2. Results
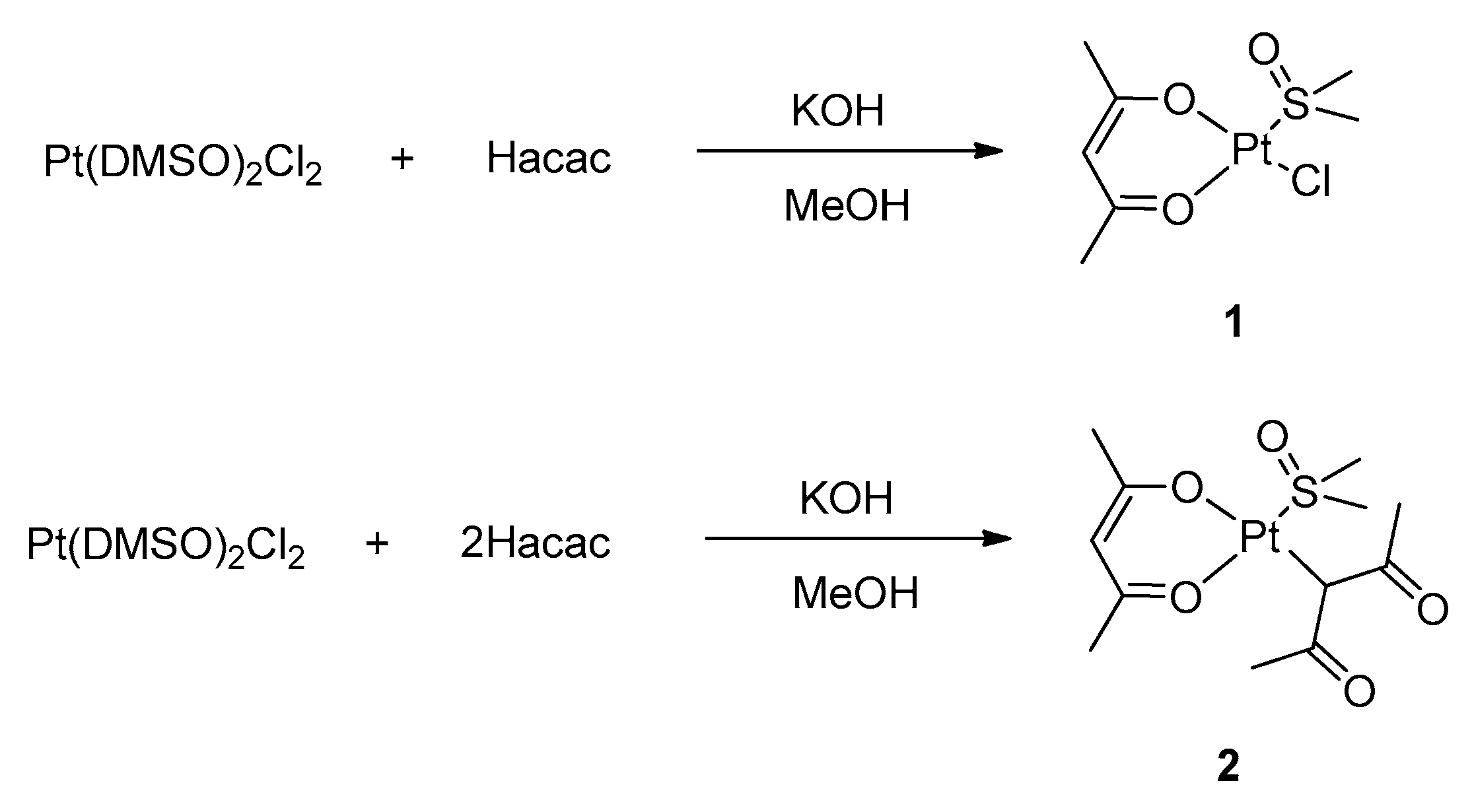
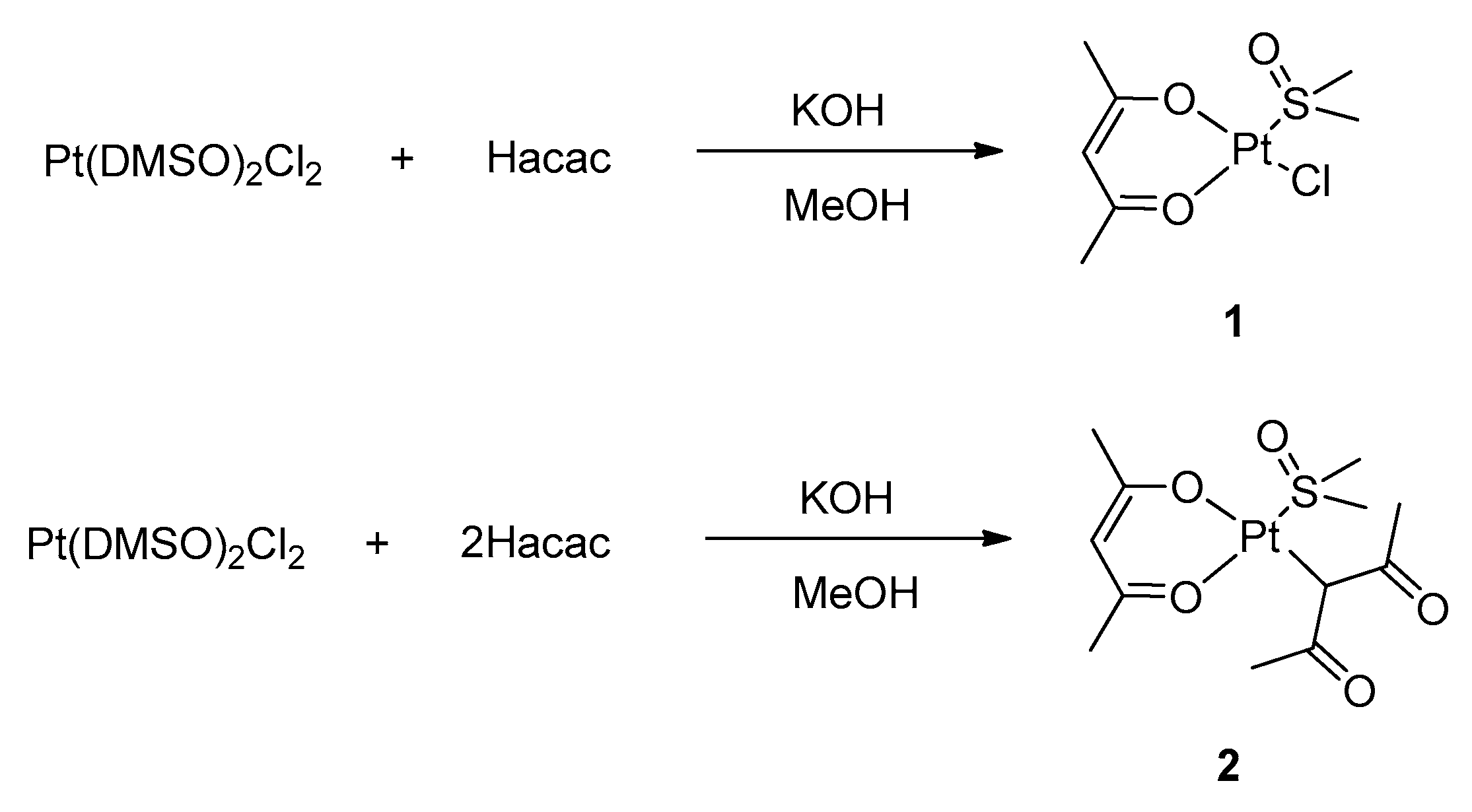
The synthesis of the target complexes moves from well-established routes that are reported in the current literature. In particular, we started by exploiting a route for the preparation of heteroleptic platinum (II) acetylacetonate complexes with DMSO (DMSO = dimethylsulfoxide), which was described by De Pascali et al., (
Scheme 1) [
20].
