
CO and CO2 Methanation over CeO2-Supported Cobalt Catalysts



Abstract
:1. Introduction
2. Results and Discussion
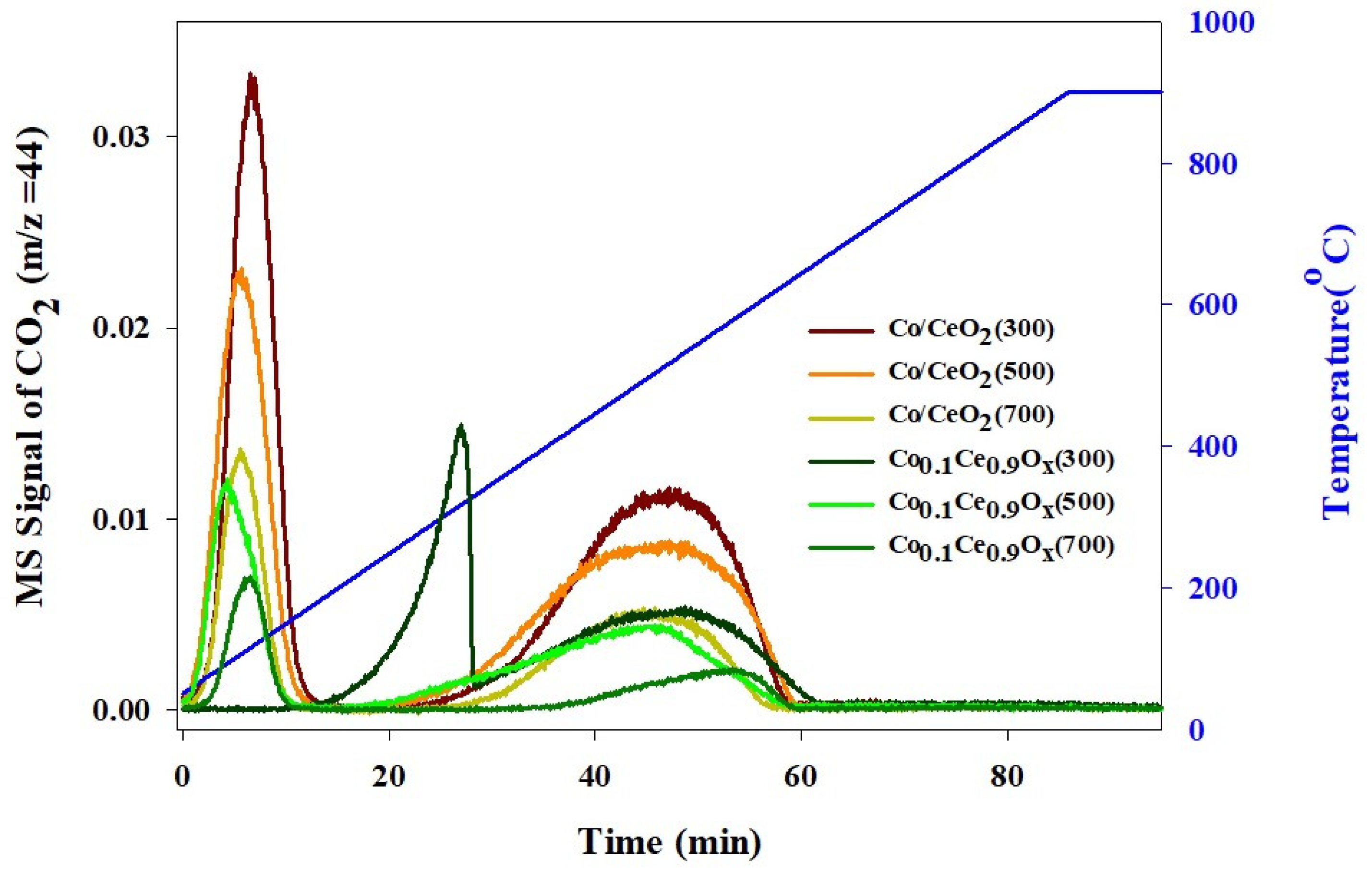
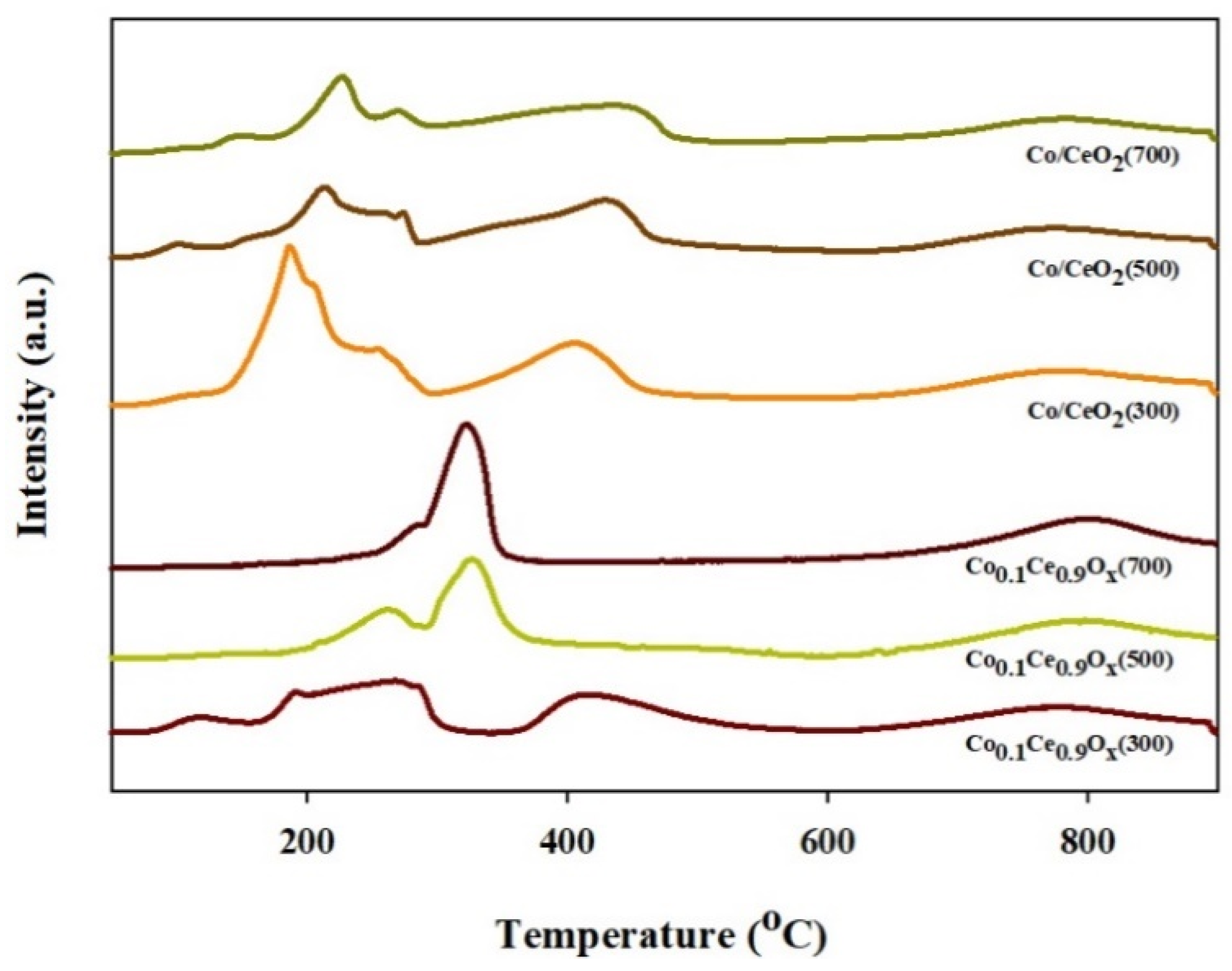
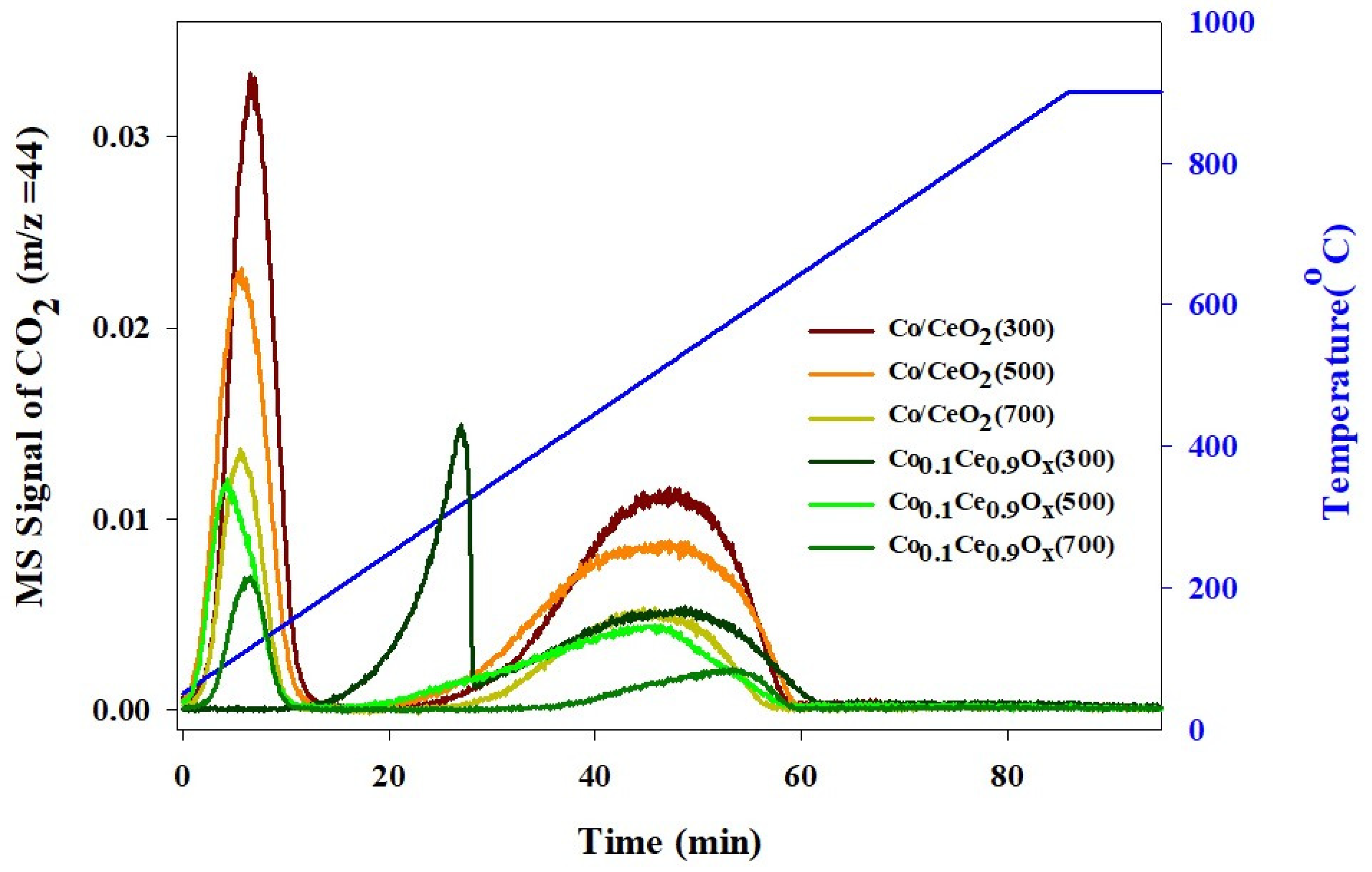
2.1. Physicochemical Properties of the Prepared Catalysts
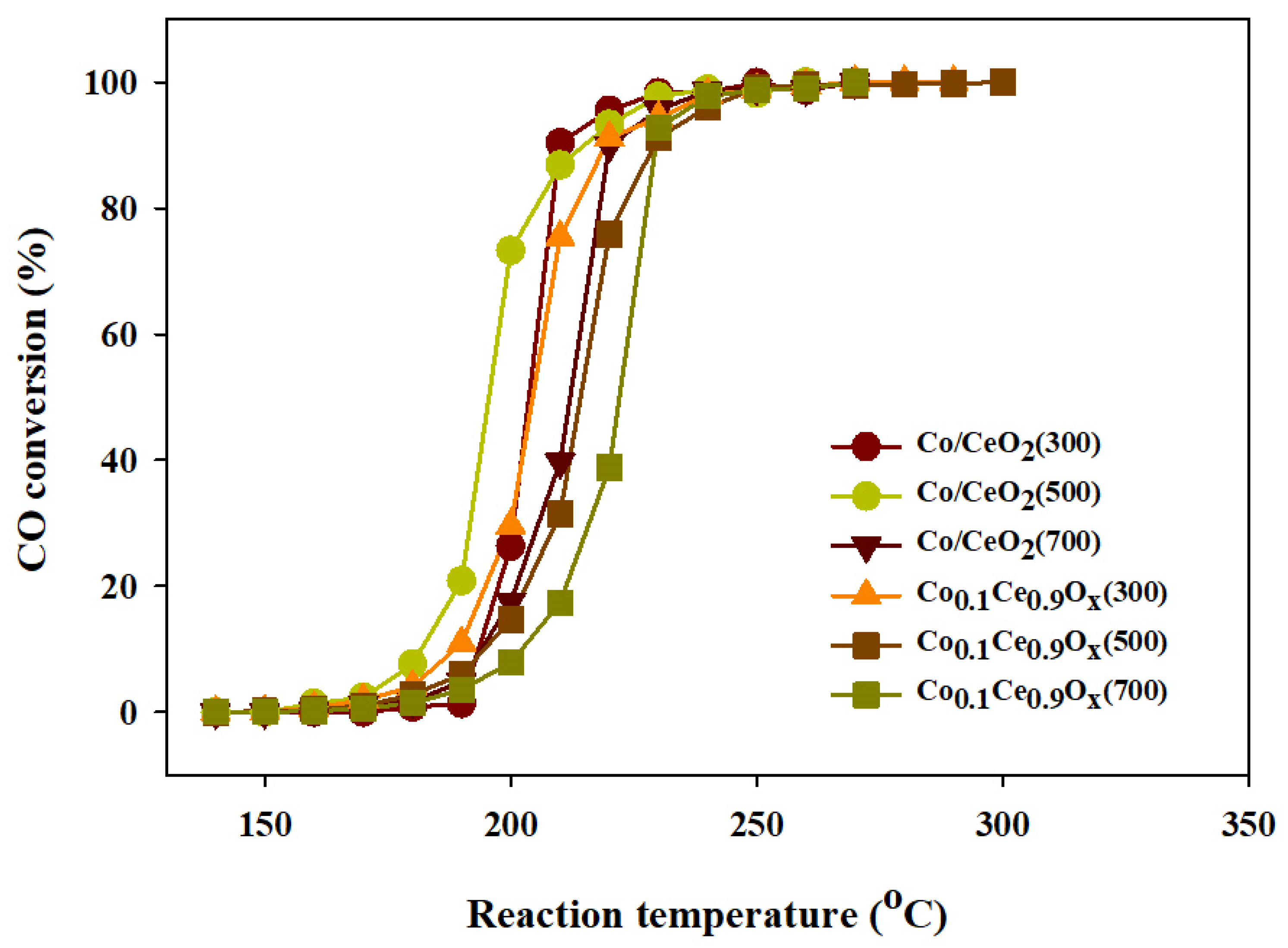
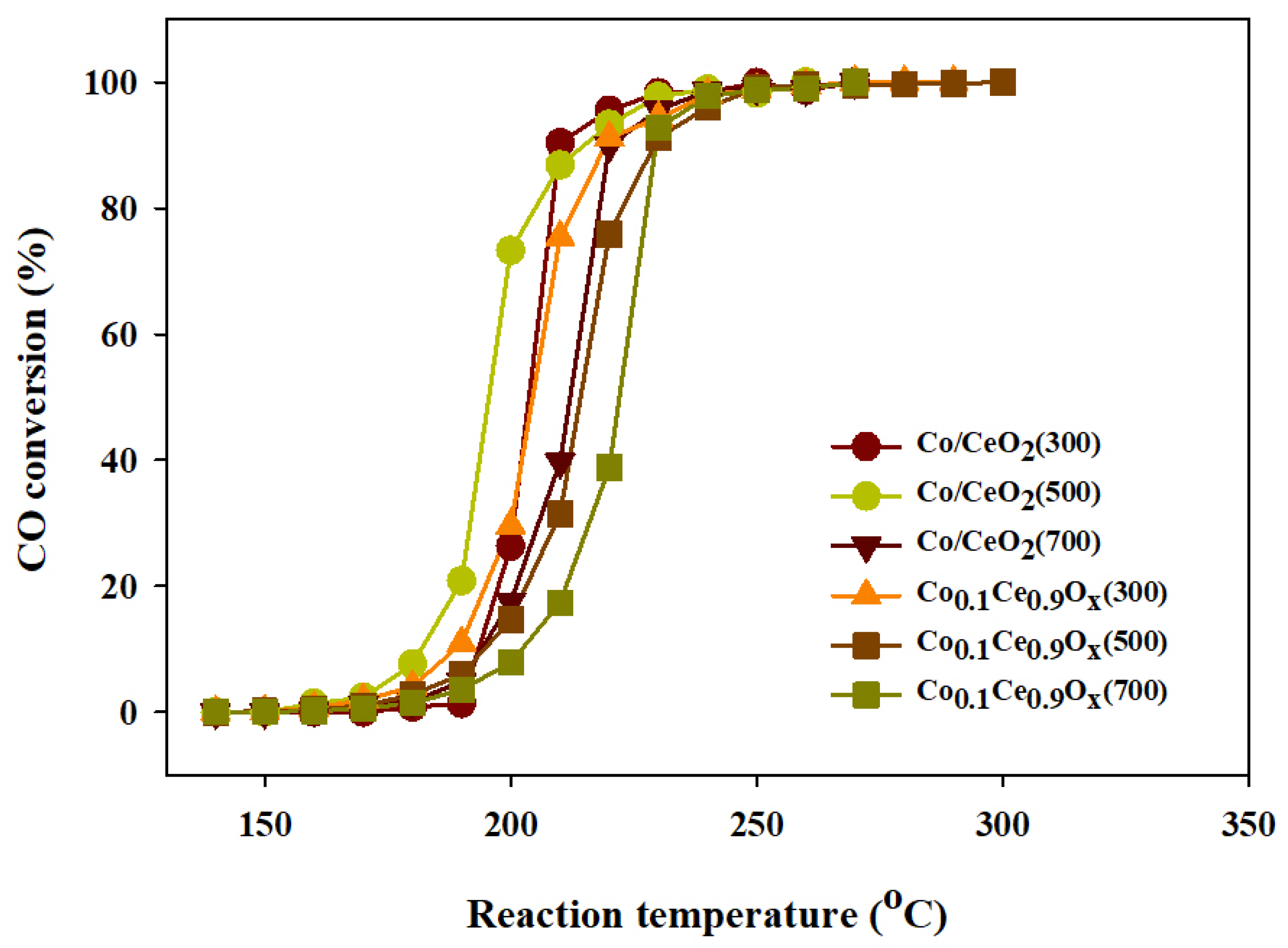
2.2. CO Methanation
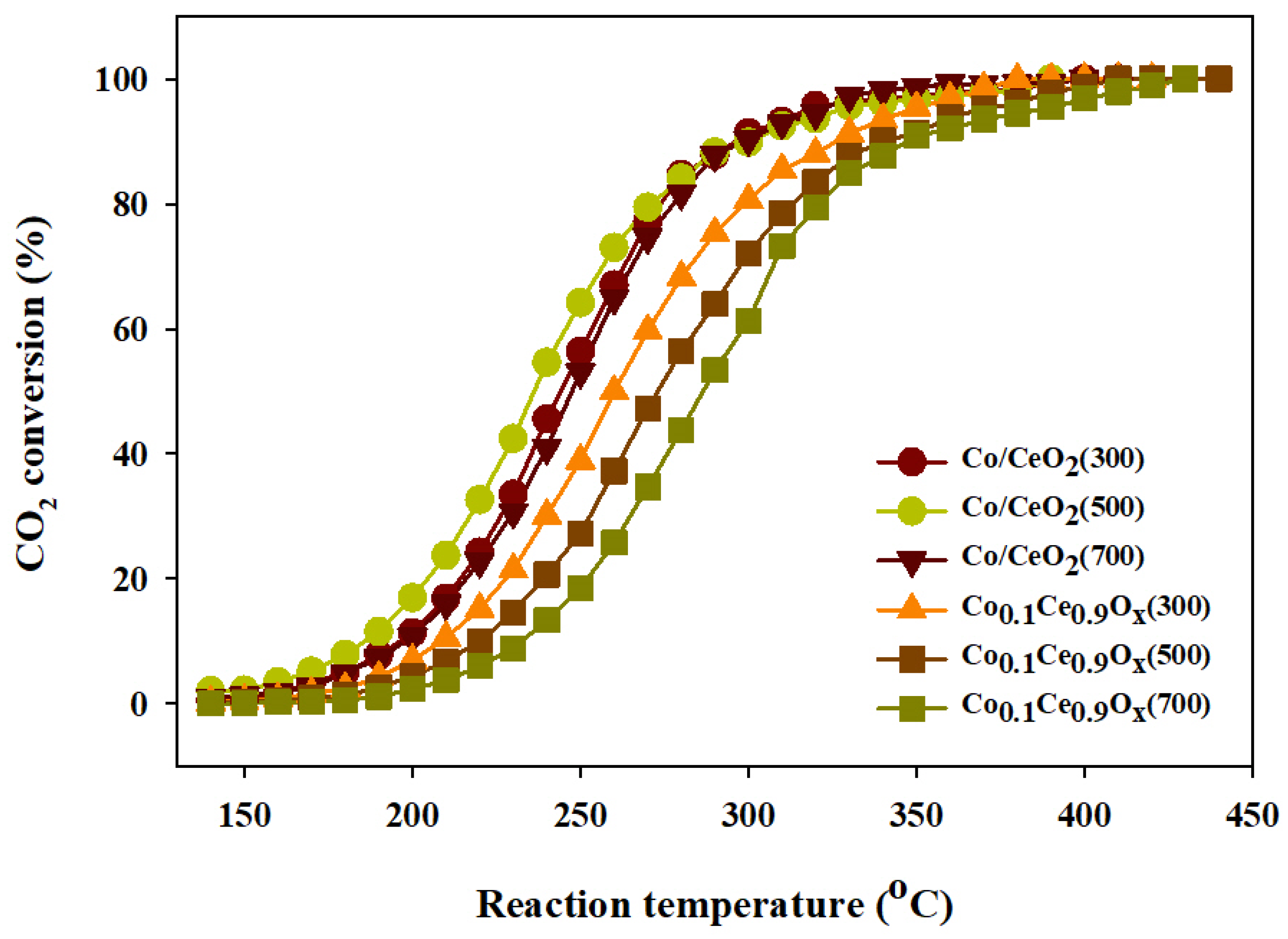
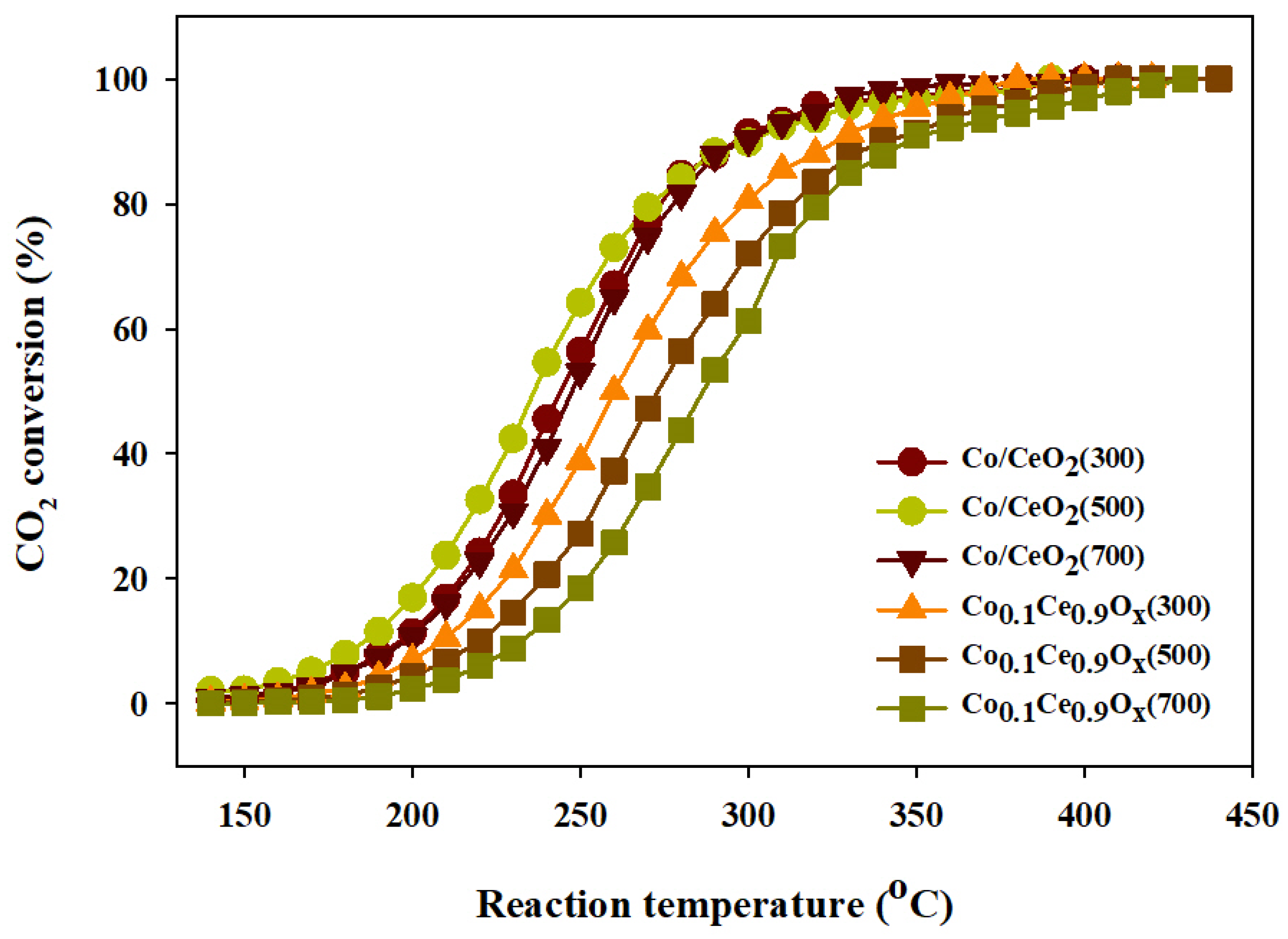
2.3. CO2 Methanation
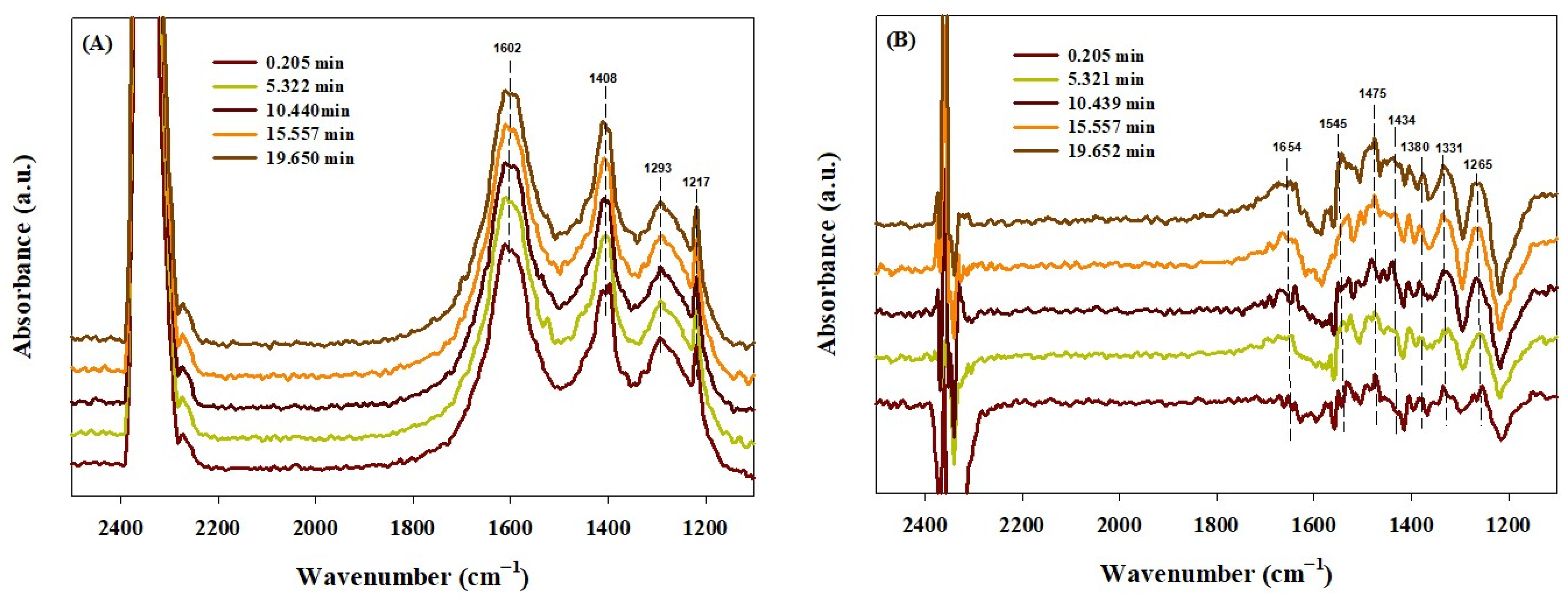
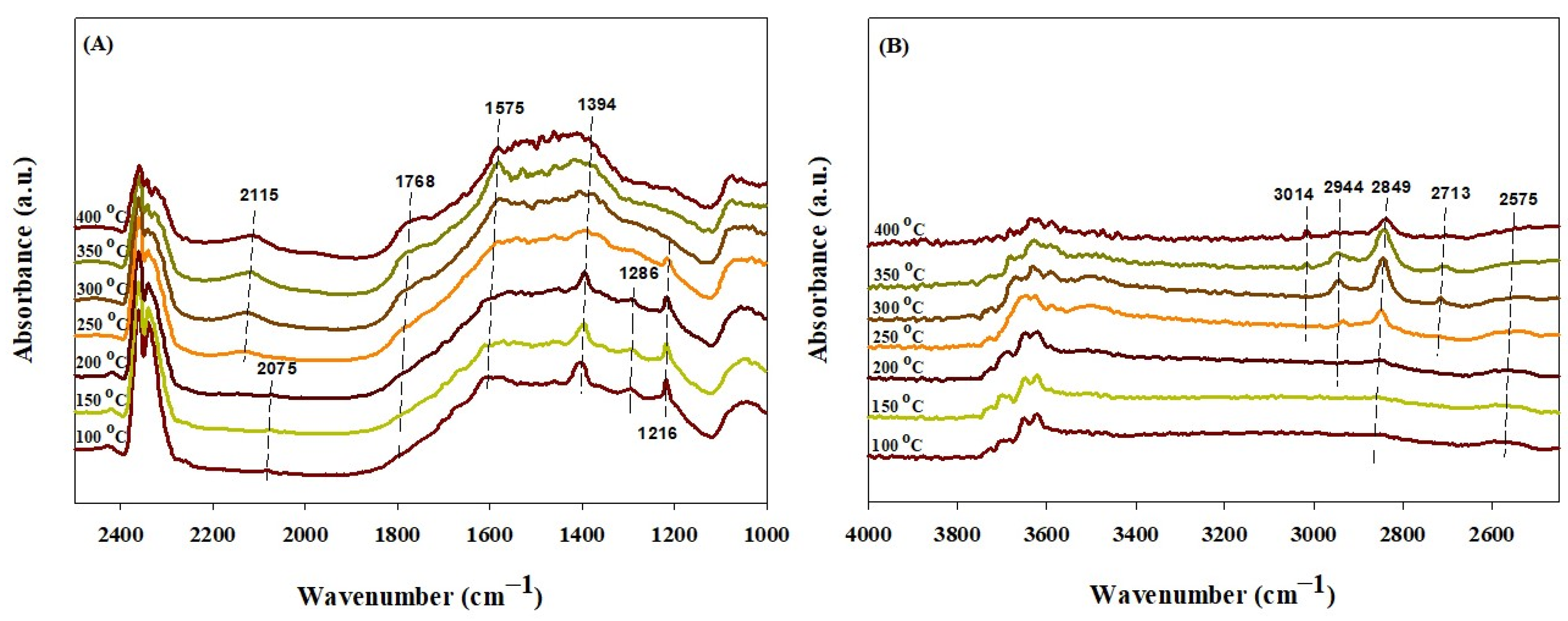
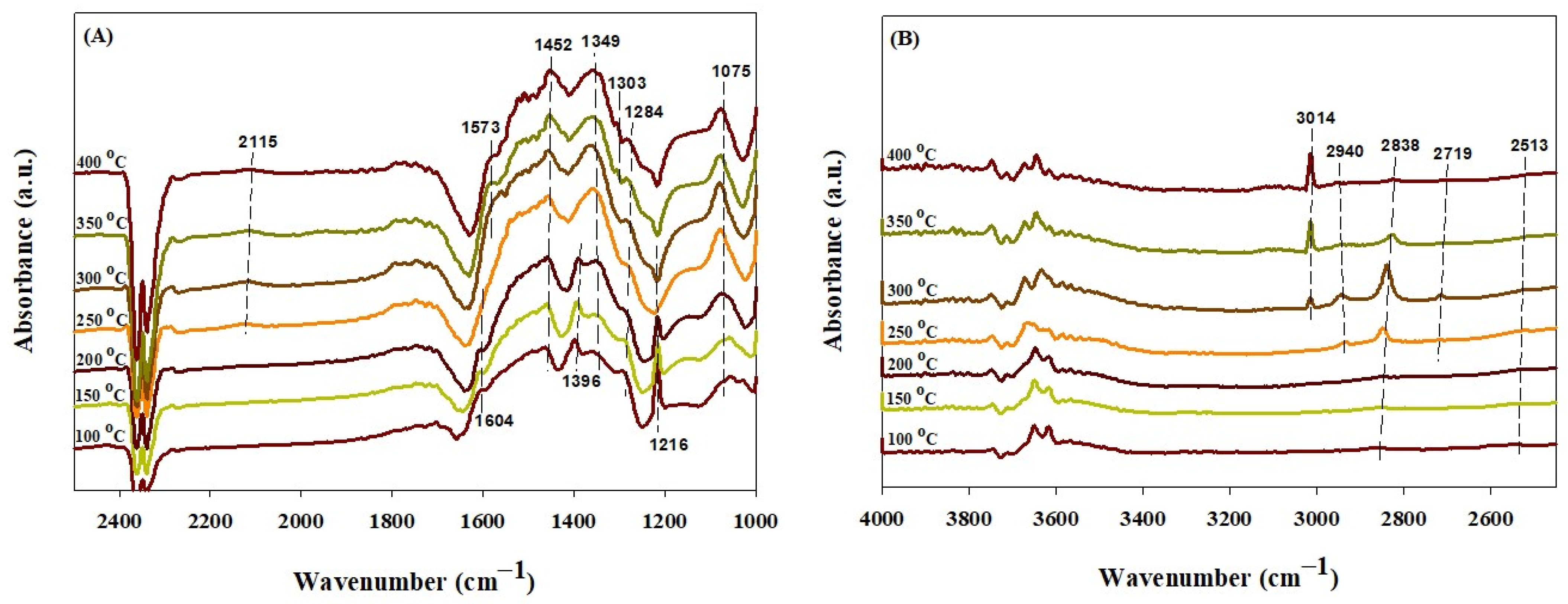
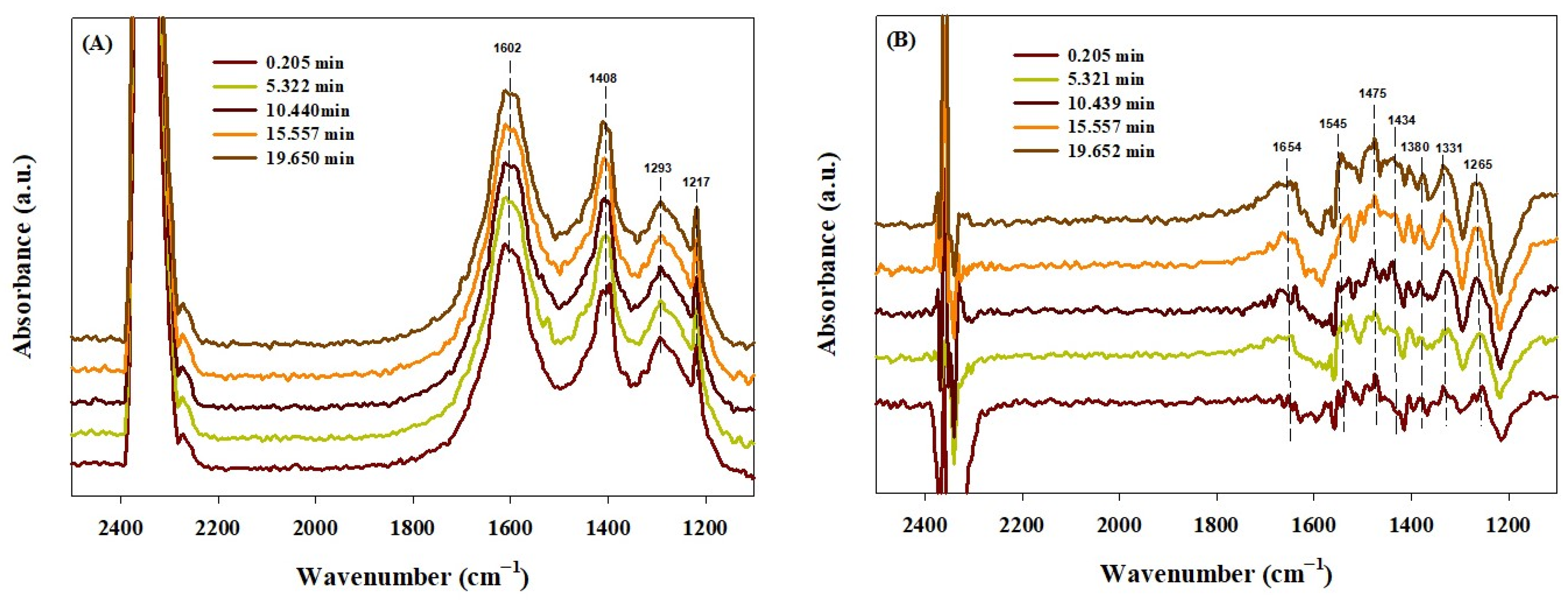
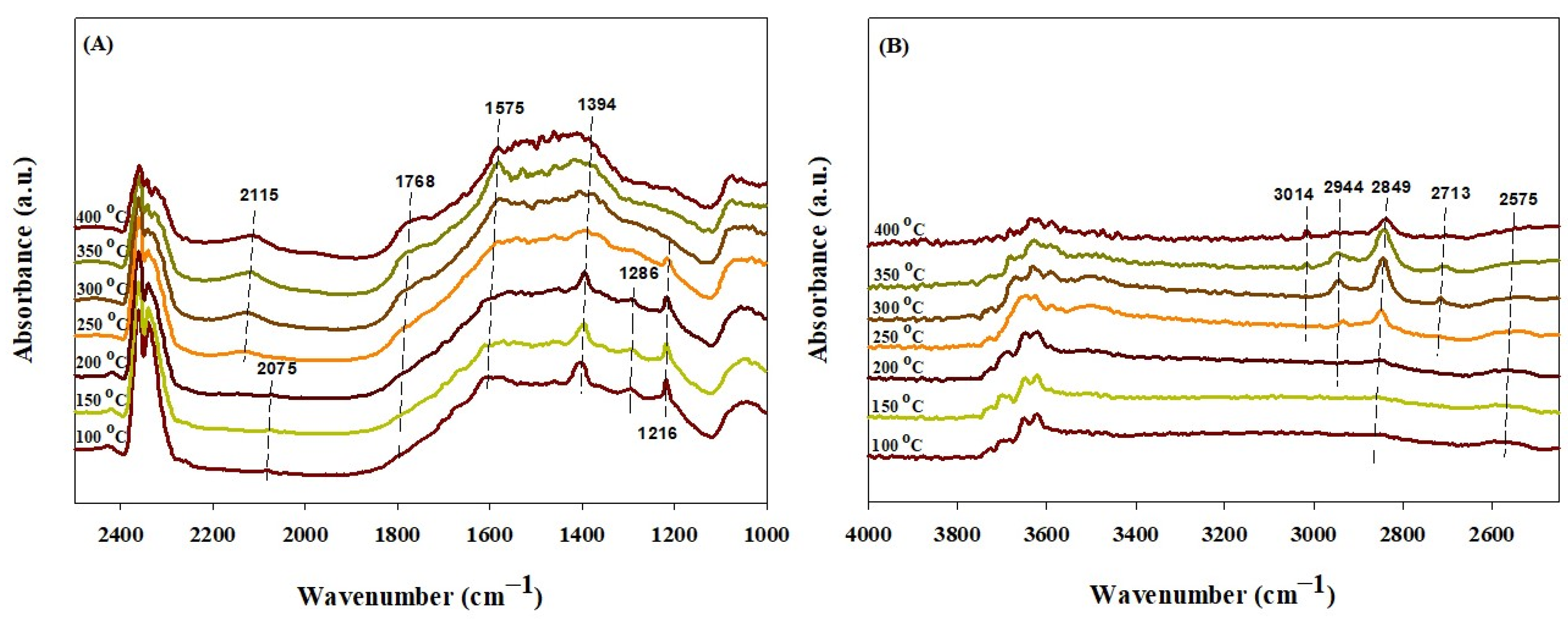
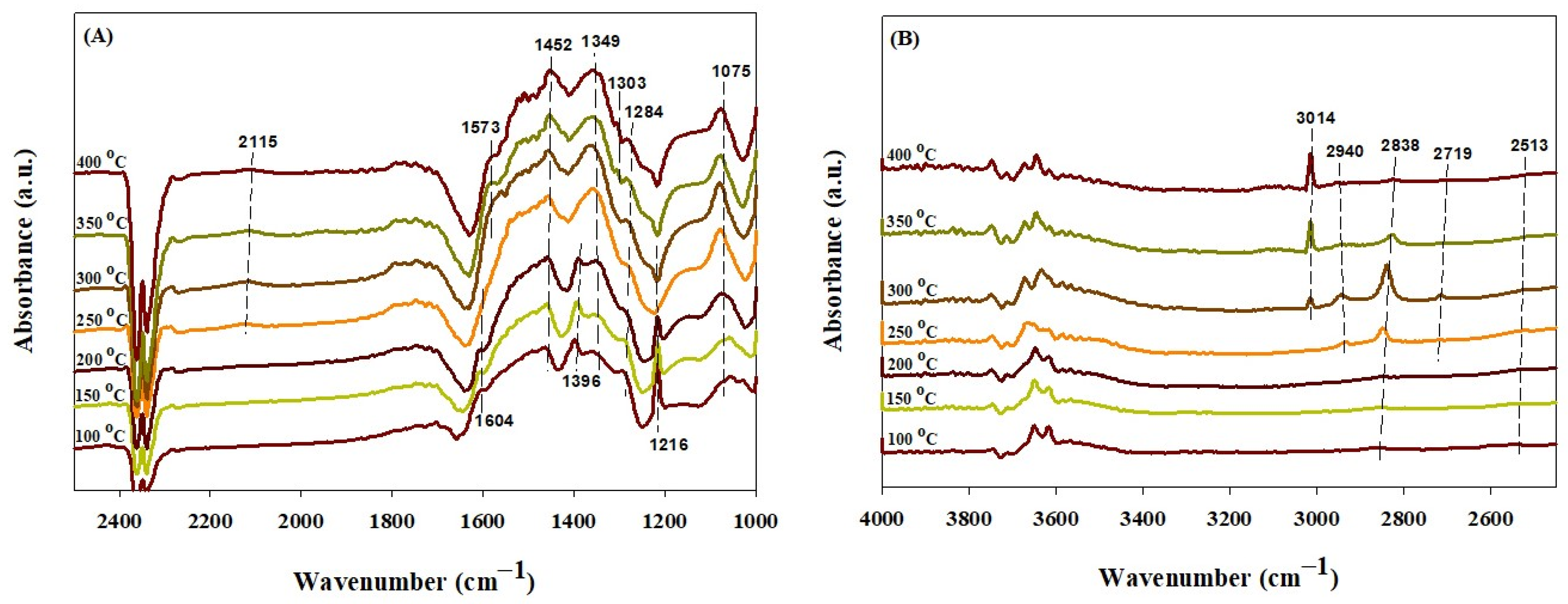
2.4. In Situ DRIFTS Study
3. Experimental Sections
3.1. Materials
3.2. Preparation of the Catalysts
3.3. Catalyst Characterizations
3.4. Catalytic Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nguyen, T.T.H.; Park, E.D. A mini-review on power-to-methane catalysts. J. Energy Eng. 2021, 30, 10–25. [Google Scholar]
- Sreedhar, I.; Varun, Y.; Singh, S.A.; Venugopal, A.; Reddy, B.M. Developmental trends in CO2 methanation using various catalysts. Catal. Sci. Technol. 2019, 9, 4478–4504. [Google Scholar] [CrossRef]
