
Birds of a Feather—Asymmetric Organocatalysis Meets Asymmetric Transition Metal Catalysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Rh-Catalyzed Asymmetric Hydrogenation
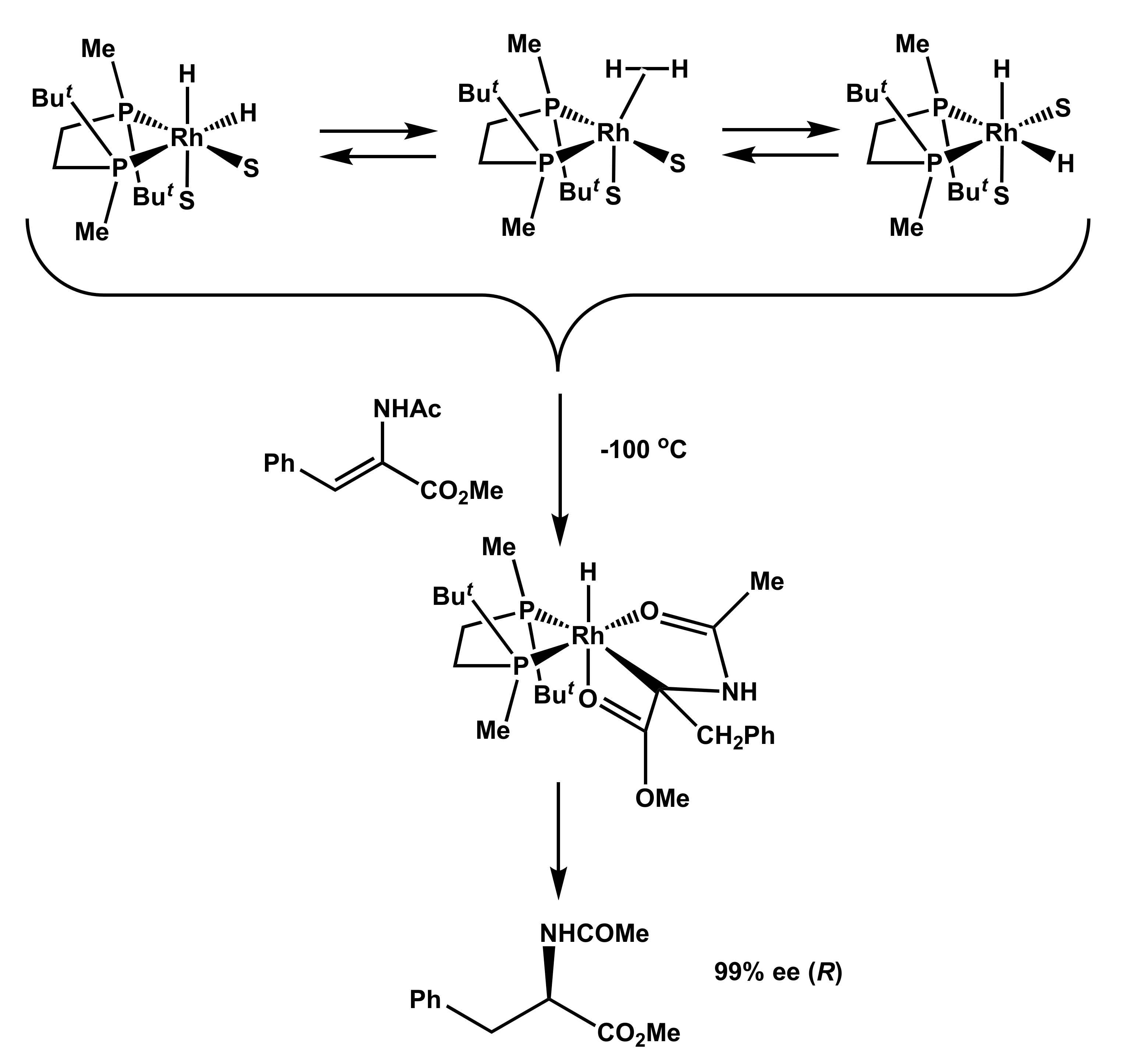
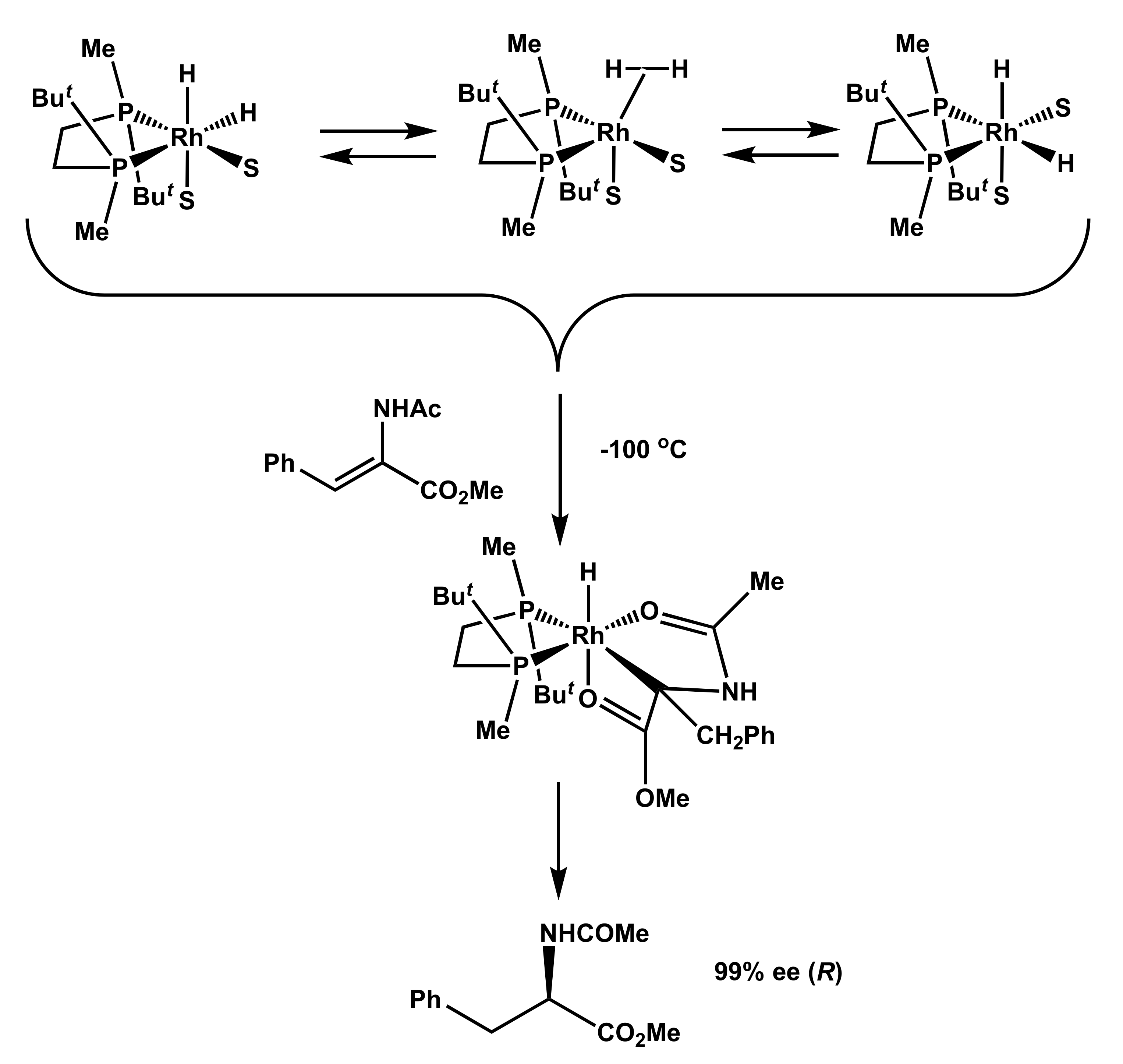
2.1. Low-Temperature Reactions of Solvate Dihydrides with Prochiral Substrates
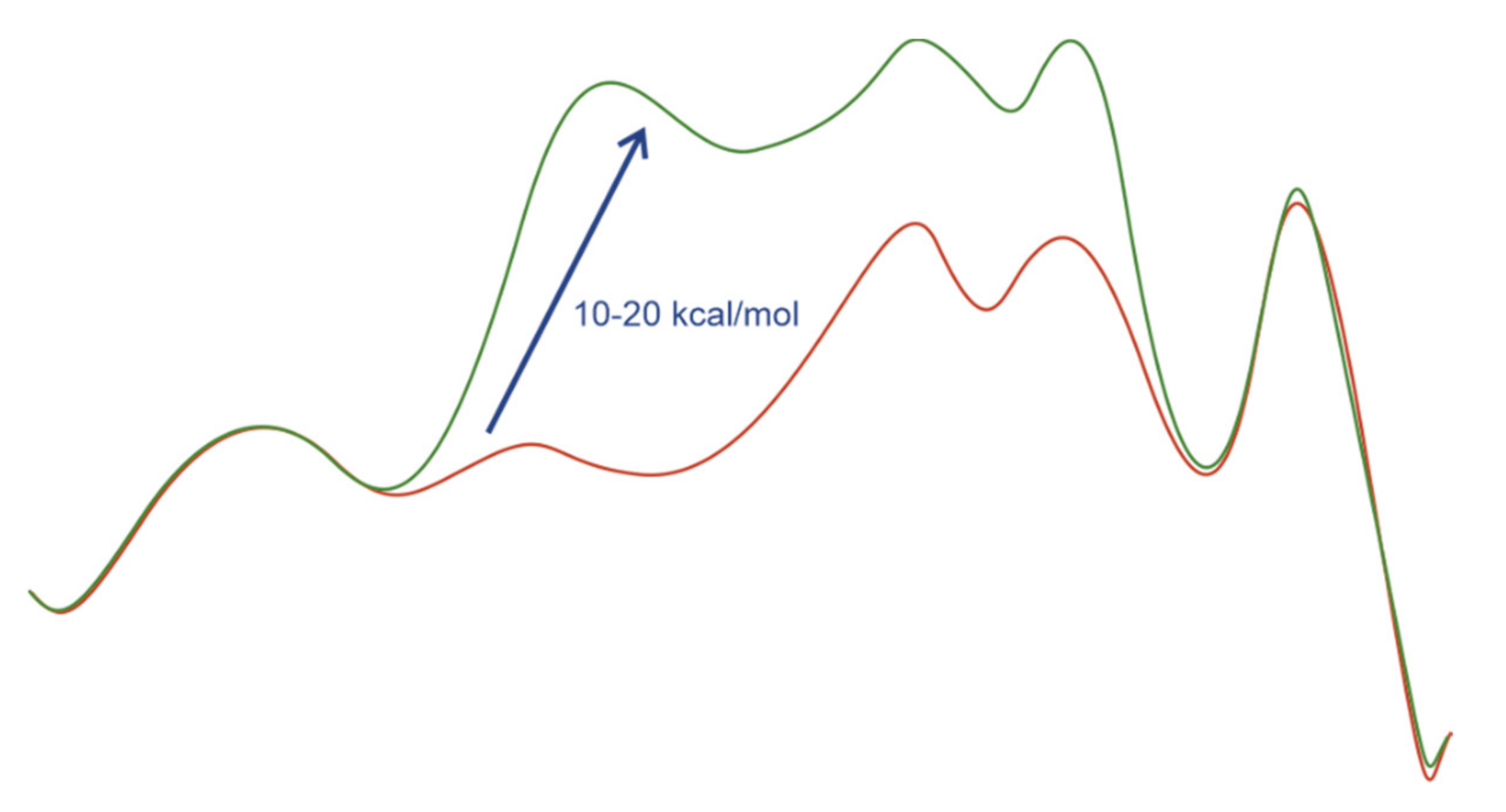
- Stereoselection is possible after hydrogen activation and kinetically viable at significantly decreased temperatures.
