
Effect of Potassium Promoter on the Performance of Nickel-Based Catalysts Supported on MnOx in Steam Reforming of Ethanol

 , , and
, , and 
Abstract
1. Introduction
2. Experimental Procedure
2.1. Catalyst Synthesis
2.2. Catalysts Characterization
2.3. Catalyst Evaluation in the SRE Process
3. Results and Discussion
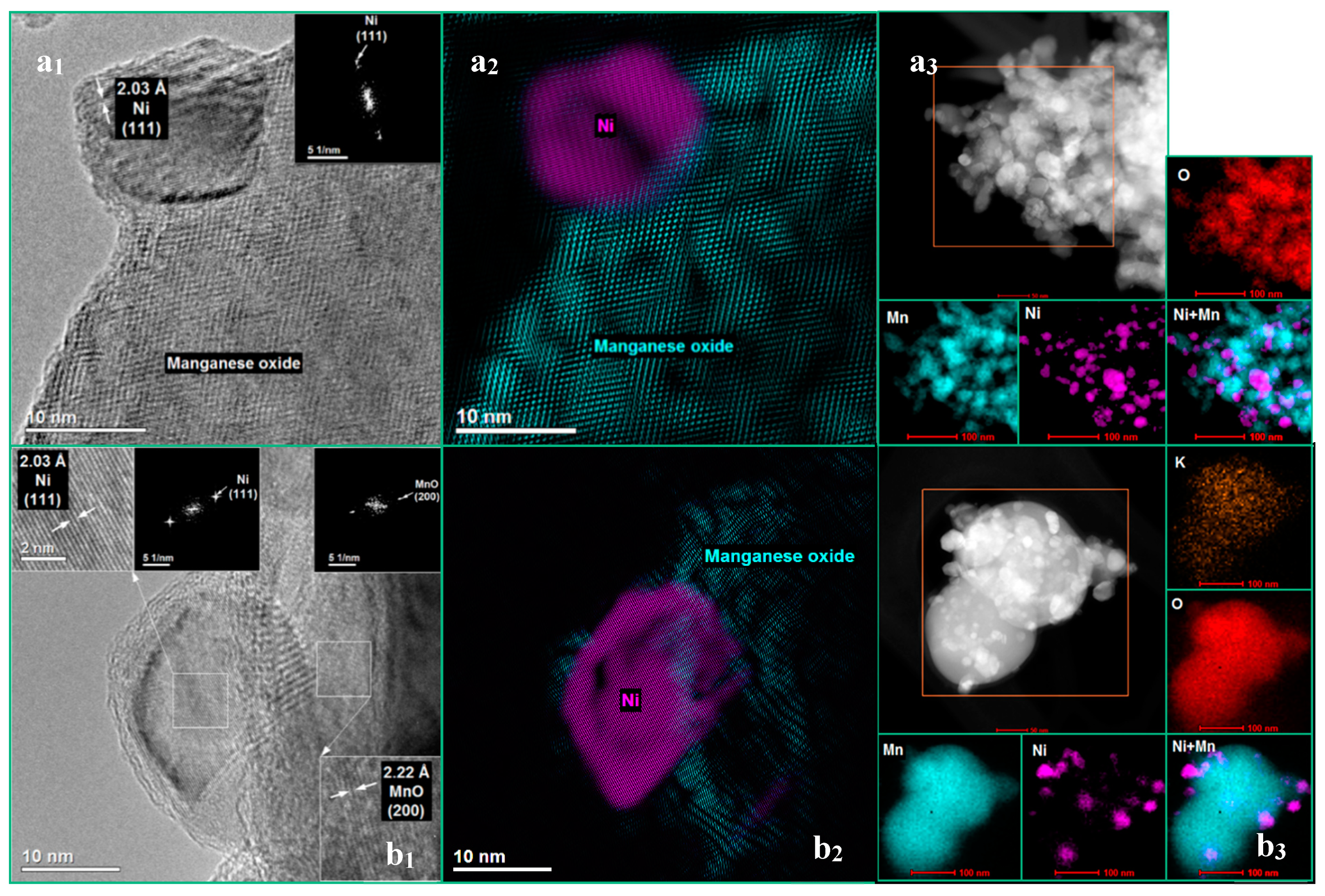
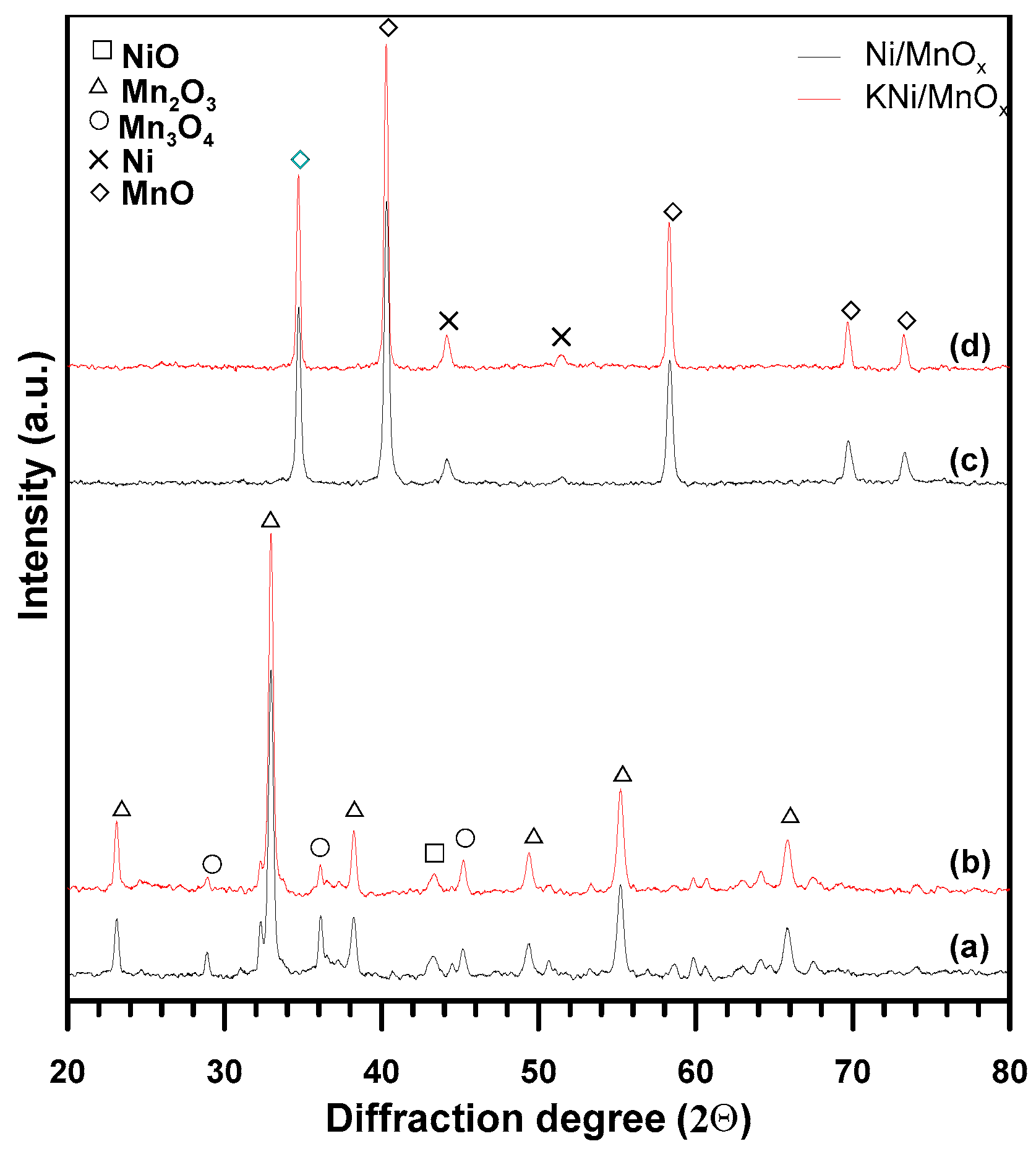
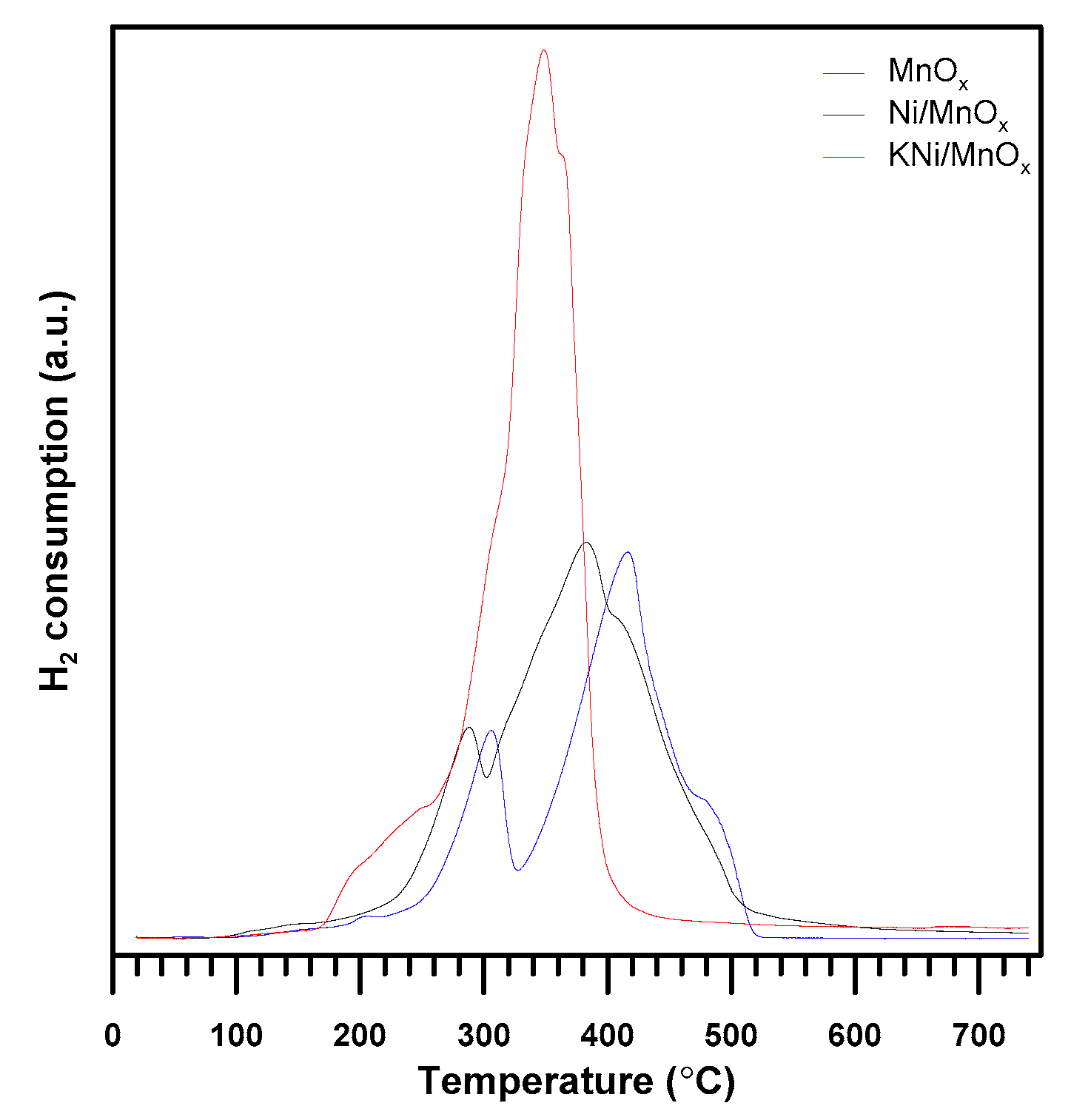
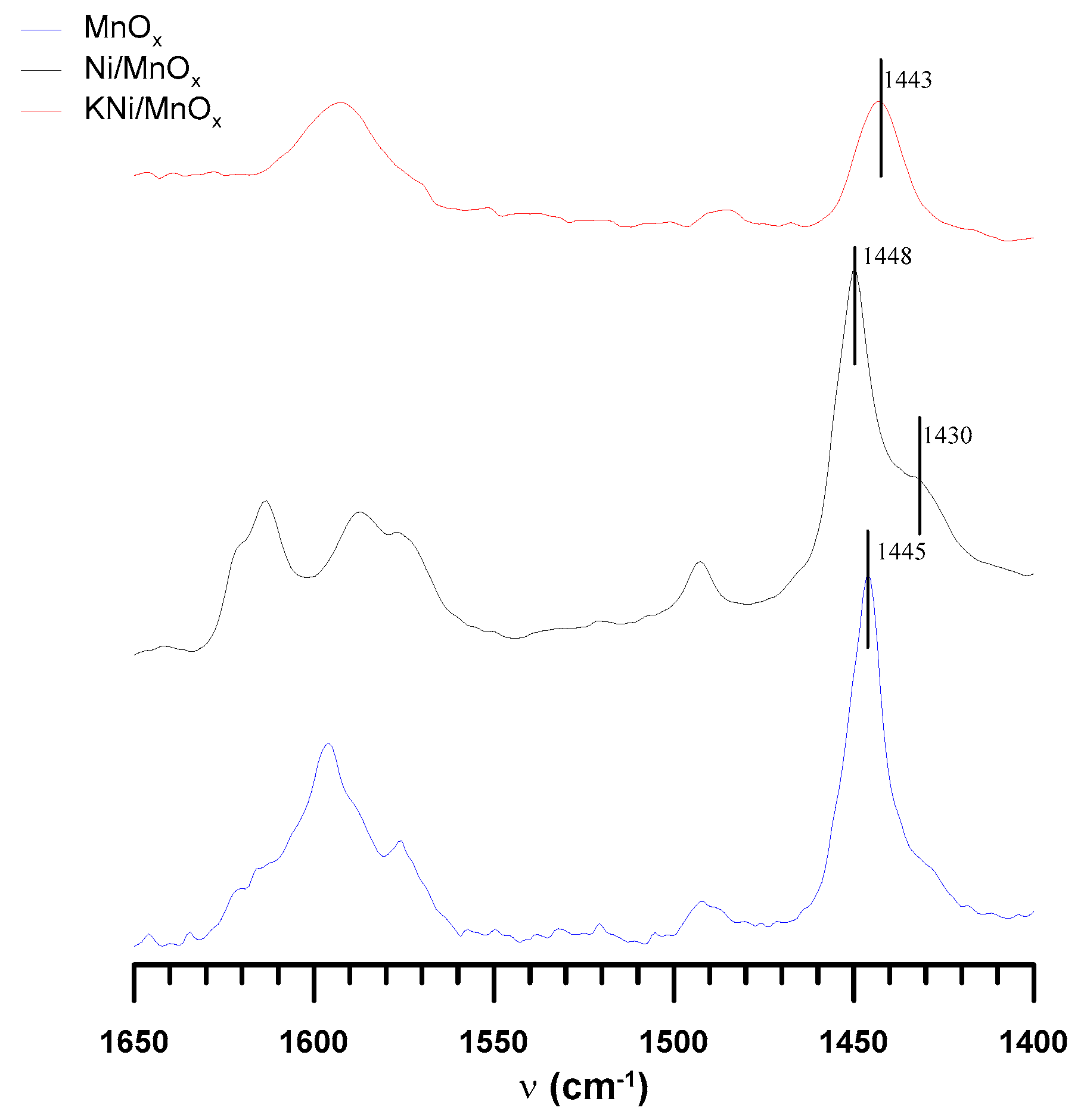
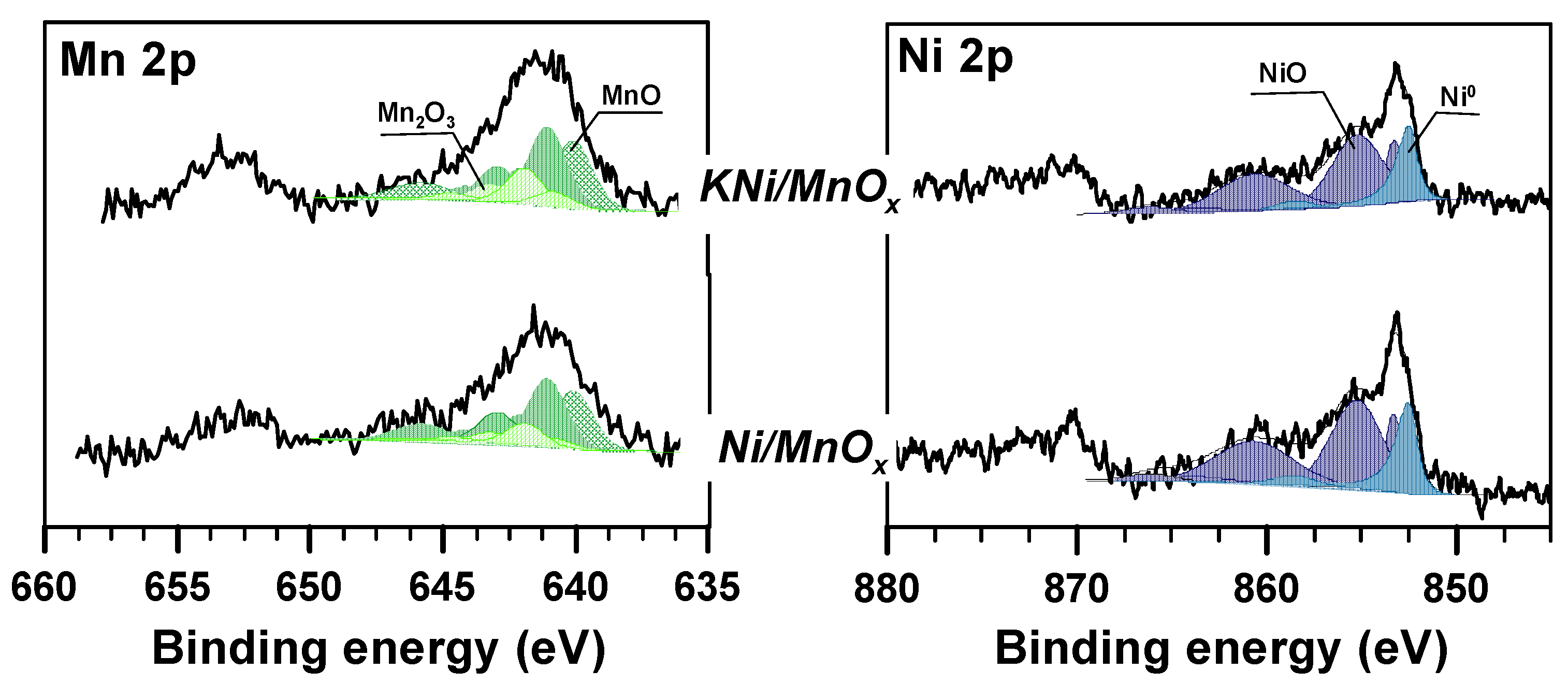
3.1. The Influence of a Potassium Promoter on the Catalysts’ Physicochemical Properties
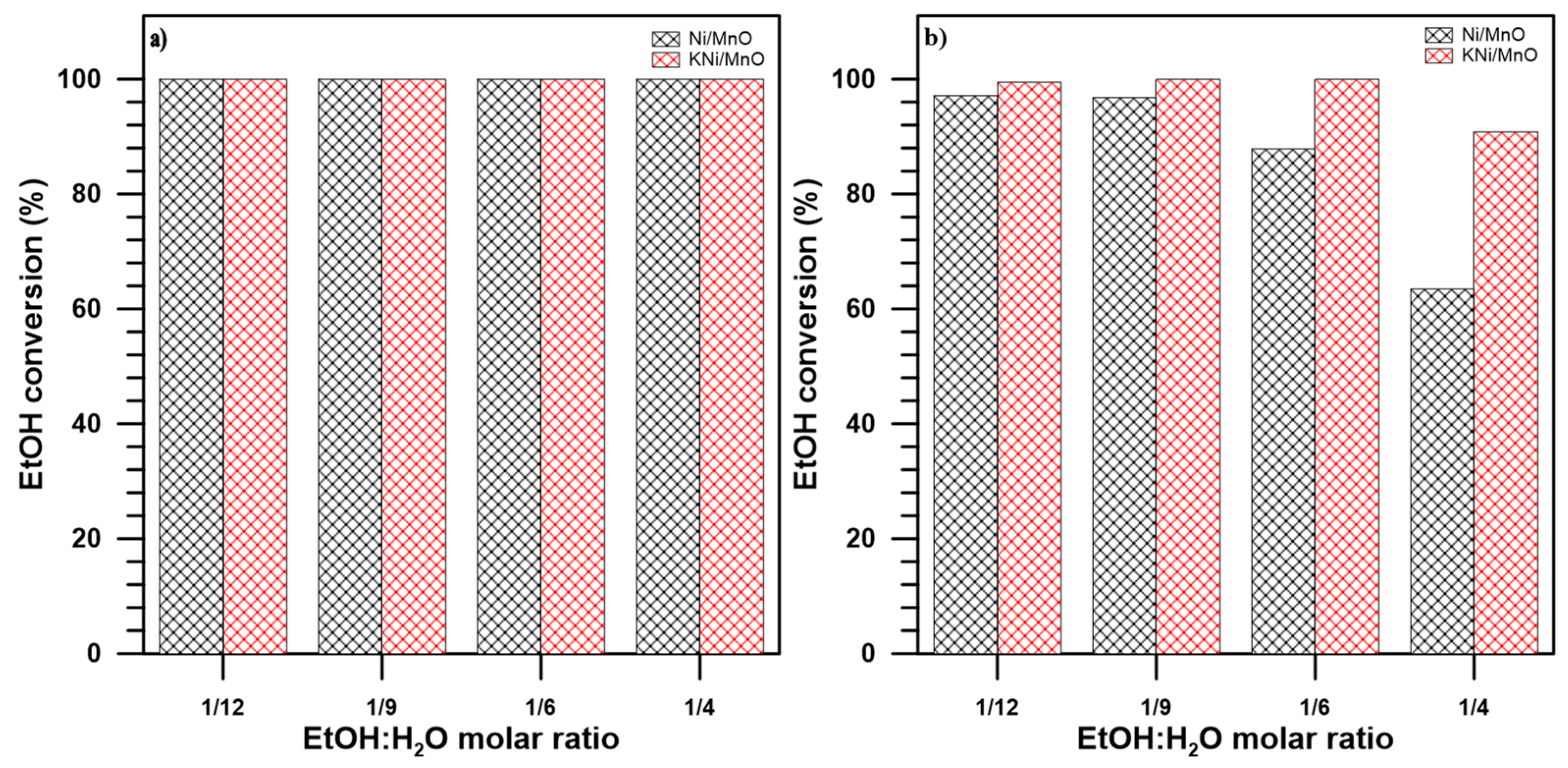
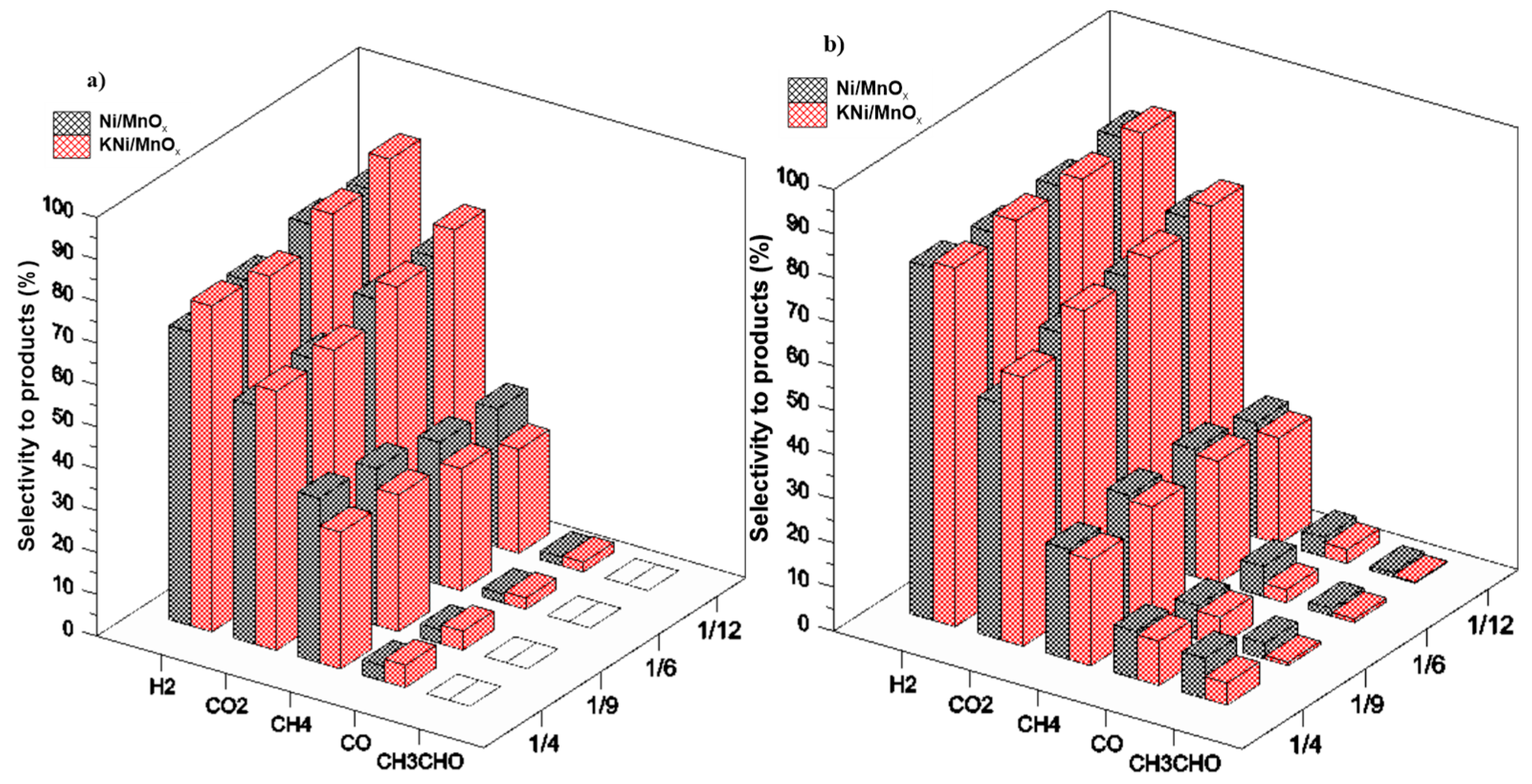
3.2. The Influence of a Potassium Promoter on the Performance of the Catalysts in Ethanol Steam Reforming Process
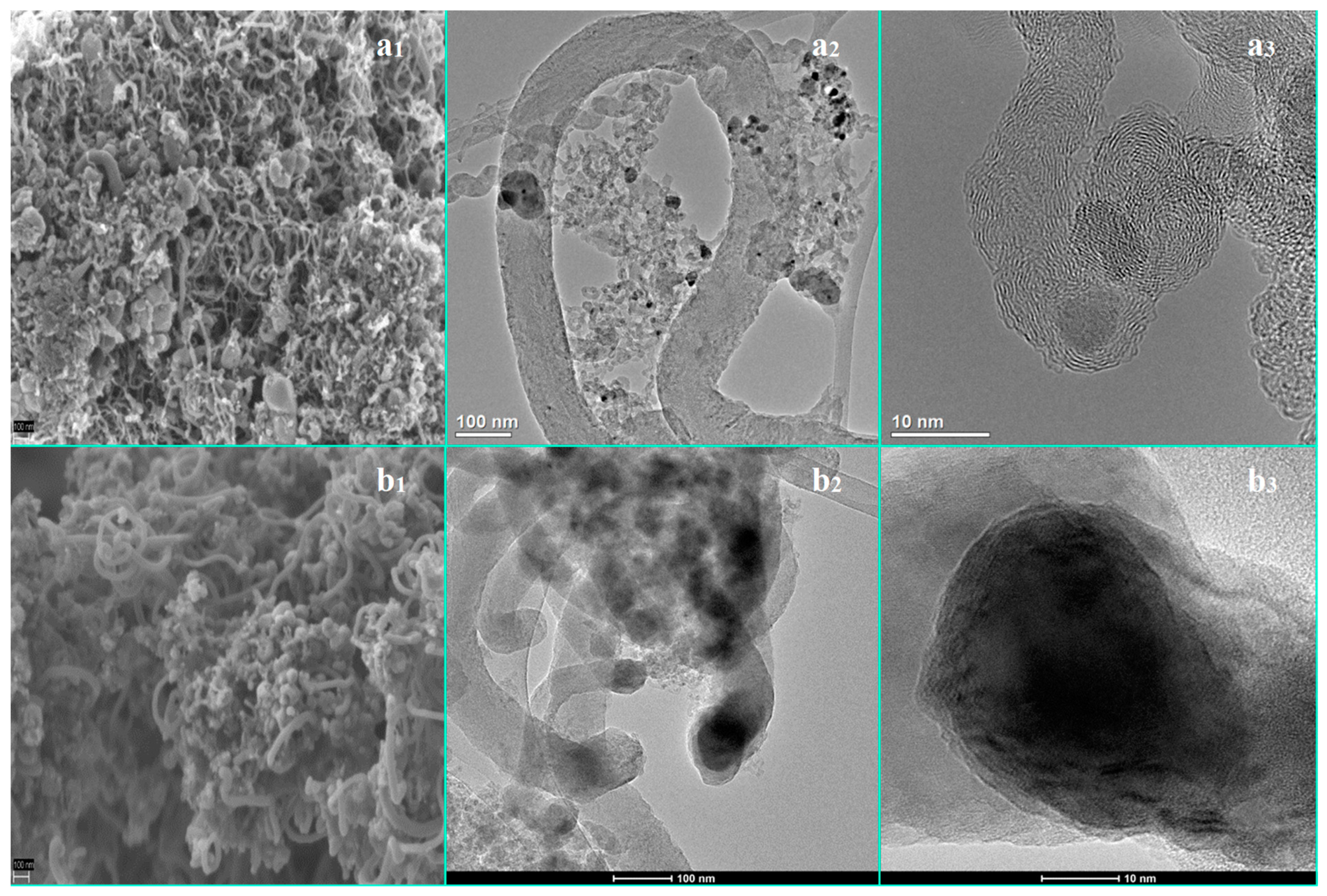
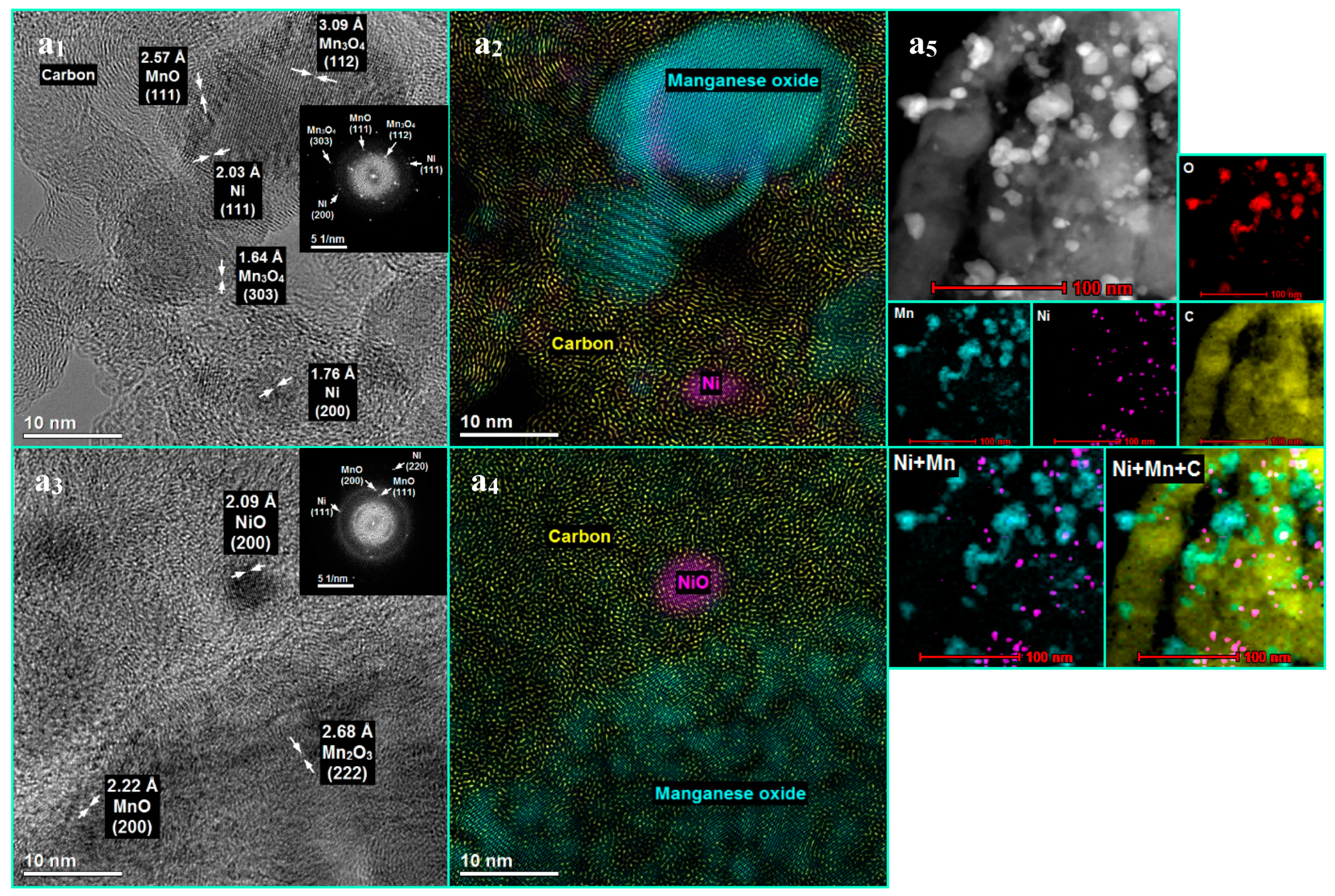
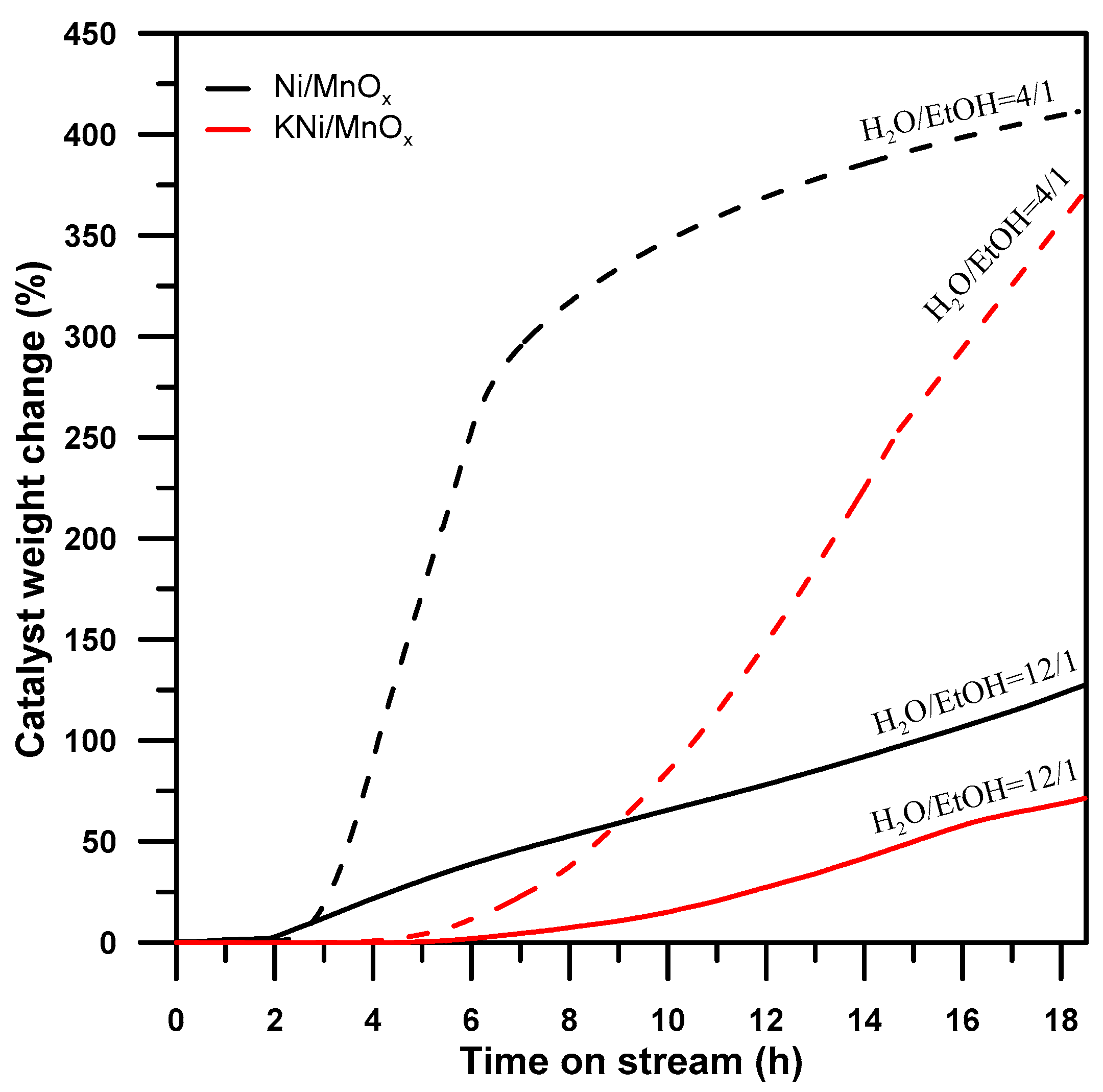
3.3. The Influence of a Potassium Promoter on Prevention of the Nickel-Based Catalyst Deactivation under SRE Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jo, W.J.; Im, Y.; Do, J.Y.; Park, N.-K.; Lee, T.J.; Lee, S.T.; Cha, M.S.; Jeon, M.-K.; Kang, M. Synergies between Ni, Co, and Mn ions in trimetallic Ni1-xCoxMnO4 catalysts for effective hydrogen production from propane steam reforming. Renew. Energy 2017, 113, 248–256. [Google Scholar] [CrossRef]
- Da Costa-Serra, J.F.; Chica, A. Catalysts based on Co-Birnessite and Co-Todorokite for the efficient production of hydrogen by ethanol steam reforming. Int. J. Hydrogen Energy 2018, 43, 16859–16865. [Google Scholar] [CrossRef]
- Manfro, R.L.; Ribeiro, N.F.P.; Souza, M.M.V.M. Production of hydrogen from steam reforming of grycerol using nickel catalysts supported on Al2O3, CeO2 and ZrO2. Catal. Sustain. Energy 2013, 1, 60–70. [Google Scholar]
- Marcos, F.C.F.; Lucrédio, A.F.; Assaf, E.M. Effects of adding basic oxides of La and/or Ce to SiO2-supported Co catalysts for ethanol steam reforming. RSC Adv. 2014, 4, 43839–43849. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Jia, P.; Dong, D.; Wang, Y.; Song, H.; Xiang, J.; Liu, Q.; Hu, X. Ethanol steam reforming over cobalt catalysts: Effect of range of additives on the catalytic behaviors. J. Energy Inst. 2020, 93, 165–184. [Google Scholar] [CrossRef]