The synthetic route proved successful and allowed for the preparation of complexes
1 and
2, depending on the stoichiometric ratio between the acetylacetonate and the platinum precursor (see
Supplementary Materials). In complex
2, the complete substitution of the chloride ligands of the precursor by acetylacetonate is achieved, although one acetylacetonate ligand coordinates as a O,O′-chelating bidentate ligand and the second one as a C-monodentate ligand through the γ carbon atom. Remarkably, simple Pt(acac)
2 cannot be synthesized through this procedure.
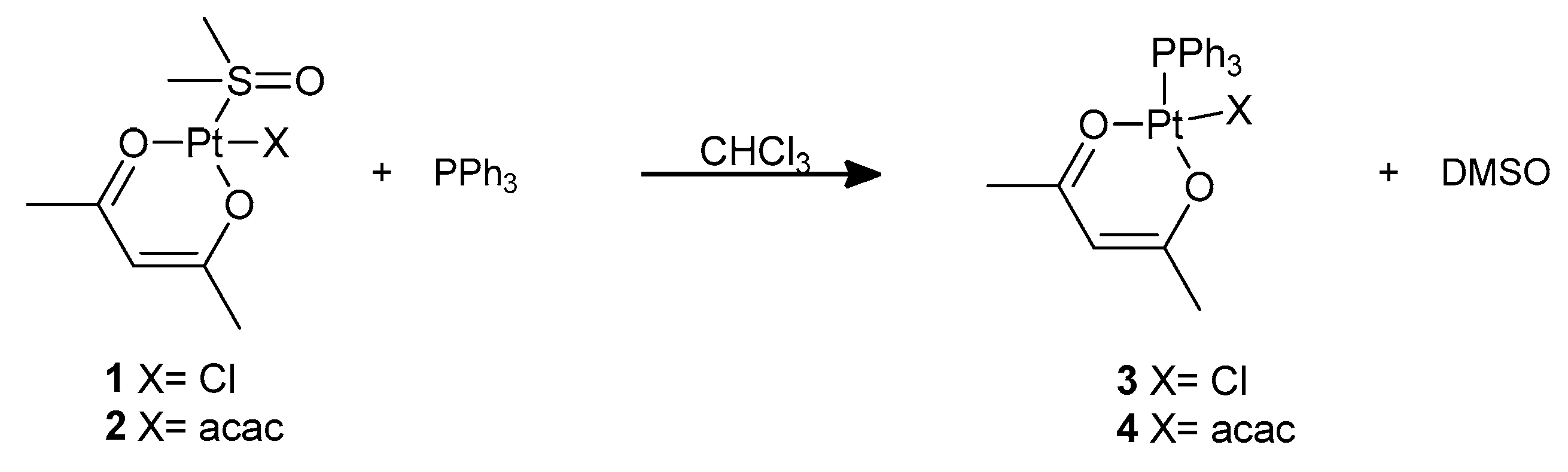
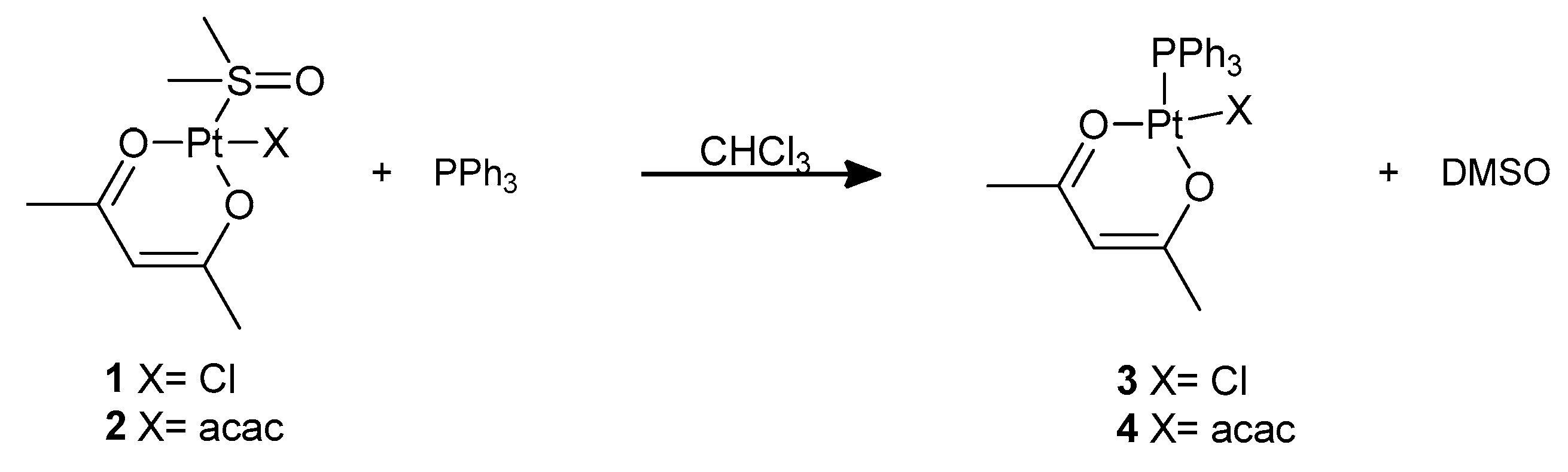
However, the DMSO ligand in complexes
1 and
2 can be substituted by softer ligands. We performed this substitution reaction at room temperature using triphenylphosphine as the incoming ligand, obtaining complexes
3 and
4 that were previously reported by De Pascali et al., (
Scheme 2, see
Supplementary Materials) [
21]. The reaction can easily be followed by monitoring the NMR signals of coordinated and free DMSO in the
1H NMR spectrum of the compound. Incidentally, complex
4 can also be prepared upon nucleophilic attack by PPh
3 on Pt(acac)
2 [
22].
When harder neutral ligands react with complexes such as
1 and
2, they reportedly substitute preferentially the chloride ligand rather than DMSO, giving rise to monocationic complexes. Our targets were instead neutral platinum complexes, since they expectedly display a higher solubility in the mixture of silicone polymers that constitute the substrate for the curing reaction. Consequently, in order to prepare complexes with harder ligands, we reverted to another published procedure that moved from preformed Pt(acac)
2 [
23]. The reaction of this complex with excess pyridine resulted again in the nucleophilic attack by the incoming ligand, causing a change in coordination of the diketonate from O,O′-chelating bidentate to C-monodentate (
Scheme 3, see
Supplementary Materials). The difficulty in this reaction is the control of the number of pyridine units entering the coordination sphere of the compound: equilibrium product mixtures containing complexes
5 and
6 are obtained, which however can be easily separated, since complex
5 is soluble in diethyl ether, whereas complex
6 is not. We attempted the same synthesis using quinoline as the base, but unfortunately the reaction proved inefficient, possibly because of the greater steric bulk of the quinoline ligand.