- Younas, M.; Loong Kong, L.; Bashir, M.J.K.; Nadeem, H.; Shehzad, A.; Sethupathi, S. Recent advancements, fundamental challenges, and opportunities in catalytic methanation of CO2. Energy Fuels 2016, 30, 8815–8831. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.; Jiang, X.; Zhu, J.; Liu, Z.; Guo, X.; Song, C. A short review of recent advances in CO2 hydrogenation to hydrocarbons over heterogeneous catalysts. RSC Adv. 2018, 8, 7651–7669. [Google Scholar] [CrossRef] [Green Version]
- Ashok, J.; Pati, S.; Hongmanorom, P.; Tianxi, Z.; Junmei, C.; Kawi, S. A review of recent catalyst advances in CO2 methanation processes. Catal. Today 2020, 356, 471–489. [Google Scholar] [CrossRef]
- Bailera, M.; Lisbona, P.; Romeo, L.M.; Espatolero, S. Power to gas projects review: Lab, pilot and demo plants for storing renewable energy and CO2. Renew. Sustain. Energy Rev. 2017, 69, 292–312. [Google Scholar] [CrossRef]
- Gao, J.; Liu, Q.; Gu, F.; Liu, B.; Zhong, Z.; Su, F. Recent advances in methanation catalysts for the production of synthetic natural gas. RSC Adv. 2015, 5, 22759–22776. [Google Scholar] [CrossRef]
- Garbarino, G.; Bellotti, D.; Riani, P.; Magistri, L.; Busca, G. Methanation of carbon dioxide on Ru/Al2O3 and Ni/Al2O3 catalysts at atmospheric pressure: Catalysts activation, behaviour and stability. Int. J. Hydrog. Energy 2015, 40, 9171–9182. [Google Scholar] [CrossRef]