- Substrate coordination and migratory insertion steps are extremely fast reactions.
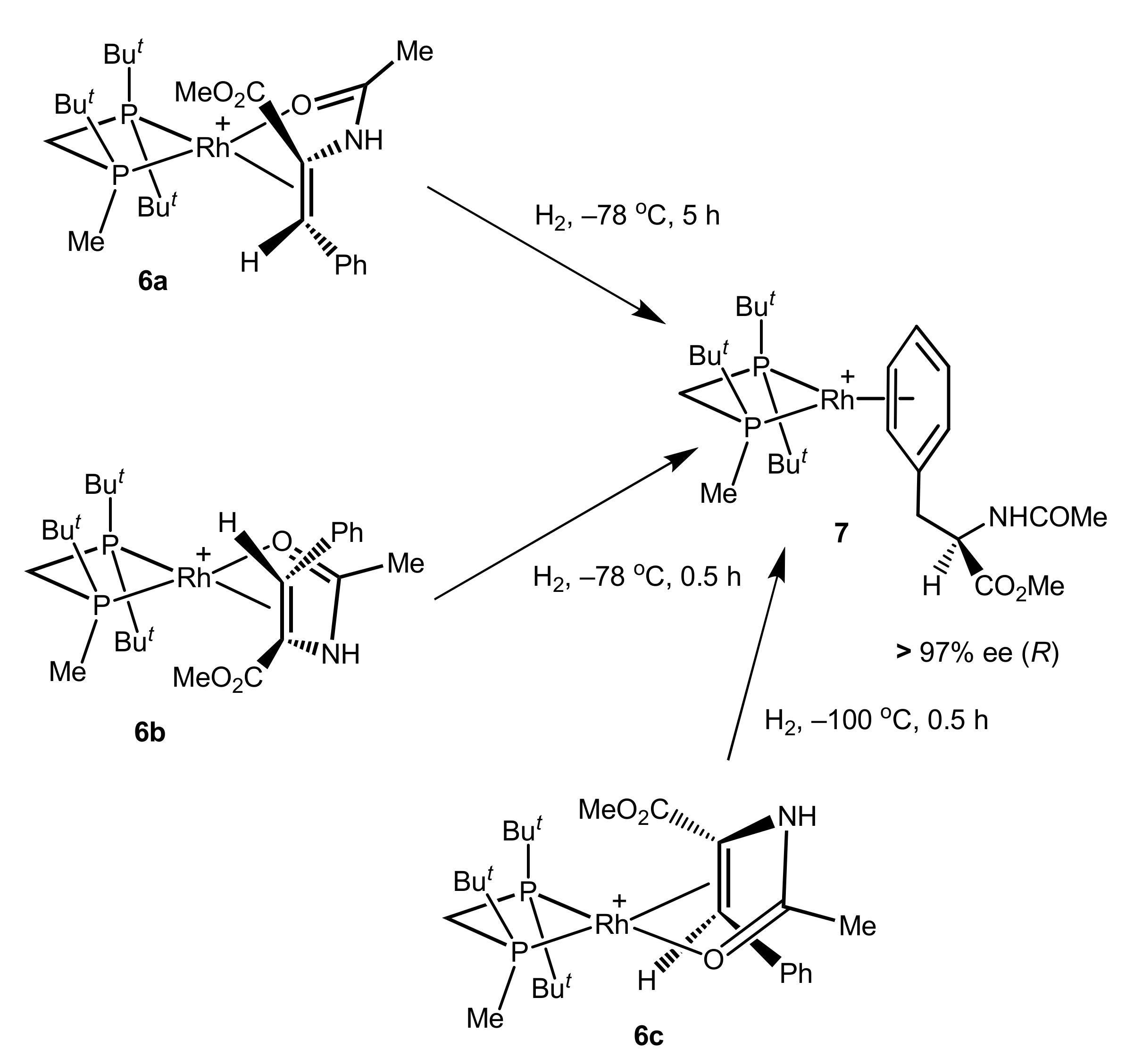
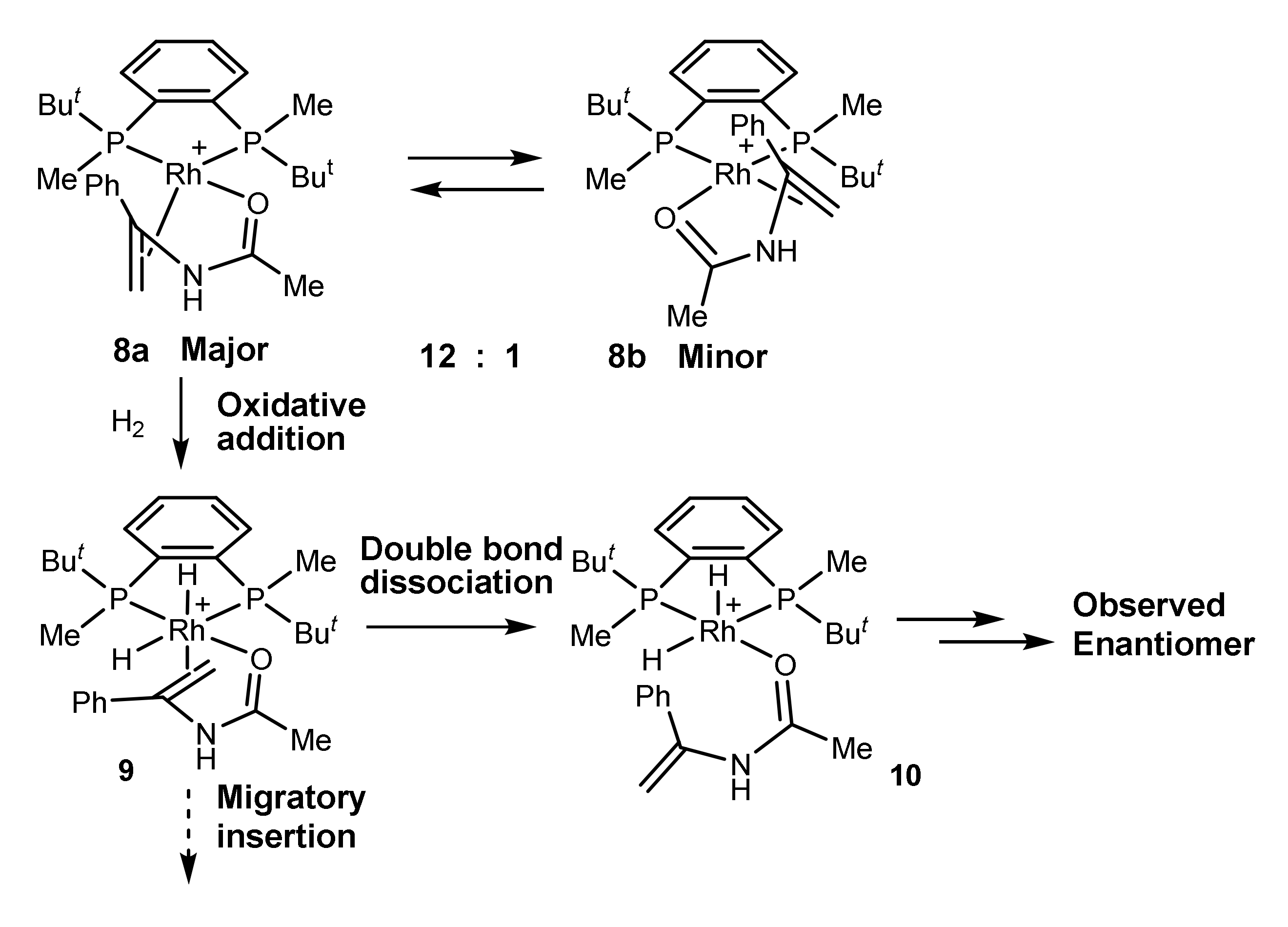
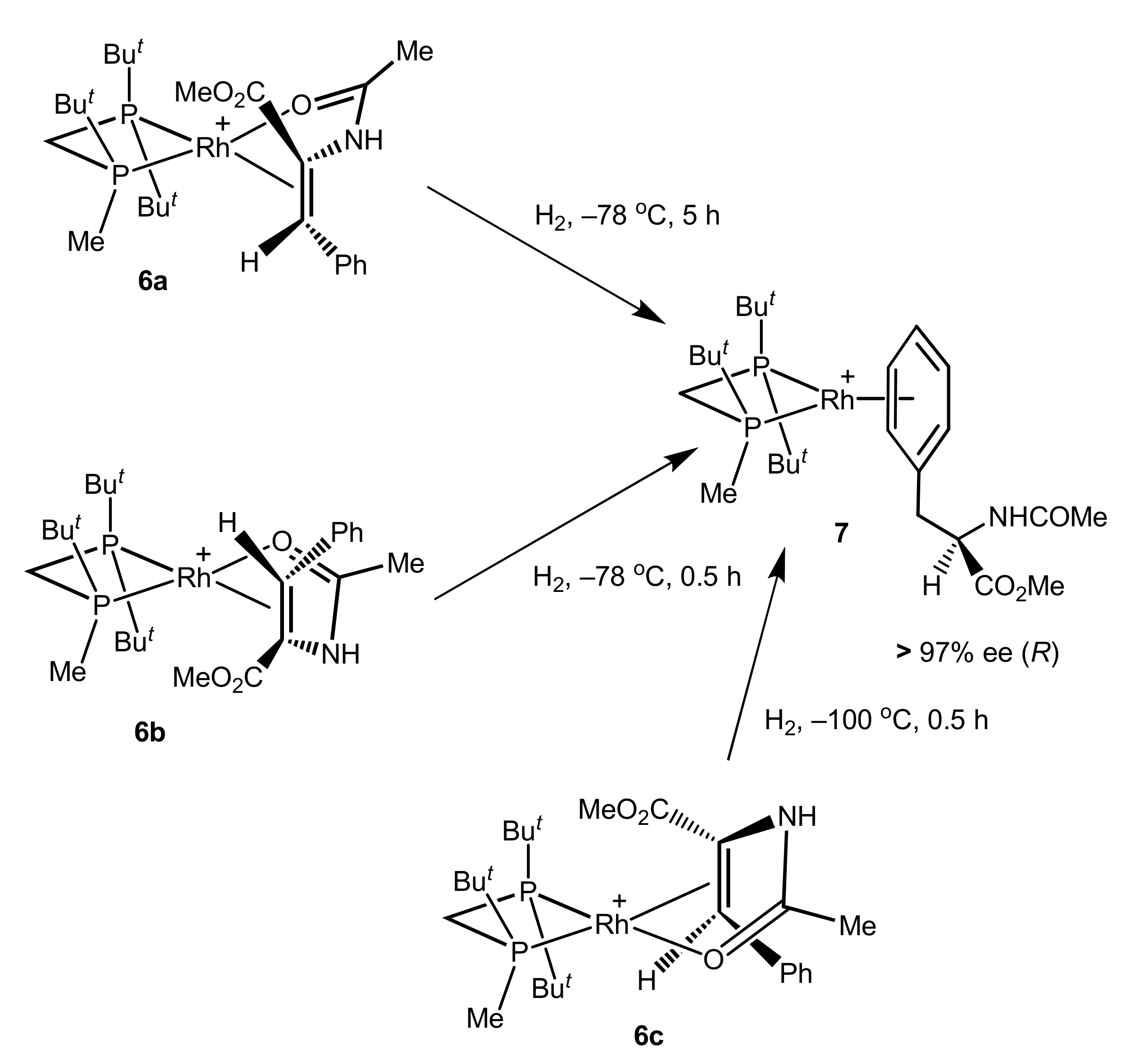
2.2. Same Handedness and Level of Enantioselection in Hydrogenations of Differently Bound Catalyst–Substrate Complexes
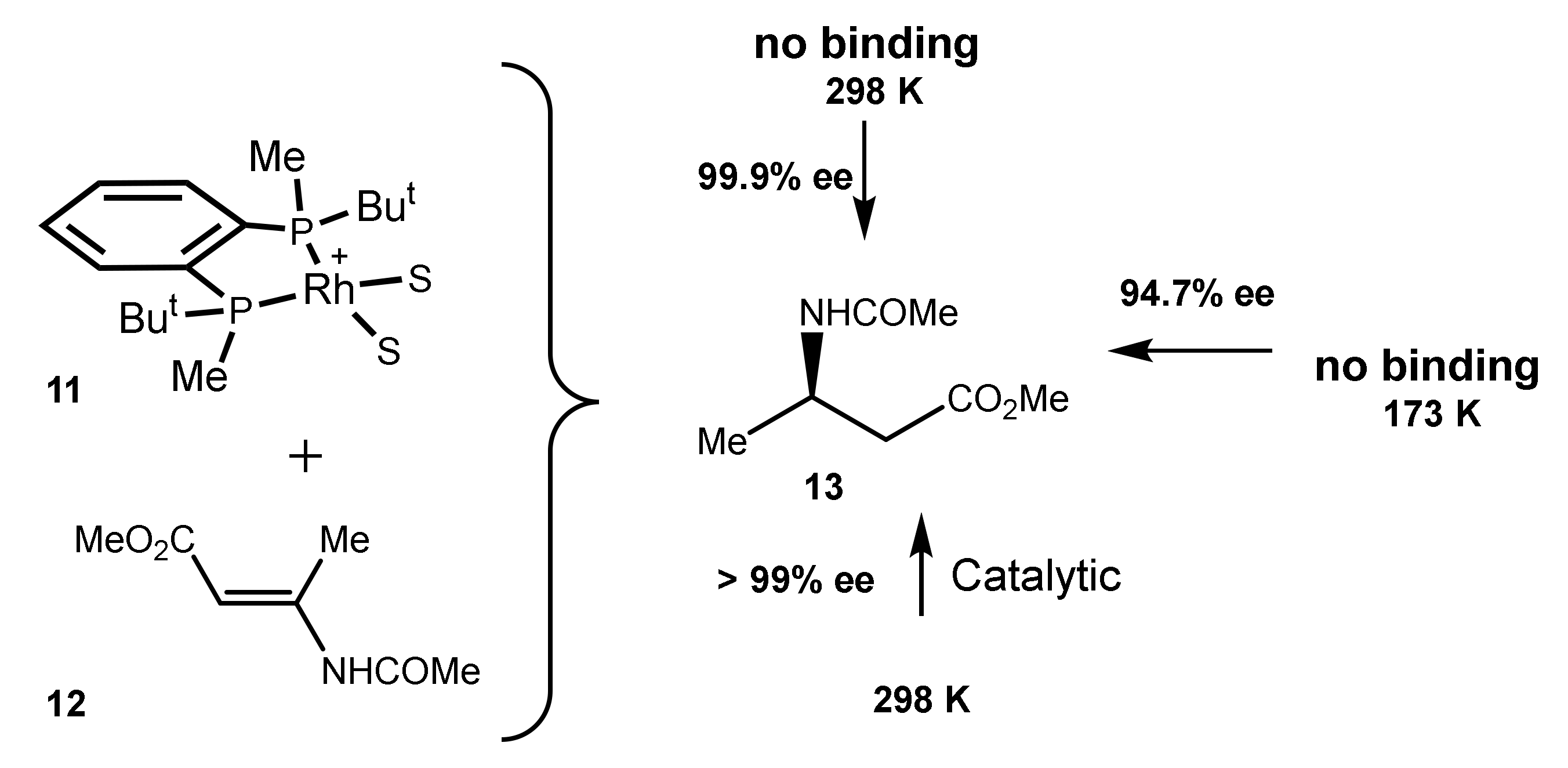
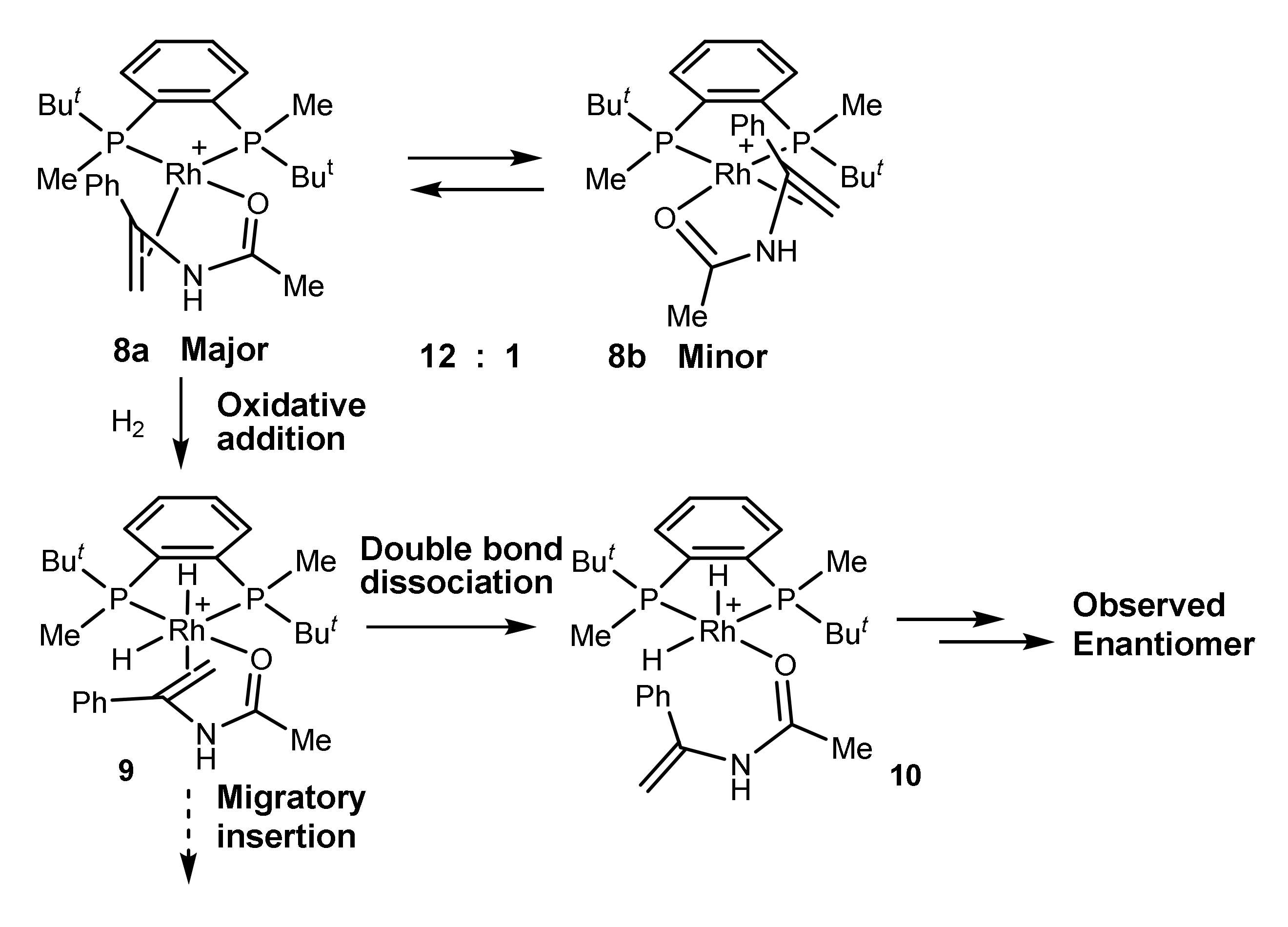
2.3. Perfect Enantioselection without Substrate Binding in Rh(I) Square Complexes
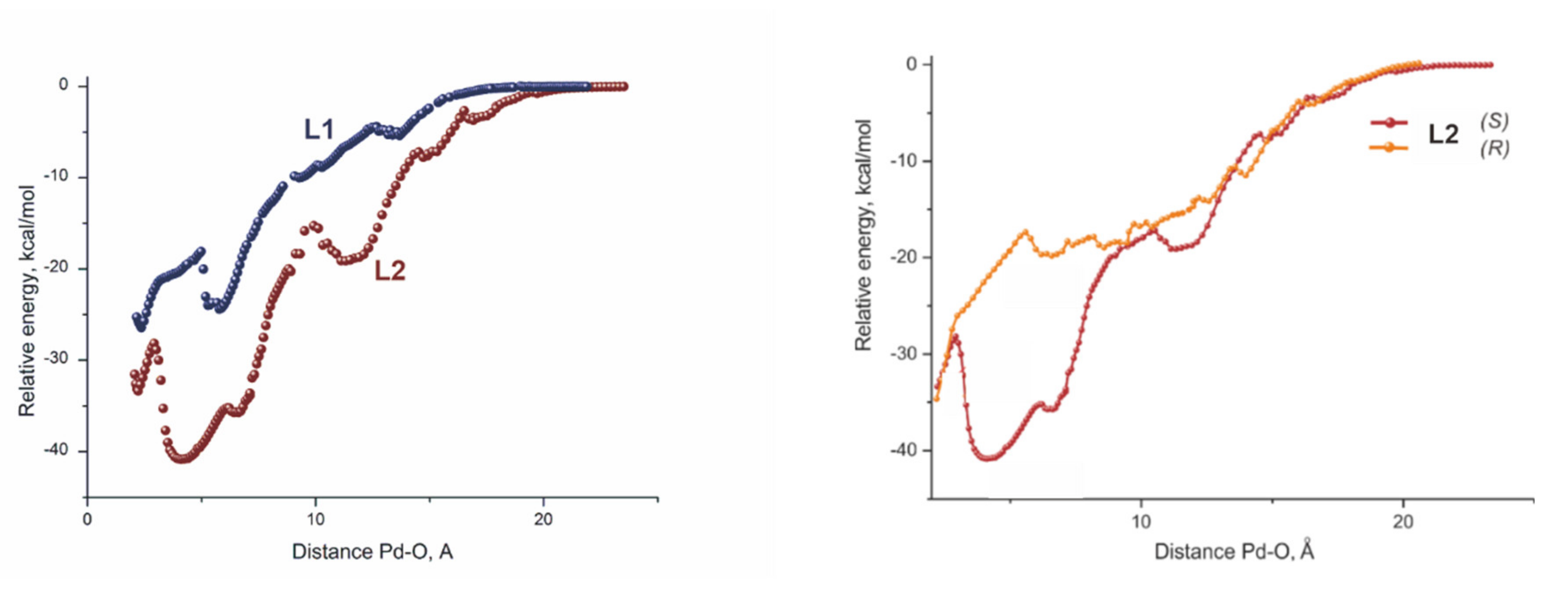
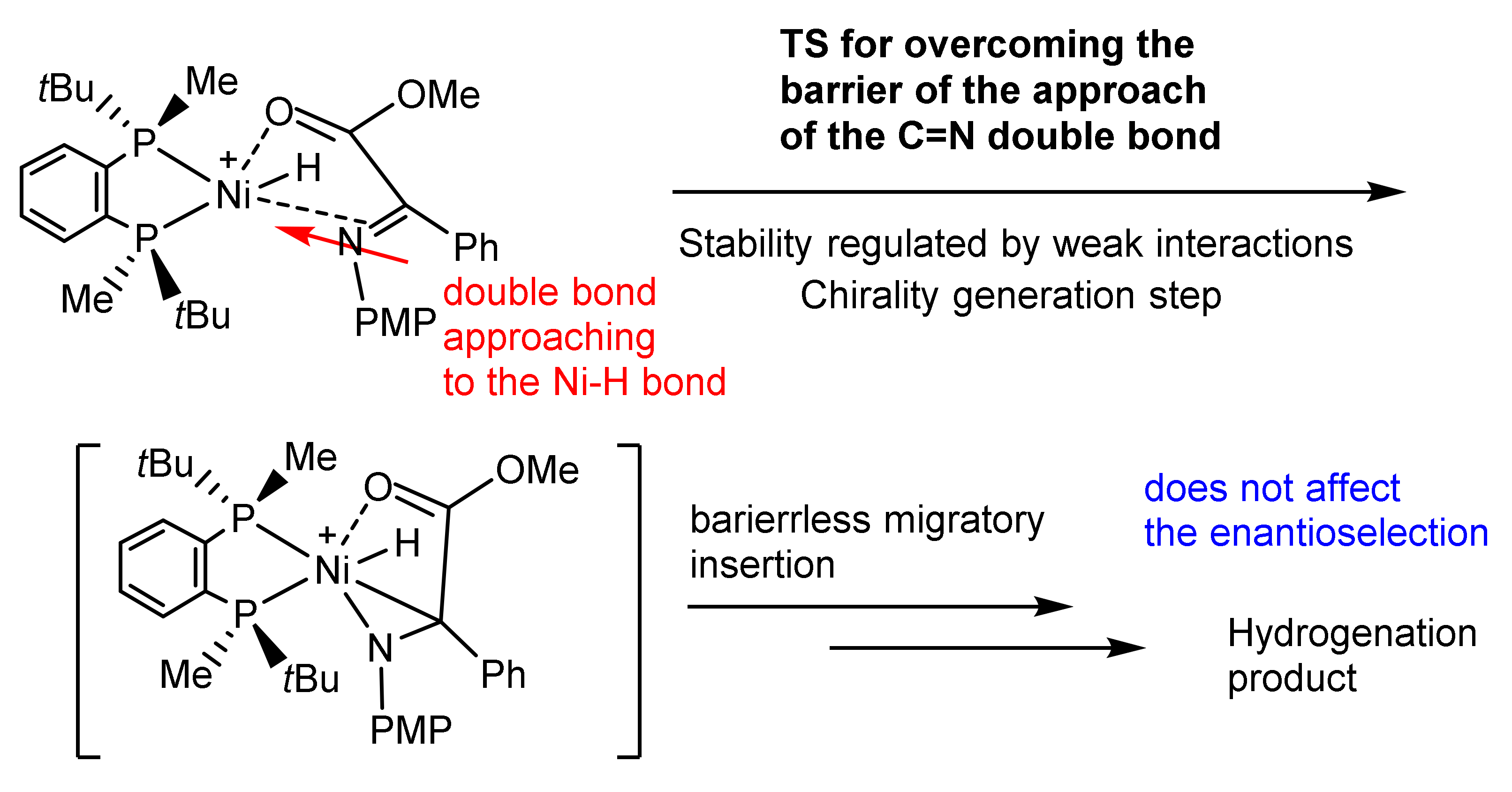
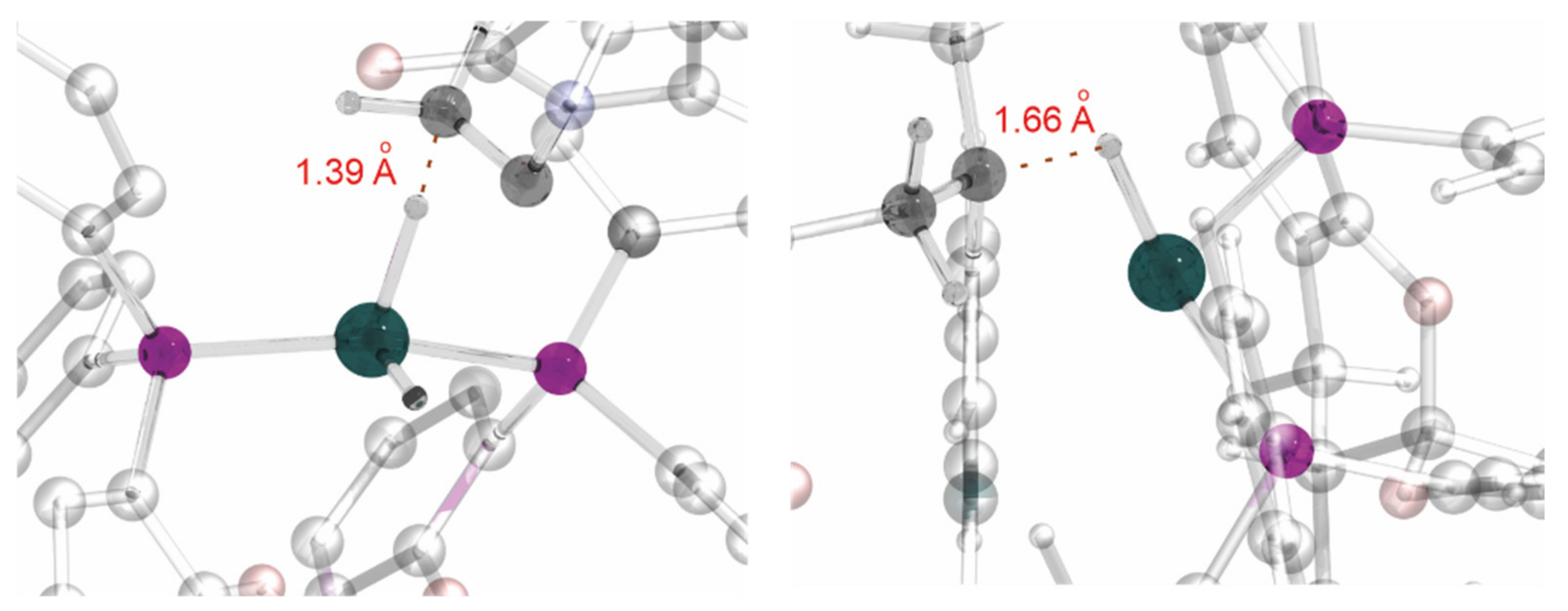
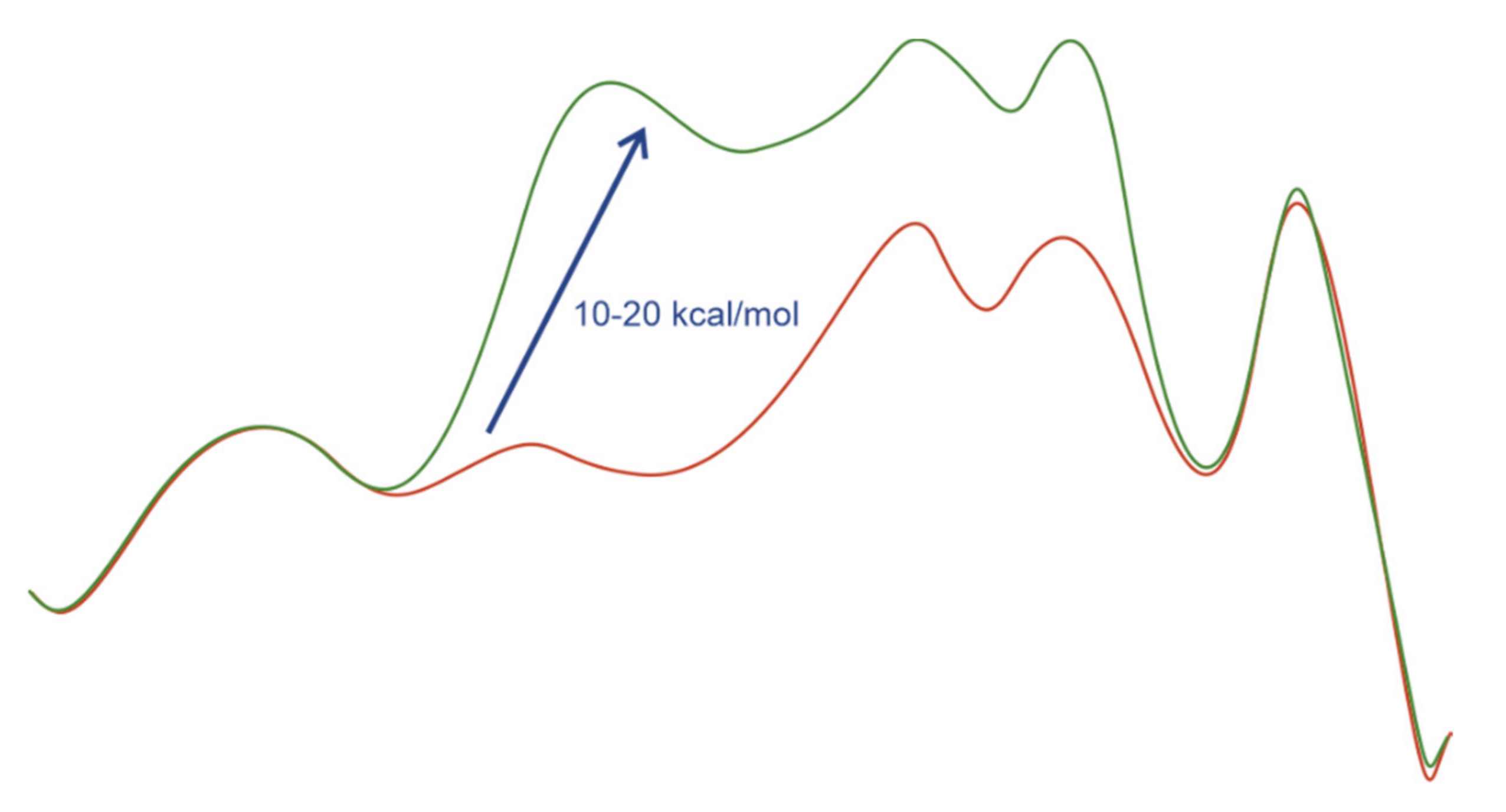
2.4. Computational Evidence
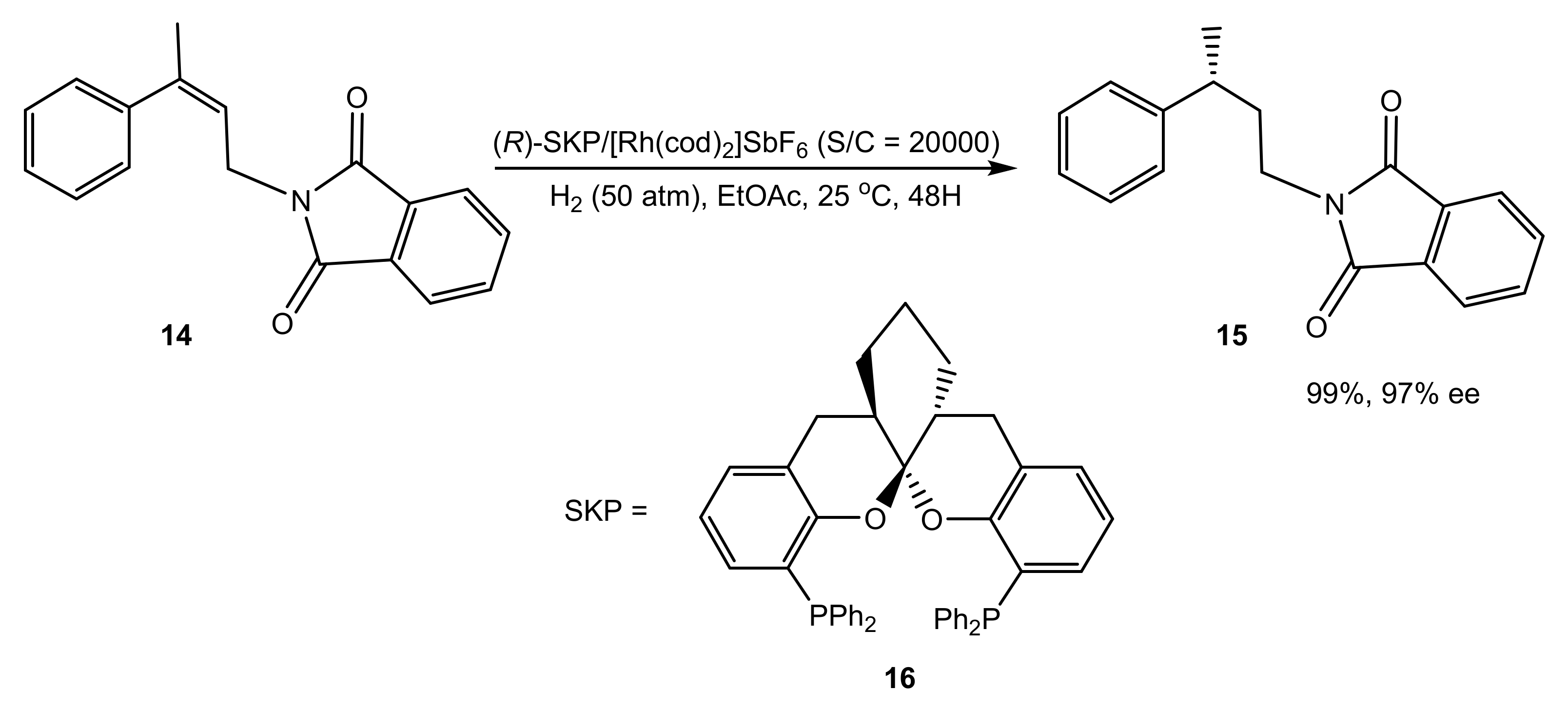
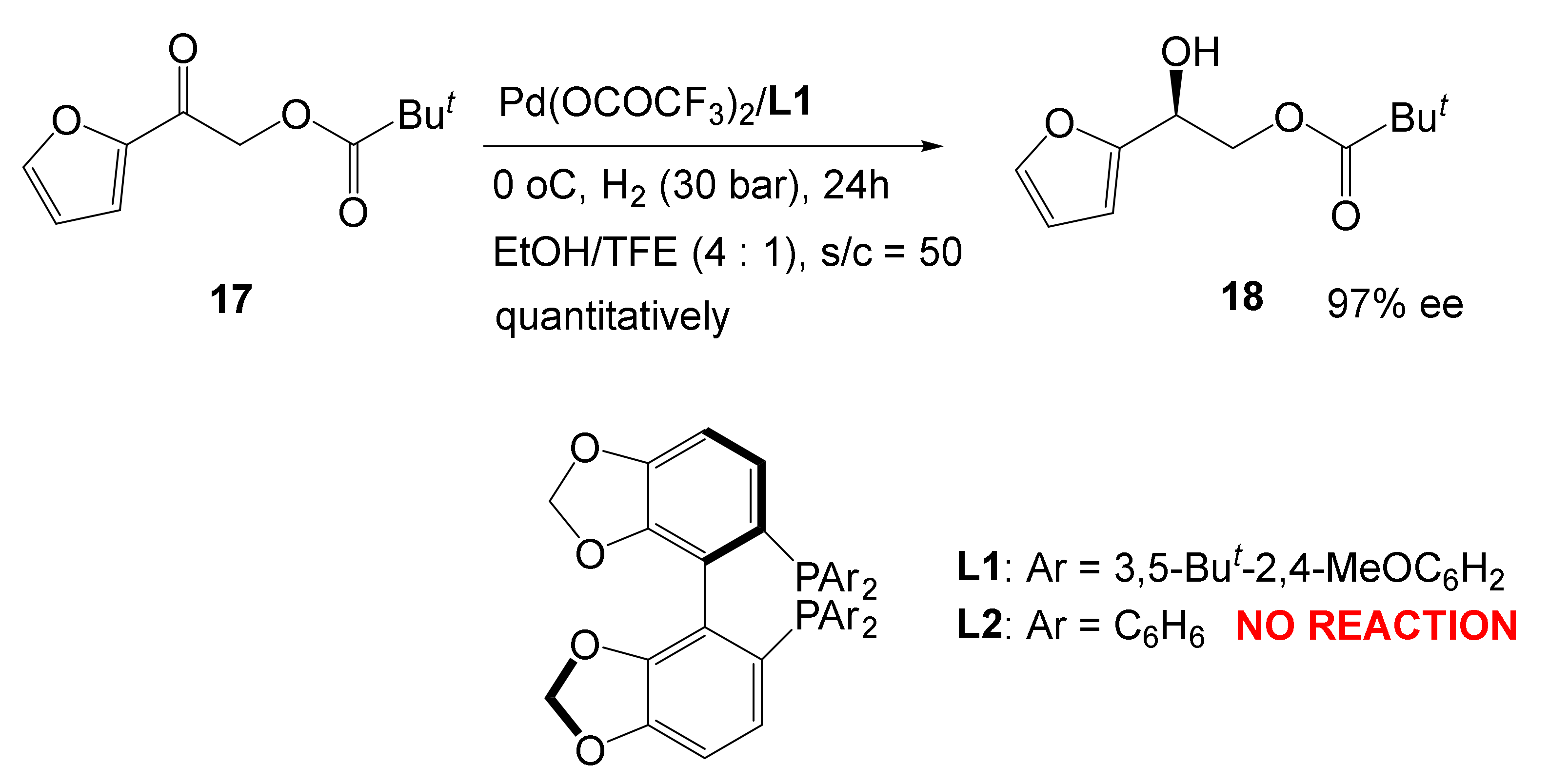
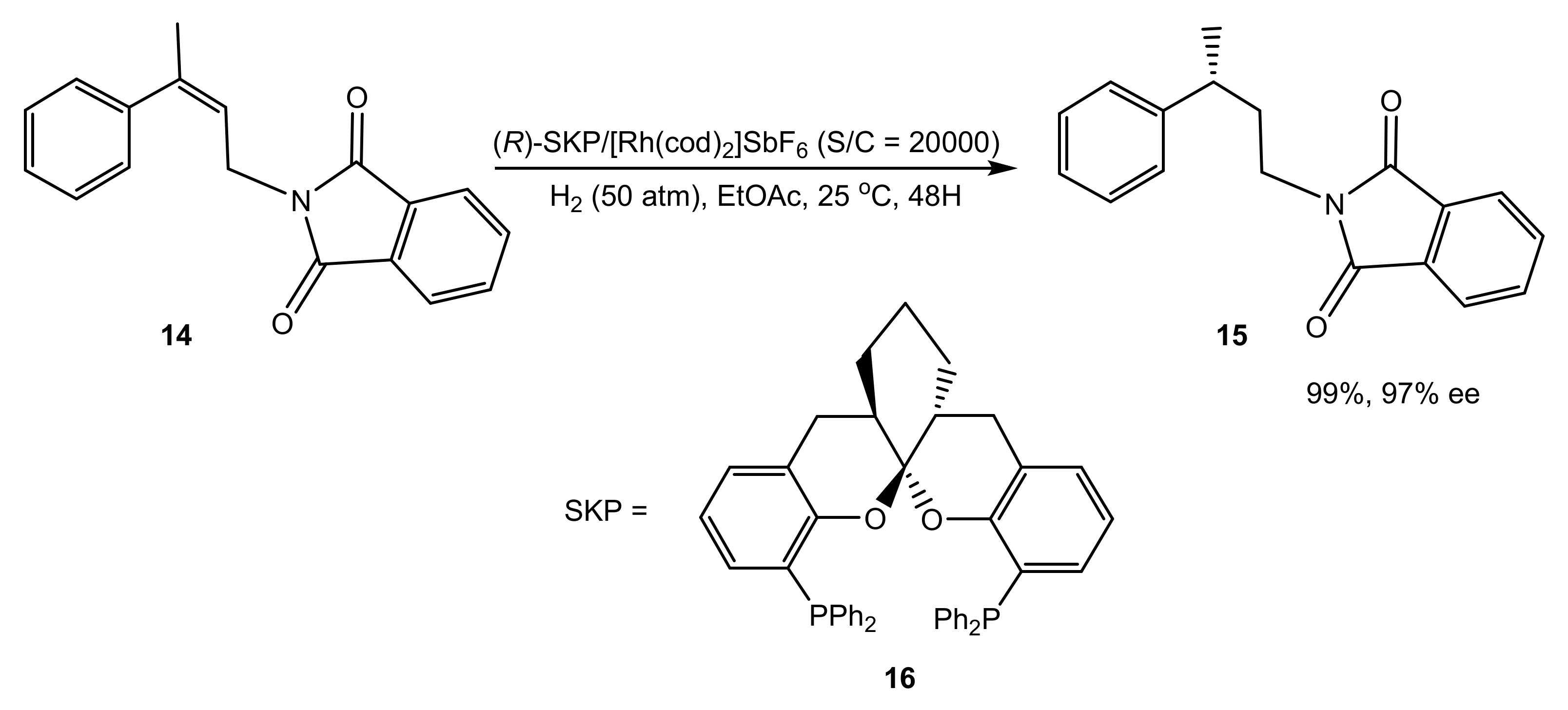
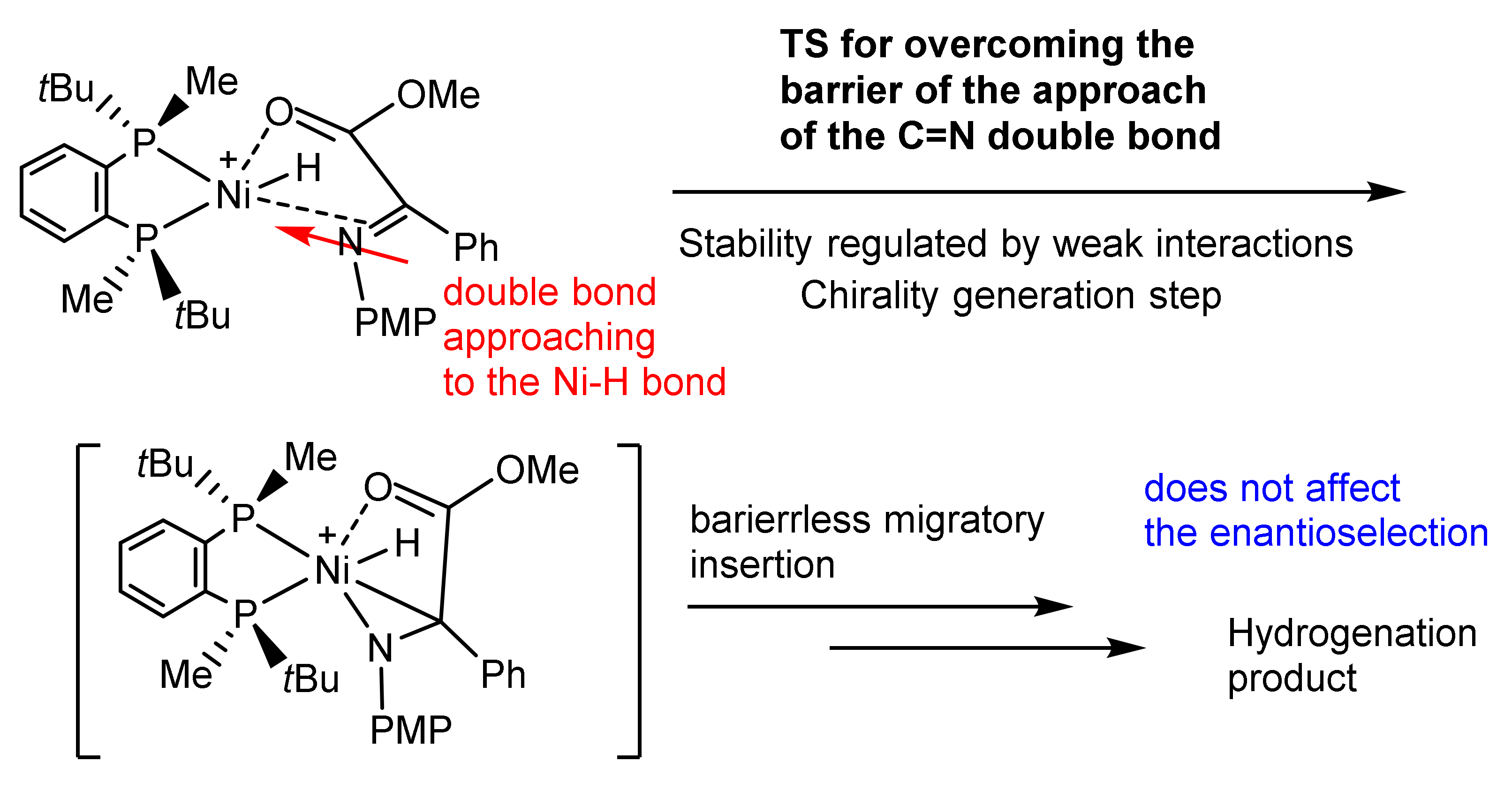
3. Asymmetric Hydrogenation Catalyzed by Co, Ni, and Pd Complexes
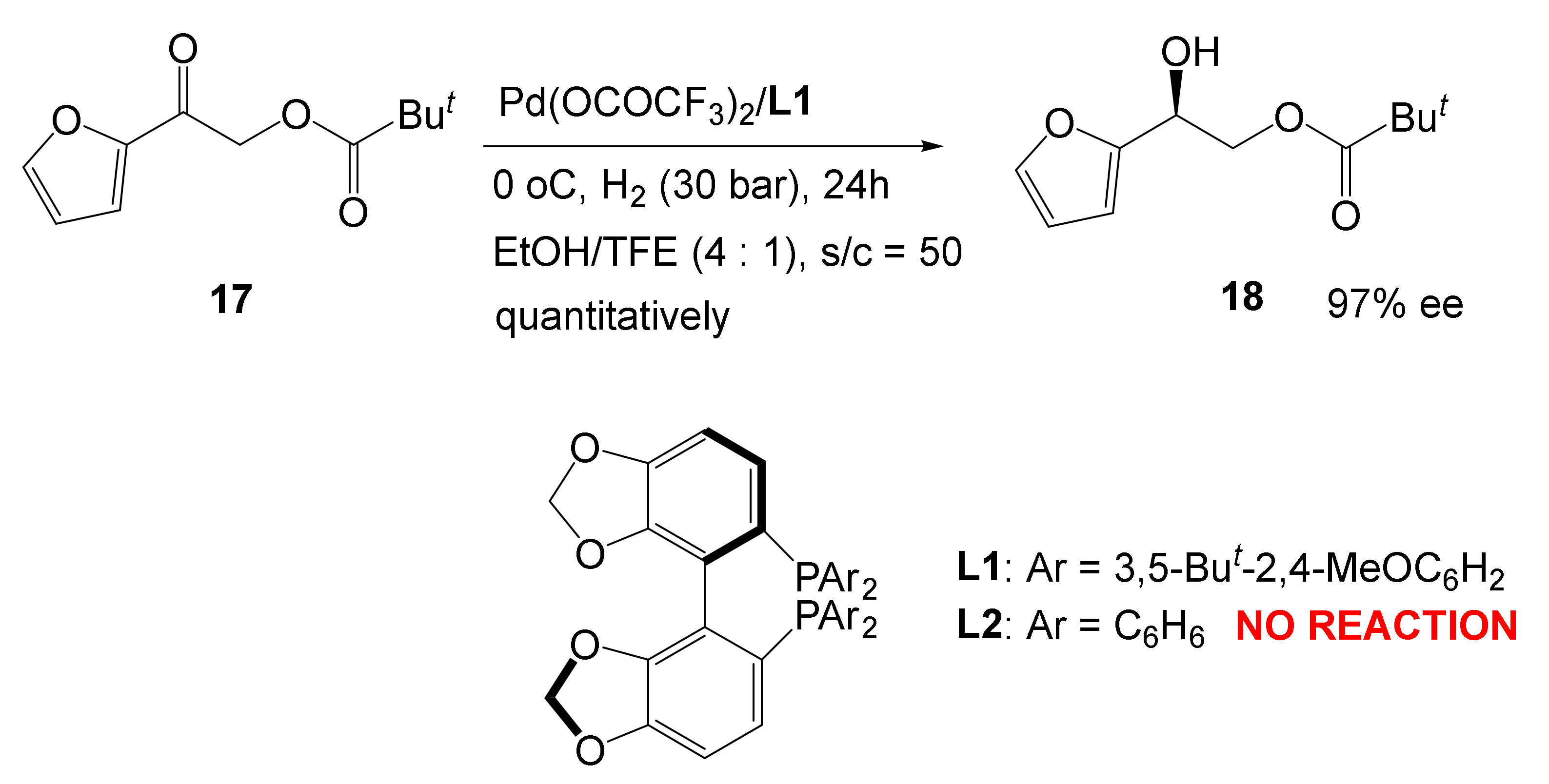
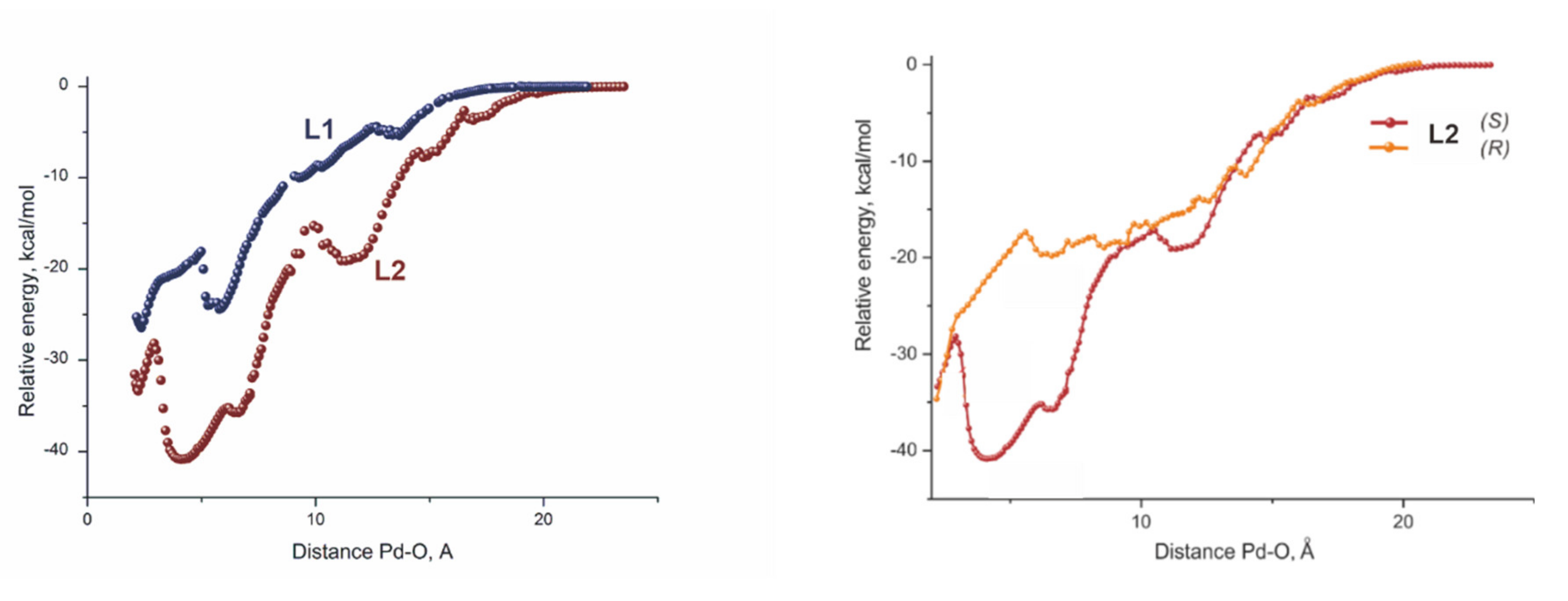
4. Artificial Catalysis Mimics Enzymatic Behavior
5. Conclusions
Funding
Conflicts of Interest
References
- Brock, W.H. The Fontana History of Chemistry; Fontana Press: London, UK, 1992; pp. 235–236. [Google Scholar]
- Sharpless, B. Searching for new reactivity (Nobel lecture). Angew. Chem. Int. Ed. 2002, 41, 2024–2032. [Google Scholar] [CrossRef]
- Knowles, W.S. Asymmetric hydrogenations (Nobel lecture). Angew. Chem. Int. Ed. 2002, 41, 1998–2007. [Google Scholar] [CrossRef]