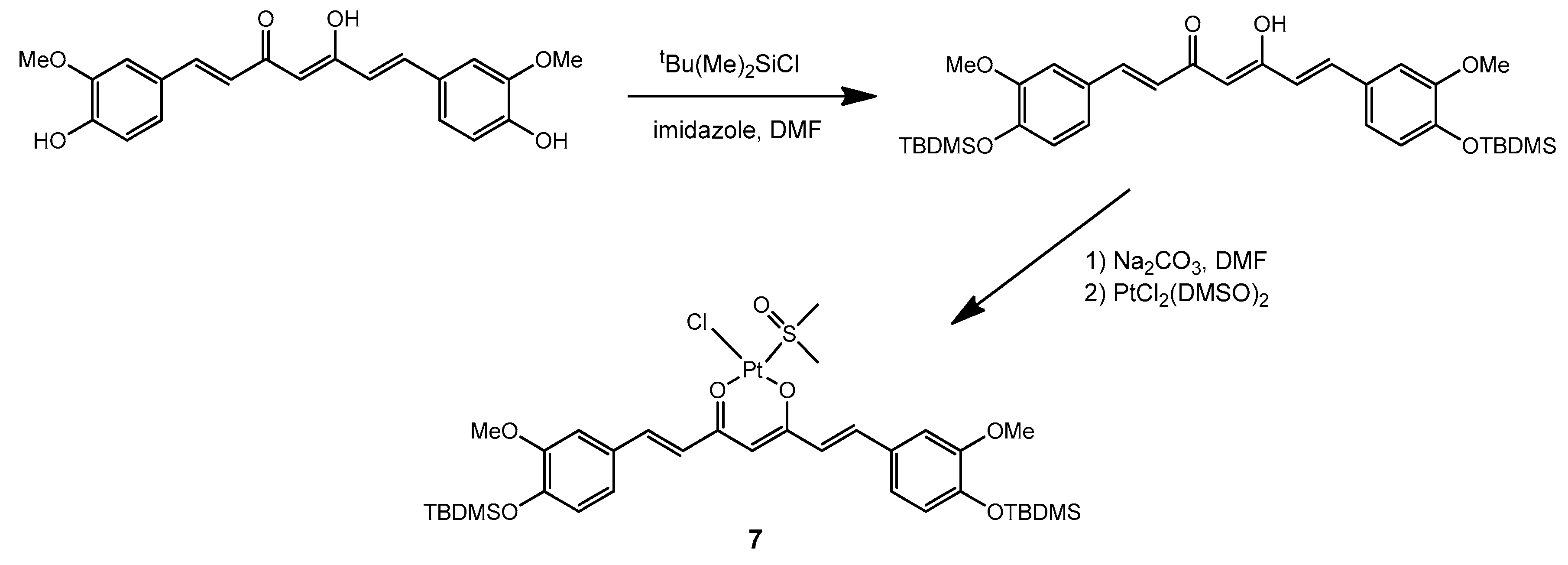
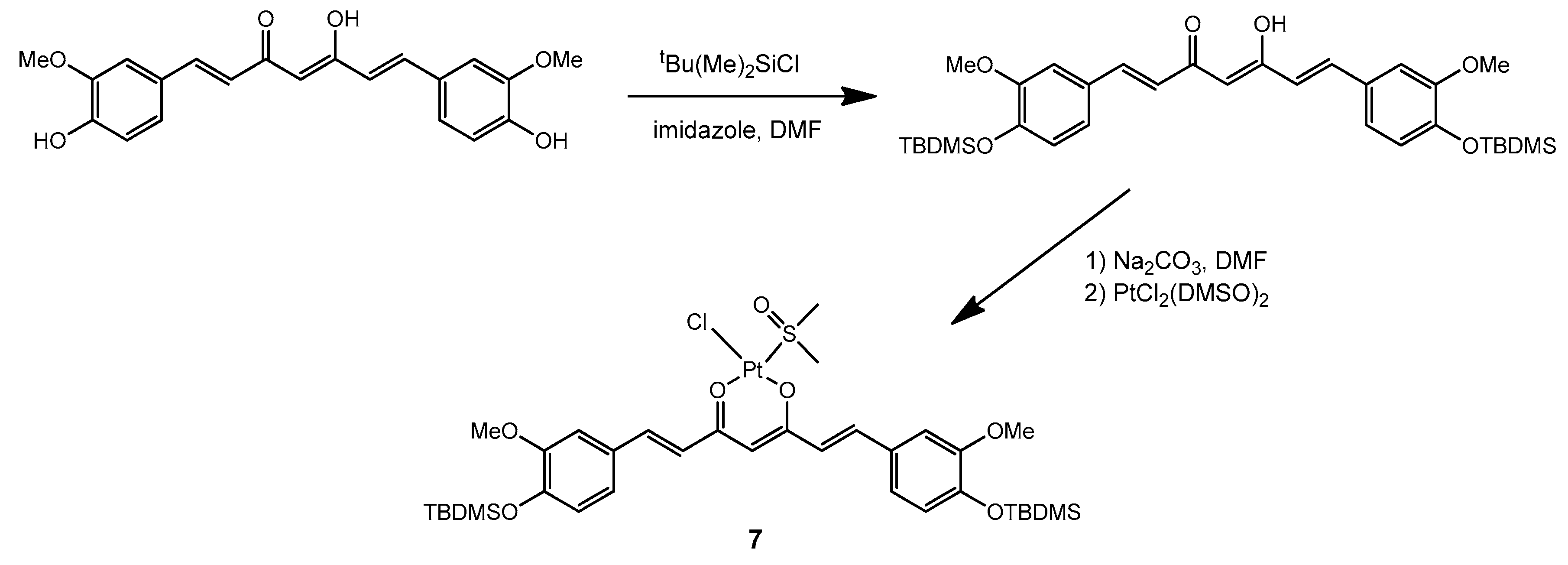
The synthetic strategy described above for the preparation of complex
1 has also been recently employed to prepare an analogous Pt complex with the peculiar β-diketone curcumin [
24]. Given the strong absorption of curcumin and its derivatives in the visible range, we became interested in including similar complexes in our study. However, in order to improve the stability of the curcumin ligand and to promote solubility of the complex in a comparatively hydrophobic medium such as a mixture of silicone polymers, we performed first a derivatization of curcumin by protecting the free hydroxyl groups on the aromatic rings. Silanization with t-butyldimethylsilyl (TBDMS) chloride proved successful in this respect and allowed us to subsequently prepare the platinum complex
7 with the derivatized ligand, using a unique procedure developed by us which avoided the use of water (
Scheme 4).
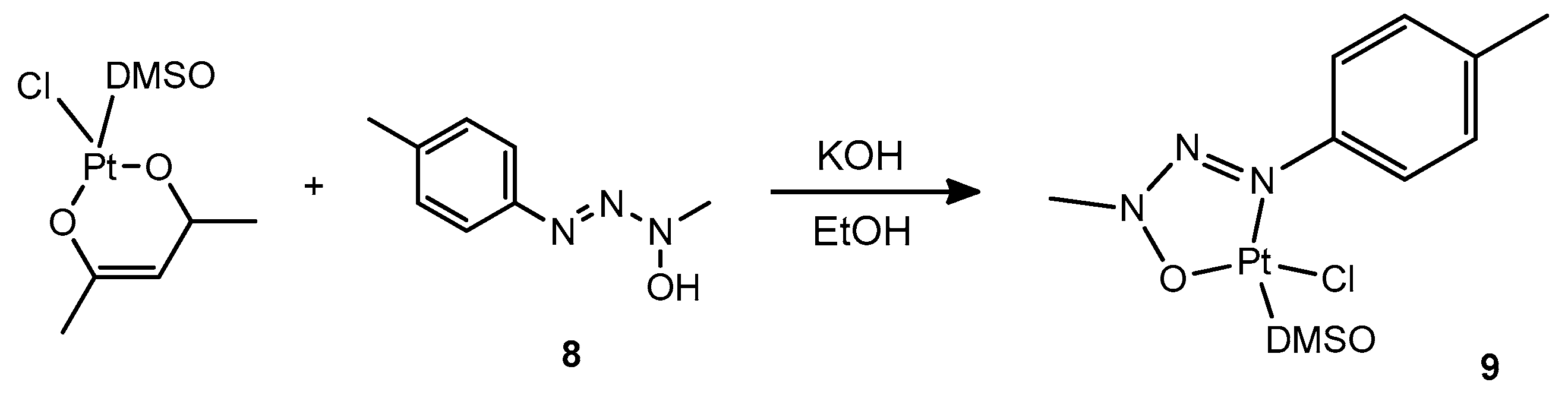
Finally, we attempted the exchange of the DMSO and of the chloride ligand in complex
1 with a triazenide
N-oxide ligand. Ligands of this kind have been previously reported in the patent literature to behave as chelating ligands towards platinum and produce homo- and heteroleptic complexes that can be conveniently photoactivated for hydrosilylation reactions by irradiation with UV light [
25]. Thus, we conveniently synthesized by literature methods the triazene oxide
8 and attempted the substitution reaction. However, the reaction had an unexpected outcome, where the triazenide
N-oxide indeed became coordinated to Pt, yet did not substitute the DMSO and chloride ligand but rather the acetylacetonate ligand, giving rise to complex
9 (
Scheme 5):
Several alternative strategies for the preparation of a complex containing both O,O′-coordinated acetylacetonate and the deprotonated proligand 8 were attempted, however all resulted in no reaction or in the production of complex 9.
