Abstract
(S)-N-Boc-3-hydroxypiperidine is an important intermediate of the anticancer drug ibrutinib and is mainly synthesized by the asymmetric reduction catalyzed by ketoreductase coupled with glucose dehydrogenase at present. In this study, the coexpression recombinant strains E. coli/pET28-K-rbs-G with single promoter and E. coli/pETDuet-K-G with double promoters were first constructed for the coexpression of ketoreductase and glucose dehydrogenase in the same cell. Then, the catalytic efficiency of E. coli/pET28-K-rbs-G for synthesizing (S)-N-Boc-3-hydroxypiperidine was found to be higher than that of E. coli/pETDuet-K-G due to the more balanced activity ratio and higher catalytic activity. On this basis, the catalytic conditions of E. coli/pET28-K-rbs-G were further optimized, and finally both the conversion of the reaction and the optical purity of the product were higher than 99%. In the end, the cell-free extract was proved to be a better catalyst than the whole cell with the improved catalytic efficiency of different recombinant strains. This study developed a better coexpression strategy for ketoreductase and glucose dehydrogenase by investigating the effect of activity ratios and forms of the biocatalysts on the catalytic efficiency deeply, which provided a research basis for the efficient synthesis of chiral compounds.
1. Introduction
Chiral alcohols are a class of important compounds that can be used as chiral synthetic blocks for a variety of drugs, natural products and agricultural chemicals. Asymmetric reduction is considered to be one of the most powerful methods for the synthesis of chiral alcohols [1,2]. Compared with the chemical reduction method, the biological asymmetric reduction method catalyzed by ketoreductase has the advantages of high selectivity, mild conditions and environmental friendliness. The theoretical yield and atomic utilization rate can reach 100%, which is more in line with the development direction of green chemistry; thus, this method becomes an efficient and green method for the preparation of chiral alcohols [3,4].
The biological asymmetric reduction catalyzed by ketoreductase requires the participation of a coenzyme (NADH or NADPH). Due to the high price of coenzymes, it is necessary to introduce the coenzyme regeneration system to drive the reaction towards the direction of product synthesis. According to whether exogenous enzymes need to be introduced in the reaction process, the coenzyme regeneration system can be divided into substrate coupling type and enzyme coupling type [5]: (1) Substrate coupling coenzyme regeneration system relies on bifunctional ketoreductase with both oxidation and reduction activities. The enzyme can simultaneously catalyze the reduction of substrate and the oxidation of cosubstrate to recycle cofactors. Common cosubstrates include isopropanol and ethanol, among which only the applicability of isopropanol in large-scale production has been verified [6]. The reaction process requires excessive addition of isopropanol, which can easily lead to a decrease in enzyme activity and an increase in production cost. Moreover, the coproduct acetone often needs to be removed from the reaction system to drive the reduction reaction and reduce the toxicity to the enzyme [7]. (2) The enzyme-coupled coenzyme regeneration system needs to introduce at least one extra exogenous enzyme which can catalyze the oxidation of cosubstrate to generate the reduced coenzyme for the asymmetric reduction reaction by ketoreductase. At present, formate dehydrogenase and glucose dehydrogenase have been successfully used in industrial production, among which formate dehydrogenase is not universal due to the low regeneration efficiency for NADPH. Glucose dehydrogenase can be used for the regeneration of NADH and NADPH with high catalytic activity, and it exhibits a wide application range [8,9,10]. The enzyme-coupled coenzyme regeneration system usually needs to construct two recombinant strains with single gene expression, which is cumbersome to operate. In addition, when asymmetric reduction reaction is carried out using two different whole cells, the reaction efficiency will be significantly affected due to the cell membrane barrier effect. The expression of multiple enzymes in the same host cell can avoid the shortcomings of the above reaction process and significantly improve the catalytic efficiency of an enzyme-coupled coenzyme regeneration system [11,12,13].
(S)-N-Boc-3-hydroxypiperidine ((S)-NBHP) is an important intermediate for the treatment of B-cell malignant tumors and is mainly synthesized by the asymmetric reductive reaction catalyzed by ketoreductase with N-Boc-3-piperidone as the substrate. At present, it has been reported that several ketoreductases from different sources can be used for the synthesis of (S)-NBHP. For example, Xu et al. accomplished the efficient biosynthesis of (S)-NBHP by using cell extract expressing recombinant ketoreductase (ChKRED03) and commercial glucose dehydrogenase pure enzyme [14]. Chen et al. reported the biosynthesis of (S)-NBHP, in which the catalysts used were wet cells expressing ketoreductase (YDR541C) and glucose dehydrogenase alone [15]. Zheng et al. used ketoreductase mutant (CgKR1-F92C/F94W) and glucose dehydrogenase to achieve asymmetric synthesis of (S)-NBHP, in which two biocatalysts were freeze-dried cells and cell-free extract, respectively [16]. In the asymmetric reductive reaction catalyzed by ketoreductase coupled with glucose dehydrogenase, the catalytic efficiency was affected not only by the catalytic activity but also by the activity ratio of two biocatalysts and the transfer efficiency of substrates and coenzymes. The biocatalysts used in the above research were mainly prepared by the expression of a single gene; the preparation process was cumbersome, and the cost was high in mass production. Moreover, the effects of activity ratio and forms of the biocatalysts on the catalytic efficiency had also been less considered.
