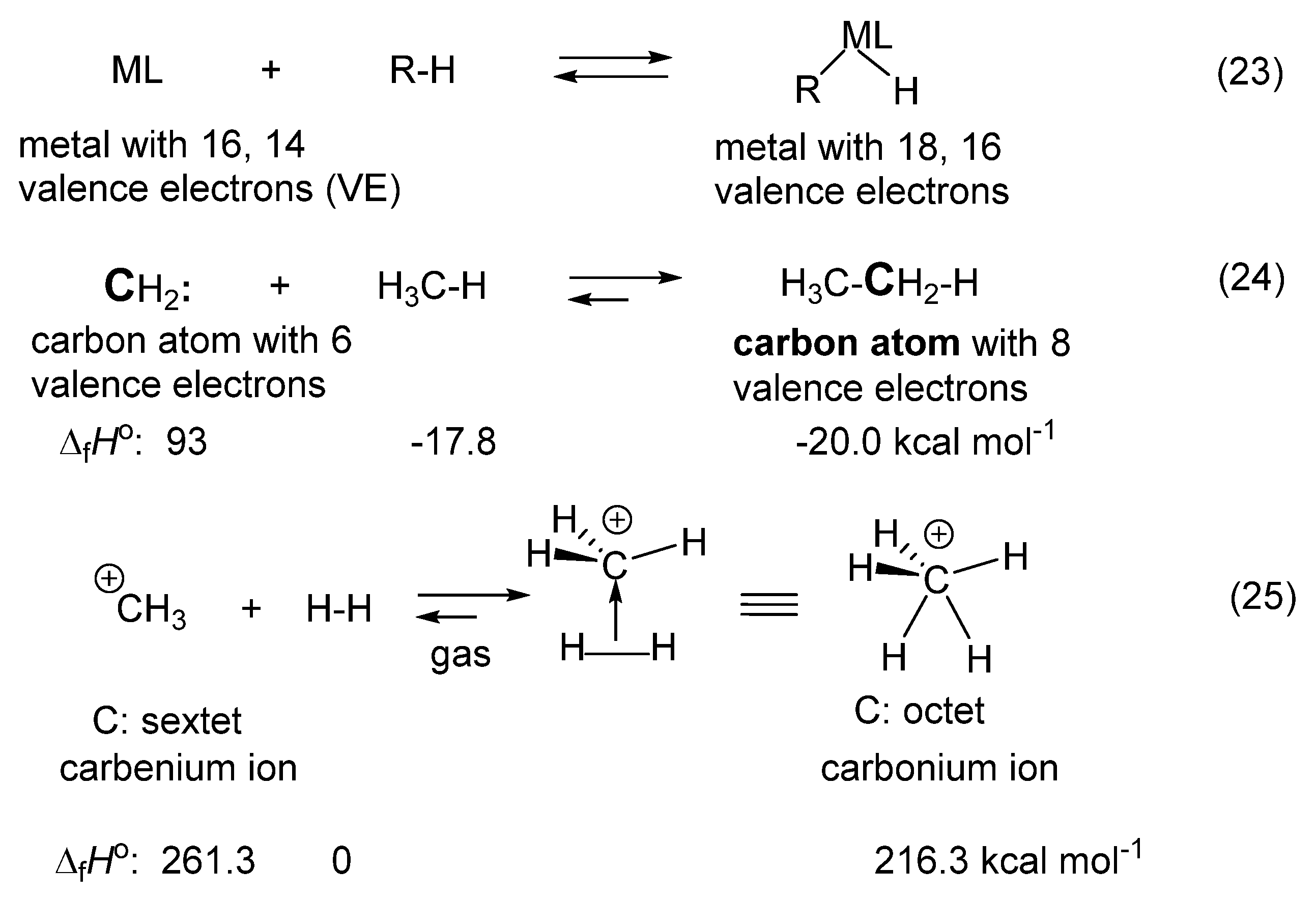
- Tolman, C.A. 16 and 18 electron rule in organometallic chemistry and homogeneous catalysis. Chem. Soc. Rev. 1972, 1, 337–353. [Google Scholar] [CrossRef]
- Carneiro, J.W.d.M.; Schleyer, P.V.R.; Saunders, M.; Remington, R.; Schaefer, H.F., III; Rauk, A.; Sorensen, T.S. Protonated ethane. A theoretical investigation of C2H7+ structures and energies. J. Am. Chem. Soc. 1994, 116, 3483–3493. [Google Scholar] [CrossRef]
- Smith, D.; Adams, N.G.; Alge, E. Isotope exchange and collisional association in the reactions of CH3+ and its deuterated analogs with H2, HD, and D2. J. Chem. Phys. 1982, 77, 1261–1268. [Google Scholar] [CrossRef]
- White, E.T.; Tang, J.; Oka, T. CH5+: The infrared spectrum observed. Science 1999, 284, 135–137. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.C.; Johnson, L.M.; Bowman, J.M.; McCoy, A.B. Deuteration effects on the structure and infrared spectrum of CH5+. J. Am. Chem. Soc. 2006, 128, 3478–3479. [Google Scholar] [CrossRef]
- Hinkle, C.E.; McCoy, A.B. Isotopic effects on the dynamics of the CH3+ + H2 → CH5+ → CH3+ + H2 reaction. J. Phys. Chem. A 2012, 116, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Asvany, O.; Yamada, K.M.T.; Bruenken, S.; Potapov, A.; Schlemmer, S. Experimental ground-state combination differences of CH5+. Science 2015, 347, 1346–1349. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M.; Vogel, P.; Hagen, E.L.; Rosenfeld, J. Evidence for protonated cyclopropane intermediates from studies of stable solutions of carbonium ions. Acc. Chem. Res. 1973, 6, 53–59. [Google Scholar] [CrossRef]
- Vogel, P. Carbocation Chemistry. In Studies in Organic Chemistry; Elsevier: Amsterdam, The Netherlands, 1985; pp. 1–596. ISBN 0444425225. [Google Scholar]