- Noyori, R. Asymmetric catalysis: Science and opportunities (Nobel lecture). Angew. Chem. Int. Ed. 2002, 41, 2008–2022. [Google Scholar] [CrossRef]
- Suzuki, A. Cross-coupling reactions of organoboranes: An easy way to construct C–C bonds (Nobel lecture). Angew. Chem. Int. Ed. 2011, 50, 6722–6737. [Google Scholar] [CrossRef] [PubMed]
- Negishi, E.-I. Magical power of transition metals: Past, present, and future (Nobel lecture). Angew. Chem. Int. Ed. 2011, 50, 6738–6764. [Google Scholar] [CrossRef] [PubMed]
- List, B.; Lerner, R.A.; Barbas, C.F.J. Proline-catalyzed direct asymmetric aldol reactions. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Jen, W.S.; Wiener, J.J.M.; McMillan, D.W.C.J. New strategies for organic catalysis: The first enantioselective organocatalytic 1, 3-dipolar cycloaddition. Am. Chem. Soc. 2000, 122, 9874–9875. [Google Scholar] [CrossRef] [Green Version]
- Antenucci, A.; Dughera, S.; Renzi, P. Green chemistry meets asymmetric organocatalysis: A critical overview on catalysts synthesis. ChemSusChem 2021, 14, 2785–2853. [Google Scholar] [CrossRef]
- Shibasaki, M.; Kanai, M.; Matsunaga, S.; Kumagai, N. Recent progress in asymmetric bifunctional catalysis using multimetallic systems. Acc. Chem. Res. 2009, 42, 1117–1127. [Google Scholar] [CrossRef]
- Xu, H.; Zuend, S.J.; Woll, M.G.; Tao, Y.; Jacobsen, E.N. Asymmetric cooperative catalysis of strong Brønsted acid–promoted reactions using chiral ureas. Science 2010, 327, 986–990. [Google Scholar] [CrossRef] [Green Version]