In this study, two kinds of recombinant E.coli coexpressing ketoreductase from Candida glabrata (KRED) [16] and glucose dehydrogenase from Bacillus sp. (GDH) [17] were constructed by different expression strategies, and the effects of different expression strategies on the synthesis of (S)-NBHP catalyzed by KRED and GDH were compared and analyzed from two levels of whole cell and cell-free extract. In the end, a coexpression recombinant E. coli with single promoter, capable of highly efficient asymmetric synthesis of (S)-NBHP, was obtained (Scheme 1).
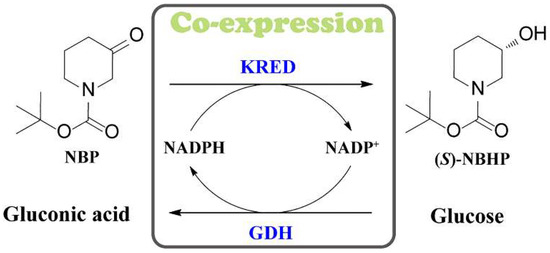
Scheme 1.
Schematic representation of the asymmetric synthesis of (S)-NBHP by coexpressing KRED and GDH.
2. Results
2.1. Construction of Recombinant Strains E. coli/pET28-K-rbs-G and E. coli/pETDuet-K-G
As shown in Figure 1c, KRED and GDH fragments were amplified using primers K1 and K2 and G1 and G2 with pET-KRED and pET-GDH as templates, respectively. Then, KRED-rbs-GDH fragments were further amplified by overlap extension PCR using KRED and GDH fragments as templates and K1 and G2 as primers, with the size of about 1950 bp. The fusion fragment was inserted into pET-28a (+) to obtain the coexpression recombinant plasmid pET28-K-rbs-G. Two fragments, about 5400 bp and 1950 bp, were obtained by the double digestion of pET28-K-rbs-G with Nco I and Xho I, the size of which was in line with expectations.
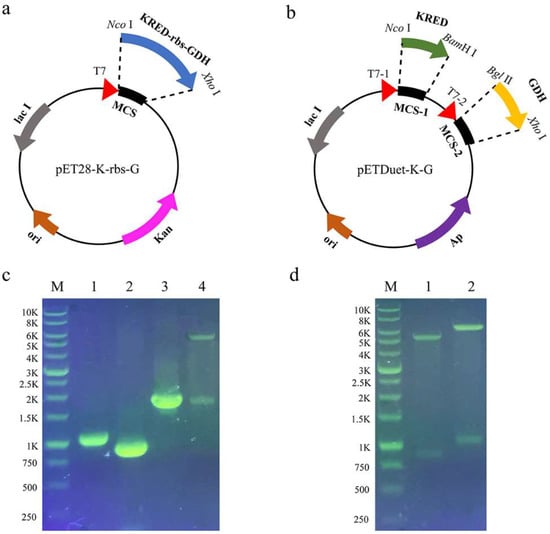
Figure 1.
Schematic representation and identification of the coexpression recombinant plasmids pET28-K-rbs-G and pETDuet-K-G by agarose gel electrophoresis. (a) Single-promotor plasmid pET28-K-rbs-G. (b) Dual-promotor plasmid pETDuet-K-G. (c) Agarose gel electrophoresis analysis of pET28-K-rbs-G. M: DNA marker; lanes 1–4: PCR product of KRED, PCR product of GDH, PCR product of KRED-rbs-GDH, fragments obtained by double digestion of pET28-K-rbs-G with Nco I and Xho I. (d) Agarose gel electrophoresis analysis of pETDuet-K-G. M: DNA marker; lanes 1–2: fragments obtained by double digestion of pETDuet-G with Bgl II and Xho I, fragments obtained by double digestion of pETDuet-K-G with Nco I and BamH I.
KRED and GDH fragments were also amplified using primers K1, KDuet2, GDuet1 and G2. The GDH fragment was inserted into pETDuet to construct the recombinant plasmid pETDuet-G, and two fragments were obtained by the double digestion with Bgl II and Xho I, which were about 5400 bp and 850 bp (Figure 1d), respectively. On this basis, the KRED fragment was inserted, and finally the coexpression recombinant plasmid pETDuet-K-G was constructed. Two fragments were obtained by the double digestion with Nco I and BamH I, and the sizes were about 6250 bp and 1050 bp (Figure 1d), respectively. The fragment size was in line with expectations. The recombinant plasmids pET28-K-rbs-G and pETDuet-K-G were sequenced. After the sequence was confirmed correctly, they were transformed into the expression host E. coli BL21(DE3) to construct the single-promotor coexpression strain E. coli/pET28-K-rbs-G and dual-promotor coexpression strain E. coli/pETDuet-K-G.