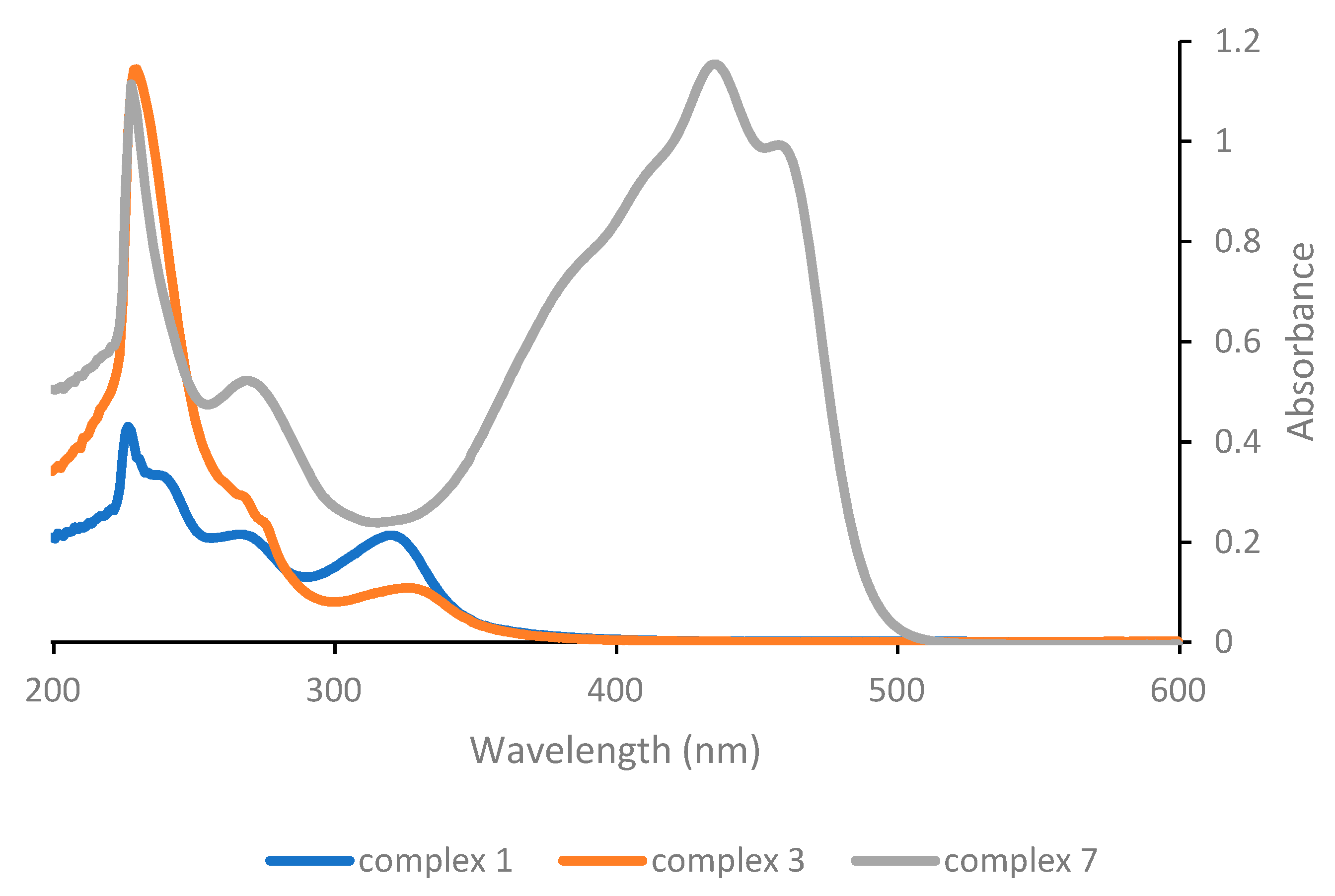
The UV-Vis spectra of the various complexes were recorded in order to determine their absorption wavelengths. Dichloromethane solutions 5 × 10
−5 M were employed in all cases. The UV-Vis absorption spectrum of the parent Pt(acac)
2 has been reported in the literature and exhibits absorption bands at 265, 290 and 350 nm [
18]. Complex
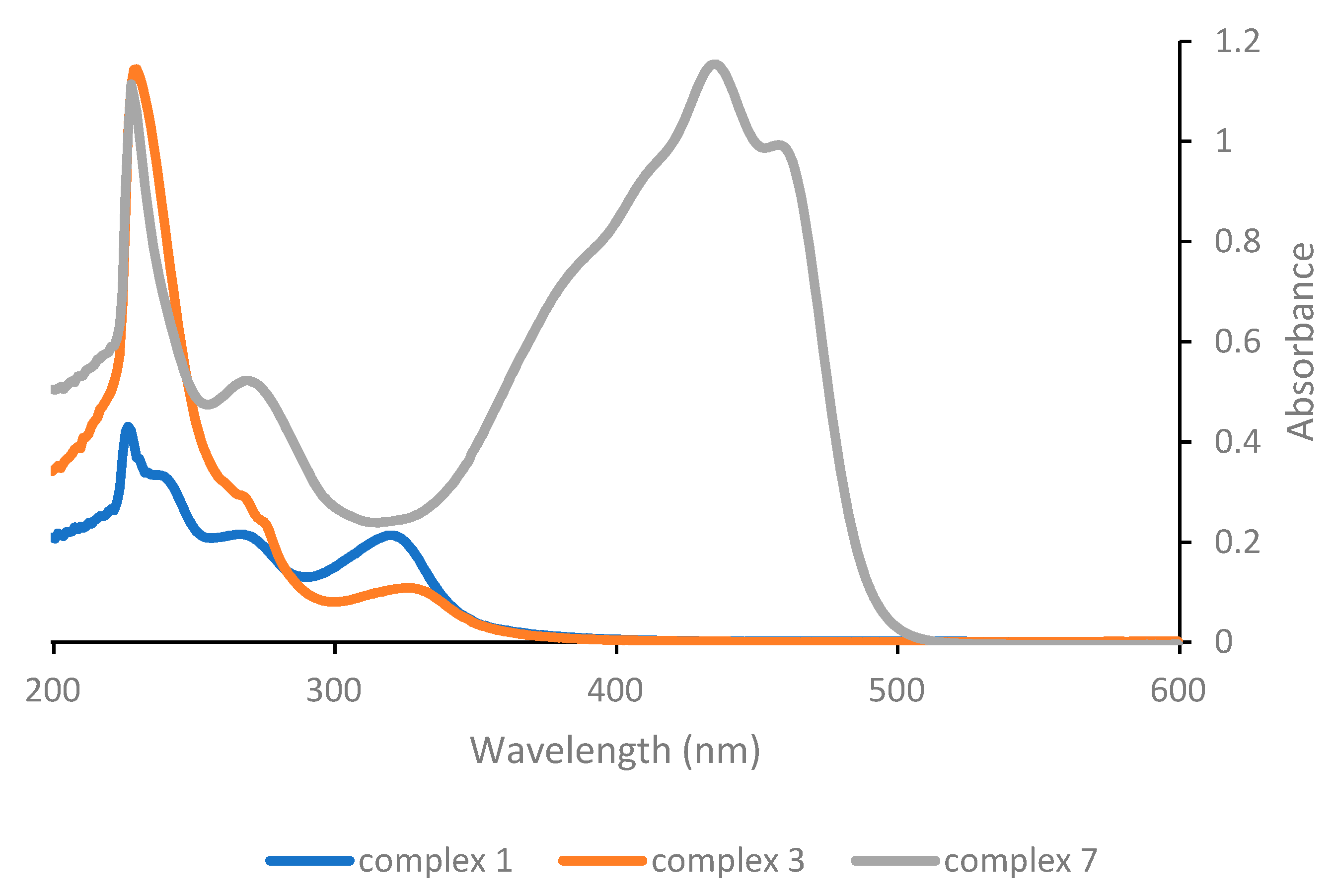
1 exhibits the same three bands, but shifted at 240, 270 and 321 nm (
Figure 1), although the low energy band is more intense compared with the one of the homoleptic acetylacetonate complex. This is relevant, as the absorption features of the compound in the spectral region above 300 nm will expectedly influence the efficiency of the photoactivation process, which in our study will be carried out in the near-UV as well as in the visible region. The low energy band in the UV-Vis of Pt(acac)
2 has been proposed on the basis of a semi-empirical molecular orbital treatment to arise from a π-π* transition, involving the acetylacetonate ligand [
26] and we assume that the analogous low energy bands displayed by all other complexes reported herein (with the only obvious exception of complex
6) originate from an electronic transition of the same nature.