- Riani, P.; Garbarino, G.; Canepa, F.; Busca, G. Cobalt nanoparticles mechanically deposited on α-Al2O3: A competitive catalyst for the production of hydrogen through ethanol steam reforming. J. Chem. Technol. Biotechnol. 2019, 94, 538–546. [Google Scholar] [CrossRef]
- Yu, S.-W.; Huang, H.-H.; Tang, C.-W.; Wang, C.-B. The effect of accessible oxygen over Co3O4-CeO2 catalysts on the steam reforming of ethanol. Int. J. Hydrogen Energy 2014, 39, 20700–20711. [Google Scholar] [CrossRef]
- Ibrahim, S.h.M.; El-Shobaky, G.A.; Mohamed, G.M.; Hassan, N.A. Effects of ZnO and MoO3 Doping on surface and catalytic properties of manganese oxide supported on alumina system. Open Catal. J. 2011, 4, 27–35. [Google Scholar] [CrossRef]
- Comas, J.; Mariňo, F.; Laborde, M.; Amadeo, N. Bio-ethanol steam reforming on Ni/Al2O3 catalyst. Chem. Eng. J. 2004, 98, 61–68. [Google Scholar] [CrossRef]
- Bshish, A.; Yaakob, A.; Ebshish, A.; Alhasan, F.H. Hydrogen production via ethanol steam reforming over Ni/Al2O3 catalysts: Effect of Ni loading. J. Energy Resour. Technol. 2014, 136, 012601. [Google Scholar] [CrossRef]
- Alberton, A.L.; Souza, M.M.V.M.; Schmal, M. Carbon formation and its influence on ethanol steam reforming over Ni/Al2O3 catalysts. Catal. Today 2007, 123, 257–264. [Google Scholar] [CrossRef]
- Garcia, S.R.; Assaf, J.M. Effect of the preparation method on Co/Al2O3 catalyst applied to ethanol steam reforming reaction production of hydrogen. Mod. Res. Catal. 2012, 1, 52–57. [Google Scholar] [CrossRef][Green Version]
- Lucredio, A.F.; Bellido, J.D.A.; Zawadzki, A.; Assaf, E.M. Co catalysts supported on SiO2 and γ-Al2O3 applied to ethanol steam reforming: Effect of the solvent used in the catalyst preparation method. Fuel 2011, 90, 1424–1430. [Google Scholar] [CrossRef]
- Kaddouri, A.; Mazzocchia, C. A study of the influence of the synthesis conditions upon the catalytic properties of Co/SiO2 or Co/Al2O3 catalysts used for ethanol steam reforming. Catal. Commun. 2014, 5, 339–345. [Google Scholar] [CrossRef]
- Carvalho, F.L.S.; Asencios, Y.J.O.; Rego, A.M.B.; Assaf, E.M. Hydrogen production by steam reforming of ethanol over Co3O4/La2O3/CeO2 catalysts synthesized by one-step polymerization method. Appl. Catal. A Gen. 2014, 483, 52–62. [Google Scholar] [CrossRef]