2.2. Effects of Different Expression Strategies on the Expression and Activities of KRED and GDH
The coexpression recombinant strains E. coli/pET28-K-rbs-G and E. coli/pETDuet-K-G were induced by 0.1 mmol/L IPTG at 24 °C for 16 h, and the cells were collected and disrupted. The cell-free extract was analyzed by SDS-PAGE, and the strains expressing KRED and GDH alone were used as control (Figure 2). The results showed that the theoretical size of KRED protein in this study was about 38 KDa, and the theoretical size of GDH protein was about 32 KDa due to its N-terminal with His and T7 tags, which were consistent with the size of the target bands in SDS-PAGE. When KRED and GDH were expressed alone, the concentration of target bands was similar. The results of activity determination showed that the activity of KRED against N-Boc-3-piperidone was 31.6 U·g−1 wet cells, and that of GDH against glucose was 40.3 U·g−1 wet cells. In the two coexpression strains, KRED and GDH can be expressed in soluble form, and the target protein size was consistent with that of the protein expressed alone. In the dual-promoter coexpression strain E. coli/pETDuet-K-G, the expression levels of the two enzymes were similar, while in the single-promoter coexpression strain E. coli/pET28-K-rbs-G, the expression level of GDH far from the T7 promoter was significantly lower than that of KRED close to the T7 promoter. The results of activity determination showed that the activity of KRED against N-Boc-3-piperidone in E. coli/pET28-K-rbs-G was 24.1 U·g−1 wet cells, and that of GDH against glucose was 17.9 U·g−1 wet cells. In E. coli/pETDuet-K-G, the enzyme activity of KRED to N-Boc-3-piperidone was 16.8 U·g−1 wet cells, and that of GDH to glucose was 25.4 U·g−1 wet cells.
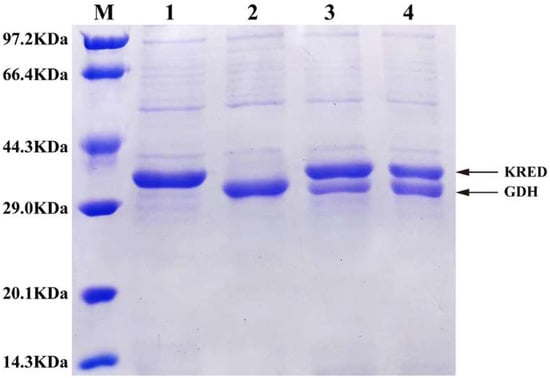
Figure 2.
SDS-PAGE analysis of KRED and GDH obtained by different expression strategies. M: protein marker; lanes 1–4: cell-free extract of the strain expressing KRED alone, cell-free extract of the strain expressing GDH alone, cell-free extract of E. coli/pET28-K-rbs-G, cell-free extract of E. coli/pETDuet-K-G.
2.3. Effect of Activity Ratio on the Asymmetric Synthesis of (S)-NBHP Catalyzed by KRED and GDH
According to the activity of wet cells expressing KRED and GDH alone, on the basis of constant total amount of two enzymes added, the catalytic reaction was carried out according to the activity ratio of 10:1–1:10, and the reaction conversion and the optical purity of the product were determined. As shown in Figure 3, when the activity ratio of KRED to GDH was higher than 1:1, the reaction conversion was gradually improved with the increase in the activity ratio of GDH. When the activity ratios of KRED to GDH were 2.5:1 and 1:1, respectively, the conversions at 24 h were all higher than 95%; when the activity ratio of KRED to GDH was lower than 1:1, the reaction conversion decreased significantly with the increase in the activity ratio of GDH. When the ratio of the two enzymes was 1:2.5, the conversion was only 62.8% at 24 h. The activity ratios of KRED to GDH in the coexpression recombinant strains E. coli/pET28-K-rbs-G and E. coli/pETDuet-K-G were 1.35:1 and 1:1.51, respectively. Under the same conditions, the conversion of E. coli/pET28-K-rbs-G at 24 h was 99.2%, significantly higher than that of E. coli/pETDuet-K-G (87.9%). In addition, the reaction with different activity ratios had no significant effect on the optical purity of the product, and the ee values of the product were all higher than 99%.
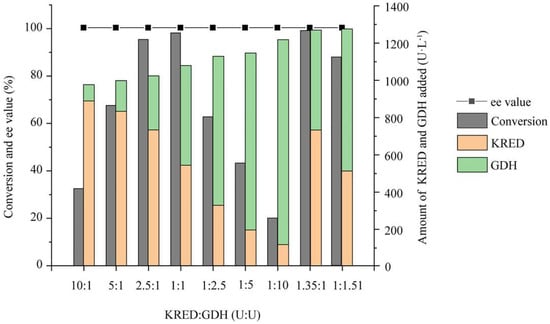
Figure 3.
Asymmetric synthesis of (S)−NBHP catalyzed by various ratios of KRED to GDH. The activity ratios of KRED to GDH in E. coli/pET28−K−rbs−G and E. coli/pETDuet−K−G were 1.35:1 and 1:1.51, respectively. All values of conversion are means from three individual measurements and standard deviations did not exceed 5%.
2.4. Optimization of the Catalytic Conditions of E. coli/pET28-K-rbs-G
In order to further improve the catalytic efficiency of the coexpression strain E. coli/pET28-K-rbs-G, the effects of coenzyme NADP+ concentration, glucose concentration, temperature and pH on the asymmetric reduction to prepare (S)-NBHP were investigated.