- Kim, A.; Sanchez, C.; Haye, B.; Boissière, C.; Sassoye, C.; Debecker, D.P. Mesoporous TiO2 support materials for Ru-based CO2 methanation catalysts. ACS Appl. Nano Mater. 2019, 2, 3220–3230. [Google Scholar] [CrossRef]
- Chen, S.; Abdel-Mageed, A.M.; Dyballa, M.; Parlinska-Wojtan, M.; Bansmann, J.; Pollastri, S.; Olivi, L.; Aquilanti, G.; Behm, R.J. Raising the COx methanation activity of a Ru/gamma-Al2O3 catalyst by activated modification of metal-support interactions. Angew. Chem. Int. Ed. Eng. 2020, 59, 22763–22770. [Google Scholar] [CrossRef]
- Quindimil, A.; de la Torre, U.; Pereda-Ayo, B.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; González-Marcos, J.A.; Bueno-López, A.; González-Velasco, J.R. Effect of metal loading on the CO2 methanation: A comparison between alumina supported Ni and Ru catalysts. Catal. Today 2020, 356, 419–432. [Google Scholar] [CrossRef]
- Tan, J.; Wang, J.; Zhang, Z.; Ma, Z.; Wang, L.; Liu, Y. Highly dispersed and stable Ni nanoparticles confined by MgO on ZrO2 for CO2 methanation. Appl. Surf. Sci. 2019, 481, 1538–1548. [Google Scholar] [CrossRef]
- Bacariza, M.C.; Amjad, S.; Teixeira, P.; Lopes, J.M.; Henriques, C. Boosting Ni Dispersion on zeolite-supported catalysts for CO2 methanation: The influence of the impregnation solvent. Energy Fuels 2020, 34, 14656–14666. [Google Scholar] [CrossRef]