Substitution of the DMSO ligand in complex 1 with triphenylphosphine in complex 3 has significant consequences on the position and, most notably, on the intensity of the low energy band; while the maximum shifts slightly towards higher wavelengths (to 325 nm), the intensity decreases considerably. On the other hand, substitution of the acetylacetonate ligand with the protected curcuminate ligand in complex 7 results in the appearance of a very intense and broad absorption band in the visible range, which is obviously to be attributed to the peculiar properties of the curcuminate ligand.
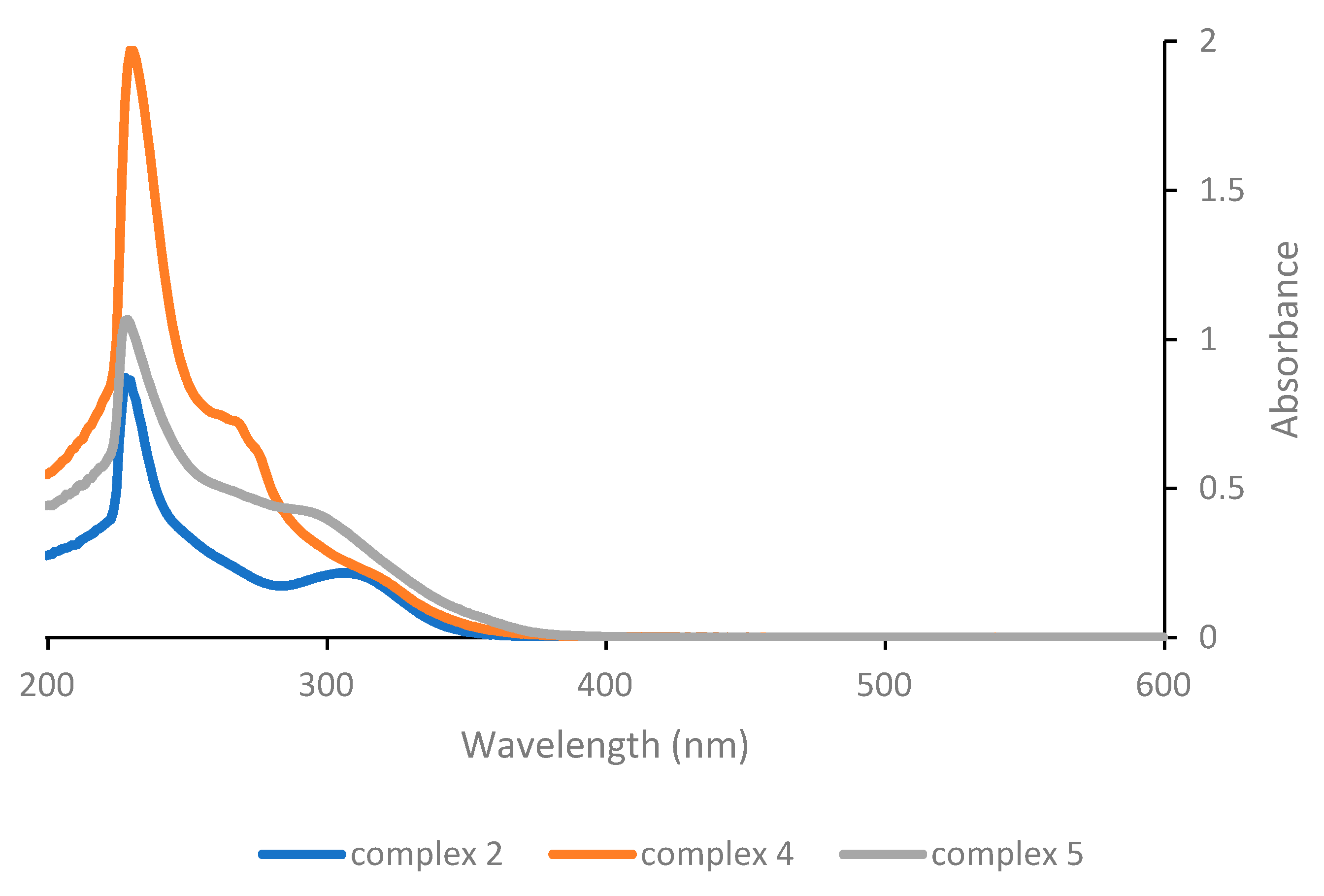
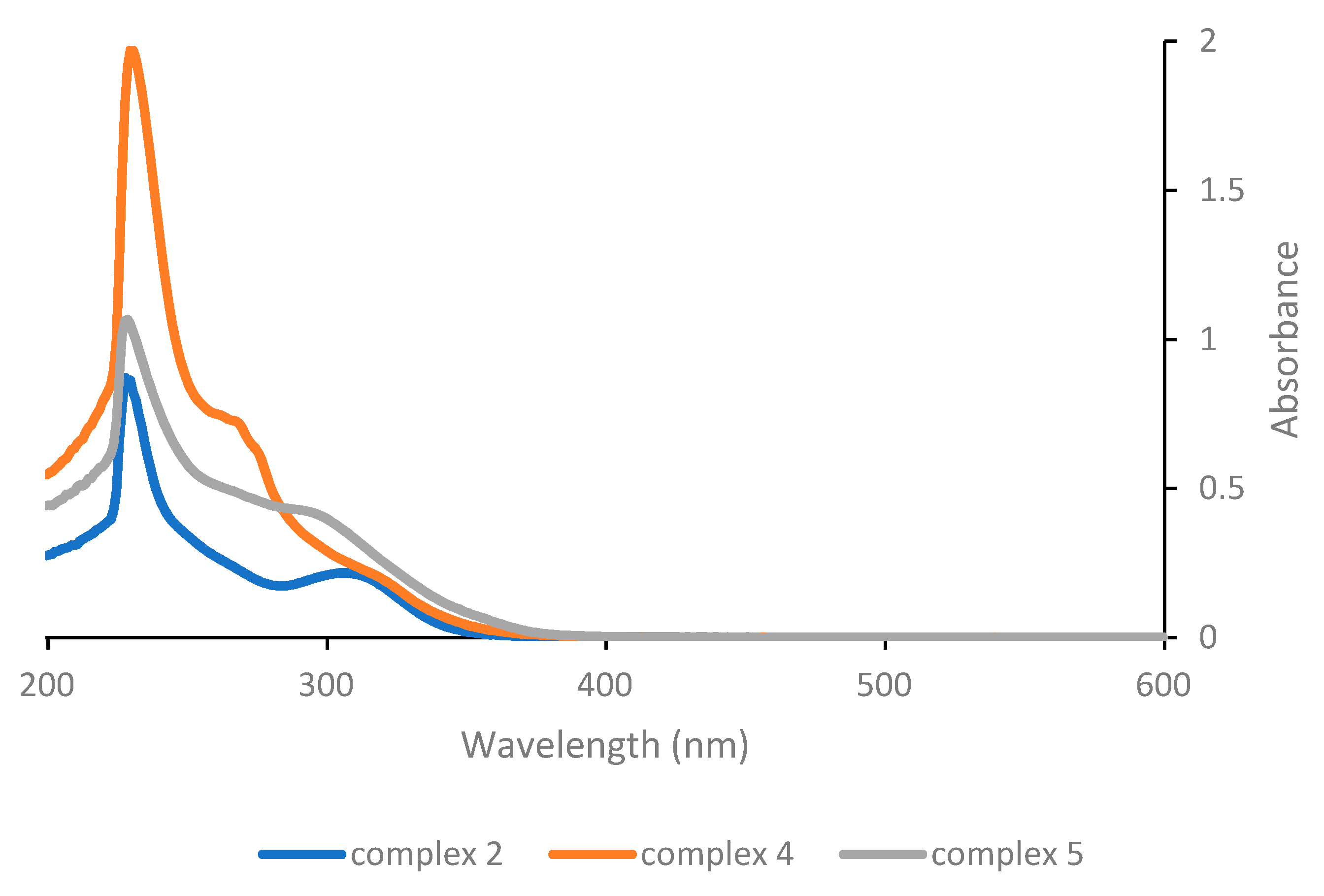
Considering instead the complexes of the general structure [Pt(γ-acac)(O,O′-acac)L] (
Figure 2), the low energy band is found at 307 nm for the parent complex
2, hence again at a much lower wavelength compared with Pt(acac)
2 and also at significantly lower wavelengths compared with complex
1. The effect of the change in neutral ligand L is somewhat more significant here, and although the absorption maximum of the low energy band remains almost unchanged (for complex
4 with L = PPh
3) or results even slightly shifted to shorter wavelengths (for complex
5 with L = pyridine), it turns out that in the latter case both the intensity of the absorption and the tailing towards higher wavelengths increase slightly, potentially resulting in a more efficient photoactivation of complex
5.