When the exogenous coenzyme was not added and only a small amount of coenzyme existed in the cell itself, the catalytic efficiency was low with the conversion at 24 h of 54.7%, and the addition of NADP+ can significantly improve the reaction efficiency. In the range of 0–0.2 g·L−1, the reaction efficiency was improved significantly with the increase in NADP+ concentration. Further increase in NADP+ concentration led to a slow increase in the reaction efficiency (Figure 4a). Therefore, 0.2 g·L−1 was selected as the optimal amount of NADP+ added. At 20–35 °C, the conversion increased significantly with the increase in temperature, and the conversion was 99.1% at 35 °C for 24 h. The conversion decreased gradually over 35 °C, and the conversion was only 39.6% at 45 °C for 24 h (Figure 4b). Therefore, 35 °C was selected as the optimal temperature for the reaction. During the reaction, the reaction conversion first increased and then decreased with the increase in pH. The conversion was the highest at pH = 6.5, reaching 99.3% after 24 h (Figure 4c). Therefore, 6.5 was selected as the optimal pH for the reaction.
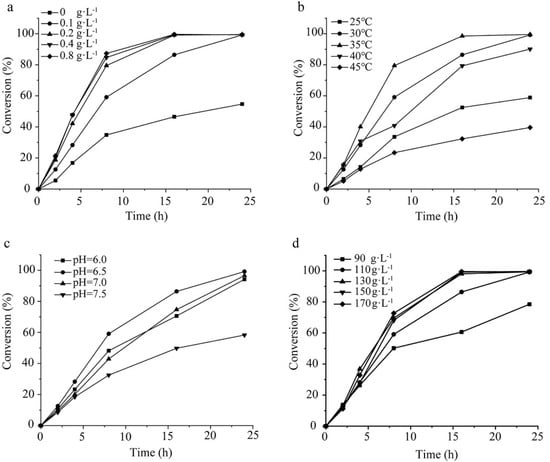
Figure 4.
Optimization of the catalytic conditions of E. coli/pET28−K−rbs−G. (a) NADP+ concentration. (b) Temperature. (c) pH. (d) Glucose concentration. All values are means from three individual measurements and standard errors of mean did not exceed 5%.
The addition of cosubstrates has an important influence on the reaction conversion, and it is usually necessary to add excessive cosubstrates to ensure that the substrate can be converted as much as possible. However, the addition of excessive cosubstrates will increase the cost of production on one hand and affect the postextraction of products on the other hand. Therefore, we optimized the concentration of cosubstrate glucose in this study to determine the optimal amount added. The results (Figure 4d) showed that the reaction rate increased significantly with the increase in glucose concentration in the range of 90–130 g·L−1. When the glucose concentration was 130 g·L−1, the conversions at 8 h and 24 h were 69.6% and 99.4%, respectively. Upon a further increase in the concentration of glucose, the catalytic efficiency did not increase significantly. Therefore, 130 g·L−1 was selected as the optimal amount of glucose added.
2.5. Asymmetric Reduction Reaction under the Optimized Conditions
The whole cell and cell-free extract of E. coli/pET28-K-rbs-G were used as the catalysts (coexpression group), and the transformation experiments were carried out under the optimized conditions to determine their application effect in the preparation of (S)-NBHP by asymmetric reduction. The recombinant strains expressing KRED and GDH alone were selected as the control (monoexpression group). As shown in Figure 5, when the cell-free extract was used as the catalyst, it was found that the catalytic efficiency was significantly higher than that of whole cell whether in the coexpression group or the monoexpression group. In the coexpression group, the conversion at 8 h reached 90.1% with the cell-free extract as the catalyst, and that of the whole cell was only 44.5% under the same conditions. After 12 h of reaction, the conversion reached 99.2% with the cell-free extract as the catalyst, which was much higher than that of the whole cell (69.8%). The same trend was also found in the monoexpression group. After 8 h and 20 h of reaction, the conversions of monoexpression group were 62.5% and 99.1%, respectively, with the cell-free extract as the catalyst, while those of the whole cell were only 22.1% and 65.6%. Comparing the catalytic efficiency of different expression strategies of KRED and GDH, it was found that under the premise of 30 g·L−1 wet cells added, with the conversion of 99% as the standard, the coexpression group and monoexpression group needed 12 h and 20 h, respectively, with cell-free extract as the catalyst, and the former advanced the completion time of the reaction by 8 h.
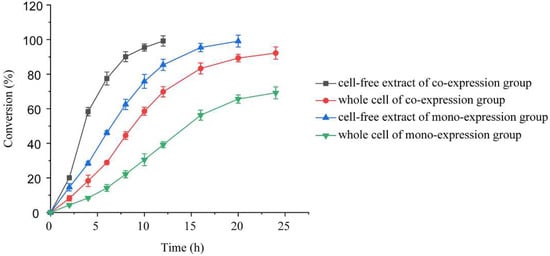
Figure 5.
Time courses of asymmetric synthesis of (S)-NBHP by whole cell and cell-free extract of E. coli/pET28-K-rbs-G (coexpression group) and strains expressing KRED and GDH alone (monoexpression group). All reactions were performed in triplicate, and error bars represent the standard error of the mean.