- Simmons, B.; Walji, A.M.; MacMillan, D.W.C. Cycle-specific organocascade catalysis: Application to olefin hydroamination, hydro-oxidation, and amino-oxidation, and to natural product synthesis. Angew. Chem. Int. Ed. 2009, 48, 4349–4353. [Google Scholar] [CrossRef] [Green Version]
- Allen, A.E.; McMillan, D.W.C. Synergistic catalysis: A powerful synthetic strategy for new reaction development. Chem. Sci. 2012, 3, 633–658. [Google Scholar] [CrossRef]
- Qin, Y.; Zhu, L.; Luo, S. Organocatalysis in inert C–H bond functionalization. Chem. Rev. 2017, 117, 9433–9520. [Google Scholar] [CrossRef]
- Bo, L.; Dang, Y.; Houk, K.N. Dispersion and Steric Effects on Enantio-/Diastereoselectivities in Synergistic Dual Transition-Metal Catalysis. J. Am. Chem. Soc. 2022, 144, 1971–1985. [Google Scholar] [CrossRef]
- Liu, D.; Chen, J.; Gridnev, I.D.; Yan, D.; Zhang, W.B. Construction of chiral α-tert-amine scaffolds via amine-catalyzed asymmetric Mannich reactions of alkyl-substituted ketimines. Nat. Commun. 2020, 10, 1699. [Google Scholar]
- Halpern, J. Mechanism and stereoselectivity of asymmetric hydrogenation. Science 1982, 217, 401–407. [Google Scholar] [CrossRef]
- Brown, J.M. Hydrogenation of Functionalized Carbon-Carbon Double Bonds; Jacobsen, E.N., Pfalz, A., Yamamoto, H., Eds.; Springer: Berlin/Heidelberg, Germany, 1999; Volume 1, pp. 119–182. [Google Scholar]
