
Dimethyl Ether Hydrolysis over WO3/γ-Al2O3 Supported Catalysts



Abstract
:
1. Introduction
2. Results
2.1. Catalyst Characterization
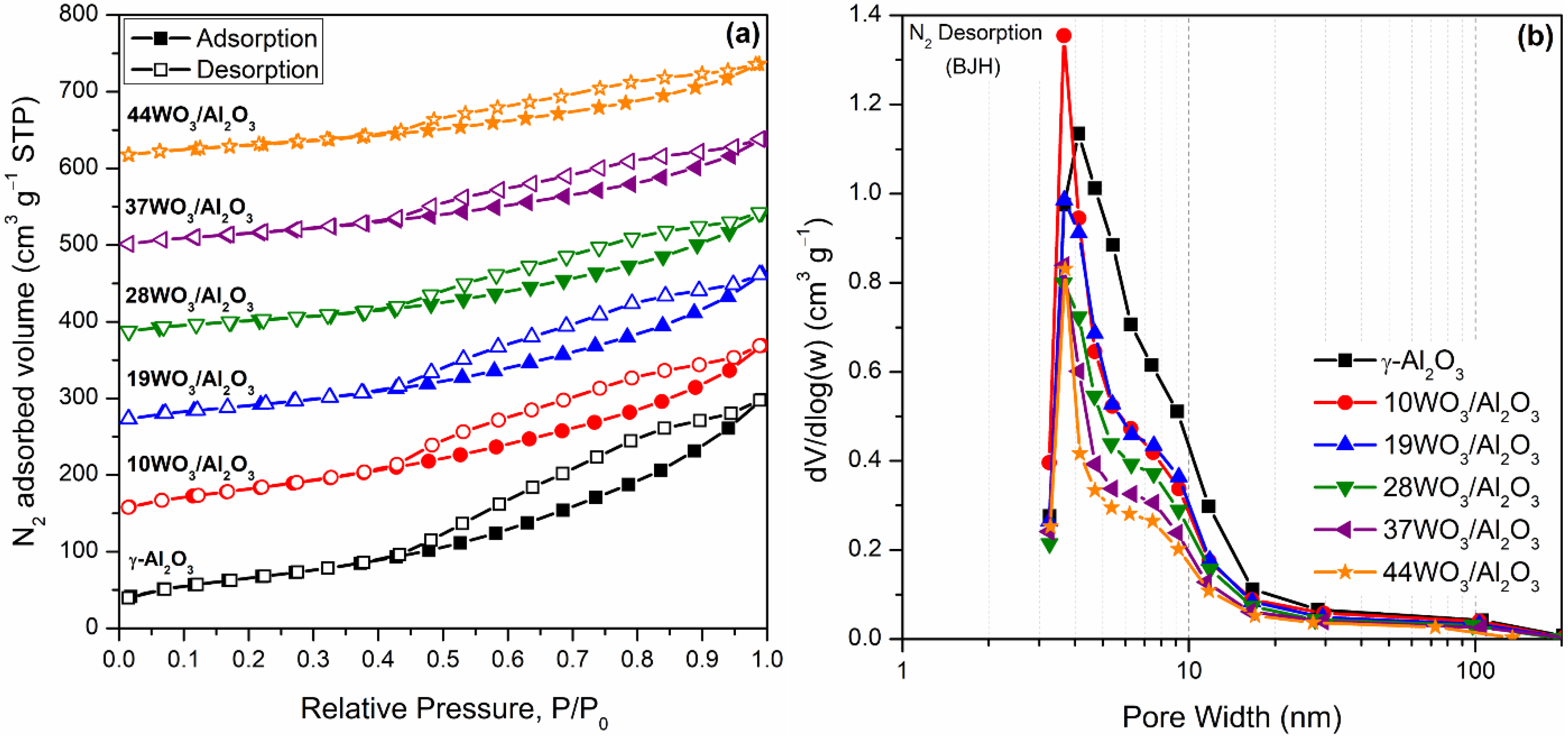
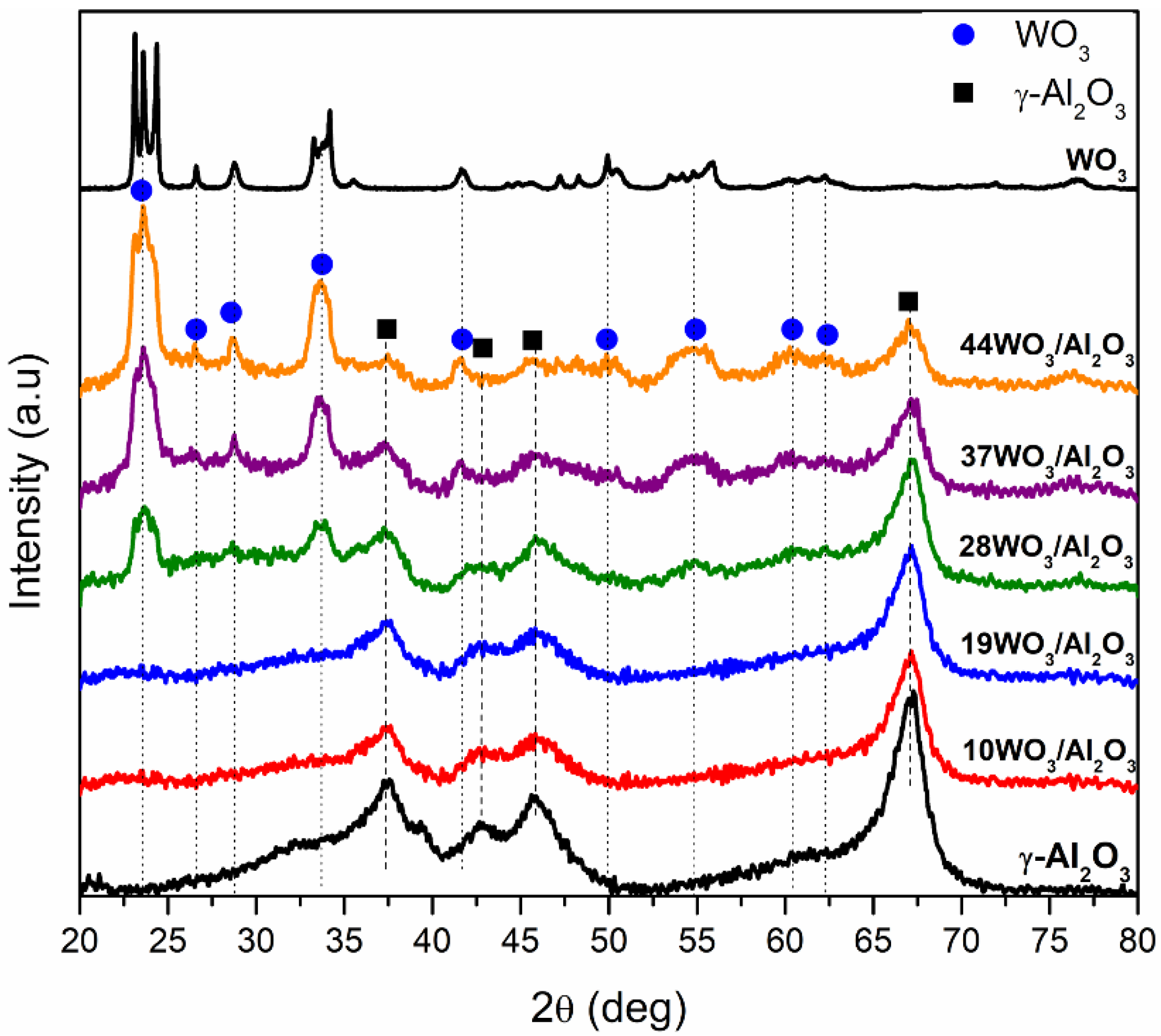
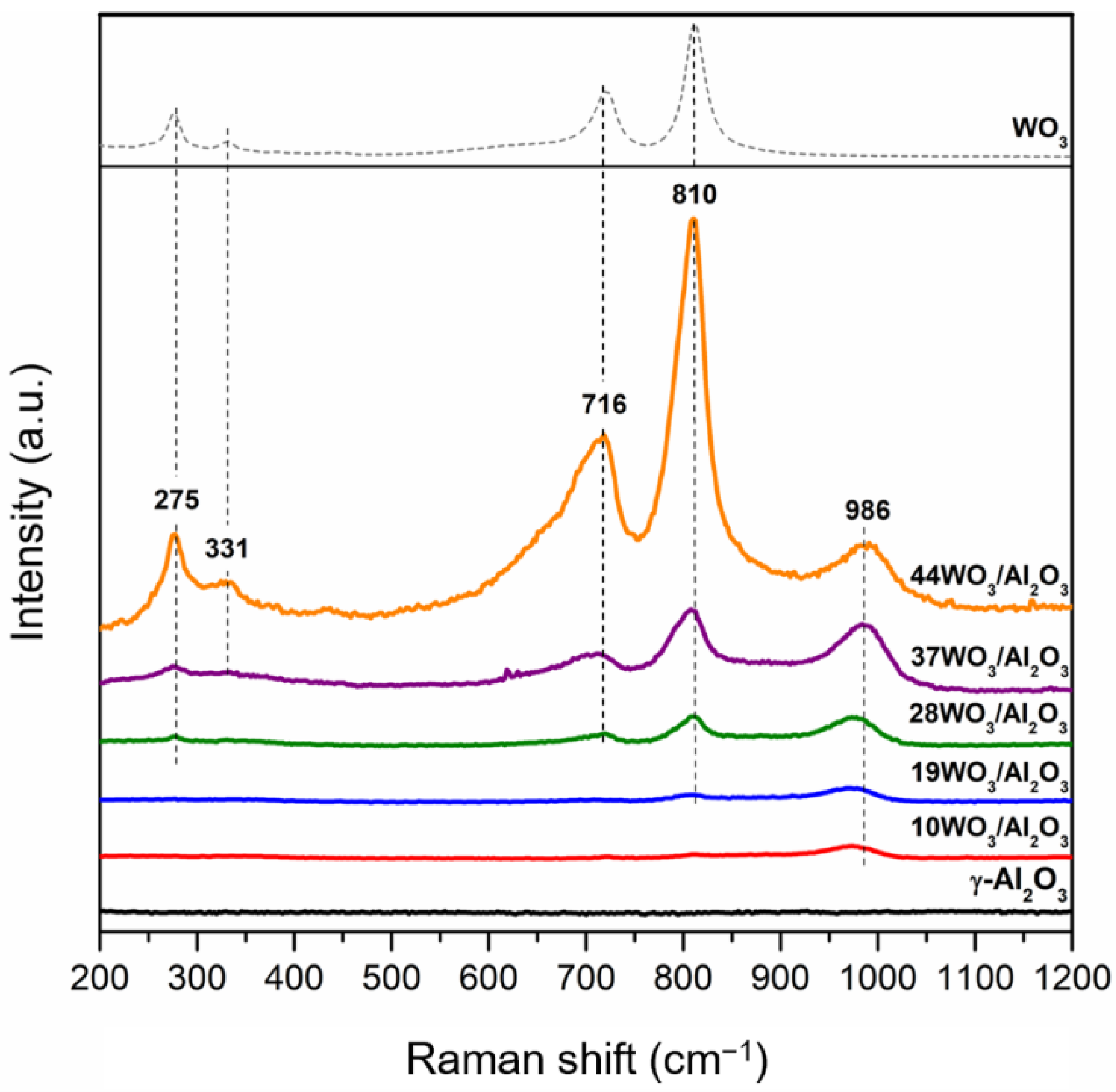
2.1.1. Textural and Structural Properties
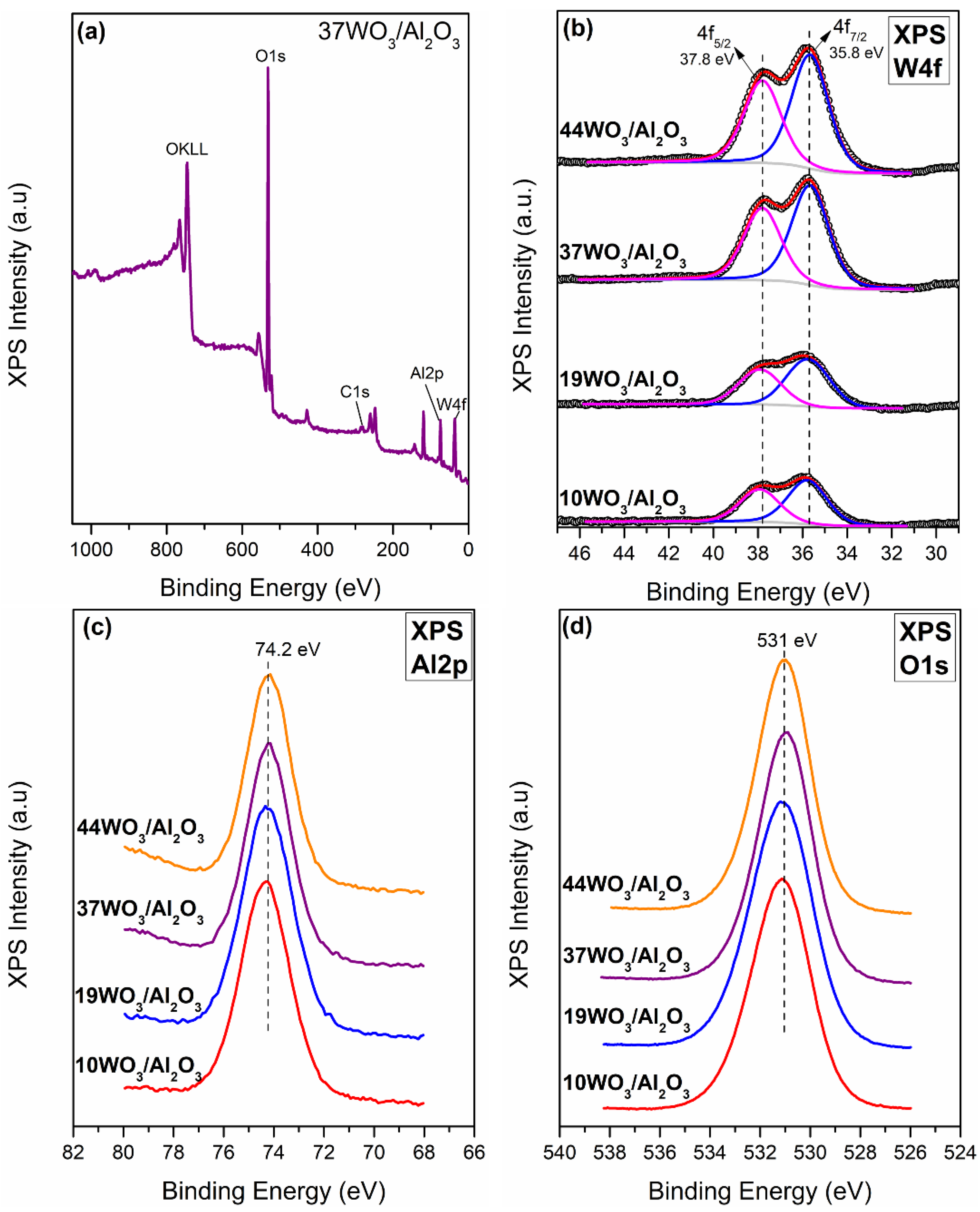
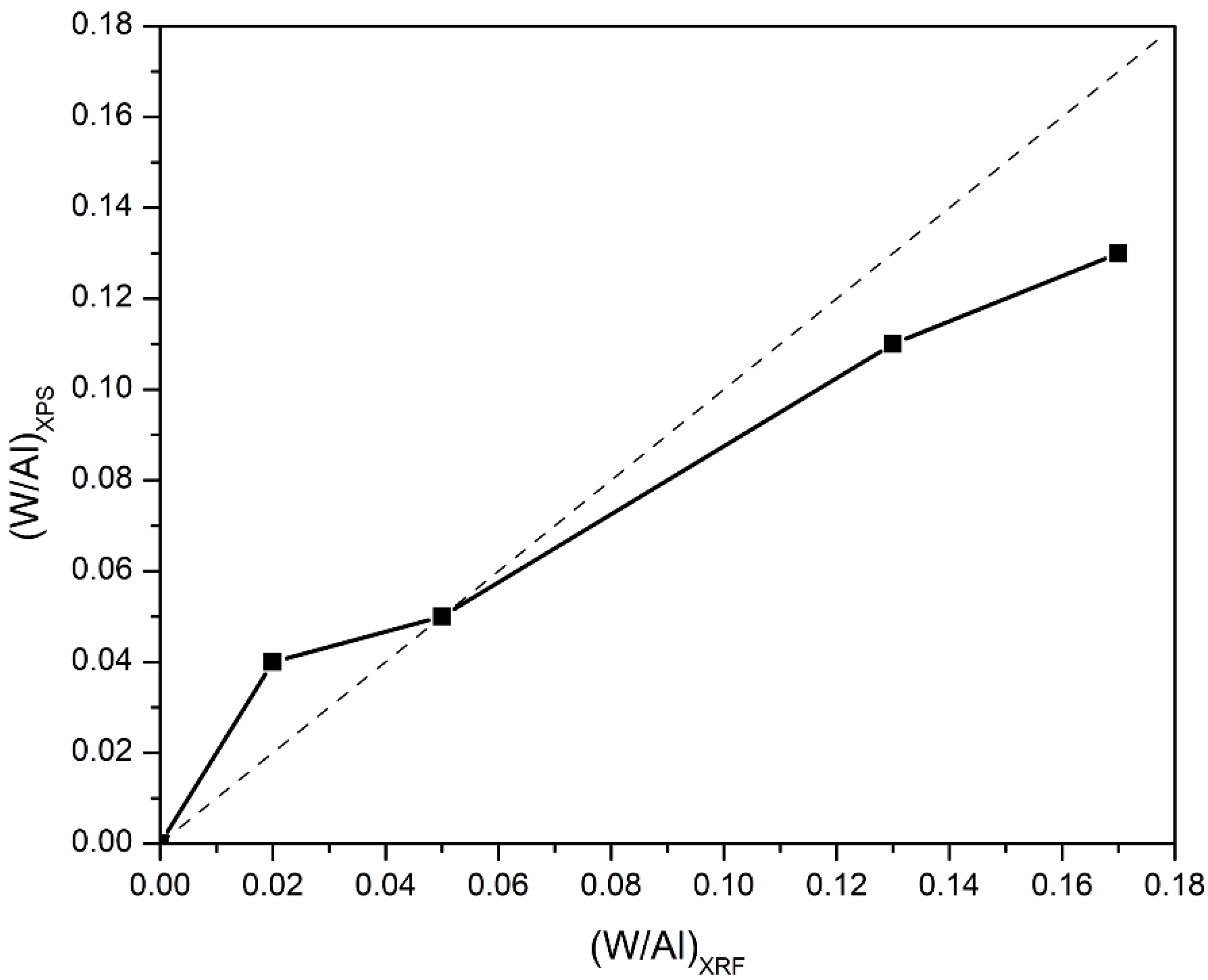
2.1.2. XPS
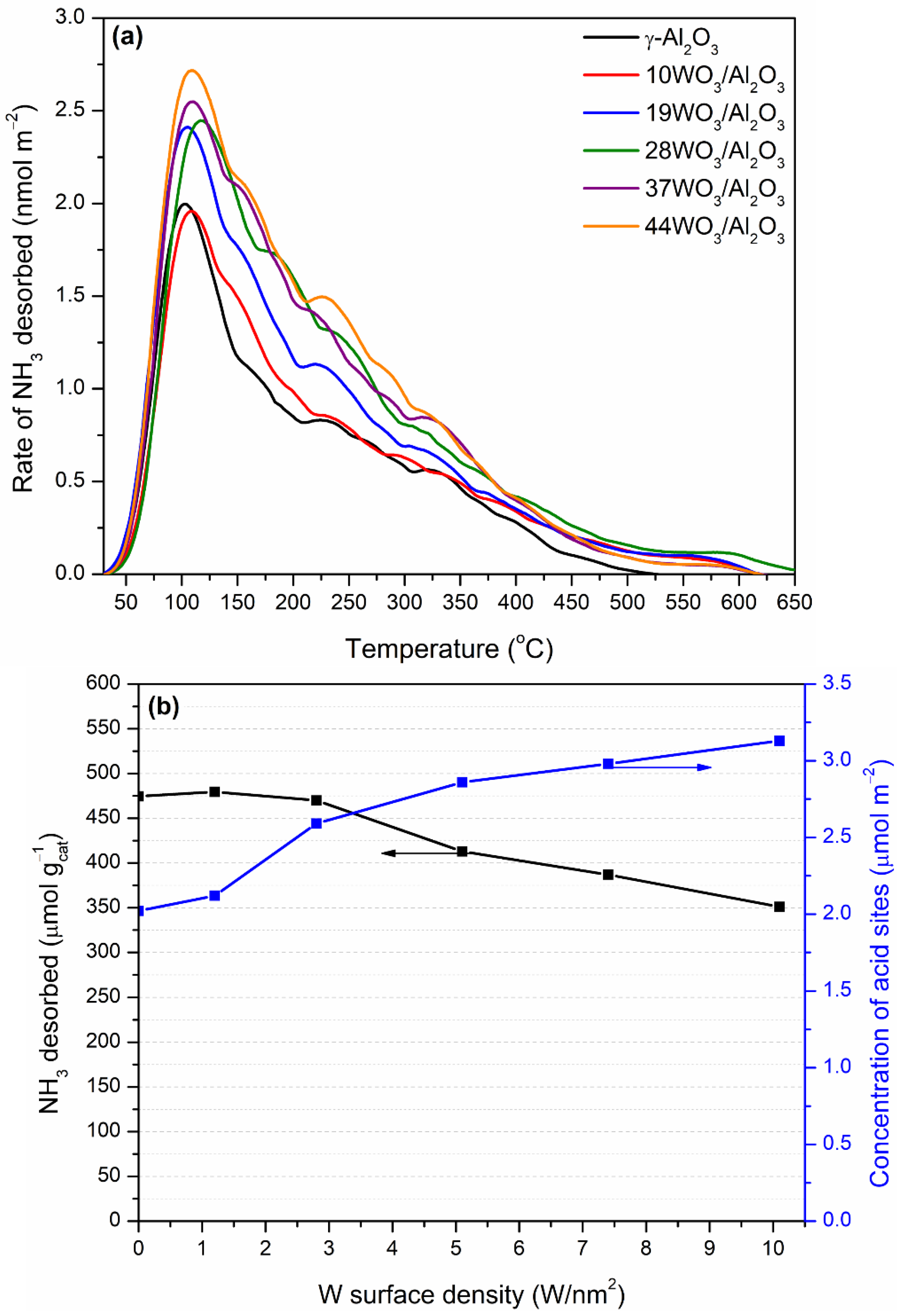
2.1.3. Acidity of the Catalysts
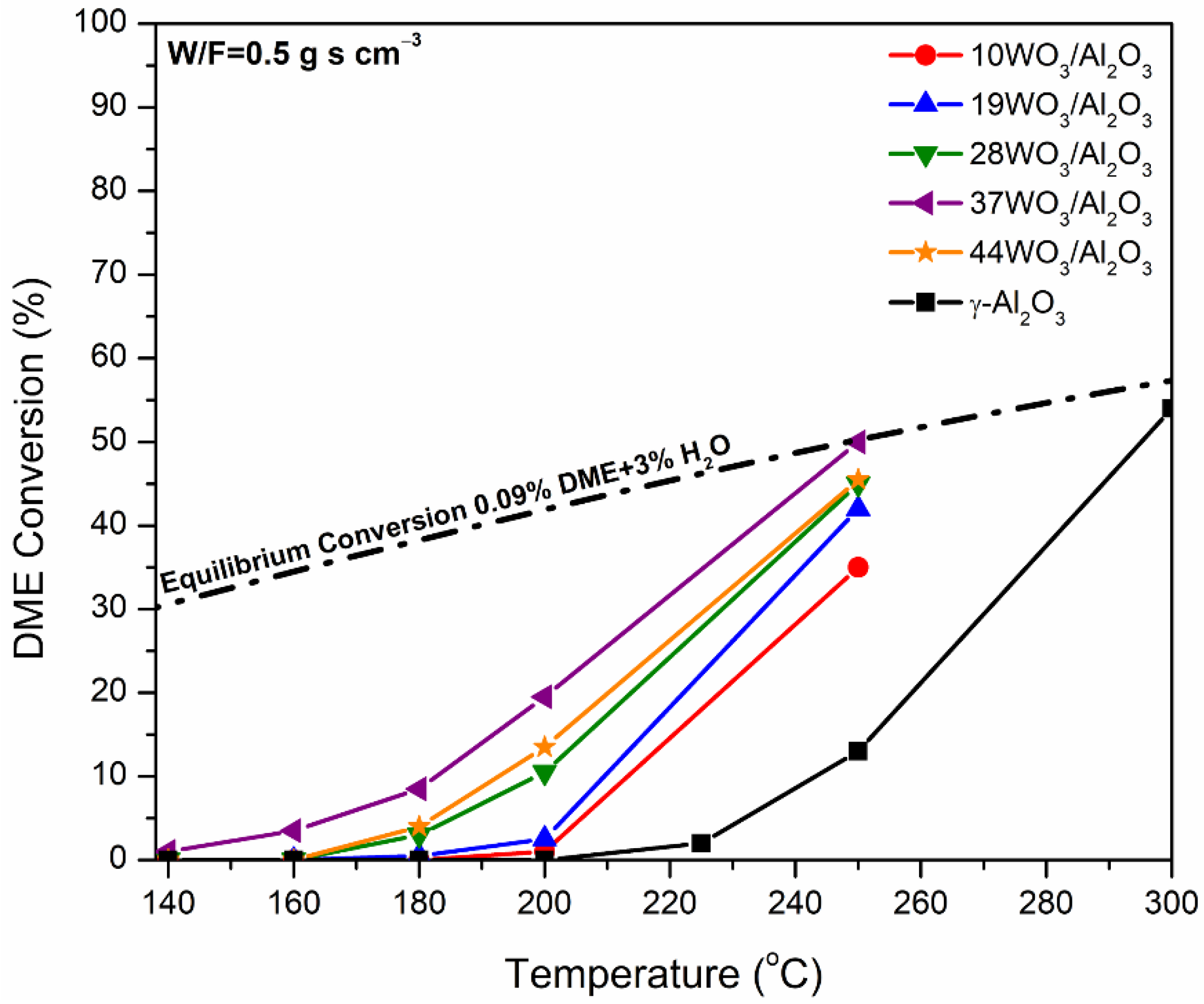
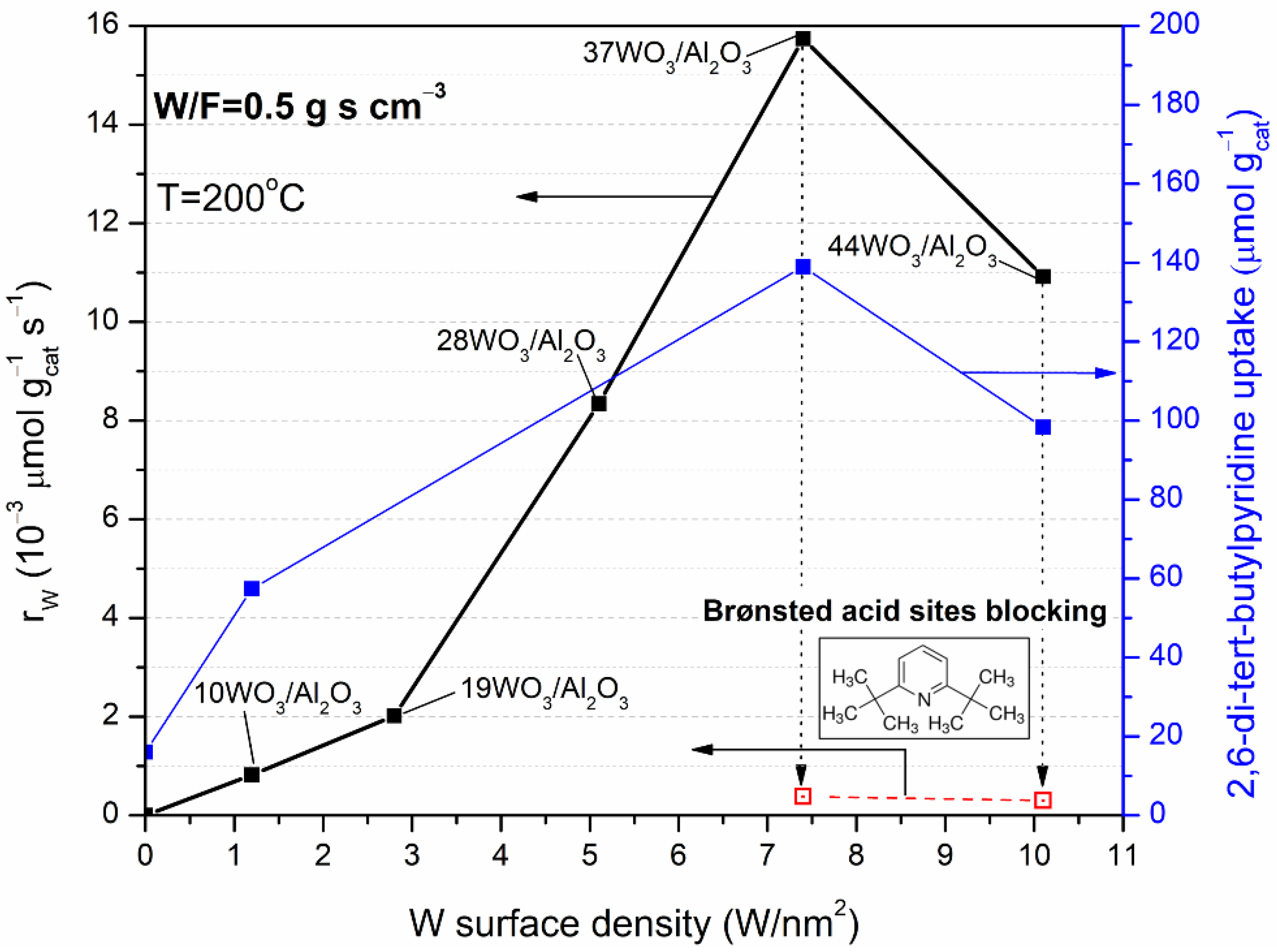
2.2. Catalytic Evaluation in DME Hydrolysis
3. Materials and Methods
3.1. Catalysts Synthesis
3.2. Catalyst Characterization
3.3. Catalyst Evaluation Tests
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Namuangruk, S.; Meeprasert, J.; Khemthong, P.; Faungnawakij, K. A Combined Experimental and Theoretical Study on the Hydrolysis of Dimethyl Ether over H-ZSM-5. J. Phys. Chem. C 2011, 115, 11649–11656. [Google Scholar] [CrossRef]
- Hirunsit, P.; Faungnawakij, K.; Namuangruk, S.; Luadthong, C. Catalytic Behavior and Surface Species Investigation over γ-Al2O3 in Dimethyl Ether Hydrolysis. Appl. Catal. Gen. 2013, 460–461, 99–105. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Chemical Recycling of Carbon Dioxide to Methanol and Dimethyl Ether: From Greenhouse Gas to Renewable, Environmentally Carbon Neutral Fuels and Synthetic Hydrocarbons. J. Org. Chem. 2009, 74, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Faungnawakij, K.; Kikuchi, R.; Matsui, T.; Fukunaga, T.; Eguchi, K. A Comparative Study of Solid Acids in Hydrolysis and Steam Reforming of Dimethyl Ether. Appl. Catal. Gen. 2007, 333, 114–121. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Ott, K.C.; Borup, R.L.; Greene, H.L. Role of Acidity on the Hydrolysis of Dimethyl Ether (DME) to Methanol. Appl. Catal. B Environ. 2005, 61, 281–287. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Borup, R.L.; Greene, H.L. Dimethyl Ether (DME) as an Alternative Fuel. J. Power Sources 2006, 156, 497–511. [Google Scholar] [CrossRef]
- Fleisch, T.H.; Basu, A.; Gradassi, M.J.; Masin, J.G. Dimethyl Ether: A Fuel for the 21st Century. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1997; Volume 107, pp. 117–125. ISBN 978-0-444-82352-6. [Google Scholar]