- Guilera, J.; del Valle, J.; Alarcón, A.; Díaz, J.A.; Andreu, T. Metal-oxide promoted Ni/Al2O3 as CO2 methanation micro-size catalysts. J. CO2 Util. 2019, 30, 11–17. [Google Scholar] [CrossRef]
- Song, F.; Zhong, Q.; Yu, Y.; Shi, M.; Wu, Y.; Hu, J.; Song, Y. Obtaining well-dispersed Ni/Al2O3 catalyst for CO2 methanation with a microwave-assisted method. Int. J. Hydrog. Energy 2017, 42, 4174–4183. [Google Scholar] [CrossRef]
- Daroughegi, R.; Meshkani, F.; Rezaei, M. Enhanced activity of CO2 methanation over mesoporous nanocrystalline Ni–Al2O3 catalysts prepared by ultrasound-assisted co-precipitation method. Int. J. Hydrog. Energy 2017, 42, 15115–15125. [Google Scholar] [CrossRef]
- Kesavan, J.K.; Luisetto, I.; Tuti, S.; Meneghini, C.; Iucci, G.; Battocchio, C.; Mobilio, S.; Casciardi, S.; Sisto, R. Nickel supported on YSZ: The effect of Ni particle size on the catalytic activity for CO2 methanation. J. CO2 Util. 2018, 23, 200–211. [Google Scholar] [CrossRef]
- Vogt, C.; Groeneveld, E.; Kamsma, G.; Nachtegaal, M.; Lu, L.; Kiely, C.J.; Berben, P.H.; Meirer, F.; Weckhuysen, B.M. Unravelling structure sensitivity in CO2 hydrogenation over nickel. Nat. Catal. 2018, 1, 127–134. [Google Scholar] [CrossRef]
- Danaci, S.; Protasova, L.; Lefevere, J.; Bedel, L.; Guilet, R.; Marty, P. Efficient CO2 methanation over Ni/Al2O3 coated structured catalysts. Catal. Today 2016, 273, 234–243. [Google Scholar] [CrossRef]