- Gridnev, I.D.; Imamoto, T. On the mechanism of stereoselection in Rh-catalyzed asymmetric hydrogenation: A general approach for predicting the sense of enantioselectivity. Acc. Chem. Res. 2004, 37, 633–644. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Imamoto, T. Mechanism of enantioselection in Rh-catalyzed asymmetric hydrogenation. The origin of utmost catalytic performance. Chem. Commun. 2009, 48, 7447–7464. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Higashi, N.; Asakura, K.; Imamoto, T. Mechanism of Asymmetric Hydrogenation Catalyzed by a Rhodium Complex of (S,S)-1,2-Bis(tert-butylmethylphosphino)ethane. Dihydride Mechanism of Asymmetric Hydrogenation. J. Am. Chem. Soc. 2000, 122, 7183–7194. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Yasutake, M.; Higashi, N. Imamoto, T. Asymmetric hydrogenation of enamides with Rh-BisP* and Rh-MiniPHOS catalysts. Scope, limitations, and mechanism. J. Am. Chem. Soc. 2001, 123, 5268–5276. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Higashi, N.; Imamoto, T. Interconversion of monohydride intermediates in Rh (I)-catalyzed asymmetric hydrogenation of dimethyl 1-benzoyloxyethenephosphonate. J. Am. Chem. Soc. 2001, 123, 4631–4632. [Google Scholar] [CrossRef] [PubMed]
- Gridnev, I.D.; Yasutake, M.; Imamoto, T.; Beletskaya, I.P. Asymmetric Hydrogenation of α,β-umsaturated phosphonates with Rh-BisP* and Rh-MiniPHOS catalysts: Scope and mechanism of the reaction. Proc. Natl. Acad. Sci. USA 2004, 101, 