- Cabrera, J.M.; Krische, M.J. Total synthesis of clavosolide A via asymmetric alcohol-mediated carbonyl allylation: Beyond protecting groups or chiral auxiliaries in polyketide construction. Angew. Chem. Int. Ed. 2019, 58, 10718–10722. [Google Scholar] [CrossRef]
- Della-Felice, F.; Sarotti, A.M.; Krische, M.J.; Pilli, R.A. Total synthesis and structural validation of phosdiecin A via asymmetric alcohol-mediated carbonyl reductive coupling. J. Am. Chem. Soc. 2019, 141, 13778–13782. [Google Scholar] [CrossRef]
- McLafferty, F.W. Mass Spectrometry of Organic Ions; eBook; Academic Press: London, UK; New York, NY, USA, 1963; p. 700. ISBN 9780323142779. [Google Scholar]
- Gerlich, D. Probing the structure of CH5+ ions and deuterated variants via collisions. Phys. Chem. Chem. Phys. 2005, 7, 1583–1591. [Google Scholar] [CrossRef]
- Collins, C.J. The pinacol rearrangement. Quart. Rev. 1960, 14, 357–377. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Rzepa, H.S. Gaseous ions. 4. MINDO-3 calculations for some simple organic cations and for their hydrogen elimination-reactions. J. Am. Chem. Soc. 1977, 99, 7432–7439. [Google Scholar] [CrossRef]
- Lischka, H.; Köhler, H.-J. Structure and stability of carbocations C2H3+ and C2H4X+, X = H, F, Cl, Cl, and CH3 –ab initio investigations including electron correlation and comparison with MINDO-3 results. J. Am. Chem. Soc. 1978, 100, 5297. [Google Scholar] [CrossRef]
- Andrei, H.-S.; Solca, N.; Dopfer, O. IR spectrum of the ethyl cation: Evidence for the nonclassical structure. Angew. Chem. Int. Ed. 2008, 47, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Ricks, A.M.; Douberly, G.E.; Schleyer, P.v.R.; Duncan, M.A. Infrared spectroscopy of protonated ethylene: The nature of proton binding in the non-classical structure. Chem. Phys. Lett. 2009, 480, 17–20. [Google Scholar] [CrossRef]