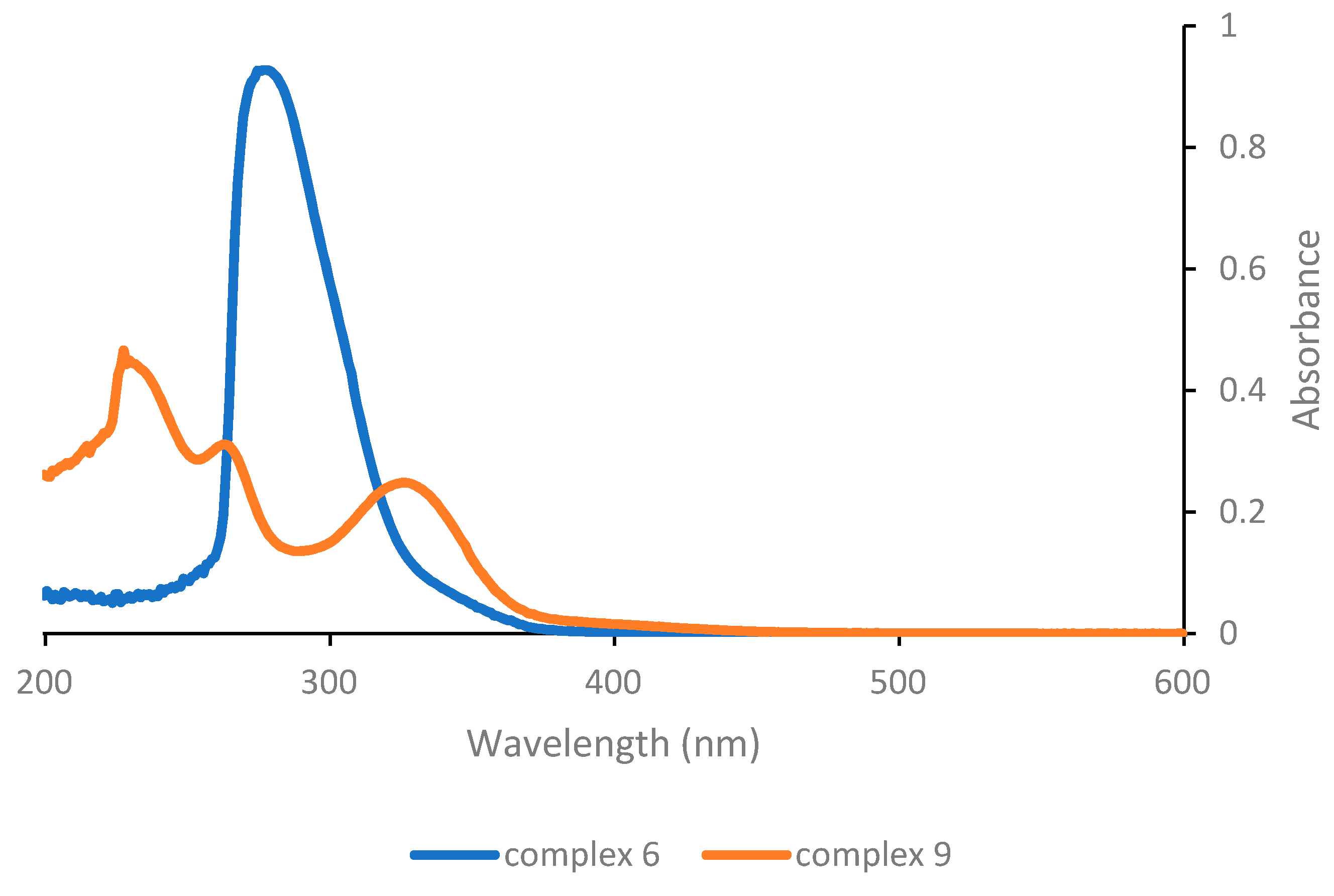
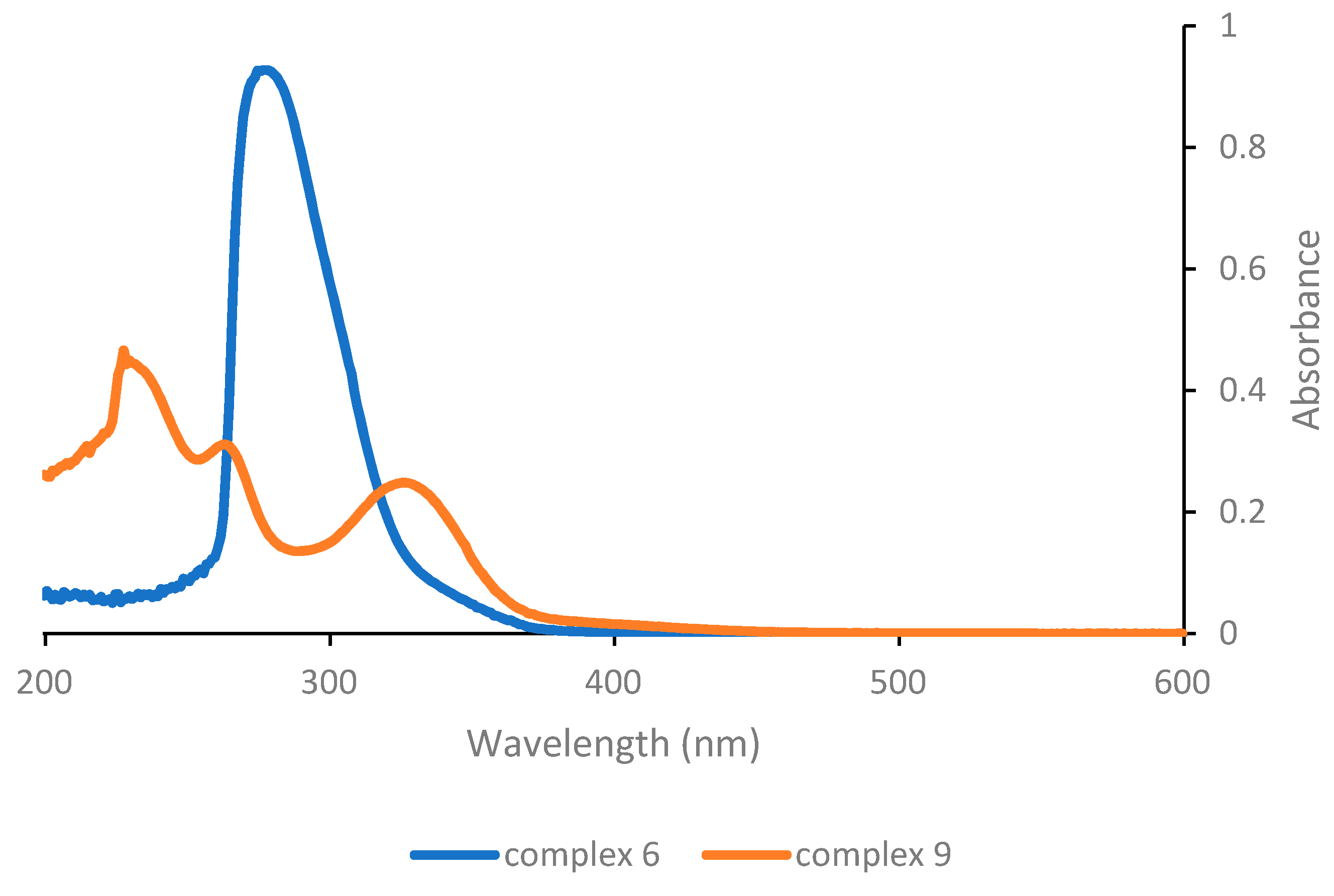
Finally, considering the last complexes in the series (
Figure 3), it can be appreciated that complex
6 that lacks any O,O′-β-diketonate ligand exhibits a single absorption band at a low wavelength (278 nm) and consequently appears to not be very suitable for photoactivation in the near-UV or visible range. On the other hand, complex
9 with the triazenide-
N-oxide ligand shows absorption features similar to the complexes with structure [PtCl(O,O′-β-diketonate)L]. In particular, a low energy band with appreciable intensity is detected at 326 nm, i.e., at a slightly higher wavelength compared with complex
1.
Following their UV-Vis characterization, the complexes were tested for their ability to catalyze the curing of a standard mixture of silicone polymers. The mixture was composed of a vinyl-terminated poly (dimethylsiloxane), with a viscosity of 204 cP and a content of vinyl groups equal to 1.24 mmol/g, as well as a poly (methylsiloxane) with a viscosity of 11.6 cP and a content of hydride groups equal to 5.19 mmol/g. Samples of the two polymers were mixed in order to ensure a 1:1 molar ratio between Si-H bonds and vinyl groups. The catalyst (100 ppm Pt) was pre-dissolved in 1,2-dichloroethane, quickly mixed with the silicone mixture in the dark and then subjected to irradiation or to reaction in the dark; the time needed until the mixture solidified (tsol) was monitored.
Two systems were employed for irradiation: a standard UV lamp for nail reconstruction featuring 4 × 9 W bulbs irradiating in the range 340–380 nm and a blue LED lamp irradiating at 455 nm with an output power at 1.4 W. Samples were placed at a fixed 2 cm or 3 cm distance from the light source, respectively (
Figure 4). Tests were run at room temperature or, in the case of the tests in the dark, in an oven thermostated at 50 °C to speed up the process.
The results from these tests are reported in
Table 1. It can be appreciated that some of the complexes with general stoichiometry [PtCl(O,O′-β-diketonate)L], e.g., complexes
1 and
7, display good catalytic performance under UV irradiation, although the activity appears to be significantly lower compared with the parent Pt(acac)
2. It must be remarked that since complex
1 presents the low energy absorption band shifted to lower wavelength compared to Pt(acac)
2, its absorption capability in the range covered by the employed UV lamp, exemplified by ε
360nm reported in
Table 1, is also markedly lower. Under blue light, where nearly all the complexes show negligible absorption (ε
455nm ~10
1 M
−1 cm
−1), complex
1 becomes markedly less active, whereas the performance of complex
7 is maintained and under these conditions approaches that of the reference compound. This can be explained with the exceptional light absorption capability of complex
7 at 455 nm (ε
455nm 19,800 M
−1 cm
−1), which turns into an efficient activation of the catalyst even at higher wavelengths. On the other hand, and at variance with Pt(acac)
2, these complexes are also able to efficiently promote the curing reaction under dark conditions. Apparently, the substitution of a chelating bidentate ligand such as the second O,O′-β-diketonate with two monodentate ligands is detrimental for the stability of these catalyst precursors under dark conditions. The substitution of DMSO with PPh
3 (i.e., complexes
1 vs.