- Liu, H.; Iglesia, E. Selective One-Step Synthesis of Dimethoxymethane via Methanol or Dimethyl Ether Oxidation on H3+nVnMo12-nPO40 Keggin Structures. J. Phys. Chem. B 2003, 107, 10840–10847. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Tan, Y.; Yang, C.; Han, Y. MnCl2 Modified H4SiW12O40/SiO2 Catalysts for Catalytic Oxidation of Dimethy Ether to Dimethoxymethane. J. Mol. Catal. Chem. 2007, 263, 149–155. [Google Scholar] [CrossRef]
- Cheung, P.; Bhan, A.; Sunley, G.; Law, D.; Iglesia, E. Site Requirements and Elementary Steps in Dimethyl Ether Carbonylation Catalyzed by Acidic Zeolites. J. Catal. 2007, 245, 110–123. [Google Scholar] [CrossRef]
- Zhou, W.; Kang, J.; Cheng, K.; He, S.; Shi, J.; Zhou, C.; Zhang, Q.; Chen, J.; Peng, L.; Chen, M.; et al. Direct Conversion of Syngas into Methyl Acetate, Ethanol, and Ethylene by Relay Catalysis via the Intermediate Dimethyl Ether. Angew. Chem. Int. Ed. 2018, 57, 12012–12016. [Google Scholar] [CrossRef]
- Liu, H.; Cheung, P.; Iglesia, E. Zirconia-Supported MoOxCatalysts for the Selective Oxidation of Dimethyl Ether to Formaldehyde: Structure, Redox Properties, and Reaction Pathways. J. Phys. Chem. B 2003, 107, 4118–4127. [Google Scholar] [CrossRef] [Green Version]
- Peláez, R.; Marín, P.; Ordóñez, S. Synthesis of Formaldehyde from Dimethyl Ether on Alumina-Supported Molybdenum Oxide Catalyst. Appl. Catal. Gen. 2016, 527, 137–145. [Google Scholar] [CrossRef]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol Steam Reforming for Hydrogen Production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef] [PubMed]
- Sá, S.; Silva, H.; Brandão, L.; Sousa, J.M.; Mendes, A. Catalysts for Methanol Steam Reforming—A Review. Appl. Catal. B Environ. 2010, 99, 43–57. [Google Scholar] [CrossRef]
- Papavasiliou, J.; Avgouropoulos, G.; Ioannides, T. Steam Reforming of Methanol over Copper–Manganese Spinel Oxide Catalysts. Catal. Commun. 2005, 6, 497–501. [Google Scholar] [CrossRef]
- Liu, Y.; Hayakawa, T.; Suzuki, K.; Hamakawa, S.; Tsunoda, T.; Ishii, T.; Kumagai, M. Highly Active Copper/Ceria Catalysts for Steam Reforming of Methanol. Appl. Catal. Gen. 2002, 223, 137–145. [Google Scholar] [CrossRef]