- Saunders, M.; Hagen, E.L. Rearrangement reactions of secondary carbonium Ions. Isopropyl cation. J. Am. Chem. Soc. 1968, 90, 6881–6882. [Google Scholar] [CrossRef]
- Olah, G.A.; White, A.M. Stable carbonium ions. XCI: Carbon-13 nuclear magnetic resonance spectroscopy study of carbonium ions. J. Am. Chem. Soc. 1969, 91, 5801–5810. [Google Scholar] [CrossRef]
- McAdoo, D.J.; McLafferty, F.W.; Bente, P.F., III. Ion cyclotron resonance spectroscopy in structure determination. II. Propyl ions. J. Am. Chem Soc. 1972, 94, 2027–2033. [Google Scholar] [CrossRef]
- Chiavarino, B.; Crestoni, M.E.; Fornarini, S.; Lemaire, J.; Mac Aleese, L.; Maître, P. Infrared absorption features of gaseous isopropyl carbocations. ChemPhysChem 2004, 5, 1679–1685. [Google Scholar] [CrossRef]
- Saunders, M.; Hagen, E.L.; Rosenfeld, J. Rearrangement reactions of secondary carbonium ions. Protonated cyclopropane intermediates formed from sec-butyl cation. J. Am. Chem. Soc. 1968, 90, 6882–6884. [Google Scholar] [CrossRef]
- Lee, C.C.; Zohdi, H.F. Solvolytic studies with 2-methyl-[1-13C]-1-propyl tosylate. Can. J. Chem. 1983, 61, 2092–2094. [Google Scholar] [CrossRef]
- Fiaux, A.; Smith, D.L.; Futrell, J.H. Ion-molecule reaction of CH3+, C2H5+, C3H5+ and C3H7+ with unsaturated C2, C3 and C4 hydrocarbons. Int. J. Mass Spectrom. Ion Phys. 1977, 25, 281–294. [Google Scholar] [CrossRef]
- Saunders, M.; Hagen, E.L. Rapid rearrangements in t-amyl cation and relative sign of coupling constants. J. Am. Chem. Soc. 1968, 90, 2436–2437. [Google Scholar] [CrossRef]
- Saunders, M.; Budiansky, S.P. Computer modeling of multistep carbonium-ion rearrangements-Applications to methylcyclopentyl and tert-amyl. Tetrahedron 1979, 35, 929–932. [Google Scholar] [CrossRef]