5228–5235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gridnev, I.D.; Hoge, G.; Kouchi, T.M.; Takahashi, H.; Imamoto, T. Asymmetric Hydrogenation Catalyzed by a Rhodium Complex of (R)-(tert-Butylmethylphosphino)(di-tert-butylphosphino)methane: Scope of Enantioselectivity and Mechanistic Study. J. Am. Chem. Soc. 2008, 130, 2560–2572. [Google Scholar] [CrossRef] [PubMed]
- Gridnev, I.D.; Imamoto, T. Challenging the major/minor concept in Rh-catalyzed asymmetric hydrogenation. ACS Catal. 2015, 5, 2911–2915. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Liu, Y.; Imamoto, T. Mechanism of asymmetric hydrogenation of β-Dehydroamino acids catalyzed by rhodium complexes: Large-scale experimental and computational study. ACS Catal. 2014, 4, 203–219. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, T.; Wang, Y.; Zhou, F.; Zhang, Z.; Gridnev, I.D.; Zhang, W. Asymmetric hydrogenation for the synthesis of 2-substituted chiral morpholines. Nat. Sci. 2021, 1, e10021. [Google Scholar]
- Imamoto, T.; Tamura, K.; Zhang, Z.; Horiuchi, Y.; Sugiya, M.; Yoshida, K.; Yanagisawa, A.; Gridnev, I.D. Rigid P-Chiral Phosphine Ligands with tert-Butylmethylphosphino Groups for Rhodium-Catalyzed Asymmetric Hydrogenation of Functionalized Alkenes. J. Am. Chem. Soc. 2012, 134, 1754–1769. [Google Scholar] [CrossRef] [PubMed]
- Gridnev, I.D.; Imamoto, T. Enantioselection mechanism in Rh-catalyzed asymmetric hydrogenation. Russ. Chem. Bull. 2016, 65, 1514–1534. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Dub, P.A. Enantioselection in Asymmetric Catalysis; CRC Press: Boca Raton, FL, USA, 2016; 234p. [Google Scholar]