- Zhou, G.; Wu, T.; Xie, H.; Zheng, X. Effects of structure on the carbon dioxide methanation performance of Co-based catalysts. Int. J. Hydrog. Energy 2013, 38, 10012–10018. [Google Scholar] [CrossRef]
- Li, W.; Liu, Y.; Mu, M.; Ding, F.; Liu, Z.; Guo, X.; Song, C. Organic acid-assisted preparation of highly dispersed Co/ZrO2 catalysts with superior activity for CO2 methanation. Appl. Catal. B Environ. 2019, 254, 531–540. [Google Scholar] [CrossRef]
- Razzaq, R.; Li, C.; Usman, M.; Suzuki, K.; Zhang, S. A highly active and stable Co4N/γ-Al2O3 catalyst for CO and CO2 methanation to produce synthetic natural gas (SNG). Chem. Eng. J. 2015, 262, 1090–1098. [Google Scholar] [CrossRef]
- Yu, W.-Z.; Fu, X.-P.; Xu, K.; Ling, C.; Wang, W.-W.; Jia, C.-J. CO2 methanation catalyzed by a Fe-Co/Al2O3 catalyst. J. Environ. Chem. Eng. 2021, 9, 105594. [Google Scholar] [CrossRef]
- Li, W.; Nie, X.; Jiang, X.; Zhang, A.; Ding, F.; Liu, M.; Liu, Z.; Guo, X.; Song, C. ZrO2 support imparts superior activity and stability of Co catalysts for CO2 methanation. Appl. Catal. B Environ. 2018, 220, 397–408. [Google Scholar] [CrossRef]
- Li, W.; Zhang, G.; Jiang, X.; Liu, Y.; Zhu, J.; Ding, F.; Liu, Z.; Guo, X.; Song, C. CO2 hydrogenation on unpromoted and m-promoted Co/TiO2 catalysts (M = Zr, K, Cs): Effects of crystal phase of supports and metal-support interaction on tuning product distribution. ACS Catal. 2019, 9, 2739–2751. [Google Scholar] [CrossRef]