3. Discussion
In 2020, the global sales of anticancer drug ibrutinib reached USD 9442 million, and it has entered the world’s top five selling drugs. The market demand for its key chiral intermediate (S)-NBHP is increasing rapidly. The asymmetric synthesis of (S)-NBHP by ketoreductase has attracted the attention of global researchers, and several ketoreductases from different sources can be used for the synthesis of (S)-NBHP, in which the coenzyme regeneration is mainly achieved by coupling glucose dehydrogenase reaction [18,19,20]. The cycle efficiency of coenzyme in the glucose dehydrogenase coupled coenzyme regeneration system has an important influence on the enzymatic reaction effect. In the study of asymmetric synthesis of (S)-NBHP by ketoreductase, single gene expression is generally used to prepare ketoreductase and glucose dehydrogenase. If the whole cell is used as the catalyst, the coenzyme needs to cross two layers of cell membrane, and the reaction efficiency is bound to be affected. If cell-free extract or pure enzyme is used as the catalyst, the two enzymes need to be prepared and proportioned separately. The process is cumbersome and the cost is high. In view of this, in this study, ketoreductase from Candida glabrata and glucose dehydrogenase from Bacillus sp. were used as the research objects, and two coexpression systems of single promoter and dual promoters were constructed by different coexpression strategies, and then the efficient coexpression of KRED and GDH was achieved in the same cell.
The results showed that in the asymmetric synthesis of (S)-NBHP, the single-promoter coexpression strain E. coli/pET28-K-rbs-G showed higher catalytic efficiency than the dual-promoter coexpression strain E. coli/pETDuet-K-G. By comparing the effects of different activity ratios of KRED to GDH on the catalytic efficiency, it was found that when the activity ratio of KRED to GDH was between 2.5:1 and 1:1, the reaction conversion was higher than 95%. Under the same amount of wet cell added, although the total activities of KRED and GDH in the two coexpression recombinant strains were close (1260 U·L−1 and 1266 U·L−1, respectively (Figure 3)), the activity ratio of KRED to GDH in E. coli/pET28-K-rbs-G was 1.35:1, and the activity ratio was significantly better than that of E. coli/pETDuet-K-G (1:1.51). In the study of (S)-ketoreductase II and glucose dehydrogenase cocatalyzed synthesis of (S)-phenylethylene glycol, Jiang et al. reported that the optimal ratio of two enzymes was 1:1 [21]. Zhou et al. constructed a coexpression system of (R)-ketoreductase and glucose dehydrogenase and applied it to the asymmetric synthesis of (R)-1-phenyl-1,2-ethylene glycol [22]. Optimizing the protein expression of the two enzymes resulted in the catalytic efficiencies of the two enzymes being close, so the coenzyme cycle tended to balance. In theory, when the activity ratio of the two enzymes is 1:1, the ratio should be more balanced, which is more conducive to the coenzyme cycle in the reaction system. However, in this study, the enzyme activity of KRED and GDH was measured with the low substrate concentration in a short time, conditions which were significantly different from the conditions of high substrate concentration and long reaction time in the actual transformation process. The differences in the stability and substrate tolerance of KRED and GDH lead to the optimal ratio of the two enzymes in this study being not absolute 1:1.
The optimal concentrations of coenzyme and glucose and the optimal temperature and pH of the reaction system were determined by optimizing the reaction conditions of the single-promoter coexpression strain E. coli/pET28-K-rbs-G with a more balanced ratio of KRED and GDH. Under this condition, when the cell-free extract was selected as the catalyst, the reaction efficiency was the highest, and the conversion at 12 h reached 99.2%. Under the same conditions, the conversion was only 69.8% with the whole cell as the catalyst. The cell membrane is the natural barrier of cells and has selective permeability. In the reaction, substrates, coenzymes and cosubstrates will be hindered from entering the cells quickly to participate in the reaction. Disrupted cells can remove the barrier effect of the cell membrane and significantly enhance the mass transfer effect, thus improving the reaction efficiency [23,24]. In addition, although the amount of wet cells added (30 g·L−1) and the activity ratio of KRED to GDH (1.35:1) were both the same, the catalytic efficiency in the group E. coli/pET28-K-rbs-G was significantly higher than that in the group of strains expressing KRED and GDH alone. It was found that due to the difference in the enzyme activity per unit cell caused by different expression strategies, the total activity of KRED and GDH in the group of E. coli/pET28-K-rbs-G was 1260 U·L−1, which was significantly higher than that in the group of strains expressing KRED and GDH alone (1044 U·L−1), so the catalytic efficiency exhibited a significant difference between the two groups.
The above results indicated that the single-promoter coexpression strain E. coli/pET28-K-rbs-G exhibited not only higher catalytic efficiency than the dual-promoter coexpression strain E. coli/pETDuet-K-G due to the more balanced activity ratio, but also better application effect than the strains expressing KRED and GDH alone owing to the higher catalytic activity. Moreover, the cell-free extract was further validated to be a better catalyst than the whole cell with the improved catalytic efficiency of different recombinant strains in this study. At present, the recombinant enzymes used in this study are mainly obtained by small-scale culture in the shake flasks. In industrial production, the recombinant enzymes are mainly prepared by high-density fermentation in which the culture conditions are quite different from those in the shake flasks. It is not clear whether it will affect the activity ratio of KRED and GDH in E. coli/pET28-K-rbs-G. Subsequent studies will further optimize the high-density fermentation process of the single-promoter coexpression recombinant strain E. coli/pET28-K-rbs-G and verify its application effect in the asymmetric synthesis of (S)-NBHP.