- Arena, F.; Torre, T.; Raimondo, C.; Parmaliana, A. Structure and redox properties of bulk and supported manganese oxide catalysts. Phys. Chem. Chem. Phys. 2001, 3, 1911–1917. [Google Scholar] [CrossRef]
- Li, J.; Li, L.; Cheng, W.; Wu, F.; Lu, X.; Li, Z. Controlled synthesis of diverse manganese based catalysts for complete oxidation of toluene and carbon monoxide. Chem. Eng. J. 2014, 22, 59–67. [Google Scholar] [CrossRef]
- Lee, G.; Kim, D.; Kwak, B.S.; Kang, M. Hydrogen rich production by ethanol steam reforming reaction over Mn/Co10Si90MCM-48 catalysts. Catal. Today 2014, 232, 139–150. [Google Scholar] [CrossRef]
- Kwak, B.S.; Lee, G.; Park, S.-M.; Kang, M. Effect of MnOx in the catalytic stabilization of Co2MnO4 spinel during the ethanol steam reforming reaction. Appl. Catal. A Gen. 2015, 503, 165–175. [Google Scholar] [CrossRef]
- Fuertes, A.; Da Costa-Serra, J.F.; Chica, A. New catalysts based on Ni-Birnessite and Ni-Todorokite for the efficient production of hydrogen by bioethanol steam reforming. Energy Procedia 2012, 29, 181–191. [Google Scholar] [CrossRef][Green Version]
- Gac, W.; Greluk, M.; Słowik, G.; Turczyniak-Surdacka, S. Structural and surface changes of cobalt modified manganese oxide during activation and ethanol steam reforming reaction. Appl. Surf. Sci. 2018, 440, 1047–1062. [Google Scholar] [CrossRef]
- Sohrabi, S.; Irankhah, A. Synthesis, characterization, and catalytic activity of Ni/CeMnO2 catalysts promoted by copper, cobalt, potassium and iron for ethanol steam reforming. Int. J. Hydrogen Energy 2021, 46, 12846–12856. [Google Scholar] [CrossRef]
- Greluk, M.; Rybak, P.; Słowik, G.; Rotko, M.; Machocki, A. Comparative study on steam and oxidative steam reforming of ethanol over 2KCo/ZrO2 catalyst. Catal. Today 2015, 242, 50–59. [Google Scholar] [CrossRef]
- Banach, B.; Machocki, A. Effect of potassium addition on a long term performance of Co-ZnO-Al2O3 catalysts in the low-temperature steam reforming of ethanol: Co-precipitation vs citrate method of catalysts synthesis. Appl. Catal. A Gen. 2015, 505, 173–182. [Google Scholar] [CrossRef]