- Ranganathan, E.S.; Bej, S.K.; Thompson, L.T. Methanol Steam Reforming over Pd/ZnO and Pd/CeO2 Catalysts. Appl. Catal. Gen. 2005, 289, 153–162. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kikuchi, R.; Takeguchi, T.; Eguchi, K. Steam Reforming of Dimethyl Ether over Composite Catalysts of γ-Al2O3 and Cu-Based Spinel. Appl. Catal. B Environ. 2005, 57, 211–222. [Google Scholar] [CrossRef]
- Faungnawakij, K.; Tanaka, Y.; Shimoda, N.; Fukunaga, T.; Kikuchi, R.; Eguchi, K. Hydrogen Production from Dimethyl Ether Steam Reforming over Composite Catalysts of Copper Ferrite Spinel and Alumina. Appl. Catal. B Environ. 2007, 74, 144–151. [Google Scholar] [CrossRef]
- Faungnawakij, K.; Shimoda, N.; Fukunaga, T.; Kikuchi, R.; Eguchi, K. Cu-Based Spinel Catalysts CuB2O4 (B=Fe, Mn, Cr, Ga, Al, Fe0.75Mn0.25) for Steam Reforming of Dimethyl Ether. Appl. Catal. Gen. 2008, 341, 139–145. [Google Scholar] [CrossRef]
- Faungnawakij, K.; Shimoda, N.; Fukunaga, T.; Kikuchi, R.; Eguchi, K. Crystal Structure and Surface Species of CuFe2O4 Spinel Catalysts in Steam Reforming of Dimethyl Ether. Appl. Catal. B Environ. 2009, 92, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Pan, X.; Lin, R.; Kou, S.; Zou, W.; Ma, J.-X. Steam Reforming of Dimethyl Ether over Cu–Ni/γ-Al2O3 Bi-Functional Catalyst Prepared by Deposition–Precipitation Method. Int. J. Hydrogen Energy 2010, 35, 4060–4068. [Google Scholar] [CrossRef]
- Takeishi, K.; Akaike, Y. Hydrogen Production by Dimethyl Ether Steam Reforming over Copper Alumina Catalysts Prepared Using the Sol–Gel Method. Appl. Catal. Gen. 2016, 510, 20–26. [Google Scholar] [CrossRef]
- Shimoda, N.; Faungnawakij, K.; Kikuchi, R.; Fukunaga, T.; Eguchi, K. Catalytic Performance Enhancement by Heat Treatment of CuFe2O4 Spinel and γ-Alumina Composite Catalysts for Steam Reforming of Dimethyl Ether. Appl. Catal. Gen. 2009, 365, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, T.; Matsuoka, H.; Shishido, T.; Li, D.; Tian, Y.; Sano, T.; Takehira, K. Steam Reforming of Dimethyl Ether over ZSM-5 Coupled with Cu/ZnO/Al2O3 Catalyst Prepared by Homogeneous Precipitation. Appl. Catal. Gen. 2006, 308, 82–90. [Google Scholar] [CrossRef]