- Gridnev, I.D. Attraction versus Repulsion in Rhodium-Catalyzed Asymmetric Hydrogenation. ChemCatChem 2016, 8, 3463–3465. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, J.; Zeng, X.; Wang, Y.; Zhang, Z.; Gridnev, I.D.; Zhang, W. Chemo- and Enantioselective Hydrogenation of α-Formyl Enamides: An Efficient Access to Chiral α-Amido Aldehydes. Angew. Chem. Int. Ed. 2019, 58, 11505–11512. [Google Scholar] [CrossRef]
- Fan, D.; Zhang, J.; Hu, Y.; Zhang, Z.; Gridnev, I.D.; Zhang, W. Asymmetric Hydrogenation of α-Boryl Enamides Enabled by Nonbonding Interactions. ACS Catal. 2020, 10, 3232–3240. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Z.; Li, B.; Li, F.; Wang, Y.; Zhao, M.; Gridnev, I.D.; Imamoto, T.; Zhang, W. Pd (OAc)2-catalyzed asymmetric hydrogenation of sterically hindered N-tosylimines. Nat. Commun. 2018, 9, 5000. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, Z.; Zhang, J.; Liu, Y.; Gridnev, I.D.; Zhang, W. Cobalt-Catalyzed Asymmetric Hydrogenation of C= N Bonds Enabled by Assisted Coordination and Nonbonding Interactions. Angew. Chem. Int. Ed. 2019, 58, 15767–15771. [Google Scholar] [CrossRef]
- Li, B.; Chen, Z.; Zhang, Z.; Gridnev, I.D.; Zhang, W. Nickel-Catalyzed Asymmetric Hydrogenation of N-Sulfonyl Imines. Angew. Chem. Int. Ed. 2019, 131, 7407–7412. [Google Scholar] [CrossRef]
