
C−H Methylation Using Sustainable Approaches


 ,
, 
 and
and
Abstract
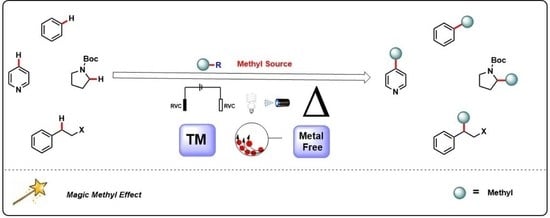
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Photo-Redox-Redox Catalyzed C(sp2)−H Methylation
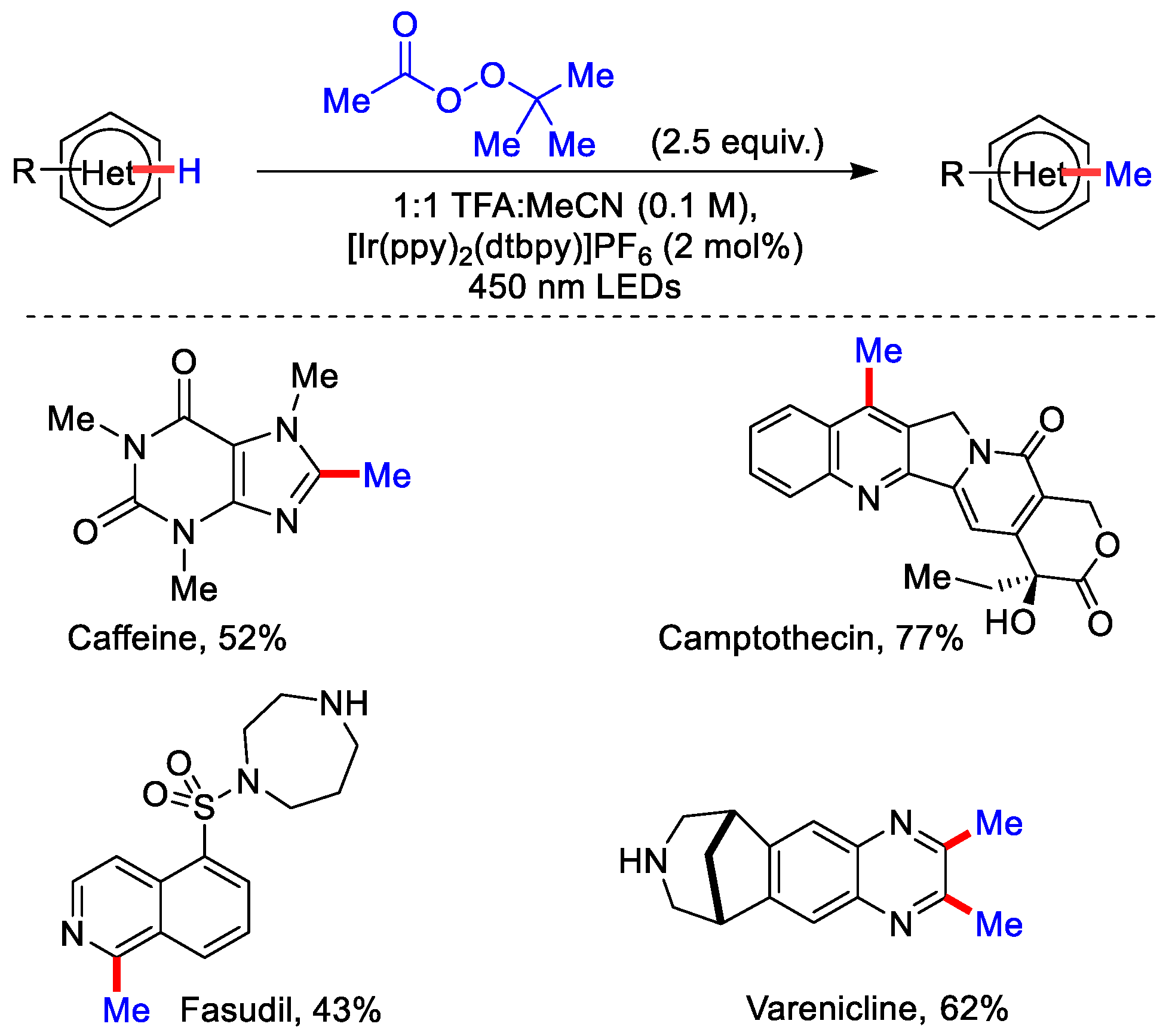
2.1. Ir(III)-Catalysed Methylation Using Peroxides
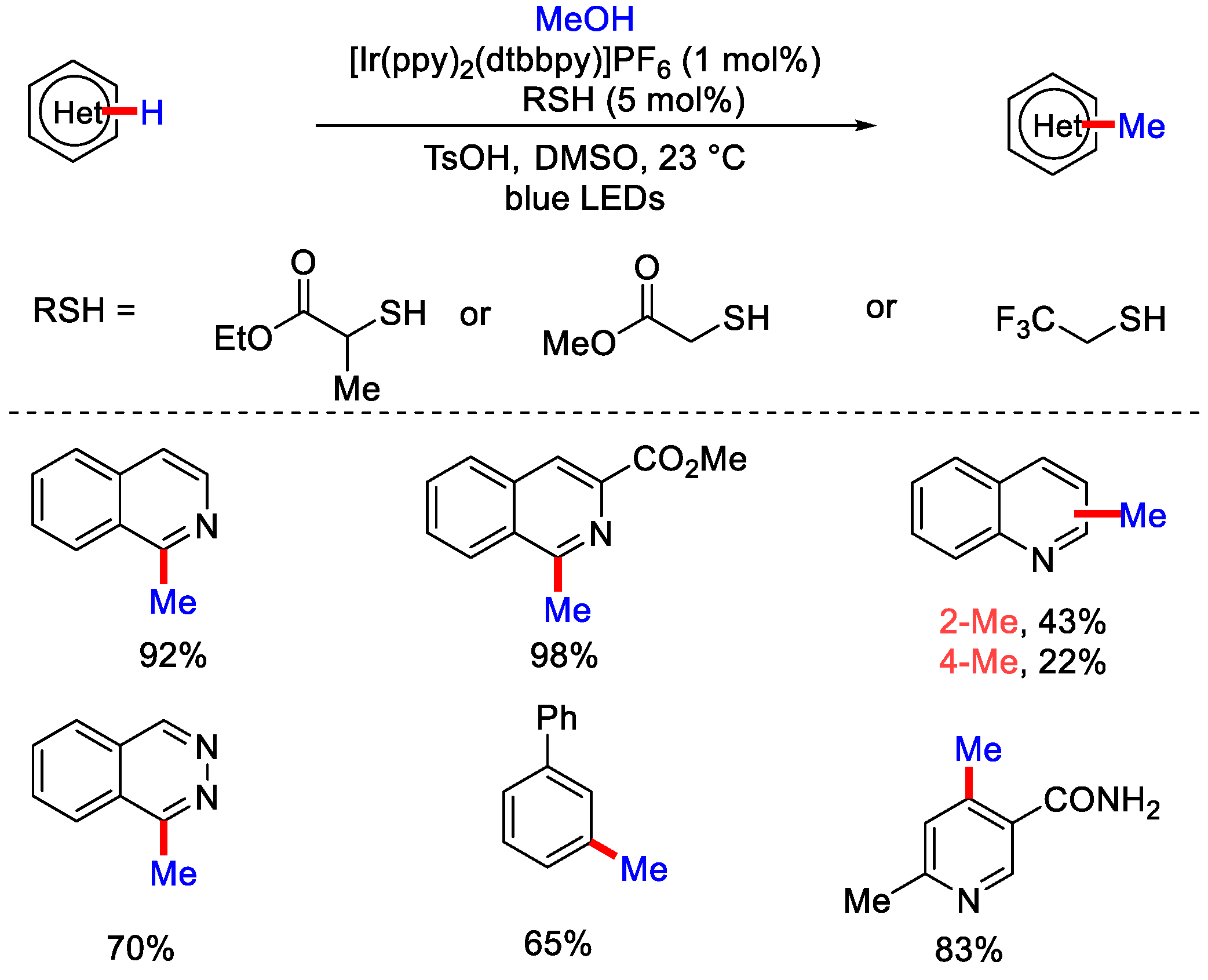
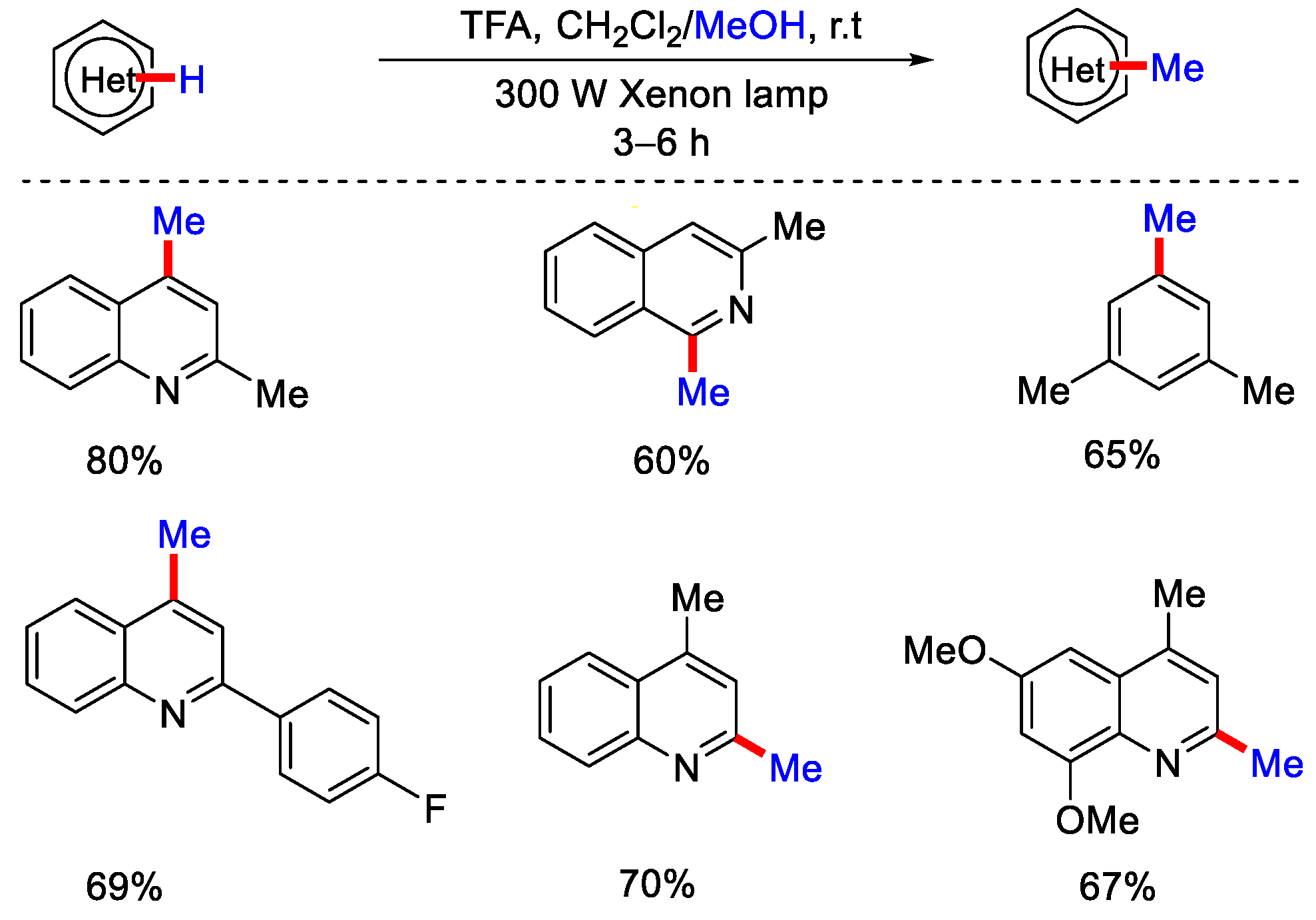
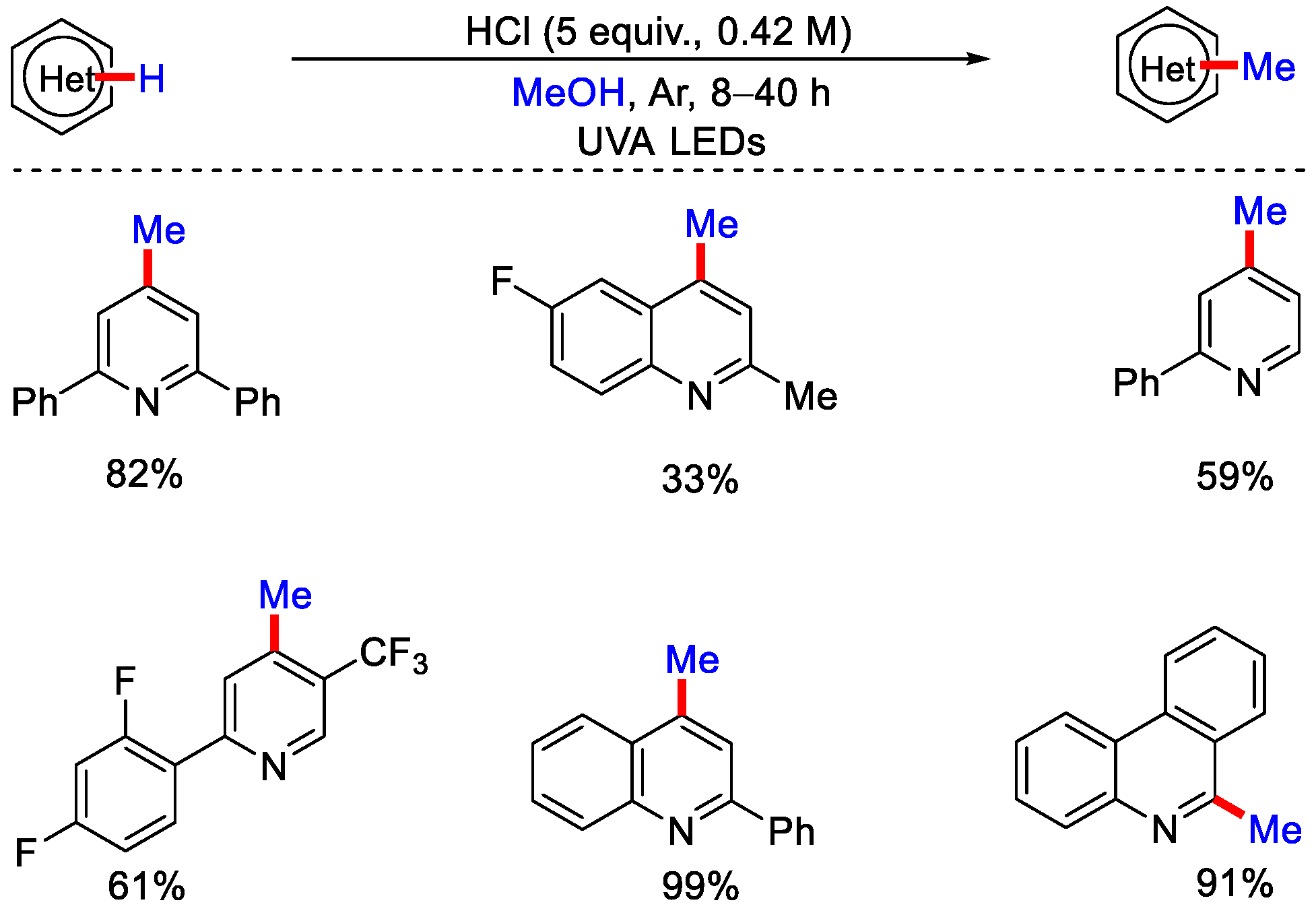
2.2. Methylation Using CH3OH
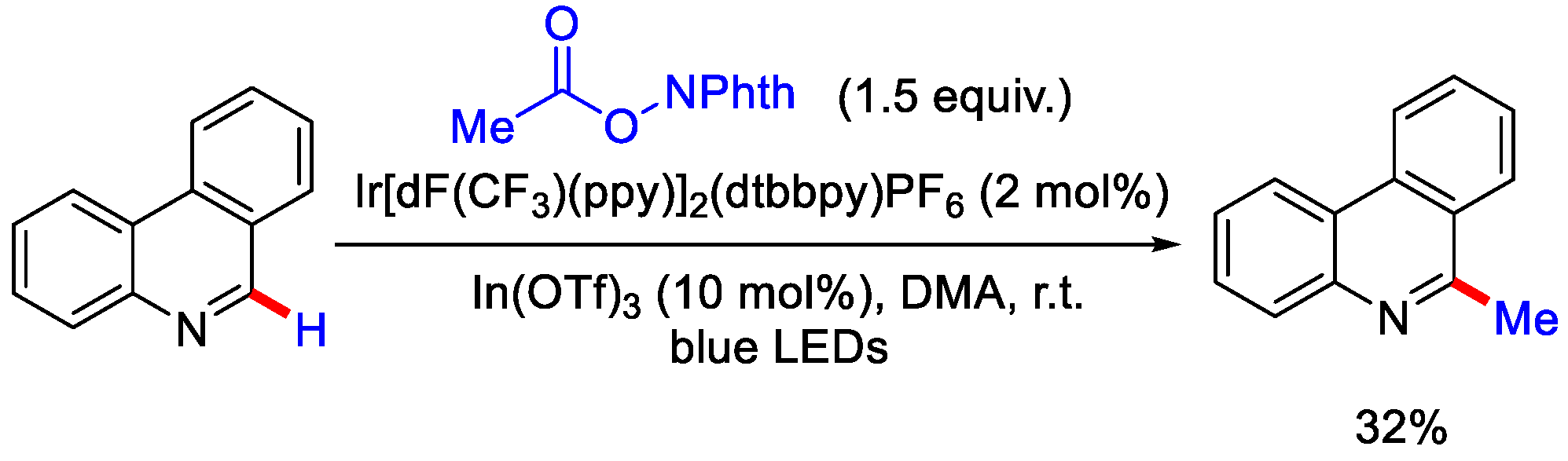
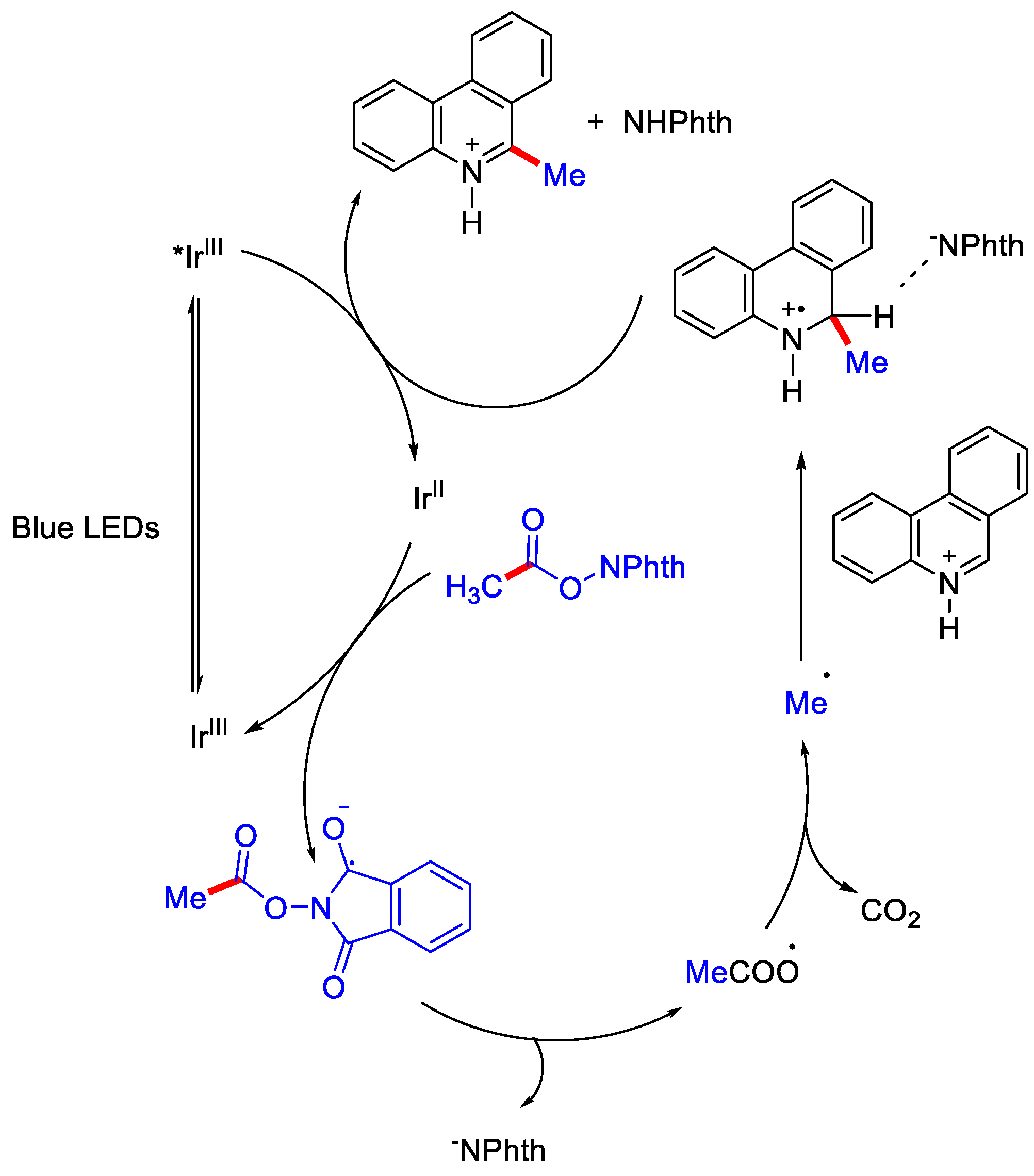
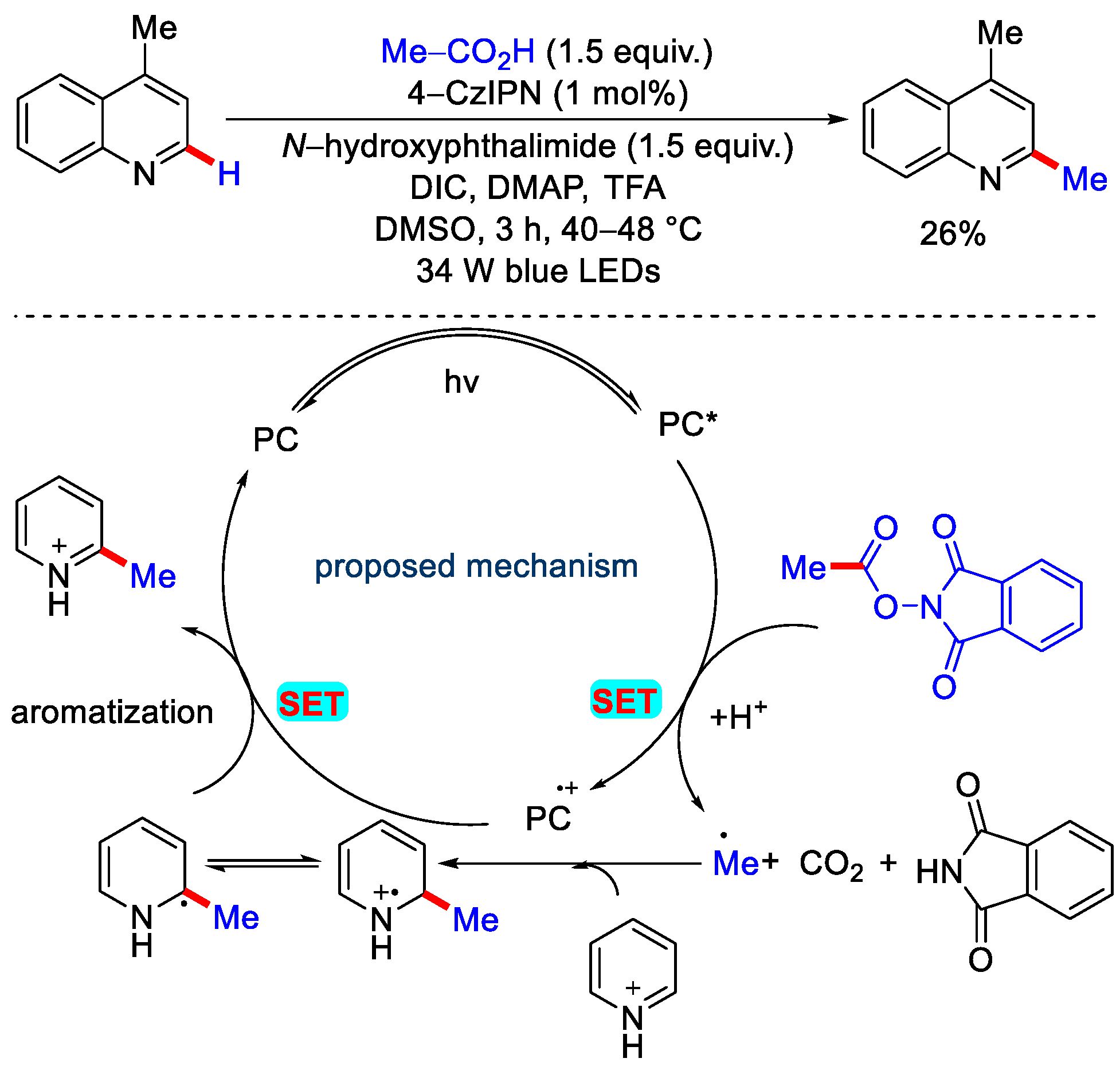
2.3. Methylation Using Acetic Acid and Derivatives
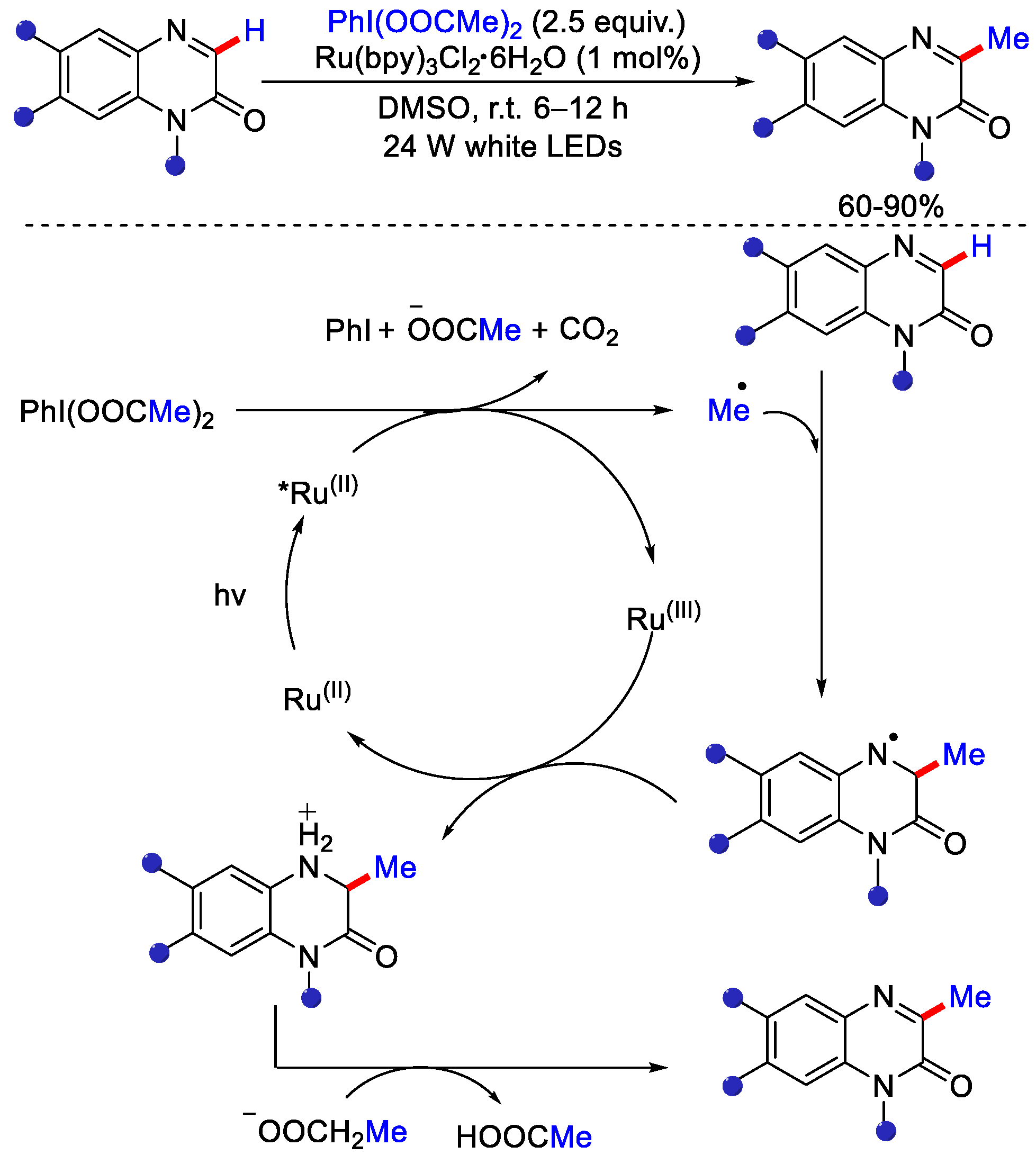
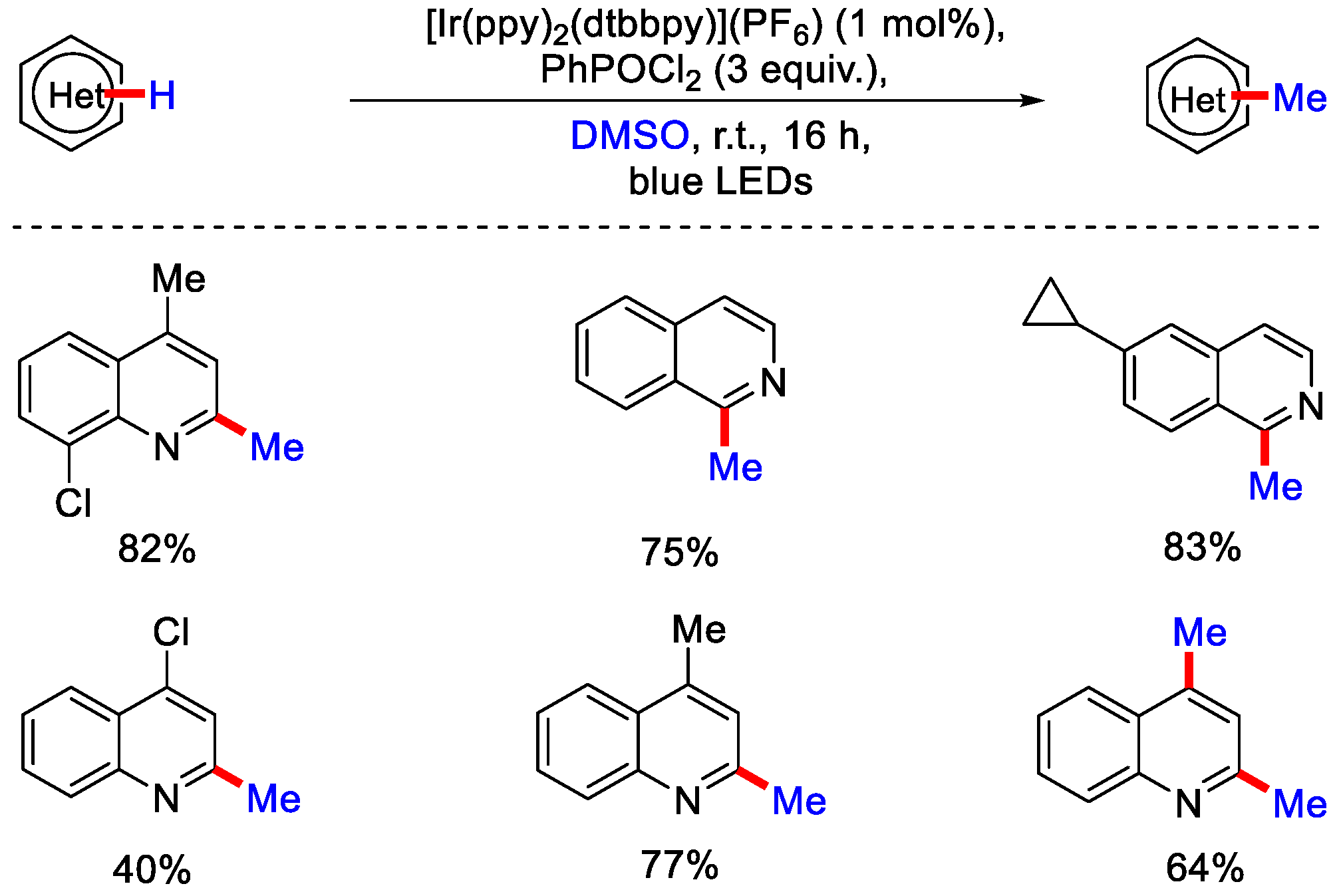
2.4. Methylation Using Dimethylsulfoxide (DMSO)
2.5. Ru-Catalysed Methylation Using Methyl Boronic Acids
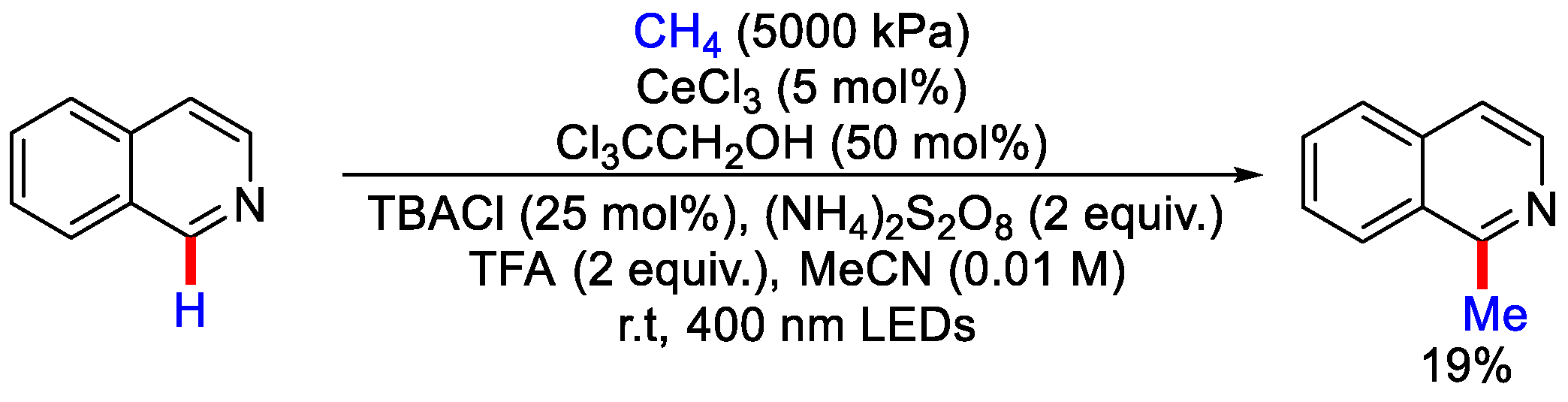
2.6. Methylation Using Methane Gas
3. Photo-Redox Catalysed C(sp3)−H Methylation
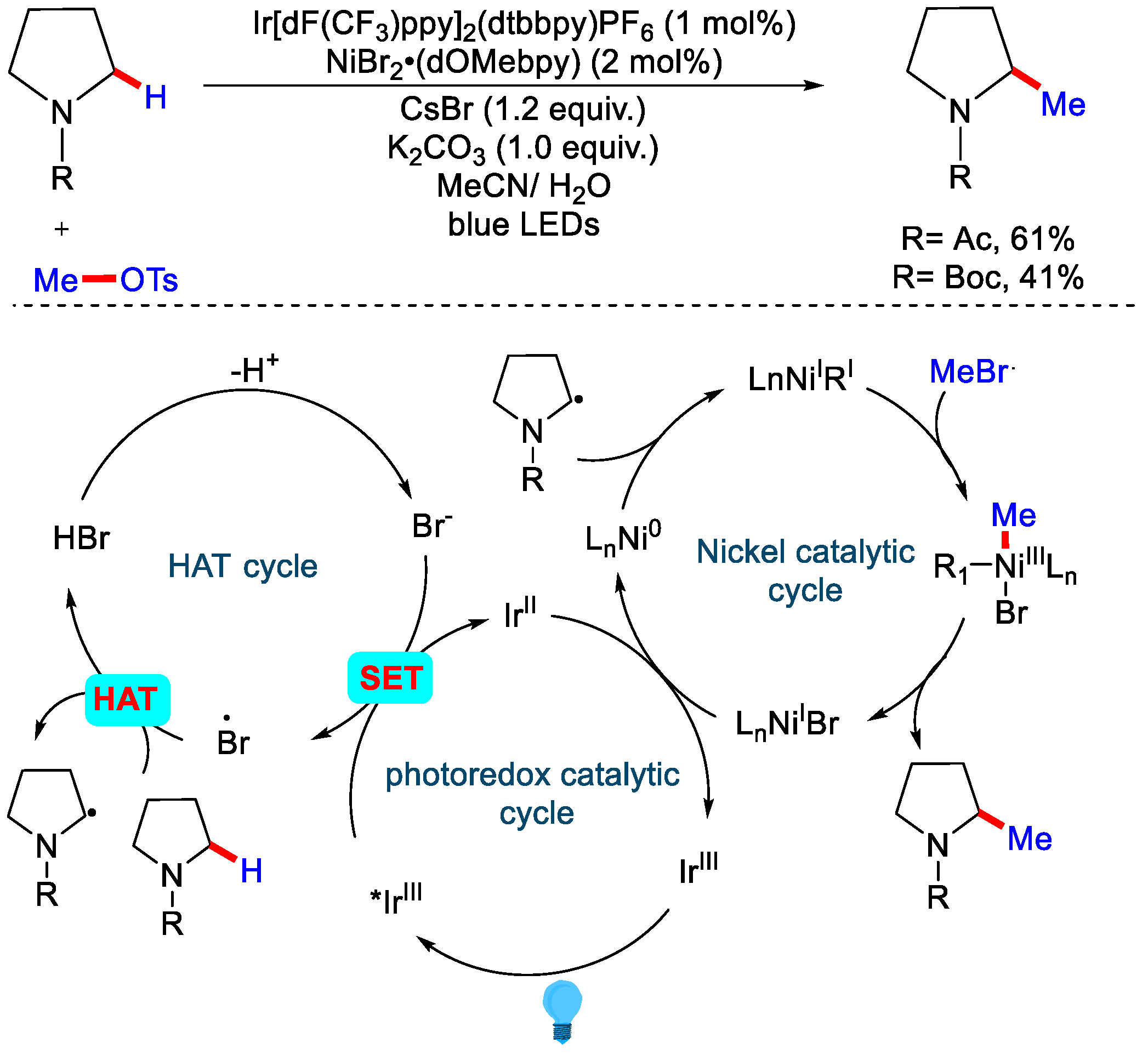
3.1. Oxidative C(sp3)−H Methylation
3.2. Formation of Radical via Peroxides
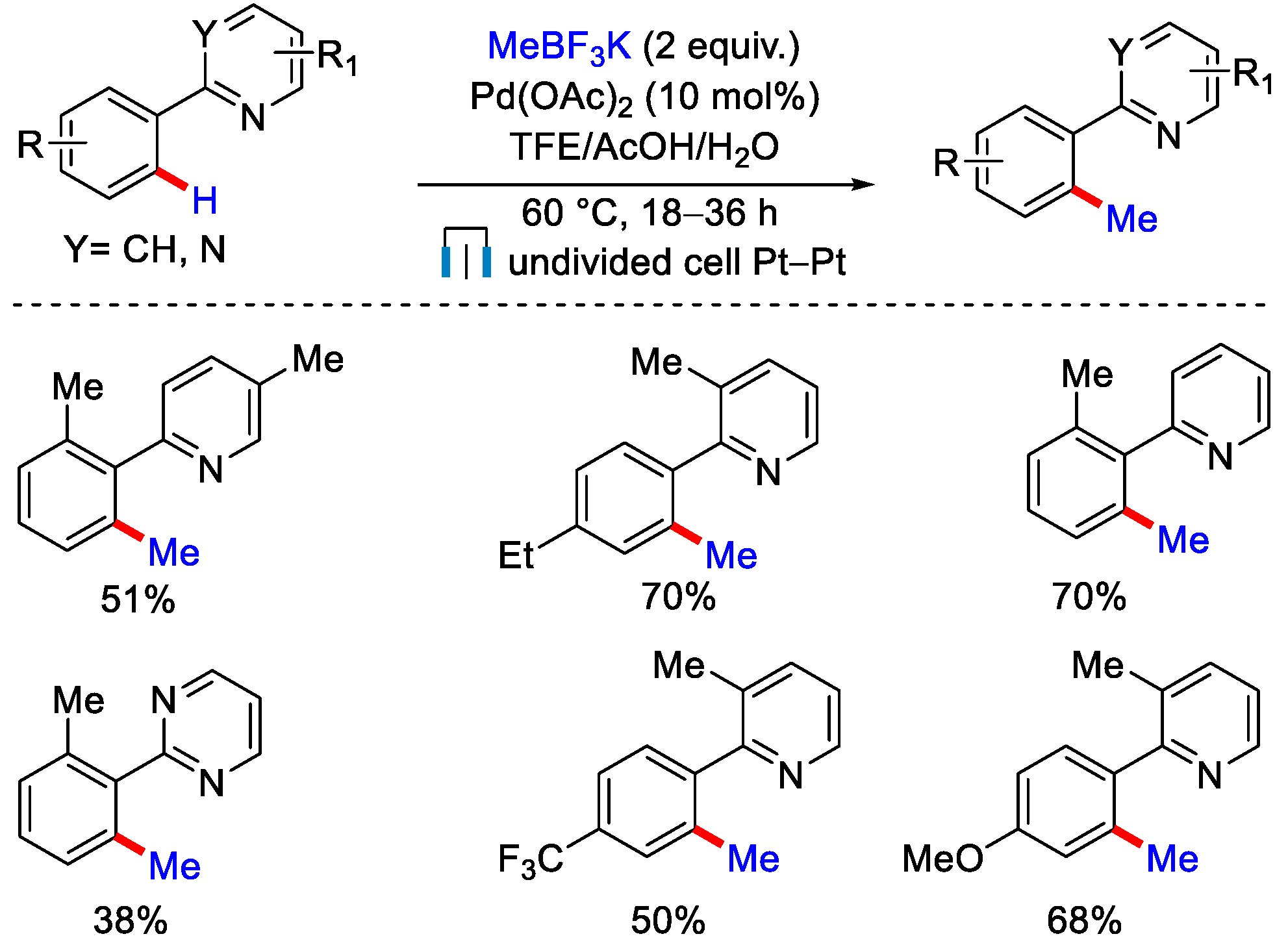
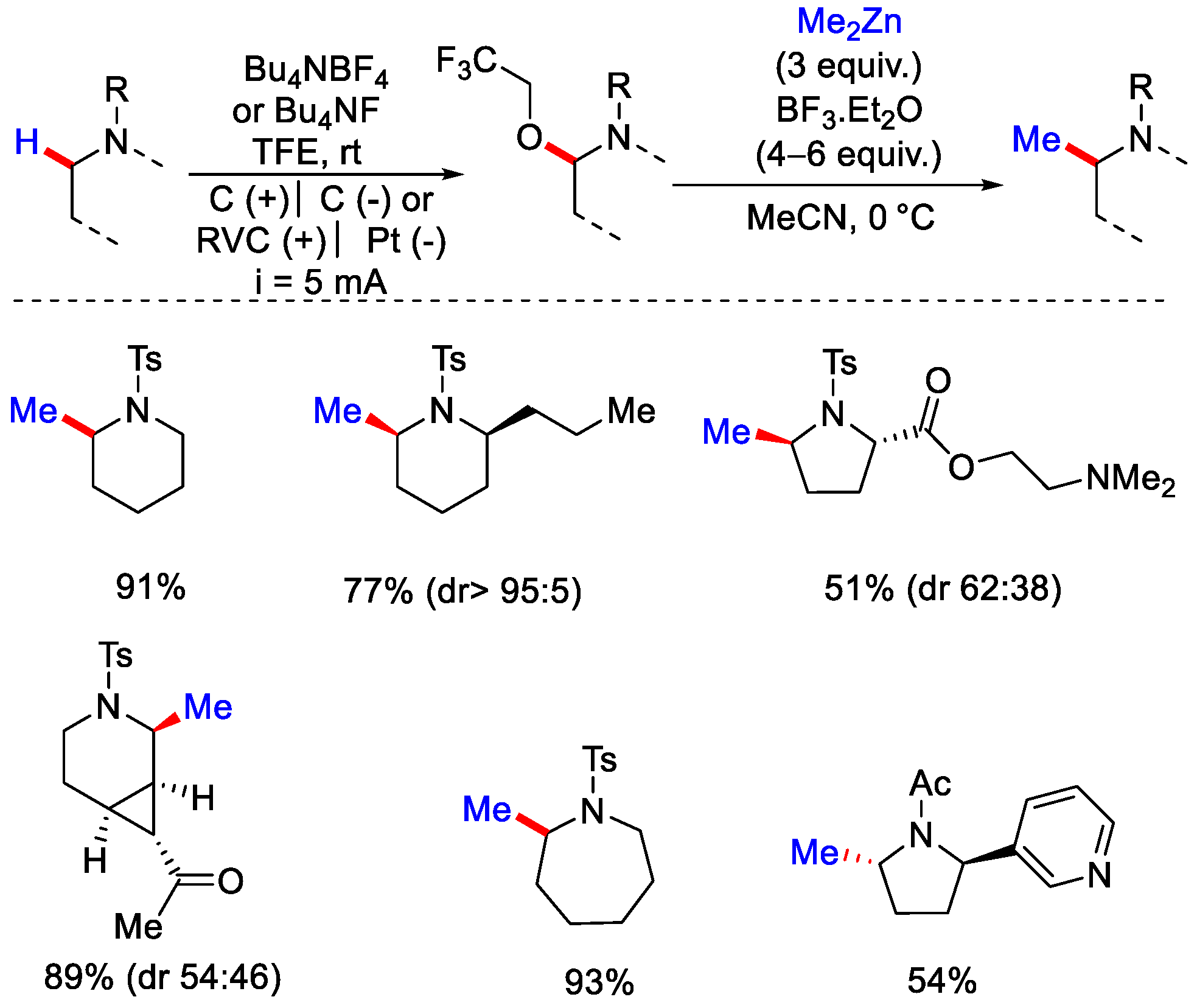
4. C−H Methylation by Electrochemical Oxidation
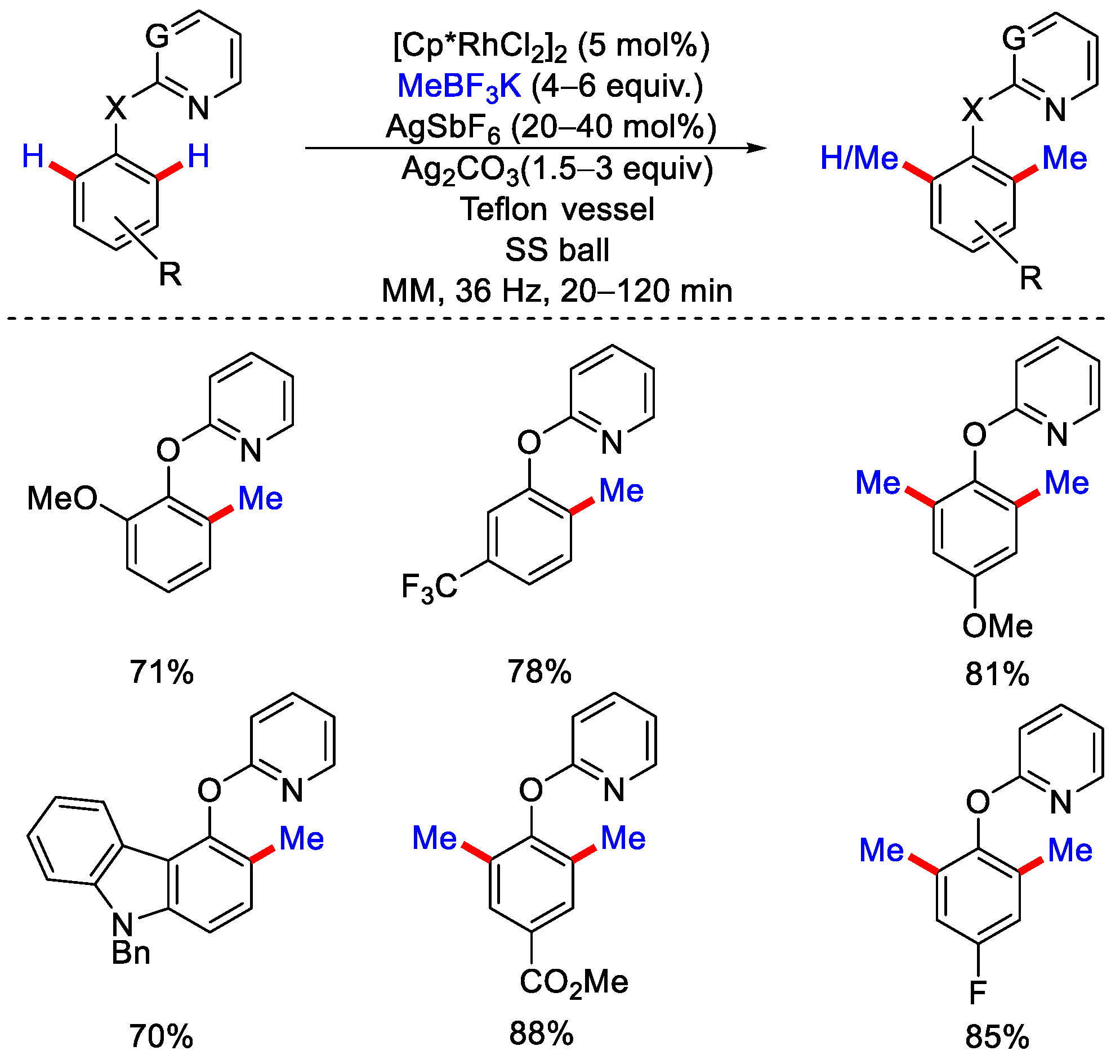
5. Rh-Catalyzed Mechanochemical C−H Methylation
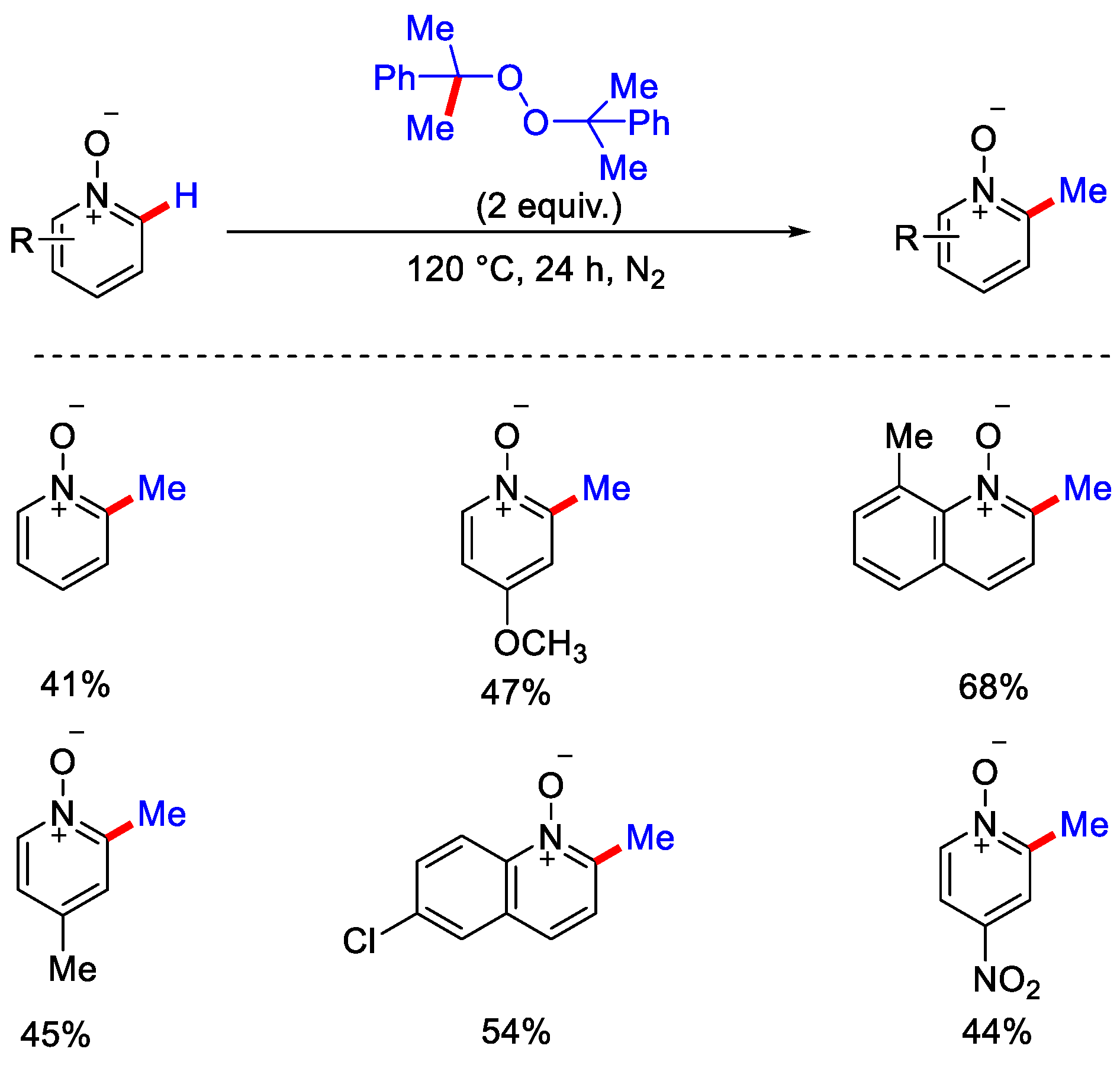
6. Metal-Free Approaches for C−H Methylation
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moir, M.; Danon, J.J.; Reekie, T.A.; Kassiou, M. An overview of late-stage functionalization in today’s drug discovery. Expert Opin. Drug Discov. 2019, 14, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Jana, R.; Begam, H.M.; Dinda, E. The emergence of the C–H functionalization strategy in medicinal chemistry and drug discovery. Chem. Commun. 2021, 57, 10842–10866. [Google Scholar] [CrossRef]
- Dutta, U.; Maiti, S.; Bhattacharya, T.; Maiti, D. Arene Diversification through Distal C(sp2)−H Functionalization. Science 2021, 372, eabd5992. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Bois, J.D.; Yu, J.-Q. C–H Functionalization in Organic Synthesis. Chem. Soc. Rev. 2011, 40, 1855–1856. [Google Scholar] [CrossRef]