- Greluk, M.; Rotko, M.; Machocki, A. Conversion of ethanol over Co/CeO2 and KCo/CeO2 catalysts for hydrogen production. Catal. Lett. 2016, 146, 163–173. [Google Scholar] [CrossRef]
- Słowik, G.; Greluk, M.; Machocki, A. Microscopic characterization of changes in the structure of KCo/CeO2 catalyst used in the steam reforming of ethanol. Mater. Chem. Phys. 2016, 173, 219–237. [Google Scholar] [CrossRef]
- Greluk, M.; Słowik, G.; Rotko, M.; Machocki, A. Steam reforming and oxidative steam reforming of ethanol over PtKCo/CeO2 catalyst. Fuel 2016, 183, 518–530. [Google Scholar] [CrossRef]
- Słowik, G.; Greluk, M.; Rotko, M.; Machocki, A. Evolution of the structure of unpromoted and potassium-promoted ceria-supported nickel catalysts in the steam reforming of ethanol. Appl. Catal. B Environ. 2018, 221, 490–509. [Google Scholar] [CrossRef]
- Grzybek, G.; Greluk, M.; Indyka, P.; Góra-Marek, K.; Legutko, P.; Słowik, G.; Turczyniak-Surdacka, S.; Rotko, M.; Sojka, Z.; Kotarba, A. Cobalt catalyst for steam reforming of ethanol–Insights into the promotional role of potassium. Int. J. Hydrogen Energy 2020, 45, 22658–22673. [Google Scholar] [CrossRef]
- Grzybek, G.; Góra-Marek, K.; Patulski, P.; Greluk, M.; Rotko, M.; Słowik, G.; Kotarba, A. Optimization of the potassium promotion of the Co|α-Al2O3 catalyst for the effective hydrogen production via ethanol steam reforming. Appl. Catal. A Gen. 2021, 614, 1188051. [Google Scholar] [CrossRef]
- Llorca, J.; Homs, N.; Sales, J.; Fierro, J.L.G.; Ramirez de la Piscina, P. Effect of sodium addition on the performance of Co–ZnO-based catalysts for hydrogen production from bioethanol. J. Catal. 2004, 222, 470–480. [Google Scholar] [CrossRef]
- Oktaviano, H.S.; Trisunaryanti, W. Sol-gel derived Co and Ni based catalysts: Application for steam reforming of ethanol. Indones. J. Chem. 2008, 8, 47–53. [Google Scholar] [CrossRef]
- Vizcaíno, A.J.; Carrero, A.; Calles, J.A. Ethanol steam reforming on Mg- and Ca-modified Cu–Ni/SBA-15 catalysts. Catal. Today 2009, 146, 63–70. [Google Scholar] [CrossRef]
- Ogo, S.; Shimizu, T.; Nakazawa, Y.; Mukawa, K.; Mukai, D.; Sekine, Y. Steam reforming of ethanol over K promoted Co catalyst. Appl. Catal. A Gen. 2015, 495, 30–38. [Google Scholar] [CrossRef]