4. Materials and Methods
4.1. Strains, Plasmids and Main Reagents
The strains E. coli JM109 and BL21 (DE3) and plasmids pET28a (+) and pETDuet-1 were preserved in our laboratory. The recombinant plasmids pET-KRED and pET-GDH were constructed and preserved in our laboratory. The primers were synthesized by Genescript (Nanjing, China), as shown in Table 1.

Table 1.
Primers used in this work.
PrimeSTAR HS DNA polymerase, T4 DNA ligase, Nco I, BamH I, Bgl II, Xho I, IPTG, DNA Marker and Protein Marker were purchased from Takara (Dalian, China); plasmid extraction kit and PCR product recovery kit were purchased from Sangon Biotech (Shanghai, China); N-Boc-3-piperidone (NBP), N-Boc-3-hydroxypiperidine, (S)-NBHP and NADP+ were purchased from Aladdin Reagent (Shanghai, China).
4.2. Construction of Coexpression Recombinant Strains
Using plasmid pET-KRED and pET-GDH as templates, the genes encoding KRED and GDH were amplified by primers K1 and K2 and G1 and G2, respectively. Using these two fragments as templates, KRED-rbs-GDH fragment was further amplified by overlap extension PCR with primers K1 and G2. The KRED-rbs-GDH fragment was inserted between Nco I and Xho I in pET-28a (+) after double digestion, and the recombinant plasmid pET28-K-rbs-G was constructed (Figure 1a). The GDH fragment was also amplified by using primers GDuet1 and G2 with plasmid pET-GDH as the template. After double digestion, it was inserted between Bgl II and Xho I downstream of the T7-2 promoter in pETDuet-1 to construct the recombinant plasmid pETDuet-G. The KRED fragment was then amplified by using primers K1 and KDuet2 with plasmid pET-KRED as the template. After double digestion, it was inserted between Nco I and BamH I downstream of the T7-1 promoter in pETDuet-G to construct the coexpression recombinant plasmid pETDuet-K-G (Figure 1b). The coexpression recombinant plasmids pET28-K-rbs-G and pETDuet-K-G were transformed into the expression host E. coli BL21 (DE3). The single-promotor coexpression strain E. coli/pET28-K-rbs-G and dual-promotor coexpression strain E. coli/pETDuet-K-G were obtained by kanamycin or ampicillin resistance plate screening.
4.3. Expression of Recombinant Enzymes and Determination of Enzyme Activity
The constructed recombinant strain was inoculated into LB liquid medium containing 50 mg·L−1 kanamycin or 100 mg·L−1 ampicillin and cultured overnight at 37 °C and 200 r/min. The cells were transferred into 1 L of the same medium with 1% inoculation amount and cultured to OD600 of 0.6–0.8. IPTG with final concentration of 0.1 mmol·L−1 was added to the medium. After induction at 24 °C for 18 h, the cells were collected by centrifugation. After being washed with phosphate buffer, the cells were resuspended and disrupted by using a high-pressure cell disruptor (ATS, Suzhou, China) at 80 MPa. The supernatant (cell-free extract) was obtained by centrifugation and used for SDS-PAGE analysis and activity determination.
In this study, KRED and GDH were both coenzyme NADP(H)-dependent enzymes, and the enzyme activity was determined based on the difference in the absorbance of oxidized and reduced coenzymes at 340 nm. KRED activity was determined as follows: The total reaction volume was 100 μL, and 100 mmol·L−1 PBS buffer (pH = 6.5), 0.5 mmol·L−1 NADPH and 10 mmol·L−1 N-Boc-3-piperidone were added. The absorbance at 340 nm was monitored after adding an appropriate amount of enzyme solution at 30 °C for 2 min. GDH activity was determined as follows: The total reaction volume was 100 μL, and 100 mmol·L−1 PBS buffer (pH = 6.5), 2 mmol·L−1 NADP+ and 100 mmol·L−1 D-glucose were added. After adding an appropriate amount of enzyme solution at 30 °C for 2 min, the change of absorbance at 340 nm was monitored. The enzyme activity unit was defined as follows: under the above conditions, the amount of enzyme required to oxidize (reduce) 1 μmol NADP(H) per minute was one enzyme activity unit.
4.4. Analysis of the Optimal Activity Ratio of KRED to GDH for Asymmetric Synthesis of (S)-NBHP
According to the activity of wet cells expressing KRED and GDH alone, the catalytic reaction was carried out according to the activity ratios of 10:1, 5:1, 2.5:1, 1:1, 1:2.5, 1:5 and 1:10, and the results were compared with those of the coexpression recombinant strains E. coli/pETDuet-K-G and E. coli/pET28-K-rbs-G. The reaction system was as follows: 100 g·L−1 N-Boc-3-piperidone, 110 g·L−1 D-glucose, 0.1 g·L−1 NADP+, 100 mmol·L−1 PBS buffer (pH = 6.5) and a suitable amount of cell-free extract prepared by high-pressure disruption of wet cells as the catalyst. Among them, the amount of KRED and GDH added in each reaction system was totally 30 g·L−1, which was calculated according to the amount of wet cells. The reaction was carried out at 30 °C for 24 h, and the pH was controlled at 6.5 with 2 mol·L−1 NaOH solution. At the end of the reaction, the same volume of ethyl acetate was added to the extract. After centrifugation, anhydrous sodium sulfate was added to the organic phase to remove water. The conversion and optical purity of the product were determined by HPLC analysis.