- Ali, W.; Prakash, G.; Maiti, D. Recent Development in Transition Metal-Catalysed C–H Olefination. Chem. Sci. 2021, 12, 2735–2759. [Google Scholar] [CrossRef] [PubMed]
- Prakash, G.; Paul, N.; Oliver, G.A.; Werz, D.B.; Maiti, D. C–H Deuteration of Organic Compounds and Potential Drug Candidates. Chem. Soc. Rev. 2022, 51, 3123–3163. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.J.; Kemmerle, A.E.; Fraga, C.A.M. The Methylation Effect in Medicinal Chemistry. Chem. Rev. 2011, 111, 5215–5246. [Google Scholar] [CrossRef] [PubMed]
- Schçnherr, H.; Cernak, T. Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions. Angew. Chem. Int. Ed. 2013, 52, 12256–12267. [Google Scholar] [CrossRef]
- Ritchie, T.J.; Macdonald, S.J.F.; Pickett, S.D. Insights into the impact of N-and O-methylation on aqueous solubility and lipophilicity using matched molecular pair analysis. Med. Chem. Comm. 2015, 6, 1787–1797. [Google Scholar] [CrossRef]
- Kuntz, K.W.; Campbell, J.E.; Keilhack, H.; Pollock, R.M.; Knutson, S.K.; Porter-Scott, M.; Richon, V.M.; Sneeringer, C.J.; Wigle, T.J.; Allain, C.J.; et al. The importance of being me: Magic methyls, methyl transferase inhibitors, and the discovery of tazemetostat. J. Med. Chem. 2016, 59, 1556–1564. [Google Scholar] [CrossRef]
- Vitaku, E.; Ilardi, E.A.; Njarðarson, J.T. Top 200 Pharmaceutical Products by US Retail Sales in 2018; University of Arizona: Tucson, AZ, USA. Available online: https://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/2018Top200PharmaceuticalRetailSalesPosterLowResFinalV2.pdf (accessed on 2 May 2018).
- McGrath, N.A.; Brichacek, M.; Njarðarson, J.T. A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ. 2010, 87, 1348. [Google Scholar] [CrossRef]
- Sinha, S.K.; Guin, S.; Maiti, S.; Biswas, J.P.; Porey, S.; Maiti, D. Toolbox for Distal C–H bond functionalizations in organic molecules. Chem. Rev. 2022, 6, 5682–5841. [Google Scholar] [CrossRef] [PubMed]