- Le, T.A.; Kim, M.S.; Lee, S.H.; Park, E.D. CO and CO2 methanation over supported cobalt catalysts. Top. Catal. 2017, 60, 714–720. [Google Scholar] [CrossRef]
- Satthawong, R.; Koizumi, N.; Song, C.; Prasassarakich, P. Comparative study on CO2 hydrogenation to higher hydrocarbons over Fe-based bimetallic catalysts. Top. Catal. 2013, 57, 588–594. [Google Scholar] [CrossRef]
- Serrer, M.-A.; Gaur, A.; Jelic, J.; Weber, S.; Fritsch, C.; Clark, A.H.; Saraçi, E.; Studt, F.; Grunwaldt, J.-D. Structural dynamics in Ni–Fe catalysts during CO2 methanation—Role of iron oxide clusters. Catal. Sci. Technol. 2020, 10, 7542–7554. [Google Scholar] [CrossRef]
- Pastor-Pérez, L.; Shah, M.; le Saché, E.; Ramirez Reina, T. Improving Fe/Al2O3 catalysts for the reverse water-gas shift reaction: On the effect of cs as activity/selectivity promoter. Catalysts 2018, 8, 608. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, S.; Rezaei, M.; Meshkani, F. Preparation of highly active nickel catalysts supported on mesoporous nanocrystalline γ-Al2O3 for CO2 methanation. J. Ind. Eng. Chem. 2014, 20, 1346–1352. [Google Scholar] [CrossRef]