- Scholz, F.; Himmel, D.; Heinemann, F.W.; Schleyer, P.V.R.; Meyer, K.; Krossing, I. Crystal structure determination of the nonclassical 2-norbornyl cation. Science 2013, 341, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A.; Lukas, J. Stable carbonium ions. 54. Protonation of and hydride ion abstraction from cycloalkanes and polycycloalkanes in fluorosulfonic acid-antimony pentafluoride. J. Am. Chem. Soc. 1968, 90, 933–938. [Google Scholar] [CrossRef]
- Olah, G.A.; Reddy, V.P.; Prakash, G.K.S. Long-lived cyclopropylcarbinyl cations. Chem. Rev. 1992, 92, 69–95. [Google Scholar] [CrossRef]
- Mo, Y.; Schleyer, P.v.R.; Jiao, H.; Lin, Z. Quantitative evaluation of hyperconjugation in the cyclopropylcarbinyl cation and in cyclopropylborane. Chem. Phys. Lett. 1997, 280, 439–443. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Reddy, V.P.; Rasul, G.; Casanova, J.; Olah, G.A. The search for persistent cyclobutylmethyl cations in superacidic media and observation of the cyclobutyldicyclopropylmethyl cation. J. Am. Chem. Soc. 1998, 120, 13362–13365. [Google Scholar] [CrossRef]
- Reddy, V.P.; Rasul, G.; Prakash, G.K.S.; Olah, G.A. Structural studies of nonclassical cyclobutylmethyl cations by the ab initio method. J. Org. Chem. 2007, 72, 3076–3080. [Google Scholar] [CrossRef]
- Franke, W.; Schwarz, H.; Thies, H.; Chandrasekhar, J.; Schleyer, P.V.R.; Hehre, W.J.; Saunders, M.; Walker, G. An experimental and theoretical-study of the degenerate carbon skeleton isomerization of the cyclopentyl cation in the gas-phase. Angew. Chem. Int. Ed. 1980, 19, 485–487. [Google Scholar] [CrossRef]
- Franke, W.; Schwarz, H.; Thies, H.; Chandrasekhar, J.; Schleyer, P.V.R.; Hehre, W.J.; Saunders, M.; Walker, G. Entartete Isomerisierung via Kohlenstoff-Platzwechsel beim Cyclopentyl-Kation in der Gasphase. Experimenteller und theoretischer Nachweis der Existenz eines pyramidalen C5H9+-Kations bei der unimolekularen Ethylen-Eliminierung. Chem. Ber. Rec. 1981, 114, 2808–2824. [Google Scholar] [CrossRef]