- Grover, J.; Prakash, G.; Goswami, N.; Maiti, D. Traditional and Sustainable Approaches for the Construction of C–C Bonds by Harnessing C–H Arylation. Nat. Commun. 2022, 13, 1085. [Google Scholar] [CrossRef]
- Uygur, M.; Mancheno, O.G. Visible light-mediated organophotocatalysed C–H bond functionalization reactions. Org. Biomol. Chem. 2019, 17, 5475–5489. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Marzo, L.; Das, A.; Shaikh, R.; König, B. Visible light-mediated Photo-redox Catalytic Arylation Reactions. Acc. Chem. Res. 2016, 49, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Fang, P.; Mei, T.-S. Recent Advances in C–H Functionalization Using Electrochemical Transition Metal Catalysis. ACS Catal. 2018, 8, 7179–7189. [Google Scholar] [CrossRef]
- Hernández, J.G. Mechanochemical Palladium-Catalysed Carbonylative Reactions Using Mo(CO)6. Chem. Eur. J. 2017, 23, 17157–17165. [Google Scholar] [CrossRef]
- Zhao, S.; Li, Y.; Liu, C.; Zhao, Y. Recent advances in mechanochemical C–H functionalization reactions. Tetrahedron Lett. 2018, 59, 317–324. [Google Scholar] [CrossRef]
- Ranu, B.C.; Ghosh, T.; Jalal, S. Recent developments in CH functionalization via CH bond activation using ball milling and transition-metal catalysts. Arkivoc 2019, 2019, 79–92. [Google Scholar] [CrossRef]
- Shamsabadi, A.; Chudasama, V. Recent advances in metal-free aerobic C–H activation. Org. Biomol. Chem. 2019, 17, 2865–2872. [Google Scholar] [CrossRef] [Green Version]
- Samanta, R.; Matcha, K.; Antonchick, A.P. Metal-Free Oxidative Carbon-Heteroatom Bond Formation Through C–H Bond Functionalization. Eur. J. Org. Chem. 2013, 2013, 5769–5804. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Dixon, J.A.; O’Hara, F.; Funder, E.D.; Dixon, D.D.; Rodriguez, R.A.; Baxter, R.D.; Herle, B.; Sach, N.; Collins, M.R.; et al. C–H Methylation of Heteroarenes Inspired by Radical SAM Methyl Transferase. Nature 2012, 492, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Punta, C.; Minisci, F. Minisci Reaction: A Friedel-Crafts Type Process with Opposite Reactivity and Selectivity. Selective Homolytic Alkylation, Acylation, Carboxylation and Carbamoylation of Heterocyclic Aromatic Bases. Trends Heterocycl. Chem. 2008, 13, 1–68. [Google Scholar] [CrossRef]