- Barrientos, J.; Gonzalez, N.; Boutonnet, M.; Järås, S. Deactivation of Ni/γ-Al2O3 catalysts in CO methanation: Effect of Zr, Mg, Ba and Ca oxide promoters. Top. Catal. 2017, 60, 1276–1284. [Google Scholar] [CrossRef]
- Wang, A.; Tian, Z.; Liu, Q.; Qiao, Y.; Tian, Y. Facile preparation of a Ni/MgAl2O4 catalyst with high surface area: Enhancement in activity and stability for CO methanation. Main Group Met. Chem. 2018, 41, 73–89. [Google Scholar] [CrossRef]
- Richardson, J.T.; Scates, R.; Twigg, M.V. X-ray diffraction study of nickel oxide reduction by hydrogen. Appl. Catal. A Gen. 2003, 246, 137–150. [Google Scholar] [CrossRef]
- Sharrouf, M.; Awad, R.; Roumié, M.; Marhaba, S. Structural, optical and room temperature magnetic study of Mn2O3 nanoparticles. Mater. Sci. Appl. 2015, 6, 850–859. [Google Scholar]
- Khalaji, A.D.; Soleymanifard, M.; Jarosova, M.; Machek, P. Facile synthesis and characterization of Mn3O4, Co3O4, and NiO. Acta Phys. Pol. 2002, 137, 1043–1045. [Google Scholar] [CrossRef]
- Zhang, P.; Zhan, Y.; Cai, B.; Hao, C.; Wang, J.; Liu, C.; Meng, Z.; Yin, Z.; Chen, Q. Shape-controlled synthesis of Mn3O4 nanocrystals and their catalysis of the degradation of Methylene Blue. Nano Res. 2010, 3, 235–243. [Google Scholar] [CrossRef]
- Martinez de la Torre, C.; Bennewitz, M.F. Manganese oxide nanoparticle synthesis by thermal decomposition of manganese(II) acetylacetonate. J. Vis. Exp. 2020, 160, e61572. [Google Scholar] [CrossRef] [PubMed]
- Indra, A.; Menezes, P.W.; Schuster, F.; Driess, M. Significant role of Mn(III) sites in eg1 configuration in manganese oxide catalysts for efficient artificial water oxidation. J. Photochem. Photobiol. B Biol. 2015, 152, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Bayón, A.; de la Peña O’Shea, V.; Coronado, J.M.; Serrano, D.P. Role of the physicochemical properties of hausmannite on the hydrogen production via the Mn3O4-NaOH thermochemical cycle. Int. J. Hydrogen Energy 2016, 41, 113–122. [Google Scholar] [CrossRef]
- Shu, S.; Guo, J.; Li, J.; Fang, N.; Li, J.; Yuan, S. Effect of post-treatment on the selective catalytic reduction of NO with NH3 over Mn3O4. Mater. Chem. Phys. 2019, 237, 121845. [Google Scholar] [CrossRef]