- Shimoda, N.; Faungnawakij, K.; Kikuchi, R.; Eguchi, K. A Study of Various Zeolites and CuFe2O4 Spinel Composite Catalysts in Steam Reforming and Hydrolysis of Dimethyl Ether. Int. J. Hydrogen Energy 2011, 36, 1433–1441. [Google Scholar] [CrossRef]
- Ereña, J.; Vicente, J.; Aguayo, A.T.; Gayubo, A.G.; Olazar, M.; Bilbao, J. Effect of Combining Metallic and Acid Functions in CZA/HZSM-5 Desilicated Zeolite Catalysts on the DME Steam Reforming in a Fluidized Bed. Int. J. Hydrogen Energy 2013, 38, 10019–10028. [Google Scholar] [CrossRef]
- Feng, D.; Zuo, Y.; Wang, D.; Wang, J. Steam Reforming of Dimethyl Ether over Coupled Catalysts of CuO-ZnO-Al2O3-ZrO2 and Solid-Acid Catalyst. Chin. J. Chem. Eng. 2009, 17, 64–71. [Google Scholar] [CrossRef]
- Fukunaga, T.; Ryumon, N.; Shimazu, S. The Influence of Metals and Acidic Oxide Species on the Steam Reforming of Dimethyl Ether (DME). Appl. Catal. Gen. 2008, 348, 193–200. [Google Scholar] [CrossRef]
- Matsumoto, T.; Nishiguchi, T.; Kanai, H.; Utani, K.; Matsumura, Y.; Imamura, S. Steam Reforming of Dimethyl Ether over H-Mordenite-Cu/CeO2 Catalysts. Appl. Catal. Gen. 2004, 276, 267–273. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Ott, K.C.; Borup, R.L.; Greene, H.L. Generating Hydrogen-Rich Fuel-Cell Feeds from Dimethyl Ether (DME) Using Cu/Zn Supported on Various Solid-Acid Substrates. Appl. Catal. Gen. 2006, 309, 210–223. [Google Scholar] [CrossRef]
- Nishiguchi, T.; Oka, K.; Matsumoto, T.; Kanai, H.; Utani, K.; Imamura, S. Durability of WO3/ZrO2–CuO/CeO2 Catalysts for Steam Reforming of Dimethyl Ether. Appl. Catal. Gen. 2006, 301, 66–74. [Google Scholar] [CrossRef]
- Galvita, V.V.; Semin, G.L.; Belyaev, V.D.; Yurieva, T.M.; Sobyanin, V.A. Production of Hydrogen from Dimethyl Ether. Appl. Catal. Gen. 2001, 216, 85–90. [Google Scholar] [CrossRef]
- Busca, G. The Surface Acidity of Solid Oxides and Its Characterization by IR Spectroscopic Methods. An Attempt at Systematization. Phys. Chem. Chem. Phys. 1999, 1, 723–736. [Google Scholar] [CrossRef]
- Can, F.; Courtois, X.; Duprez, D. Tungsten-Based Catalysts for Environmental Applications. Catalysts 2021, 11, 703. [Google Scholar] [CrossRef]