- Molander, G.A.; Colombel, V.; Braz, V.A. Direct alkylation of heteroaryls using potassium alkyl-and alkoxymethyltrifluoroborates. Org. Lett. 2011, 13, 1852–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiRocco, D.A.; Dykstra, K.; Krska, S.; Vachal, P.; Conway, D.V.; Tudge, M. Late-Stage Functionalization of Biologically Active Heterocycles Through Photo-redox Catalysis. Angew. Chem. Int. Ed. 2014, 53, 4802–4806. [Google Scholar] [CrossRef]
- Jin, J.; MacMillan, D.W.C. Alcohols as alkylating agents in heteroarene C–H functionalization. Nature 2015, 525, 87–90. [Google Scholar] [CrossRef] [Green Version]
- Huff, C.A.; Cohen, R.D.; Dykstra, K.D.; Fuss, E.S.; DiRocco, D.A.; Krska, S.W.J. Photo-redox-Catalysed Hydroxymethylation of Heteroaromatic Bases. Org. Chem. 2016, 16, 6980–6987. [Google Scholar] [CrossRef]
- Liu, W.; Yang, X.; Zhou, Z.-Z.; Li, C.-J. Simple and Clean Photo-induced Methylation of Heteroarenes with MeOH. Chem 2017, 2, 688–702. [Google Scholar] [CrossRef] [Green Version]
- McCallum, T.; Pitre, S.P.; Morin, M.; Scaiano, J.C.; Barriault, L. The Photochemical Alkylation and Reduction of Heteroarenes. Chem. Sci. 2017, 8, 7412–7418. [Google Scholar] [CrossRef] [Green Version]
- Zidan, M.; Morris, A.O.; McCallum, T.; Barriault, L. The Alkylation and Reduction of Heteroarenes with Alcohols Using Photo-redox Catalysed Hydrogen Atom Transfer via Chlorine Atom Generation. Eur. J. Org. Chem. 2020, 2020, 1453–1458. [Google Scholar] [CrossRef]
- Cheng, W.-M.; Shang, R.; Fu, M.-C.; Fu, Y. Photo-redox-Catalysed Decarboxylative Alkylation of N-Heteroarenes with N-(Acyloxy)phthalimides. Chem. Eur. J. 2017, 23, 2537–2541. [Google Scholar] [CrossRef] [PubMed]
- Genovino, J.; Lian, Y.; Zhang, Y.; Hope, T.O.; Juneau, A.; Gagné, Y.; Ingle, G.; Frenette, M. Metal-Free-Visible Light C−H Alkylation of Heteroaromatics via Hypervalent Iodine-Promoted Decarboxylation. Org. Lett. 2018, 20, 3229–3232. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, T.C.; Li, N.; Yazdani, A.N.; Dhar, T.G.M.J. Organocatalysed, Visible-Light Photo-redox-Mediated, One-Pot Minisci Reaction Using Carboxylic Acids via N-(Acyloxy)phthalimides. Org. Chem. 2018, 83, 3000–3012. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Su, Y.; Wang, K.-H.; Zhang, R.; Feng, Y.; Cao, L.; Huang, D.; Hu, Y. Visible-light induced decarboxylative alkylation of quinoxalin-2(1H)-ones at the C3-position. Org. Biomol. Chem. 2019, 17, 6654–6661. [Google Scholar] [CrossRef]
- Lai, X.; Shu, X.; Song, J.; Xu, H. Electrophotocatalytic Decarboxylative C–H Functionalization of Heteroarenes. Angew. Chem. Int. Ed. 2020, 59, 10626–10632. [Google Scholar] [CrossRef]
- Garza-Sanchez, R.A.; Patra, T.; Tlahuext-Aca, A.; Strieth-Kalthoff, F.; Glorius, F. DMSO as a Switchable Alkylating Agent in Heteroarene C–H Functionalization. Chem. A Eur. J. 2018, 24, 10064–10068. [Google Scholar] [CrossRef]
- Jiang, S.; Yang, Z.; Guo, Z.; Li, Y.; Chen, L.; Zhu, Z.; Chen, X. Transition Metal-Free α−Methylation of 1,8-Naphthyridine Derivatives Using DMSO as Methylation Reagent. Org. Biomol. Chem. 2019, 17, 7416–7424. [Google Scholar] [CrossRef]
- Li, G.-X.; Morales-Rivera, C.A.; Wang, Y.; Gao, F.; He, G.; Liu, P.; Chen, G. Photo-redox-Mediated Minisci C−H Alkylation of NHeteroarenes Using Boronic Acids and Hypervalent Iodine. Chem. Sci. 2016, 7, 6407–6412. [Google Scholar] [CrossRef] [Green Version]
- Hu, A.; Guo, J.-J.; Pan, H.; Zuo, Z. Selective functionalization of methane, ethane, and higher alkanes by cerium photocatalysis. Science 2018, 361, 668–672. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, B.-J.; Shi, Z.-J. Challenge and progress: Palladium-catalyzed sp3 C–H activation. Catal. Sci. Technol. 2011, 1, 191–206. [Google Scholar] [CrossRef]
- Le, C.; Liang, Y.; Evans, R.W.; Li, X.; MacMillan, D.W.C. Selective sp3 C–H alkylation via polarity-matchbased cross-coupling. Nature 2017, 547, 79–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
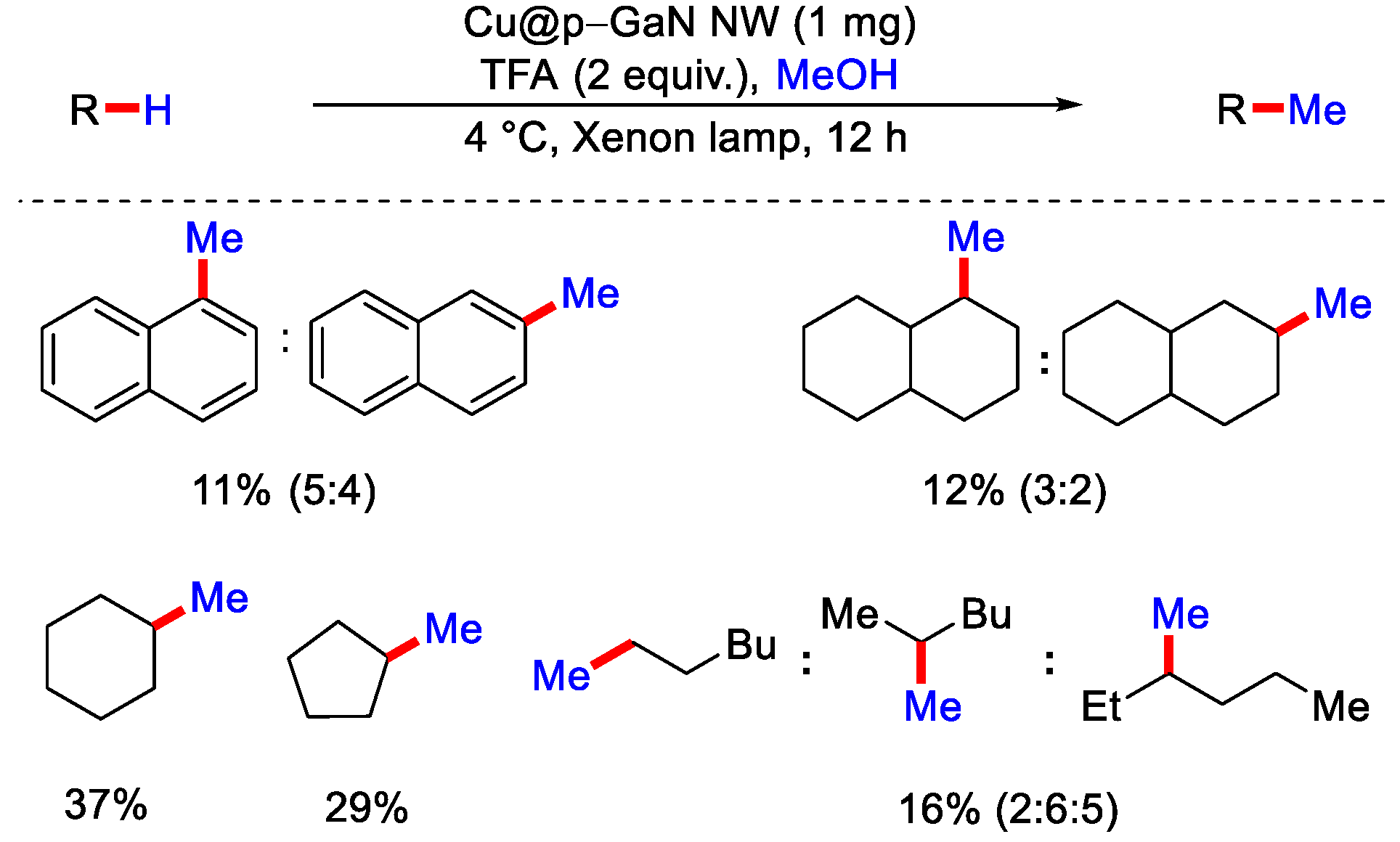