3) results instead in a dramatic drop in catalytic activity, both under irradiation and in the dark. The reasons for this poor performance need to be further investigated, although it has been suggested that strong soft σ-donor ligands at Pt increase the stability of the catalytic system (by slowing down aggregation to Pt colloids), yet depress its activity, requiring higher reaction temperatures for a good catalytic performance [
4,
5].
The catalytic performance of the complexes of general stoichiometry [Pt(O,O′-acac)(γ-acac)L] is also heavily dependent on the nature of the L ligand. Complex 2, in which L = DMSO, is markedly less efficient under both irradiation conditions compared with the related complex 1, which is coherent with its lower capability to absorb radiation in the near-UV and visible range; furthermore, complex 2 remains quite active in the dark. On the other hand, complex 4, in which L = PPh3, displays a catalytic performance that, although being poor in absolute terms, is evidently much better than the related complex 3, featuring a different stoichiometry yet also bearing a PPh3 ligand. Finally, the catalytic performance of complex 5, in which L = pyridine, is very good under UV irradiation, although it drops considerably under blue light, again in accordance with its UV-Vis absorption features. The improved activity of complex 5 compared to 2 and 4 can be related to its slightly higher absorption in the near UV range compared with the other complexes, though the main reason for this difference is probably the absence of soft ligands in the complex coordination sphere, which tend to deactivate the catalyst. In addition, complex 5 exhibits appreciable activity in the dark, although the activity is somewhat reduced compared to the other catalyst precursors described herein. Finally, complex 6 turns out to be much less efficient than complex 5, which correlates with its lack of a β-diketonate ligand, whose cleavage is facilitated by photoexcitation, whereas complex 9 exhibits a catalytic performance very similar to complex 1, which again can be linked to the similarities in the absorption spectra of the two complexes (both exhibiting a metallacyclic ring with delocalized π-electrons) and in the identical nature of the monodentate L and X ligands in the coordination sphere of the catalyst precursor.
Whereas, as it is sketched in the Introduction, the mechanism of the hydrosilylation reaction involves platinum (0) as a catalytically competent species and is well established, passing through oxidative addition of Si-H, olefin insertion and reductive elimination (Chalk–Harrod mechanism [
27]), the photoactivation mechanism of β-diketonate platinum (II) precatalysts as those shown herein is yet to be determined with precision. Studies by Lewis et al., [
18], as well as by Lavallee et al., [
19] point towards two possible mechanisms, namely: direct photodecomposition to platinum(0) or β-diketonate photoisomerization (from O,O′-dicoordinated to O- or C-monocoordinated) or photodissociation, both liberating coordination sites on the metal center and facilitating substitution reactions [
28], which, depending on the reaction conditions and on the nature of the entering ligands, may ultimately lead to chemical reduction to platinum (0). The prevalence of either mechanism is considered to depend on the reaction conditions, hence further studies are needed in order to ascertain which is the most probable mechanism under our curing conditions.