- Macht, J.; Baertsch, C.D.; May-Lozano, M.; Soled, S.L.; Wang, Y.; Iglesia, E. Support Effects on Brønsted Acid Site Densities and Alcohol Dehydration Turnover Rates on Tungsten Oxide Domains. J. Catal. 2004, 227, 479–491. [Google Scholar] [CrossRef]
- Hong, E.; Sim, H.-I.; Shin, C.-H. The Effect of Brønsted Acidity of WO3 /ZrO2 Catalysts in Dehydration Reactions of C3 and C4 Alcohols. Chem. Eng. J. 2016, 292, 156–162. [Google Scholar] [CrossRef]
- Ladera, R.; Finocchio, E.; Rojas, S.; Busca, G.; Fierro, J.L.G.; Ojeda, M. Supported WOx-Based Catalysts for Methanol Dehydration to Dimethyl Ether. Fuel 2013, 113, 1–9. [Google Scholar] [CrossRef]
- Witoon, T.; Kidkhunthod, P.; Chareonpanich, M.; Limtrakul, J. Direct Synthesis of Dimethyl Ether from CO2 and H2 over Novel Bifunctional Catalysts Containing CuO-ZnO-ZrO2 Catalyst Admixed with WOx/ZrO2 Catalysts. Chem. Eng. J. 2018, 348, 713–722. [Google Scholar] [CrossRef]
- Suwannapichat, Y.; Numpilai, T.; Chanlek, N.; Faungnawakij, K.; Chareonpanich, M.; Limtrakul, J.; Witoon, T. Direct Synthesis of Dimethyl Ether from CO2 Hydrogenation over Novel Hybrid Catalysts Containing a Cu ZnO ZrO2 Catalyst Admixed with WOx/Al2O3 Catalysts: Effects of Pore Size of Al2O3 Support and W Loading Content. Energy Convers. Manag. 2018, 159, 20–29. [Google Scholar] [CrossRef]
- Pfriem, N.; Liu, Y.; Zahn, F.; Shi, H.; Haller, G.L.; Lercher, J.A. Impact of the Local Concentration of Hydronium Ions at Tungstate Surfaces for Acid-Catalyzed Alcohol Dehydration. J. Am. Chem. Soc. 2021, 143, 20133–20143. [Google Scholar] [CrossRef] [PubMed]
- Baertsch, C.D.; Komala, K.T.; Chua, Y.-H.; Iglesia, E. Genesis of Brønsted Acid Sites during Dehydration of 2-Butanol on Tungsten Oxide Catalysts. J. Catal. 2002, 205, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Yi, L.; Deng, R.; You, K.; Song, J.; Jian, J.; Liu, P.; Ai, Q.; Luo, H. Supported WO3/γ-Al2O3 as Bifunctional Catalyst for Liquid-Phase Highly Selective Oxidation of Cyclohexylamine to Cyclohexanone Oxime under Solvent-Free Conditions. Mol. Catal. 2019, 475, 110494. [Google Scholar] [CrossRef]
- Said, A.E.-A.A.; Abd El-Wahab, M.M.M.; Abd El-Aal, M. Catalytic Dehydration of Methanol to Dimethyl Ether over Nanosized WO3/Al2O3 System under Inert and Oxidative Atmosphere. Mon. Chem. Chem. Mon. 2016, 147, 1507–1516. [Google Scholar] [CrossRef]