- Liu, M.; Qiu, Z.; Tan, L.; Rashid, R.T.; Chu, S.; Cen, Y.; Luo, Z.; Khaliullin, R.Z.; Mi, Z.; Li, C.-J. Photocatalytic Methylation of Non activated sp3 and sp2 C−H Bonds Using Methanol on GaN. ACS Catal. 2020, 10, 6248–6253. [Google Scholar] [CrossRef]
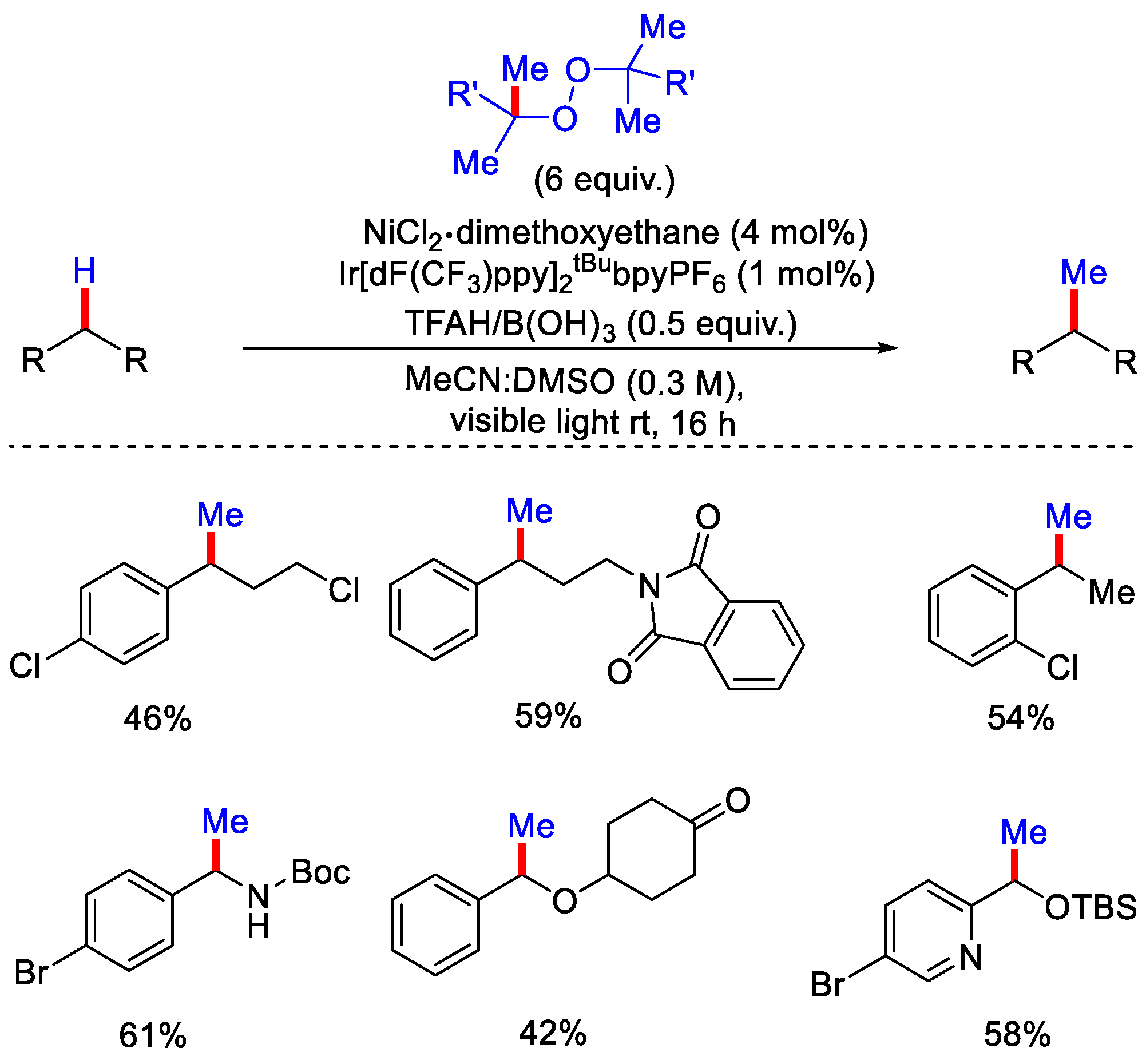
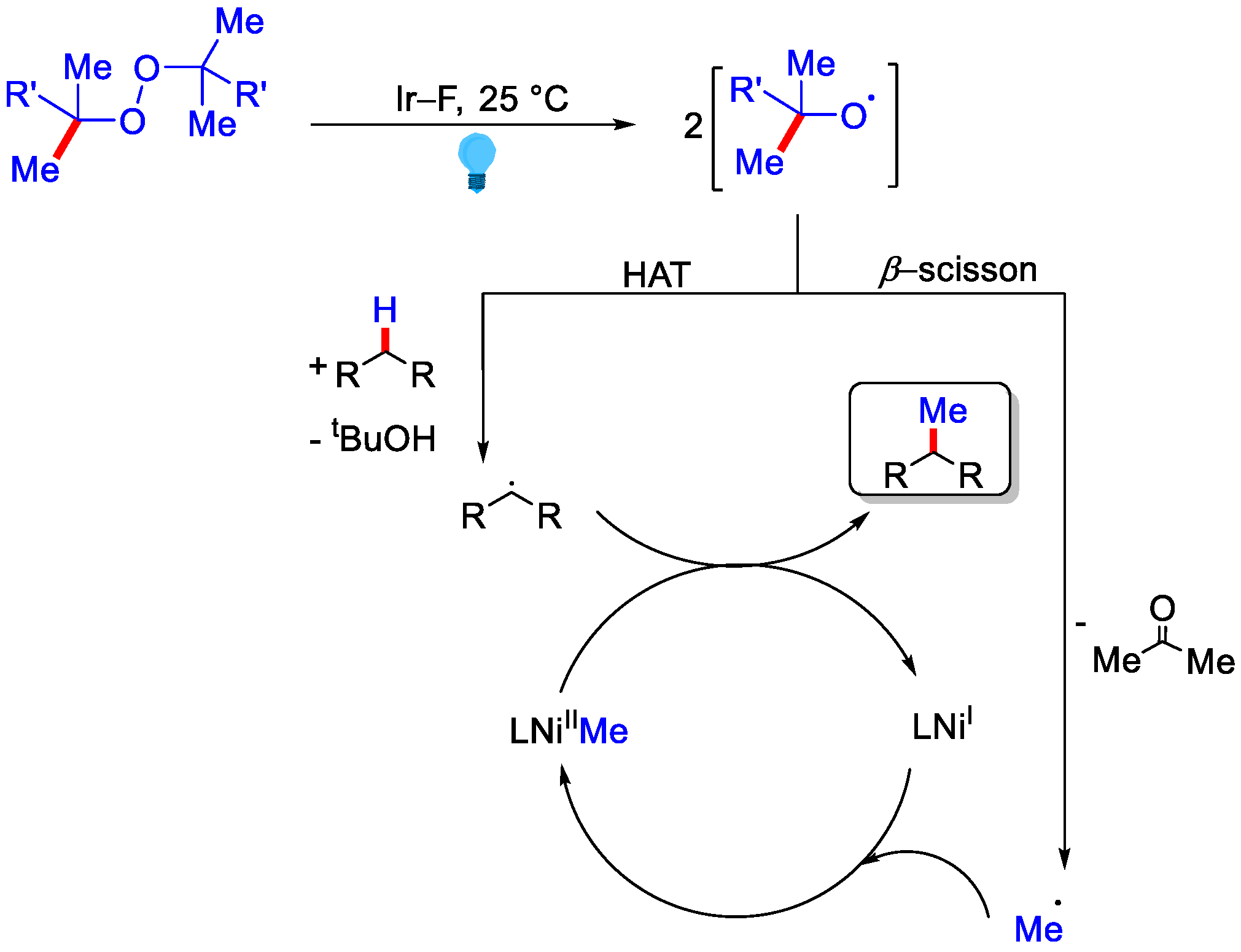
- Vasilopoulos, A.; Krska, S.W.; Stahl, S.S. C(sp3)–H methylation enabled by peroxide photosensitization and Ni-mediated radical coupling. Science 2021, 372, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Waldvogel, S.R.; Lips, S.; Selt, M.; Riehl, B.; Kampf, C.J. Electrochemical arylation reaction. Chem. Rev. 2018, 118, 6706–6765. [Google Scholar] [CrossRef] [PubMed]
- Röckl, J.L.; Pollok, D.; Franke, R.; Waldvogel, S.R. A decade of electrochemical dehydrogenative C, C-coupling of aryls. Acc. Chem. Res. 2020, 53, 45–61. [Google Scholar] [CrossRef] [PubMed]
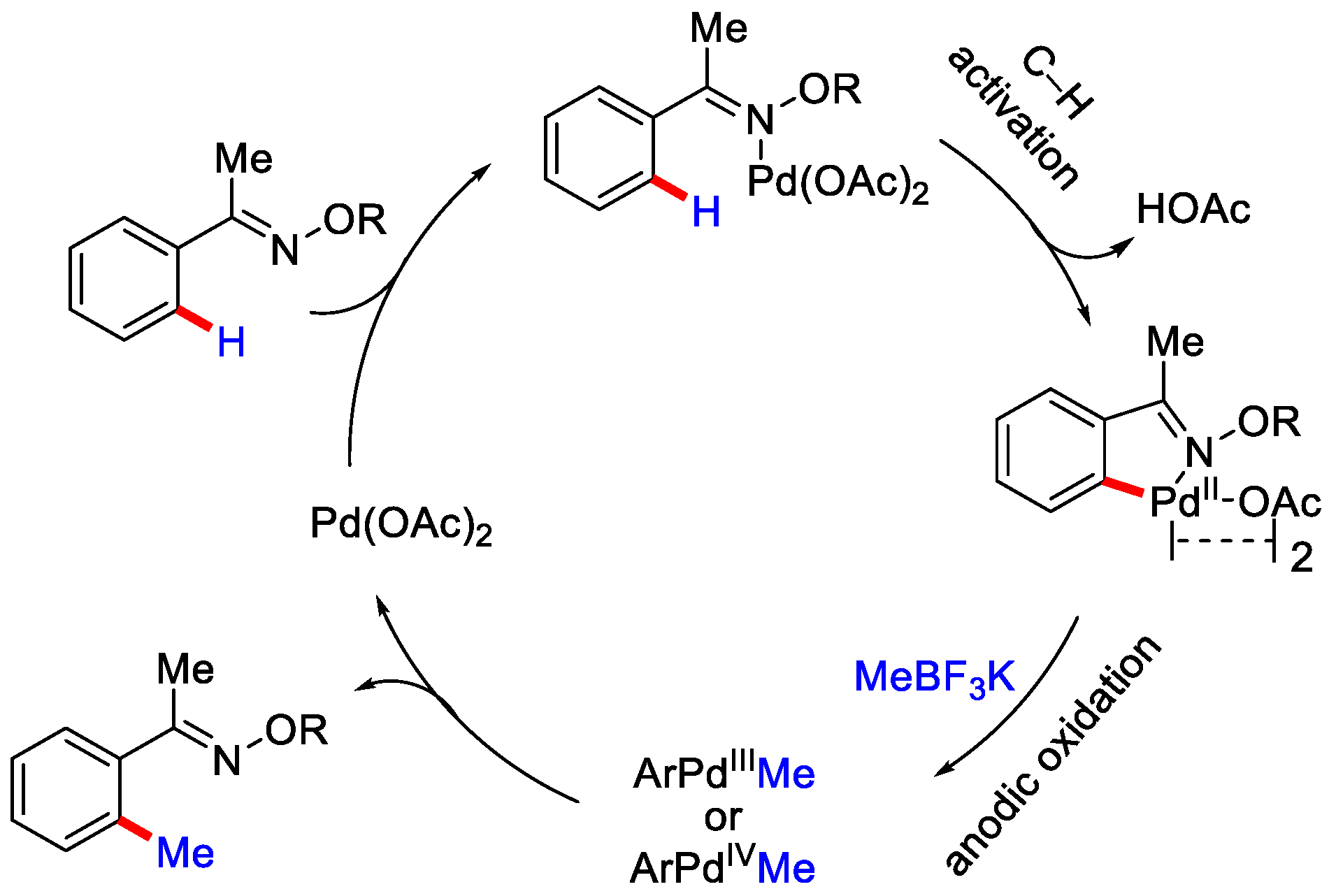
- Ma, C.; Zhao, C.-Q.; Li, Y.-Q.; Zhang, L.-P.; Xu, X.-T.; Zhang, K.; Mei, T.-S. Palladium-catalysed C–H activation/C–C cross-coupling reactions via electrochemistry. Chem. Commun. 2017, 53, 12189–12192. [Google Scholar] [CrossRef]
- Yang, Q.-L.; Li, C.-Z.; Zhang, L.-W.; Li, Y.-Y.; Tong, X.; Wu, X.-Y.; Mei, T.-S. Palladium-Catalysed Electrochemical C−H Alkylation of Arenes. Organometallics 2019, 38, 1208–1212. [Google Scholar] [CrossRef]
- Novaes, L.F.T.; Ho, J.S.K.; Mao, K.; Liu, K.; Tanwar, M.; Neurock, M.; Villemure, E.; Terrett, J.A.; Lin, S. Exploring Electrochemical C(sp3)−H Oxidation for the Late-Stage Methylation of Complex Molecules. J. Am. Chem. Soc. 2022, 3, 1187–1197. [Google Scholar] [CrossRef]
- Do, J.-L.; Friščić, T. Mechanochemistry: A force of synthesis. ACS Cent. Sci. 2017, 3, 13–19. [Google Scholar] [CrossRef] [Green Version]
- James, S.L.; Friscic, T. Mechanochemistry. Chem. Soc. Rev. 2013, 42, 7494–7496. [Google Scholar] [CrossRef]
- Ni, S.; Hribersek, M.; Baddigam, S.K.; Ingner, F.J.L.; Orthaber, A.; Gates, P.J.; Pilarski, L.T. Mechanochemical Solvent-Free Catalytic C—H Methylation. Angew. Chem. Int. Ed. 2021, 60, 6660–6666. [Google Scholar] [CrossRef] [PubMed]