In conclusion, we have demonstrated that platinum (II) complexes with general stoichiometry (O,O′-β-diketonate)PtLX can be tuned to efficiently act as precatalysts for the photoactivated curing of silicone resins at room temperature using blue light. Critical for success appears to be the absence of ligands L with a soft character and strong σ-donating properties, which are detrimental for catalyst activity at room temperature, as well as the use of a β-diketonate ligand building up with the platinum (II) center a metallacyclic system with a more extensive electron delocalization, which shifts the absorption of the complex towards the visible range and consequently enables a more efficient photoactivation. In this respect, complexes with chloride as the X ligand exhibit an absorption spectrum shifted slightly more towards higher wavelengths compared with complexes in which γ-acac is the X ligand, and appear consequently better suited for the scope. We are currently working towards a more extensive optimization of the ligands at platinum in order to develop a new generation of precatalysts with features surpassing the benchmark complex Pt(acac)2. On the other hand, platinum (II) complexes with the stoichiometry investigated herein do not ensure the stability under dark conditions that would be necessary to commercialize similar silicone mixtures as photocurable one component formulations, although it should be borne in mind that low amounts of inhibitors can be employed in this context to suppress this dark reactivity while maintaining a prompt activation under irradiation. Work aimed at the rational use of inhibitors in this context is currently underway.
3. Materials and Methods
All manipulations of air and moisture sensitive compounds were carried out using standard Schlenk techniques or in a glove box under an atmosphere of argon or dinitrogen. Silicone polymers were technical grade and were provided by Zhermack (Badia Polesine, Italy). Other chemicals were purchased from Merck (Darmstadt, Germany) as high-purity products and generally used as received; all solvents were purified and dried by standard methods. Complexes
1 and
2 [
20],
3 and
4 [
21],
5 and
6 [
23] were prepared following previously published procedures. NMR spectra were recorded at 25 °C on a Bruker (Billerica, MA, USA) Avance spectrometer working at 300 MHz (300.1 MHz for
1H, 75.5 MHz for
13C and 121.5 MHz for
31P). Chemical shifts (δ) are reported in units of ppm relative to the residual solvent signals and to external 85% H
3PO
4 (for
31P). UV-Vis analyses were carried out on a Varian Cary 100 Bio spectrophotometer. ESI-MS analyses were performed using an LCQ Duo (Thermo Finnigan, San José, CA, USA) operating in positive ion mode. Instrumental parameters were: capillary voltage 10 V, spray voltage 4.5 kV, capillary temperature 200 °C, mass scan range from 150 to 2000 amu, N
2 was used as sheath gas, the He pressure inside the trap was kept constant. The pressure directly read by an ion gauge (in the absence of the N
2 stream) was 1.33 × 10
−5 Torr. Sample solutions were prepared by dissolving the compounds in acetonitrile. Sample solutions were directly infused into the ESI source by a syringe pump at 8μL/min flow rate. Elemental analyses were carried out by the microanalytical laboratory of the Chemical Sciences Department (University of Padova) with a Fisons EA 1108 CHNS-O apparatus.
3.1. Synthesis of Complex 7
TBDMS-protected curcumin was prepared from commercial curcumin following a published procedure [
29].
0.104 g (0.18 mmol) of TBDMS-protected curcumin was stirred with 0.023 g (0.20 mmol) Na2CO3 in 4 mL dry DMF for 20 min. The obtained greenish solution was subsequently added dropwise to a solution of 0.0711 g (0.17 mmol) [PtCl2(DMSO)2] in 2 mL anhydrous DMF. The resulting solution was further stirred for 3 h at room temperature, after which the solvent was evaporated to dryness. The resulting solid was treated with 3 mL dichloromethane, solid residues were filtered off, the liquid was again evaporated to dryness and the resulting solid was recrystallized from diethyl ether/hexane. Yield 30 mg (19.5%).
1H NMR (CDCl3): 7.72 (d, 1H, vinyl-Hb, 3JHH = 14 Hz), 7.57 (d, 1H, vinyl-Hb′, 3JHH = 14 Hz), 7.07 (m, 4H, Ph), 6.86 (m, 2H, Ph), 6.53 (d, 1H, vinyl-Ha, 3JHH = 16 Hz), 6.51 (d, 1H, vinyl-Ha′, 3JHH = 16 Hz), 5.86 (s, 1H, OCCHCO), 3.86 (m, 6H, OCH3), 3.56 (m, 6H, CH3/DMSO, JHPt = 11 Hz), 1.01 (s, 18H, tBuSi), 0,19 (broad s, 12H, MeSi). 13C NMR (CDCl3): 175.91, 175.52, 150.64, 150.61, 146.80, 146.77, 141.47, 140.66, 128.59, 122.39, 121.92, 121.45, 121.38, 120.64, 110.71, 110.48, 103.51, 54.90, 44.28, 44.22, 25.09, 17.93. ESI-MS (positive ions, CH3CN); m/z: 927.00 [M + Na]+, 1830.83 [M2 + Na]+. Elemental analysis calcd. for C35H53ClO7PtSSi2: C = 46.47%, H = 5.91%, S = 3.54%; found: C = 46.77%, H = 5.68%, S = 3.69%.
3.2. Synthesis of Complex 9
Triazene oxide ligand precursor
8 was prepared using a literature procedure [
30]. 20.6 mg triazene oxide precursor
8 was suspended in 0.6 mL ethanol and 0.04 mL of a 20% solution of KOH in methanol (119.0 mg KOH in 0.6 mL MeOH) was added. The mixture was stirred for 30 min and changed in aspect from a violet suspension to a greenish-yellow solution. 0.45 mL of this solution was subsequently added to a suspension of complex
1 (23 mg, 0.064 mmol) in 1 mL ethanol and stirred for 1 h yielding a dark yellow suspension. Stirring was continued overnight, after which the solid was decanted, washed with additional ethanol and dried. Yield 15.3 mg (52%).
1H NMR (CDCl3): 7.24 (d, 2H), 7.13 (d, 2H), 3.98 (s, 3H), 3.45 (s, 6H, JH-Pt = 18 Hz), 2.34 (s, 3H). 13C NMR (CDCl3):135.47, 129.40, 128.31, 123.33, 43.87, 41.44, 20.46. ESI-MS (positive ions, CH3CN); m/z: 494.37 [M + H2O + Na]+, 510.39 [M + H2O + K]+. Elemental analysis calcd. for C10H16ClN3O2PtS: C = 25.40%, H = 3.41%, N = 8.89%, S = 6.78%; found: C = 25.56%, H = 3.66%, N = 8.55%, S = 6.59%.
3.3. Curing Tests
General procedure. Vinyl-terminated poly-dimethylsiloxane (204 cP viscosity, 1.24 mmol/g vinyl groups content) was mixed with poly-methylsiloxane (11.6 cP viscosity, 5.19 mmol/g hydride groups content) in a 22:1 weight ratio, corresponding to a 1:1 ratio between vinyl and hydride groups. The catalyst (100 ppm Pt with respect to the silicone mixture) was pre-dissolved in 1,2-dichloroethane (0.5 mL per 10 g silicone mixture) and then quickly mixed mechanically with the silicone mixture in the dark. The mixture was subsequently transferred in open low-rim cylindrical plastic containers to form an 0.5 cm thick layer, placed in a hood shielded from light and continuously irradiated using either a standard UV lamp for nail reconstruction featuring 4 × 9 W bulbs irradiating in the range 340–380 nm or a blue LED lamp irradiating at 455 nm with an output power at 1.4 W. Samples were placed at a fixed 2 cm or 3 cm distance from the light source, respectively. Alternatively, samples were placed in a light-shielded oven and either left at room temperature or thermostated at 50 °C for the dark tests. The solidification time tsol was determined by regularly checking the samples until the mixture was no longer free flowing.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}