- Wachs, I.E. Raman and IR Studies of Surface Metal Oxide Species on Oxide Supports: Supported Metal Oxide Catalysts. Catal. Today 1996, 27, 437–455. [Google Scholar] [CrossRef]
- Ross-Medgaarden, E.I.; Wachs, I.E. Structural Determination of Bulk and Surface Tungsten Oxides with UV−vis Diffuse Reflectance Spectroscopy and Raman Spectroscopy. J. Phys. Chem. C 2007, 111, 15089–15099. [Google Scholar] [CrossRef]
- Chan, S. Laser Raman Characterization of Tungsten Oxide Supported on Alumina: Influence of Calcination Temperatures. J. Catal. 1985, 92, 1–10. [Google Scholar] [CrossRef]
- Salvati, L.; Makovsky, L.E.; Stencel, J.M.; Brown, F.R.; Hercules, D.M. Surface Spectroscopic Study of Tungsten-Alumina Catalysts Using x-Ray Photoelectron, Ion Scattering, and Raman Spectroscopies. J. Phys. Chem. 1981, 85, 3700–3707. [Google Scholar] [CrossRef]
- Martín, C.; Solana, G.; Malet, P.; Rives, V. Nb2O5-Supported WO3: A Comparative Study with WO3/Al2O3. Catal. Today 2003, 78, 365–376. [Google Scholar] [CrossRef]
- Vermaire, D. The Preparation of WO3/TiO2 and WO3/Al2O3 and Characterization by Temperature-Programmed Reduction. J. Catal. 1989, 116, 309–317. [Google Scholar] [CrossRef]
- Grunert, W. Reduction Behavior and Metathesis Activity of WO3/Al2O3 Catalysts I. An XPS Investigation of WO3/Al2O3 Catalysts. J. Catal. 1987, 107, 522–534. [Google Scholar] [CrossRef]
- Rodríguez-Ramos, I.; Guerrero-Ruiz, A.; Homs, N.; de la Piscina, P.R.; Fierro, J.L.G. Reactions of Propene on Supported Molybdenum and Tungsten Oxides. J. Mol. Catal. Chem. 1995, 95, 147–154. [Google Scholar] [CrossRef]
- Wang, H.; Wu, Y.; Liu, Z.; He, L.; Yao, Z.; Zhao, W. Deposition of WO3 on Al2O3 via a Microwave Hydrothermal Method to Prepare Highly Dispersed W/Al2O3 Hydrodesulfurization Catalyst. Fuel 2014, 136, 185–193. [Google Scholar] [CrossRef]
- Li, Y.; Wang, C.; Zheng, H.; Wan, F.; Yu, F.; Zhang, X.; Liu, Y. Surface Oxygen Vacancies on WO3 Contributed to Enhanced Photothermo-Synergistic Effect. Appl. Surf. Sci. 2017, 391, 654–661. [Google Scholar] [CrossRef]
- Soled, S. Comparison of the Acidities of WO3/Al2O3 and Ultrastable Faujasite Catalysts. J. Catal. 1988, 111, 286–295. [Google Scholar] [CrossRef]