- Franke, W.; Frenking, G.; Schwarz, H.; Wolfschütz, R. Zur Kohlenstoff-Äquilibrierung in cyclischen C6H11+-Kationen in der Gasphase und zum Mechanismus der unimolekularen Ethylen-Abspaltung. Chem. Ber. Rec. 1981, 114, 3878–3895. [Google Scholar] [CrossRef]
- Cecchi, P.; Pizzabiocca, A.; Renzi, G.; Grandinetti, F.; Sparapani, C.; Buzek, P.; Schleyer, P.V.R.; Speranza, M. Gas-phase protonation of spiropentane-A novel entry into the C5H9+ potential-energy surface. J. Am. Chem. Soc. 1993, 115, 10338–10347. [Google Scholar] [CrossRef]
- Szabo, K.J.; Cremer, D. Route to a kinetically stabilized protonated spirocyclopentane with a pentacoordinated carbon-atom-the missing link between bicyclo[3.2.0]hept-3-yl and 7-norbornyl cation. J. Org. Chem. 1995, 60, 2257–2259. [Google Scholar] [CrossRef]


























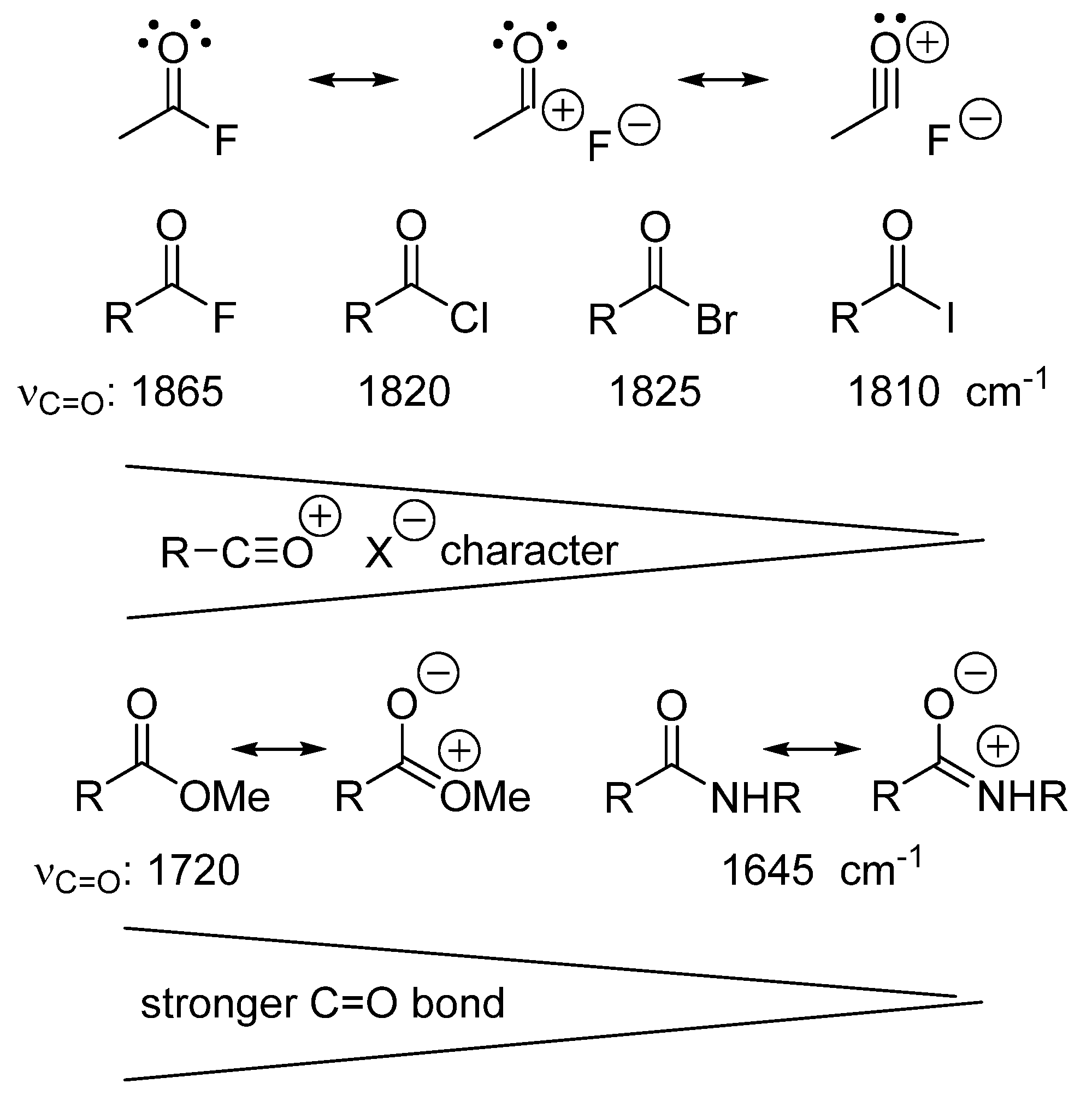{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
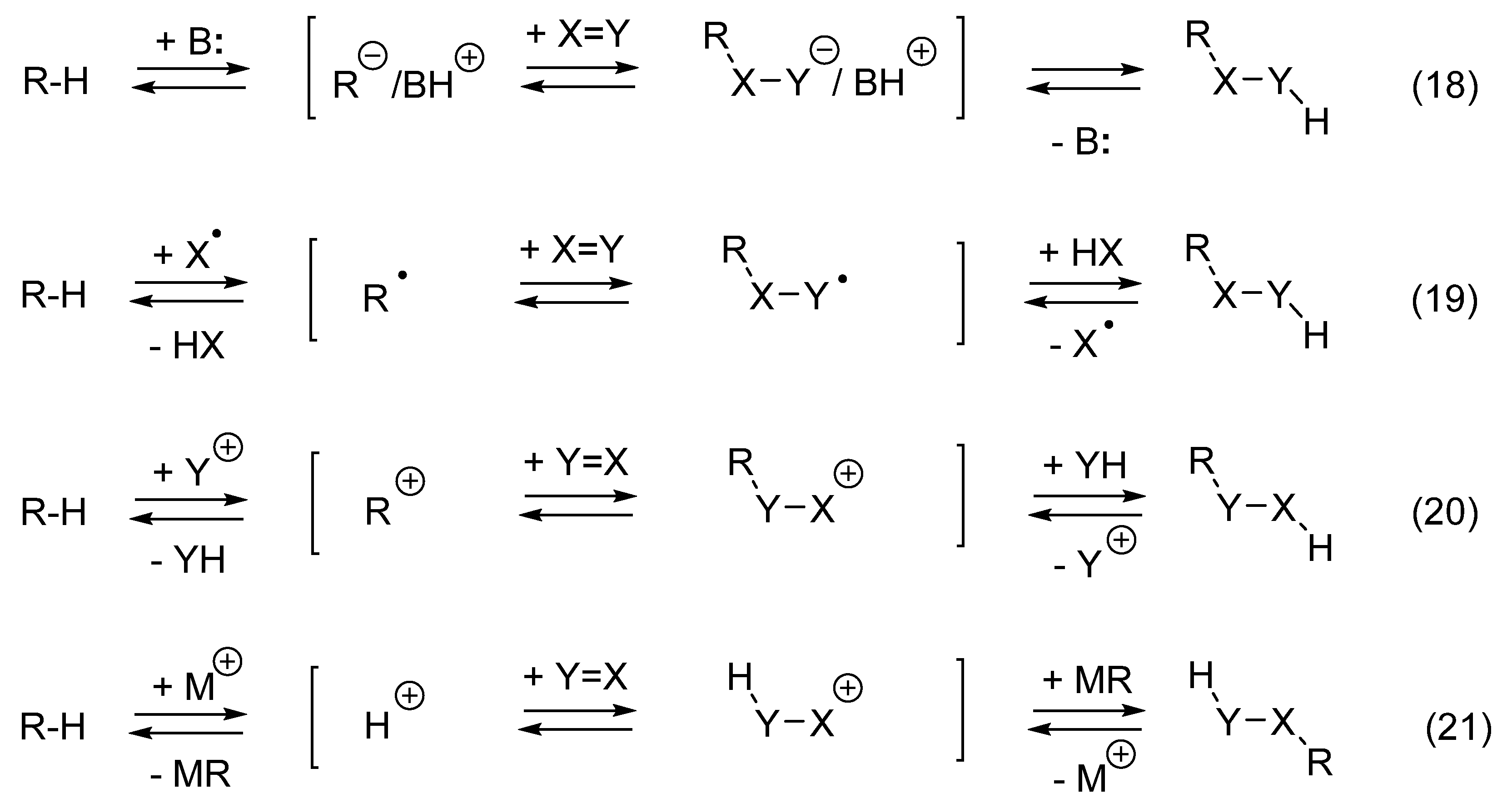{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Ligand exchange |
| No change of valence electron count and oxidation number between the metal center of reactant [M]=CO and product [M](π-alkene) by convention. |
2. Oxidative addition/reductive elimination |
| Oxidative addition requires an unsaturated metallic species (with 16, 14 VE). The metal oxidation number is increased by two and the metal of the adduct has two more VE than the metal in the starting complex. Note that unsaturated [M] can add to any kinds of X-Y bonds reversibly. The addition of carbenes R2C: (C with a sextet) to X-H bonds generates stable compounds R2C(X)-H with an octet carbon center. The opposite reaction is an α-elimination or 1,1-elimination. |