4.5. Optimization of the Catalytic Conditions of E. coli/pET28-K-rbs-G
The initial reaction system was as follows: 100 g·L−1 N-Boc-3-piperidone, 110 g·L−1 D-glucose, 0.1 g·L−1 NADP+, 100 mmol·L−1 PBS buffer and 30 g·L−1 cell-free extract as the catalyst. The reaction temperature was 30 °C, and the pH was controlled at 6.5 with 2 mol·L−1 NaOH solution. Different reaction conditions were optimized according to the following different gradients: the concentrations of NADP+ were 0, 0.1, 0.2, 0.4 and 0.8 g·L−1; the catalytic temperatures were 25, 30, 35, 40 and 45 °C; the reaction pHs were 6.0, 6.5, 7.0, 7.5, 8.0; the glucose concentrations were 90, 110, 130, 150 and 170 g·L−1. Taking samples at the specified time, the reaction conversions were measured at different NADP+ concentrations, temperatures, pHs and glucose concentrations.
4.6. Asymmetric Reduction Reaction under the Optimized Conditions
The reductive reaction of N-Boc-3-piperidone to prepare (S)-NBHP was catalyzed by whole cell and cell-free extract of E. coli/pET28-K-rbs-G (coexpression group) and strains expressing KRED and GDH alone (monoexpression group) at 100 mL scale. The reaction system was as follows: 100 g·L−1 N-Boc-3-piperidone, 130 g·L−1 D-glucose, 0.2 g·L−1 NADP +, 100 mmol·L−1 PBS buffer (pH = 6.5) and 30 g·L−1 wet cells or cell-free extract (calculated according to the amount of wet cells) as the catalyst. The activity ratio of KRED to GDH in the monoexpression group was 1.35:1, which was consistent with that of E. coli/pET28-K-rbs-G. The reaction temperature was 35 °C, and the reaction pH was controlled at 6.5 by 2 mol·L−1 NaOH solution. Taking samples at the specified time, the reaction conversion and the optical purity of the product were determined, and other operations were the same as the above.
4.7. HPLC Detection Method
For the detection of substrate and product concentrations, a TC-C18 reversed-phase chromatographic column (4.6 × 250 mm, 5 μm, Agilent, Shanghai, China) was used. The detection wavelength was 210 nm, the mobile phase was 40% acetonitrile aqueous solution (v/v), the flow rate was 1 mL/min and the column temperature was 30 °C. For the optical purity detection of the product, the chromatographic column was CHIRALPAK IE forward chromatographic column (4.6 × 250 mm, 5 μm, Daicel, Shanghai), the detection wavelength was 210 nm, the mobile phase was 5% isopropanol hexane solution (v/v), the flow rate was 0.8 mL/min and the column temperature was 30 °C.
Author Contributions
X.G. and Q.P. performed the experiments. X.G., N.Z. and S.F. conceived and designed the research. X.Z. and Y.M. analyzed the data and supervised the project. X.G. wrote the initial draft of the paper, and J.L. and S.F. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Natural Science Foundation of Jiangsu Province grant number BK20210144, National Natural Science Foundation of China grant number 22078227, Natural Science Fund for Colleges and Universities in Jiangsu Province grant number 21KJB350008 and Undergraduate Training Program for Innovation and Entrepreneurship of Taizhou University grant number 202012917010Z.
Data Availability Statement
More data can be obtained by request from authors.
Conflicts of Interest
The authors declare that they have no conflict of interest in the publication.