- Chen, X.; Clet, G.; Thomas, K.; Houalla, M. Correlation between Structure, Acidity and Catalytic Performance of WOx/Al2O3 Catalysts. J. Catal. 2010, 273, 236–244. [Google Scholar] [CrossRef]
- Zhang, R.; Jagiello, J.; Hu, J.F.; Huang, Z.-Q.; Schwarz, J.A.; Datye, A. Effect of WO3 Loading on the Surface Acidity of WO3/Al2O3 Composite Oxides. Appl. Catal. Gen. 1992, 84, 123–139. [Google Scholar] [CrossRef]
- Rorrer, J.; He, Y.; Toste, F.D.; Bell, A.T. Mechanism and Kinetics of 1-Dodecanol Etherification over Tungstated Zirconia. J. Catal. 2017, 354, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Góra-Marek, K.; Tarach, K.; Choi, M. 2,6-Di- Tert- Butylpyridine Sorption Approach to Quantify the External Acidity in Hierarchical Zeolites. J. Phys. Chem. C 2014, 118, 12266–12274. [Google Scholar] [CrossRef]
- Kitano, T.; Hayashi, T.; Uesaka, T.; Shishido, T.; Teramura, K.; Tanaka, T. Effect of High-Temperature Calcination on the Generation of Brønsted Acid Sites on WO3/Al2O3. ChemCatChem 2014, 6, 2011–2020. [Google Scholar] [CrossRef]
- Rossmedgaarden, E.; Knowles, W.; Kim, T.; Wong, M.; Zhou, W.; Kiely, C.; Wachs, I. New Insights into the Nature of the Acidic Catalytic Active Sites Present in ZrO2-Supported Tungsten Oxide Catalysts. J. Catal. 2008, 256, 108–125. [Google Scholar] [CrossRef]
- Fu, Y.; Hong, T.; Chen, J.; Auroux, A.; Shen, J. Surface Acidity and the Dehydration of Methanol to Dimethyl Ether. Thermochim. Acta 2005, 434, 22–26. [Google Scholar] [CrossRef]
- Xu, M.; Lunsford, J.H.; Goodman, D.W.; Bhattacharyya, A. Synthesis of Dimethyl Ether (DME) from Methanol over Solid-Acid Catalysts. Appl. Catal. Gen. 1997, 149, 289–301. [Google Scholar] [CrossRef]
- Sun, J.; Yang, G.; Yoneyama, Y.; Tsubaki, N. Catalysis Chemistry of Dimethyl Ether Synthesis. ACS Catal. 2014, 4, 3346–3356. [Google Scholar] [CrossRef]
- Yaripour, F.; Baghaei, F.; Schmidt, I.; Perregaard, J. Catalytic Dehydration of Methanol to Dimethyl Ether (DME) over Solid-Acid Catalysts. Catal. Commun. 2005, 6, 147–152. [Google Scholar] [CrossRef]