- Seregin, I.V.; Gevorgyan, V. Direct Transition Metal-Catalyzed Functionalization of Heteroaromatic Compounds. Chem. Soc. Rev. 2007, 36, 1173. [Google Scholar] [CrossRef] [PubMed]
- Urbina, K.; Tresp, D.; Sipps, K.; Szostak, M. Recent Advances in Metal-Catalyzed Functionalization of Indoles. Adv. Synth. Catal. 2021, 363, 2723–2739. [Google Scholar] [CrossRef]
- Dalton, T.; Faber, T.; Glorius, F. C–H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 7, 245–261. [Google Scholar] [CrossRef]
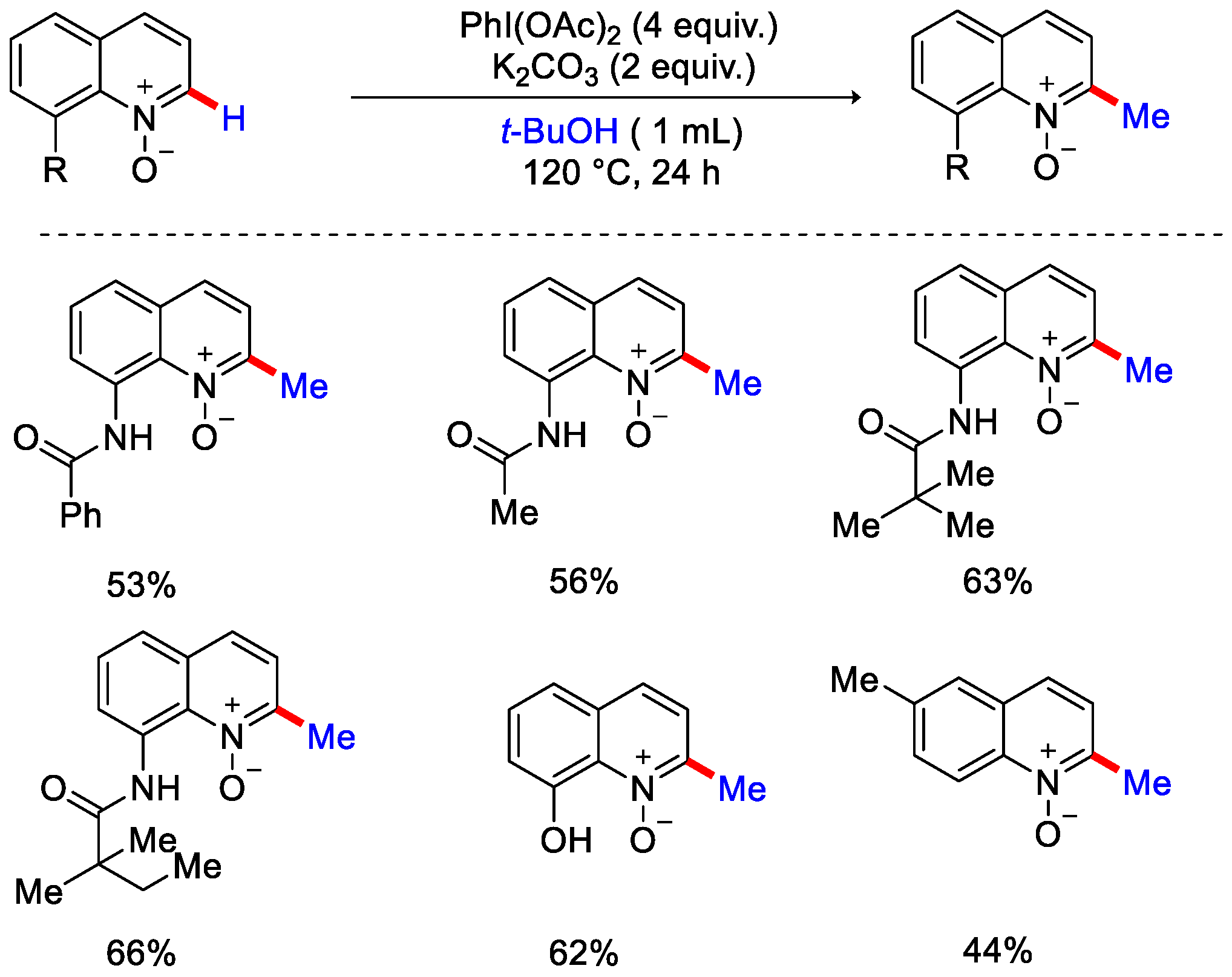
- Li, G.; Yang, S.; Lv, B.; Han, Q.; Ma, X.; Sun, K.; Wang, Z.; Zhao, F.; Lv, Y.; Wu, H. Metal-free Methylation of Pyridine N-Oxides C–H Bond by Using Peroxides. Org. Biomol. Chem. 2015, 13, 11184–11188. [Google Scholar] [CrossRef]
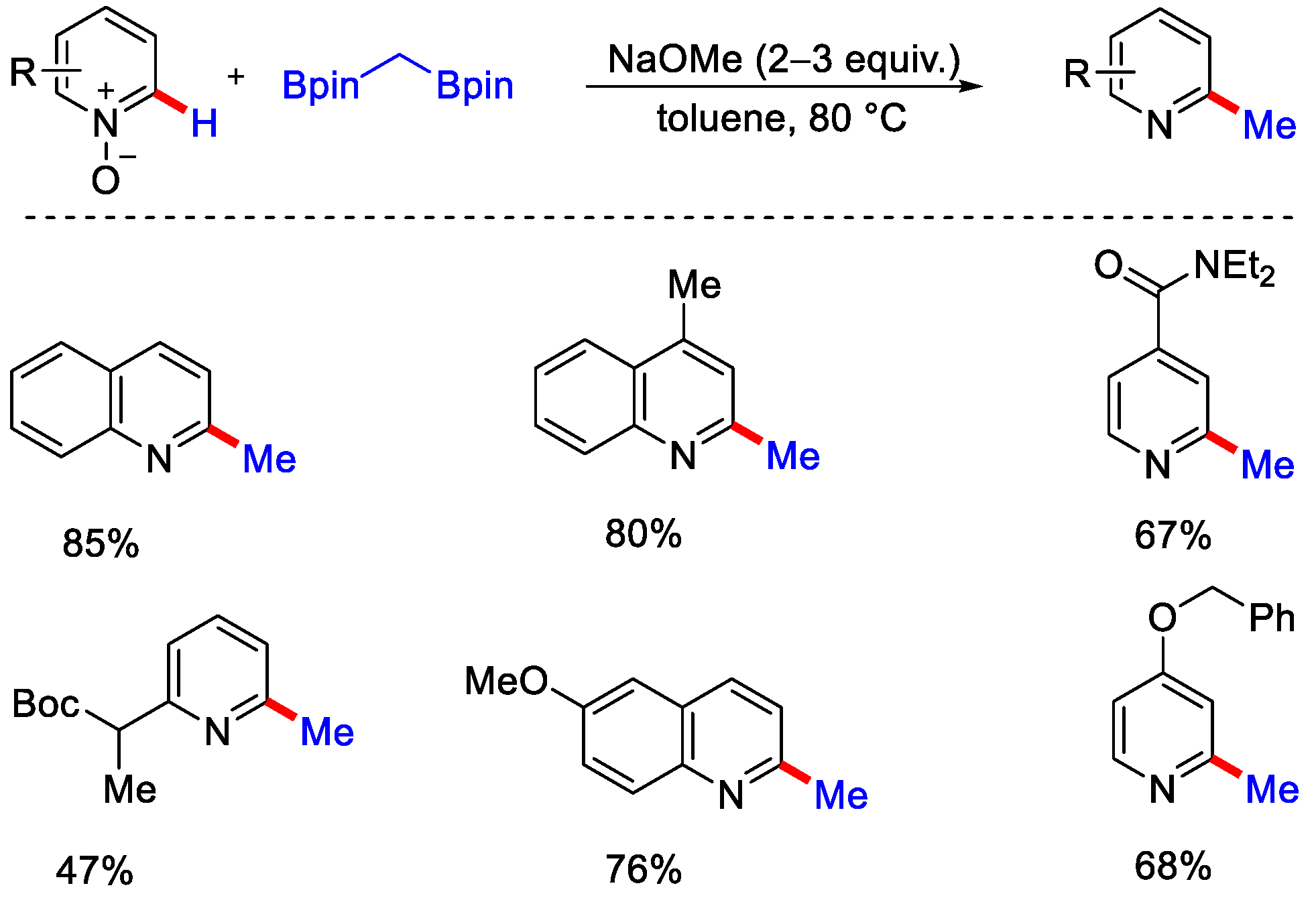
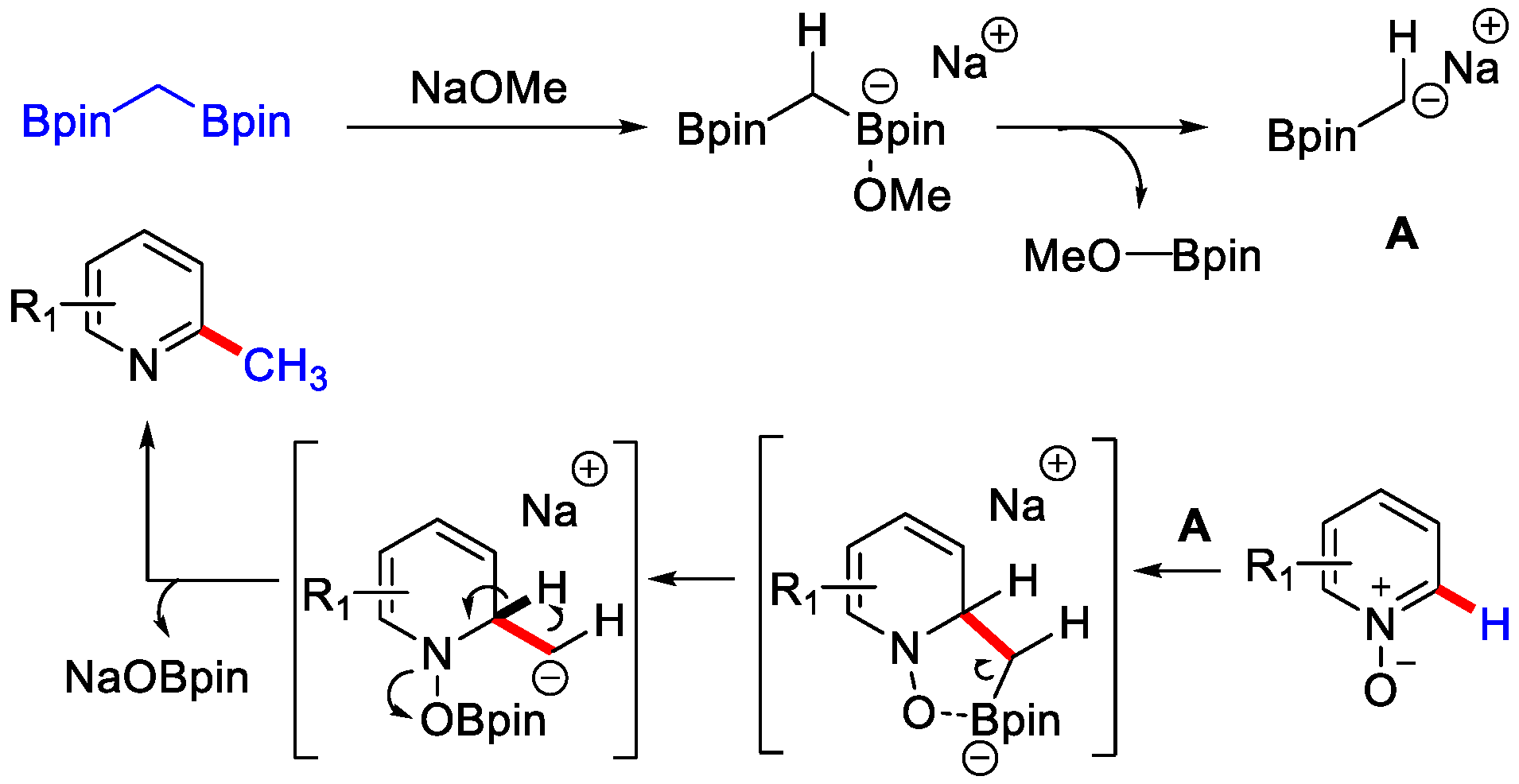
- Jo, W.; Kim, J.; Choi, S.; Cho, S.H. Transition-Metal-Free Regioselective Alkylation of Pyridine N-Oxides Using 1,1-Diborylalkanes as Alkylating Reagents. Angew. Chem. Int. Ed. 2016, 55, 9690–9694. [Google Scholar] [CrossRef]
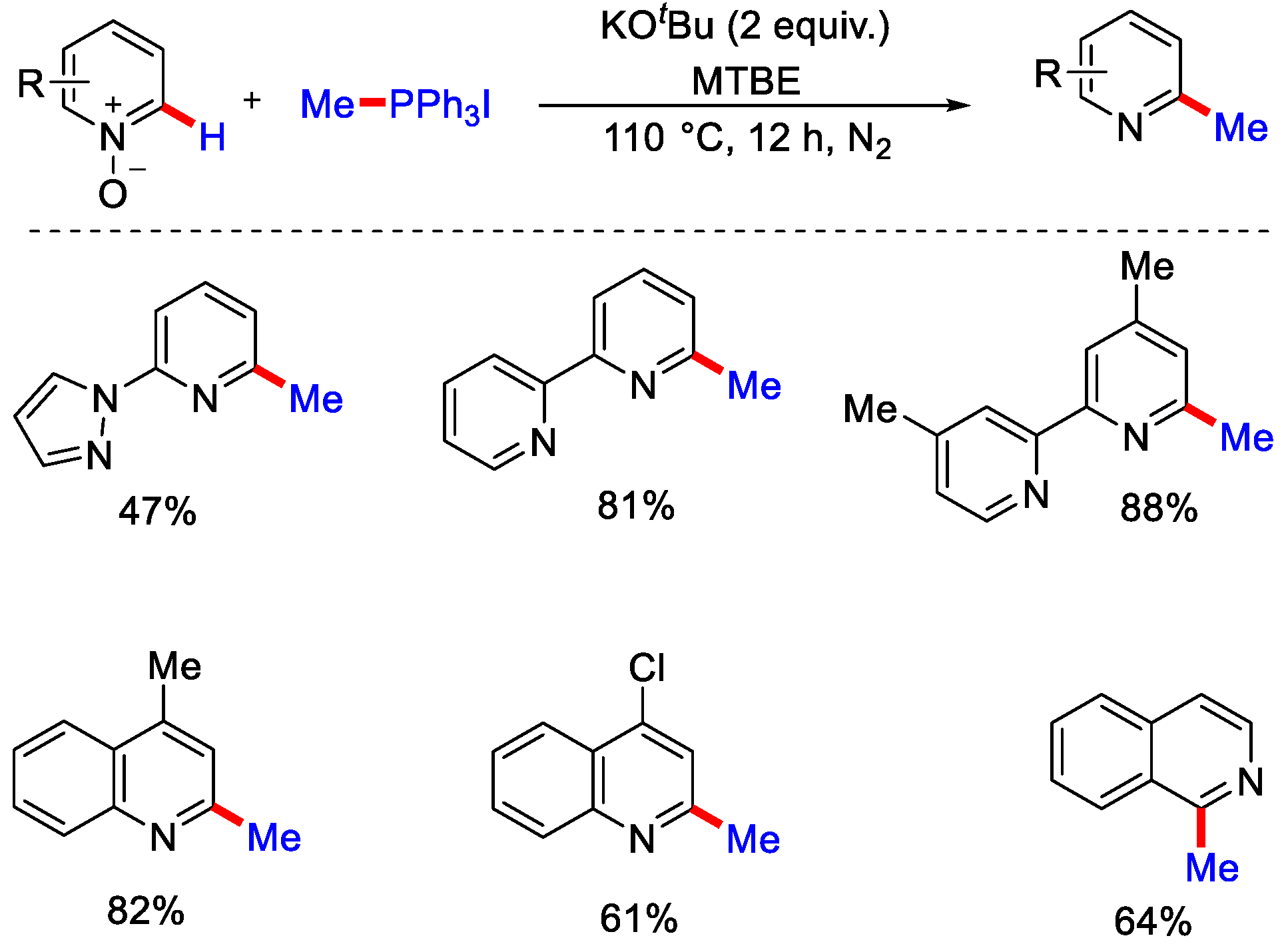
- Han, S.; Chakrasali, P.; Park, J.; Oh, H.; Kim, S.; Kim, K.; Pandey, A.K.; Han, S.H.; Han, S.B.; Kim, I.S. Reductive C2-Alkylation of Pyridine and Quinoline N-Oxides Using Wittig Reagents. Angew. Chem. Int. Ed. 2018, 57, 12737–12740. [Google Scholar] [CrossRef]
- Ghosh, P.; Kwon, N.Y.; Han, S.; Kim, S.; Han, S.H.; Mishra, N.K.; Jung, Y.H.; Chung, S.J.; Kim, I.S. Site-Selective C−H Alkylation of Diazine N-Oxides Enabled by Phosphonium Ylides. Org. Lett. 2019, 21, 6488–6493. [Google Scholar] [CrossRef]
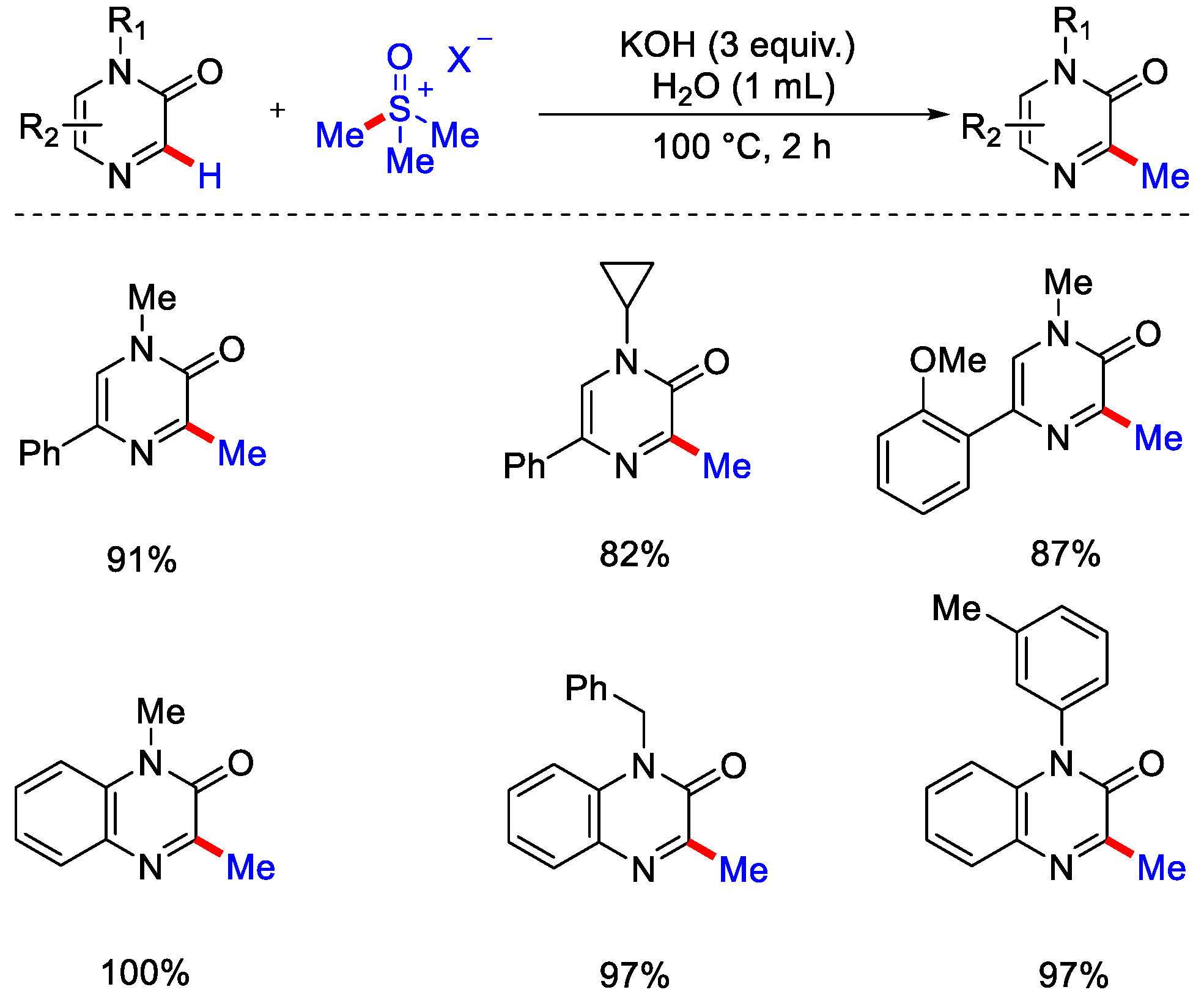
- An, W.; Choi, S.B.; Kim, N.; Kwon, N.Y.; Ghosh, P.; Han, S.H.; Mishra, N.K.; Han, S.; Hong, S.; Kim, I.S. C2-Selective C−H Methylation of Heterocyclic N-Oxides with Sulfonium Ylides. Org. Lett. 2020, 22, 9004–9009. [Google Scholar] [CrossRef]
- Ghosh, P.; Kwon, N.Y.; Kim, S.; Han, S.; Lee, S.H.; An, W.; Mishra, N.K.; Han, S.B.; Kim, I.S. C–H Methylation of Iminoamido Heterocycles with Sulfur Ylides. Angew. Chem. Int. Ed. 2021, 60, 191–196. [Google Scholar] [CrossRef]