3. α-Insertion/α-elimination |
| The number of VE is reduced by two when going from the reactant to the product in an α-insertion; the oxidation number of the metal is not changed (Z=O, NR’, C(X)Y; R=H, alkyl, alkenyl, aryl, R’O, Hal, etc.). |
4. β-Insertion/β-elimination |
| The number of VE is reduced by two when going from reactant to product in the β-insertion; the oxidation number of the metal is not changed (R = H, alkyl, alkenyl, aryl, R’O, Hal, etc.). |
5. α-cycloinsertion/α-cycloelimination |
| The number of VE of the metal is reduced by two when going from reactant to product in the α-cycloinsertion; the oxidation number of the metal is increased by two by convention. |
6. Oxidative cyclization/reductive fragmentation (β-cycloinsertion/β-cycloelimination) The number of VE of the metal is reduced by two when going from reactant to product in the oxidative cyclization and the oxidation number of the metal increases by two by convention. The metal-catalyzed cycloinsertions can combine other unsaturated reactants besides alkenes. |
7. Reactions at the coordinated ligands |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vogel, P.; Houk, K.N. Organic Chemistry and Synthesis Rely More and More upon Catalysts. Catalysts 2022, 12, 758. https://doi.org/10.3390/catal12070758
Vogel P, Houk KN. Organic Chemistry and Synthesis Rely More and More upon Catalysts. Catalysts. 2022; 12(7):758. https://doi.org/10.3390/catal12070758
Chicago/Turabian StyleVogel, Pierre, and Kendall N. Houk. 2022. "Organic Chemistry and Synthesis Rely More and More upon Catalysts" Catalysts 12, no. 7: 758. https://doi.org/10.3390/catal12070758
APA StyleVogel, P., & Houk, K. N. (2022). Organic Chemistry and Synthesis Rely More and More upon Catalysts. Catalysts, 12(7), 758. https://doi.org/10.3390/catal12070758