- Italiano, C.; Llorca, J.; Pino, L.; Ferraro, M.; Antonucci, V.; Vita, A. CO and CO2 methanation over Ni catalysts supported on CeO2, Al2O3 and Y2O3 oxides. Appl. Catal. B Environ. 2020, 264, 118494. [Google Scholar] [CrossRef]
- Visconti, C.G.; Lietti, L.; Tronconi, E.; Forzatti, P.; Zennaro, R.; Finocchio, E. Fischer–Tropsch synthesis on a Co/Al2O3 catalyst with CO2 containing syngas. Appl. Catal. A Gen. 2009, 355, 61–68. [Google Scholar] [CrossRef]
- Vile, G.; Colussi, S.; Krumeich, F.; Trovarelli, A.; Perez-Ramirez, J. Opposite face sensitivity of CeO(2) in hydrogenation and oxidation catalysis. Angew. Chem. Int. Ed. Eng. 2014, 53, 12069–12072. [Google Scholar] [CrossRef] [PubMed]
- Tada, S.; Shimizu, T.; Kameyama, H.; Haneda, T.; Kikuchi, R. Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures. Int. J. Hydrog. Energy 2012, 37, 5527–5531. [Google Scholar] [CrossRef]
- Zhou, G.; Liu, H.; Cui, K.; Xie, H.; Jiao, Z.; Zhang, G.; Xiong, K.; Zheng, X. Methanation of carbon dioxide over Ni/CeO2 catalysts: Effects of support CeO2 structure. Int. J. Hydrog. Energy 2017, 42, 16108–16117. [Google Scholar] [CrossRef]
- Dias, Y.R.; Perez-Lopez, O.W. Carbon dioxide methanation over Ni-Cu/SiO2 catalysts. Energy Convers. Manag. 2020, 203, 112214. [Google Scholar] [CrossRef]
- Ye, R.-P.; Gong, W.; Sun, Z.; Sheng, Q.; Shi, X.; Wang, T.; Yao, Y.; Razink, J.J.; Lin, L.; Zhou, Z.; et al. Enhanced stability of Ni/SiO2 catalyst for CO2 methanation: Derived from nickel phyllosilicate with strong metal-support interactions. Energy 2019, 188, 116059. [Google Scholar] [CrossRef]