- Sabour, B.; Peyrovi, M.H.; Hamoule, T.; Rashidzadeh, M. Catalytic Dehydration of Methanol to Dimethyl Ether (DME) over Al-HMS Catalysts. J. Ind. Eng. Chem. 2014, 20, 222–227. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | W Surface Density (W/nm2) | SBET (m2 g−1) | Vp (cm3 g−1) | DP (nm) | Total Acid Sites (μmol gcat−1) | Acid Sites Density (μmol m−2) | Brønsted Acid Sites (μmol gcat−1) |
|---|---|---|---|---|---|---|---|
| γ-Al2O3 | 0.0 | 235 | 0.49 | 4.11 | 475 | 2.02 | 16 |
| 10WO3/Al2O3 | 1.2 | 226 | 0.39 | 3.66 | 480 | 2.12 | 57 |
| 19WO3/Al2O3 | 2.8 | 182 | 0.36 | 3.67 | 470 | 2.59 | n.d |
| 28WO3/Al2O3 | 5.1 | 144 | 0.30 | 3.65 | 413 | 2.86 | n.d |
| 37WO3/Al2O3 | 7.4 | 130 | 0.26 | 3.66 | 387 | 2.98 | 139 |
| 44WO3/Al2O3 | 10.1 | 112 | 0.22 | 3.70 | 351 | 3.13 | 98 |
| WO3 | 2.5 | 0.002 | 3.20 | n.d. | n.d. | n.d |
| Catalyst | W/Al Atomic Ratio | BE (eV) | ||||
|---|---|---|---|---|---|---|
| XRF | XPS | W4f | Al2p | O1s | ||
| 4f5/2 | 4f7/2 | |||||
| 10WO3/Al2O3 | 0.02 | 0.04 | 37.9 | 35.8 | 74.3 | 531.1 |
| 19WO3/Al2O3 | 0.05 | 0.05 | 37.9 | 35.8 | 74.3 | 531.1 |
| 37WO3/Al2O3 | 0.13 | 0.11 | 37.8 | 35.7 | 74.2 | 530.9 |
| 44WO3/Al2O3 | 0.17 | 0.13 | 37.8 | 35.7 | 74.2 | 531 |
| Catalyst | T10 (°C) | rW | r | |
|---|---|---|---|---|
| (×10−3 μmol gcat−1 s−1) | (×10−3 μmol gWO3−1 s−1) | (×10−5 μmol m−2 s−1) | ||
| γ-Al2O3 | 243 | 0 | n.d. | 0 |
| 10WO3/Al2O3 | 213 | 0.82 | 8.2 | 0.36 |
| 19WO3/Al2O3 | 210 | 2.02 | 10.64 | 1.11 |
| 28WO3/Al2O3 | 199 | 8.34 | 29.78 | 5.77 |
| 37WO3/Al2O3 | 192 | 15.74 | 42.54 | 12.11 |
| 44WO3/Al2O3 | 183 | 10.92 | 25.10 | 9.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smyrnioti, M.; Ioannides, T. Dimethyl Ether Hydrolysis over WO3/γ-Al2O3 Supported Catalysts. Catalysts 2022, 12, 396. https://doi.org/10.3390/catal12040396
Smyrnioti M, Ioannides T. Dimethyl Ether Hydrolysis over WO3/γ-Al2O3 Supported Catalysts. Catalysts. 2022; 12(4):396. https://doi.org/10.3390/catal12040396
Chicago/Turabian StyleSmyrnioti, Maria, and Theophilos Ioannides. 2022. "Dimethyl Ether Hydrolysis over WO3/γ-Al2O3 Supported Catalysts" Catalysts 12, no. 4: 396. https://doi.org/10.3390/catal12040396
APA StyleSmyrnioti, M., & Ioannides, T. (2022). Dimethyl Ether Hydrolysis over WO3/γ-Al2O3 Supported Catalysts. Catalysts, 12(4), 396. https://doi.org/10.3390/catal12040396