- Xu, X.; Li, L.; Yu, F.; Peng, H.; Fang, X.; Wang, X. Mesoporous high surface area NiO synthesized with soft templates: Remarkable for catalytic CH4 deep oxidation. Mol. Catal. 2017, 441, 81–91. [Google Scholar] [CrossRef]
- Gil, A.; Gandía, L.M.; Korili, S.A. Effect of the temperature of calcination on the catalytic performance of manganese- and samarium-manganese-based oxides in the complete oxidation of acetone. Appl. Catal. A Gen. 2004, 274, 229–235. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, Q.; Li, H.; Li, X.; Wang, L.; Tsang, S.C. Cr–MnOx mixed-oxide catalysts for selective catalytic reduction of NOx with NH3 at low temperature. J. Catal. 2010, 276, 56–65. [Google Scholar] [CrossRef]
- Dong, Y.; Zhao, Y.; Zhang, J.-Y.; Chen, Y.; Yang, X.; Song, W.; Wei, L.; Li, W. Synergy of Mn and Ni enhanced catalytic performance for toluene combustion over Ni-doped α-MnO2 catalysts. Chem. Eng. J. 2020, 388, 124244. [Google Scholar] [CrossRef]
- Zhang, Y.; Qin, Z.; Wang, G.; Zhu, H.; Dong, M.; Li, S.; Wu, Z.; Li, Z.; Wu, Z.; Zhang, J.; et al. Catalytic performance of MnOx–NiO composite oxide in lean methane combustion at low temperature. Appl. Catal. B Environ. 2013, 129, 172–181. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Zhu, Q.; Ma, M.; Jiang, Z.; Liao, X.; He, C. Unraveling the effects of potassium incorporation routes and positions on toluene oxidation over α-MnO2 nanorods: Based on experimental and density functional theory (DFT) studies. J. Colloid Interface Sci. 2021, 598, 324–338. [Google Scholar] [CrossRef]
- Greluk, M.; Rotko, M.; Turczyniak-Surdacka, S. Comparison of catalytic performance and coking resistant behaviors of cobalt- and nickel based catalyst with different Co/Ce and Ni/Ce molar ratio under SRE conditions. Appl. Catal. A Gen. 2021, 590, 117334. [Google Scholar] [CrossRef]
- Grzona, C.B.; Lick, I.D.; Rodriguez Castellón, E.; Ponzi, M.I.; Ponzi, E.N. Cobalt and KNO3 supported on alumina catalysts for diesel soot combustion. Mater. Chem. Phys. 2010, 123, 557–562. [Google Scholar] [CrossRef]
- Morrow, B.A.; Cody, I.A.; Moran, L.E.; Palepu, R. An infrared study of the adsorption of pyridine on platinum and nickel. J. Catal. 1976, 44, 467–476. [Google Scholar] [CrossRef]