References
- Magano, J.; Dunetz, J.R. Large-scale carbonyl reductions in the pharmaceutical industry. Org. Process Res. Dev. 2012, 16, 1156–1184. [Google Scholar] [CrossRef]
- Cao, J.; Hyster, T.K. Pyridoxal-catalyzed racemization of α-aminoketones enables the stereodivergent synthesis of 1,2-amino alcohols using ketoreductases. ACS Catal. 2020, 10, 6171–6175. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Romero, E.; Jones, B.S.; Hogg, B.N.; Rué Casamajo, A.; Hayes, M.A.; Flitsch, S.L.; Turner, N.J.; Schnepel, C. Enzymatic late-stage modifications: Better late than never. Angew. Chem. Int. Ed. 2021, 60, 16824–16855. [Google Scholar] [CrossRef] [PubMed]
- Kroutil, W.; Mang, H.; Edegger, K.; Faber, K. Recent advances in the biocatalytic reduction of ketones and oxidation of sec-alcohols. Curr. Opin. Chem. Biol. 2004, 8, 120–126. [Google Scholar] [CrossRef]
- Huisman, G.W.; Liang, J.; Krebber, A. Practical chiral alcohol manufacture using ketoreductases. Curr. Opin. Chem. Biol. 2010, 14, 122–129. [Google Scholar] [CrossRef]
- Broussy, S.; Cheloha, R.W.; Berkowitz, D.B. Enantioselective, ketoreductase-based entry into pharmaceutical building blocks: Ethanol as tunable nicotinamide reductant. Org. Lett. 2009, 11, 305–308. [Google Scholar] [CrossRef]
- Kara, S.; Schrittwieser, J.H.; Hollmann, F.; Ansorge-Schumacher, M.B. Recent trends and novel concepts in cofactor-dependent biotransformations. Appl. Microbiol. Biot. 2014, 98, 1517–1529. [Google Scholar] [CrossRef]
- Huang, R.; Chen, H.; Zhou, W.; Ma, C.; Zhang, Y.H.P. Engineering a thermostable highly active glucose 6-phosphate dehydrogenase and its application to hydrogen production in vitro. Appl. Microbiol. Biot. 2018, 102, 3203–3215. [Google Scholar] [CrossRef]
- Stolarczyk, K.; Rogalski, J.; Bilewicz, R. NAD(P)-dependent glucose dehydrogenase: Applications for biosensors, bioelectrodes, and biofuel cells. Bioelectrochemistry 2020, 135, 107574. [Google Scholar] [CrossRef]
- Li, J.; Yu, L.; Li, J.; Zheng, G.; Wang, H. Construction of co-expression system of S-imine reductase and glucose dehydrogenase and synthesis of chiral amine. Biotechnol. Bull. 2019, 35, 105–111. [Google Scholar] [CrossRef]
- Yang, B.; Zheng, P.; Wu, D.; Chen, P. Efficient biosynthesis of raspberry ketone by engineered Escherichia coli coexpressing zingerone synthase and glucose dehydrogenase. J. Agric. Food Chem. 2021, 69, 2549–2556. [Google Scholar] [CrossRef] [PubMed]
- Petrovičová, T.; Markošová, K.; Hegyi, Z.; Smonou, I.; Rosenberg, M.; Rebroš, M. Co-immobilization of ketoreductase and glucose dehydrogenase. Catalysts 2018, 8, 168. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.-P.; Wang, H.-B.; Wu, Z.-L. Efficient bioreductive production of (S)-N-boc-3-hydroxypiperidine using ketoreductase ChKRED03. Process Biochem. 2016, 51, 881–885. [Google Scholar] [CrossRef]
- Chen, L.; Fan, H.; Zhang, Y.; Wu, K.; Wang, H.; Lin, J.; Wei, D. Development of a practical biocatalytic process for (S)-N-boc-3-hydroxypiperidine synthesis. Tetrahedron Lett. 2017, 58, 1644–1650. [Google Scholar] [CrossRef]
- Zheng, G.; Liu, Y.; Chen, Q.; Huang, L.; Yu, H.; Lou, W.; Li, C.; Bai, Y.; Li, A.; Xu, J. Preparation of structurally diverse chiral alcohols by engineering ketoreductase CgKR1. ACS Catal. 2017, 7, 7174–7181. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, R.; Wang, L.; Xu, Y. Gene cloning and characterization of a solvent-resistant glucose dehydrogenase from Bacillus sp. YX-1. Acta Microbiol. Sin. 2013, 53, 561–568. [Google Scholar] [CrossRef]
- Ju, X.; Tang, Y.; Liang, X.; Hou, M.; Wan, Z.; Tao, J. Development of a biocatalytic process to prepare (S)-N-boc-3-hydroxypiperidine. Org. Process Res. Dev. 2014, 18, 827–830. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, B.; Wu, H.; Li, B. A chemo-enzyme method to synthesis of (S)-t-butyl-3-hydroxypiperidine-1-carboxylate. Chin. J. Pharm. 2015, 46, 349–350. [Google Scholar] [CrossRef]
- Ying, X.; Zhang, J.; Wang, C.; Huang, M.; Ji, Y.; Cheng, F.; Yu, M.; Wang, Z.; Ying, M. Characterization of a carbonyl reductase from Rhodococcus erythropolis WZ010 and its variant Y54F for asymmetric synthesis of (S)-N-boc-3-hydroxypiperidine. Molecules 2018, 23, 3117. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, R.; Zhou, X.; Li, K.; Li, J.; Li, Y.; Xu, Y. Efficient biosynthesis of (S)-1-phenyl-1,2-ethanediol catalyzed by (S)-carbonyl reductase II and glucose dehydrogenase. Acta Microbiol. Sin. 2016, 56, 1647–1655. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, R.; Xu, Y.; Liang, H.; Jiang, J.; Xiao, R. Coupled (R)-carbonyl reductase and glucose dehydrogenase catalyzes (R)-1-phenyl-1,2-ethanediol biosynthesis with excellent stereochemical selectivity. Process Biochem. 2015, 50, 1807–1813. [Google Scholar] [CrossRef]
- Ni, Y.; Chen, R. Accelerating whole-cell biocatalysis by reducing outer membrane permeability barrier. Biotechnol. Bioeng. 2004, 87, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Chen, R. Permeability issues in whole-cell bioprocesses and cellular membrane engineering. Appl. Microbiol. Biot. 2007, 74, 730–738. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).