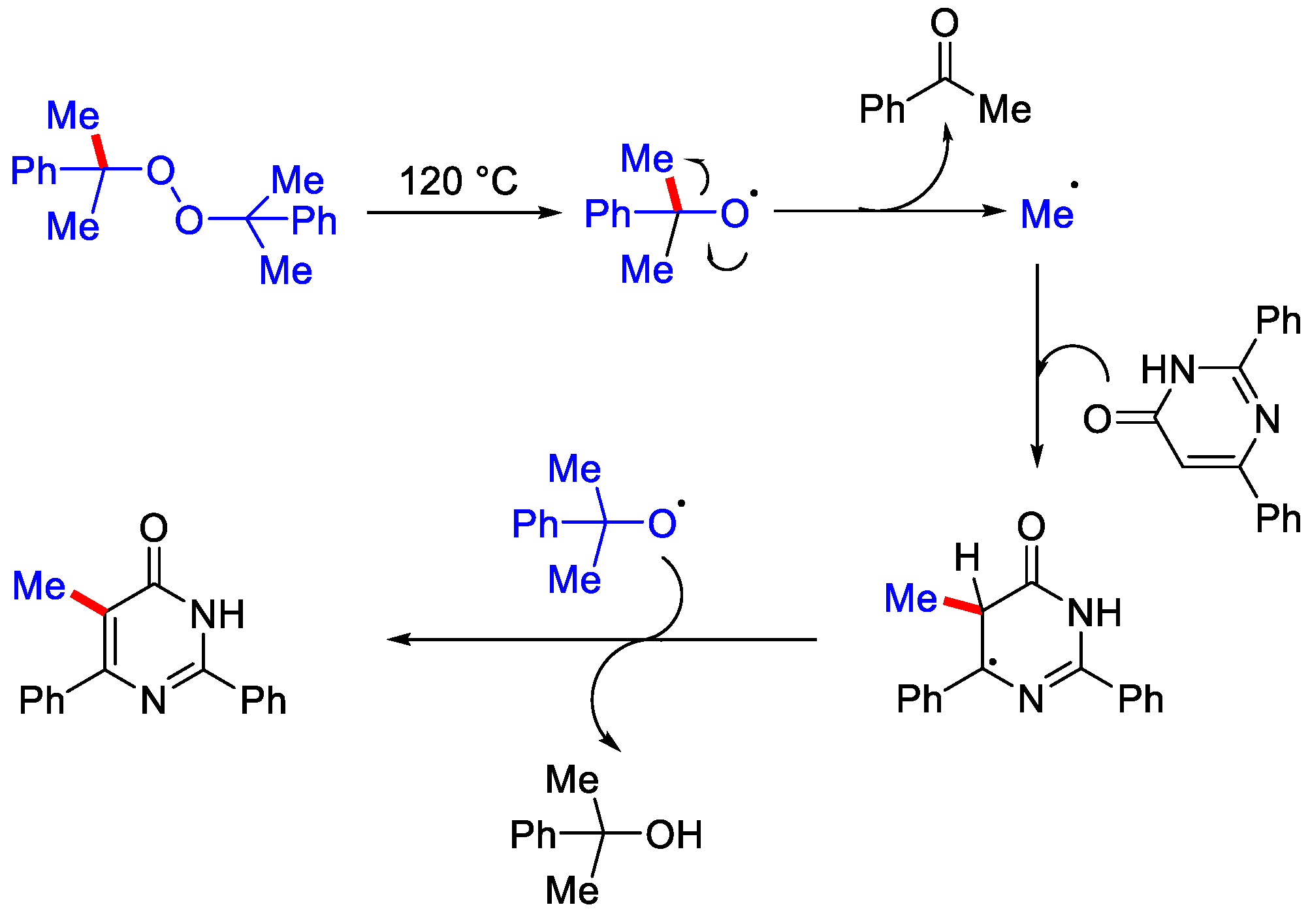
- Zhang, P.-Z.; Li, J.-A.; Zhang, L.; Shoberu, A.; Zou, J.-P.; Zhang, W. Metal-free radical C–H methylation of pyrimidinones and pyridinones with dicumyl peroxide. Green Chem. 2017, 19, 919–923. [Google Scholar] [CrossRef]
- Sen, C.; Ghosh, S.C. Transition-Metal-Free Regioselective Alkylation of Quinoline N-Oxides via Oxidative Alkyl Migration and C-C Bond Cleavage of tert-/sec-Alcohols. Adv. Synth. Catal. 2018, 360, 905–910. [Google Scholar] [CrossRef]
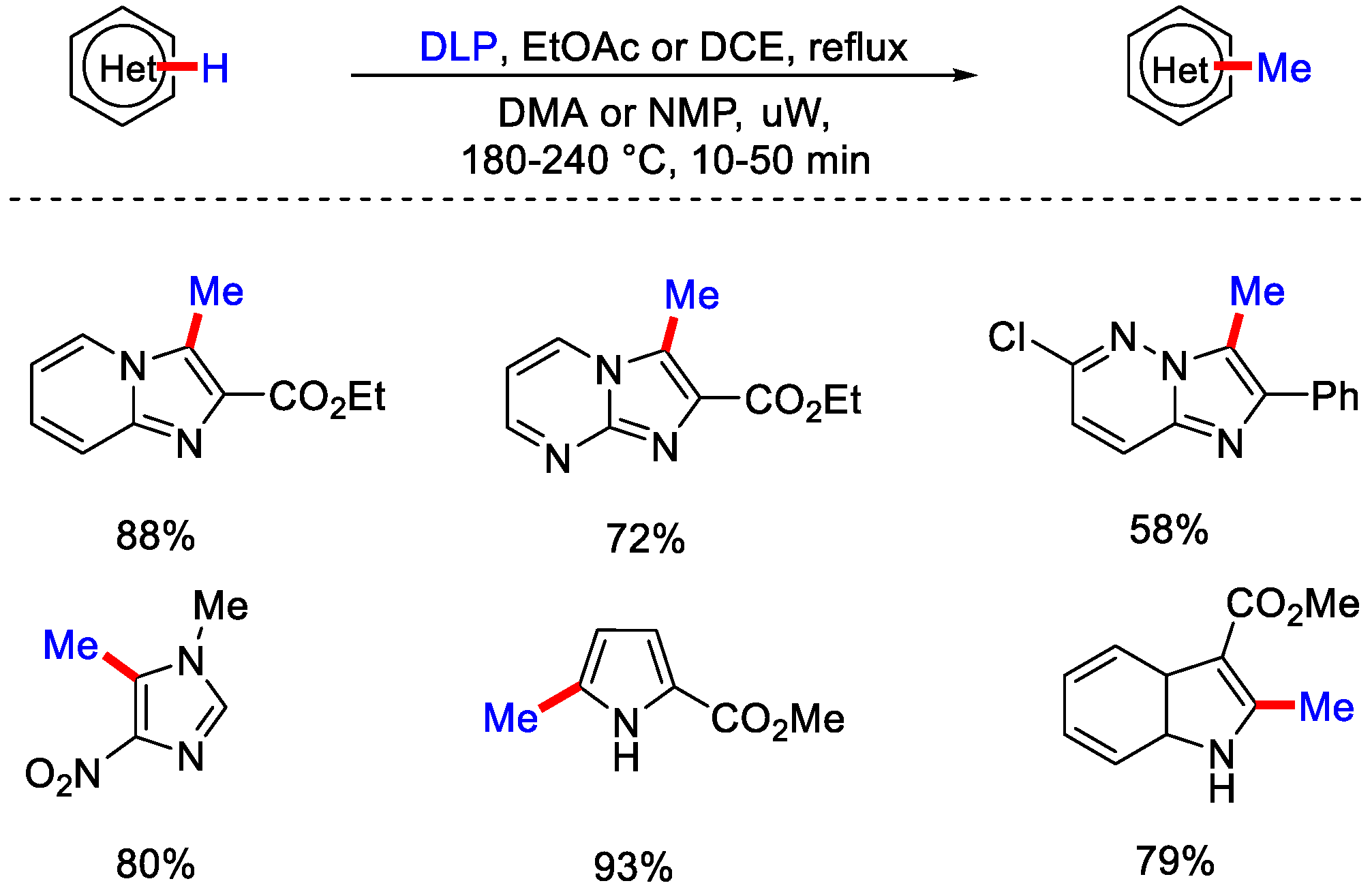
- Huang, Q.; Zard, S.Z. Inexpensive Radical Methylation and Related Alkylations of Heteroarenes. Org. Lett. 2018, 20, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
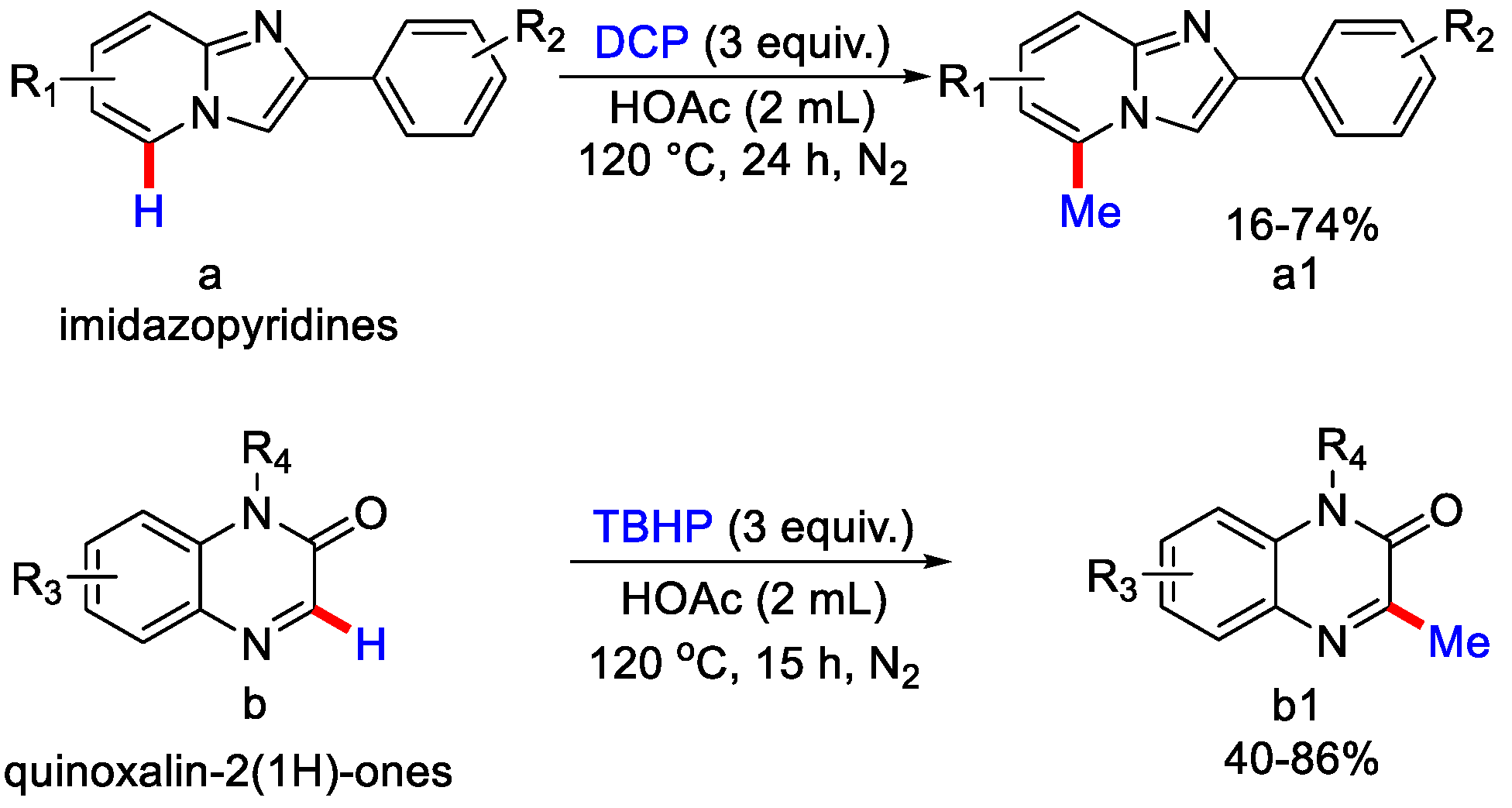
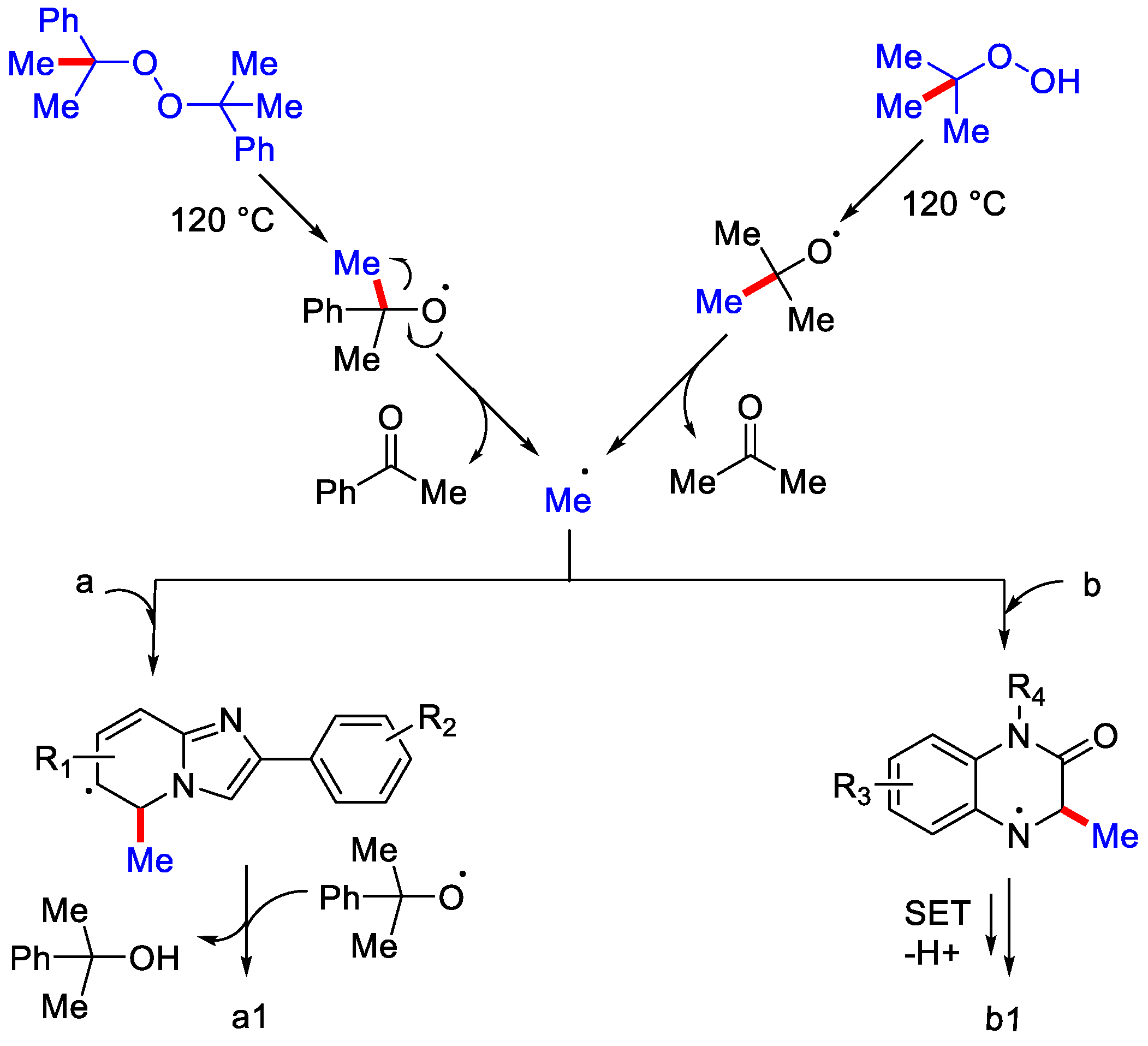
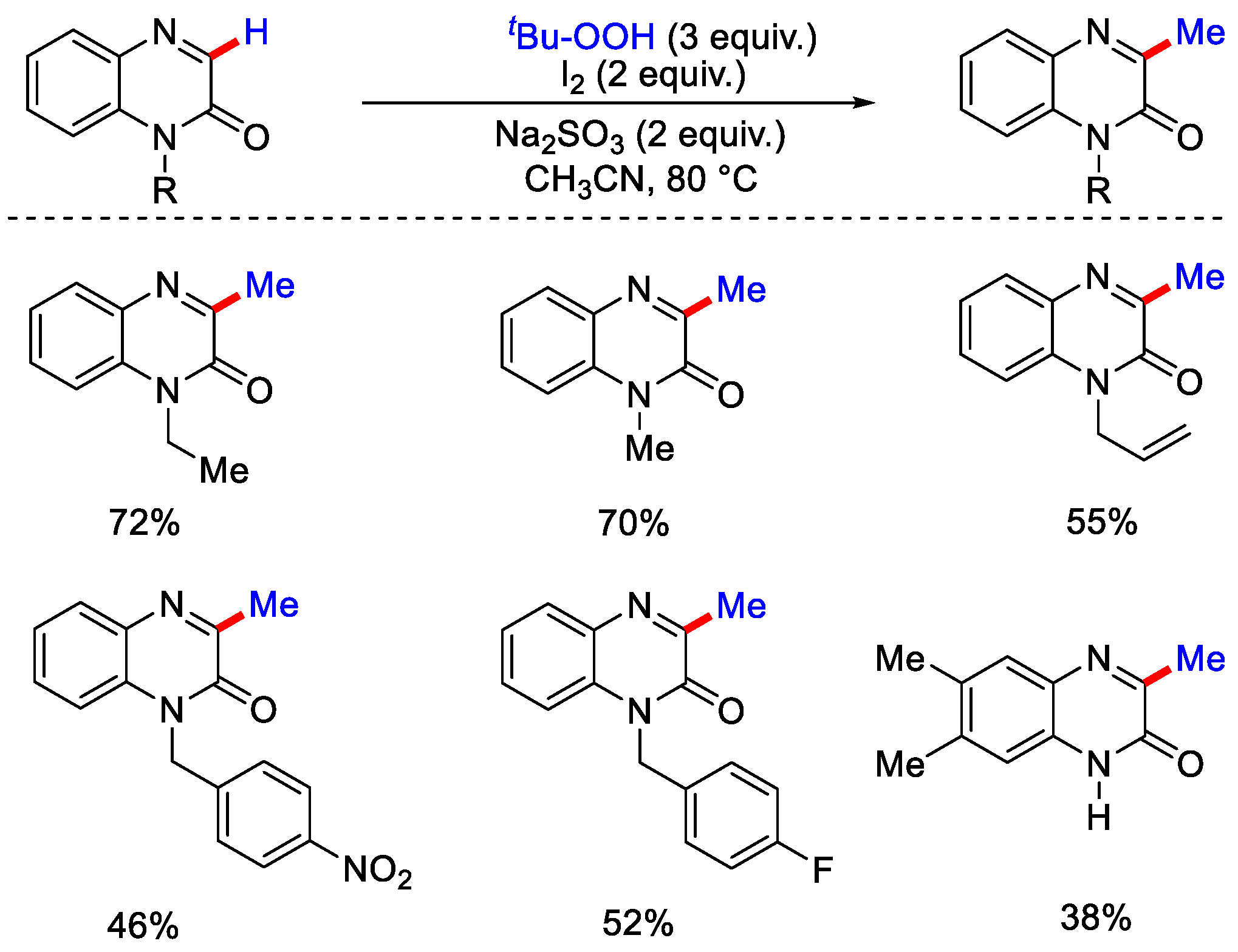
- Jin, S.; Yao, H.; Lin, S.; You, X.; Yang, Y.; Yan, Z. Peroxide-mediated site-specific C–H methylation of imidazo [1, 2-a]pyridines and quinoxalin-2(1H)-ones under metal-free conditions. Org. Biomol. Chem. 2020, 18, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.; Jin, L.; Gu, Y.; Liang, G.; Xia, Q. Transition-Metal-free Radical C–H Methylation of Quinoxalinones with TBHP. Asian J. Org. Chem. 2020, 9, 185–188. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agrawal, I.; Prakash, G.; Al-Thabaiti, S.A.; Mokhtar, M.; Maiti, D. C−H Methylation Using Sustainable Approaches. Catalysts 2022, 12, 510. https://doi.org/10.3390/catal12050510
Agrawal I, Prakash G, Al-Thabaiti SA, Mokhtar M, Maiti D. C−H Methylation Using Sustainable Approaches. Catalysts. 2022; 12(5):510. https://doi.org/10.3390/catal12050510
Chicago/Turabian StyleAgrawal, Ishika, Gaurav Prakash, Shaeel Ahmed Al-Thabaiti, Mohamed Mokhtar, and Debabrata Maiti. 2022. "C−H Methylation Using Sustainable Approaches" Catalysts 12, no. 5: 510. https://doi.org/10.3390/catal12050510
APA StyleAgrawal, I., Prakash, G., Al-Thabaiti, S. A., Mokhtar, M., & Maiti, D. (2022). C−H Methylation Using Sustainable Approaches. Catalysts, 12(5), 510. https://doi.org/10.3390/catal12050510