- Tarach, K.A.; Śrębowata, A.; Kowalewski, E.; Gołąbek, K.; Kostuch, A.; Kruczała, K.; Girman, V.; Góra-Marek, K. Nickel loaded zeolites FAU and MFI: Characterization and activity in water-phase hydrodehalogenation of TCE. Appl. Catal. A Gen. 2018, 568, 64–75. [Google Scholar] [CrossRef]
- Verhoef, R.W.; Asscherm, M. The work function of adsorbed alkalis on metals revisited: A coverage-dependent polarizability approach. Surf. Sci. 1997, 391, 11–18. [Google Scholar] [CrossRef]
- Ramírez de la Piscina, P.; Homs, N. Use of biofuels to produce hydrogen (reformation processes). Chem. Soc. Rev. 2008, 37, 2459–2467. [Google Scholar] [CrossRef]
- Espinal, R.; Taboada, E.; Molins, E.; Chimentao, R.J.; Medinac, F.; Llorca, J. Cobalt hydrotalcites as catalysts for bioethanol steam reforming. The promoting effect of potassium on catalyst activity and long-term stability. Appl. Catal. B Environ. 2012, 127, 59–67. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Akiyama, M.; Nagai, M. Steam reforming of ethanol over nickel molybdenum carbides for hydrogen production. Catal. Today 2009, 146, 87–95. [Google Scholar] [CrossRef]
- Subramani, V.; Song, C. Advances in catalysis and processes for hydrogen production from ethanol reforming. Catalysis 2007, 20, 65–106. [Google Scholar]
- Greluk, M.; Gac, W.; Rotko, M.; Słowik, G.; Turczyniak-Surdacka, S. Co/CeO2 and Ni/CeO2 catalysts for ethanol steam reforming: Effect of the cobalt/nickel dispersion on catalysts properties. J. Catal. 2021, 393, 159–178. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Azmi, R.; Trouillet, V.; Strafela, M.; Ulrich, S.; Ehrenberg, H.; Bruns, M. Surface analytical approaches to reliably characterize lithium ion battery electrodes. Surf. Interface Anal. 2018, 50, 43–51. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Lau, L.W.M.; Gerson, A.; Smart, R.S.C. X-ray photoelectron spectroscopic chemical state quantification of mixed nickel metal, oxide and hydroxide systems. Surf. Interface Anal. 2009, 41, 324–332. [Google Scholar] [CrossRef]
- Bengaard, H.S.; Alstrup, I.; Chorkendorff, I.; Ullmann, S.; Rostrup-Nielsen, J.R.; Nørskov, J.K. Chemisorption of methane on Ni(100) and Ni(111) surfaces with preadsorbed potassium. J. Catal. 1999, 187, 238–244. [Google Scholar] [CrossRef]