- Chen, J.; Liu, D.; Butt, N.; Li, C.; Fan, D.; Liu, Y.; Zhang, W. Palladium-Catalyzed Asymmetric Hydrogenation of α-Acyloxy-1-arylethanones. Angew. Chem. Int. Ed. 2013, 52, 11632–11636. [Google Scholar] [CrossRef]
- Chen, J.; Gridnev, I.D. Size is important: Artificial catalyst mimics behavior of natural enzymes. iScience 2020, 23, 100960. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, M.; La Manna, P.; Talotta, C.; Soriente, A.; Gaeta, C.; Neri, P. Supramolecular Organocatalysis in Water Mediated by Macrocyclic Compounds. Front. Chem. 2018, 6, 84. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.P.; Schreiner, P.R. London dispersion in molecular chemistry—Rreconsidering steric effects. Angew. Chem. Int. Ed. 2015, 54, 12274–12296. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gridnev, I.D. Birds of a Feather—Asymmetric Organocatalysis Meets Asymmetric Transition Metal Catalysis. Catalysts 2022, 12, 214. https://doi.org/10.3390/catal12020214
Gridnev ID. Birds of a Feather—Asymmetric Organocatalysis Meets Asymmetric Transition Metal Catalysis. Catalysts. 2022; 12(2):214. https://doi.org/10.3390/catal12020214
Chicago/Turabian StyleGridnev, Ilya D. 2022. "Birds of a Feather—Asymmetric Organocatalysis Meets Asymmetric Transition Metal Catalysis" Catalysts 12, no. 2: 214. https://doi.org/10.3390/catal12020214
APA StyleGridnev, I. D. (2022). Birds of a Feather—Asymmetric Organocatalysis Meets Asymmetric Transition Metal Catalysis. Catalysts, 12(2), 214. https://doi.org/10.3390/catal12020214