- Snoeck, J.-W.; Froment, G.F. Steam/CO2 reforming of methane. Carbon formation and gasification on catalysts with various potassium contents. Ind. Eng. Chem. Res. 2002, 41, 3548–3556. [Google Scholar] [CrossRef]
- Juan-Juan, J.; Román-Martínez, M.C.; Illán-Gómez, M.J. Effect of potassium contenet in the activity of K-promotes Ni/Al2O3 catalysts for the dry reforming of methane. Appl. Catal A Gen. 2006, 301, 9–15. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Metal Content (wt.%) | SBET (m2 g−1) | Ni0 Particle Size (nm) * | ||
|---|---|---|---|---|---|
| Ni | K | By XRD | By TEM | ||
| Ni/MnO | 9.7 | - | 16.7 | 19 | 14 |
| KNi/MnO | 9.1 | 1.6 | 10.1 | 23 | 20 |
| MnOx | - | - | 12.0 | - | - |
| Sample | Lewis Acid Sites Concentration (mmol g−1) |
|---|---|
| MnOx | 54 |
| Ni/MnOx | 50 |
| KNi/MnOx | 13 |
| Sample | Ni Particle Size (nm) | |
|---|---|---|
| H2O:EtOH = 12/1 | H2O:EtOH = 4/1 | |
| Ni/MnO | 8 | 18 |
| KNi/MnO | 10 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greluk, M.; Rotko, M.; Słowik, G.; Turczyniak-Surdacka, S.; Grzybek, G.; Góra-Marek, K.; Kotarba, A. Effect of Potassium Promoter on the Performance of Nickel-Based Catalysts Supported on MnOx in Steam Reforming of Ethanol. Catalysts 2022, 12, 600. https://doi.org/10.3390/catal12060600
Greluk M, Rotko M, Słowik G, Turczyniak-Surdacka S, Grzybek G, Góra-Marek K, Kotarba A. Effect of Potassium Promoter on the Performance of Nickel-Based Catalysts Supported on MnOx in Steam Reforming of Ethanol. Catalysts. 2022; 12(6):600. https://doi.org/10.3390/catal12060600
Chicago/Turabian StyleGreluk, Magdalena, Marek Rotko, Grzegorz Słowik, Sylwia Turczyniak-Surdacka, Gabriela Grzybek, Kinga Góra-Marek, and Andrzej Kotarba. 2022. "Effect of Potassium Promoter on the Performance of Nickel-Based Catalysts Supported on MnOx in Steam Reforming of Ethanol" Catalysts 12, no. 6: 600. https://doi.org/10.3390/catal12060600
APA StyleGreluk, M., Rotko, M., Słowik, G., Turczyniak-Surdacka, S., Grzybek, G., Góra-Marek, K., & Kotarba, A. (2022). Effect of Potassium Promoter on the Performance of Nickel-Based Catalysts Supported on MnOx in Steam Reforming of Ethanol. Catalysts, 12(6), 600. https://doi.org/10.3390/catal12060600