- Park, J.-N.; McFarland, E.W. A highly dispersed Pd–Mg/SiO2 catalyst active for methanation of CO2. J. Catal. 2009, 266, 92–97. [Google Scholar] [CrossRef]
- Liu, C.; Wang, W.; Xu, Y.; Li, Z.; Wang, B.; Ma, X. Effect of zirconia morphology on sulfur-resistant methanation performance of MoO3/ZrO2 catalyst. Appl. Surf. Sci. 2018, 441, 482–490. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, W.; Li, Z. Highly efficient Ni/ZrO2 catalysts prepared via combustion method for CO2 methanation. J. CO2 Util. 2016, 16, 236–244. [Google Scholar] [CrossRef]
- Xu, X.; Tong, Y.; Huang, J.; Zhu, J.; Fang, X.; Xu, J.; Wang, X. Insights into CO2 methanation mechanism on cubic ZrO2 supported Ni catalyst via a combination of experiments and DFT calculations. Fuel 2021, 283, 118867. [Google Scholar] [CrossRef]
- Ren, J.; Qin, X.; Yang, J.-Z.; Qin, Z.-F.; Guo, H.-L.; Lin, J.-Y.; Li, Z. Methanation of carbon dioxide over Ni–M/ZrO2 (M = Fe, Co, Cu) catalysts: Effect of addition of a second metal. Fuel Process. Technol. 2015, 137, 204–211. [Google Scholar] [CrossRef]
- Chai, S.; Men, Y.; Wang, J.; Liu, S.; Song, Q.; An, W.; Kolb, G. Boosting CO2 methanation activity on Ru/TiO2 catalysts by exposing (001) facets of anatase TiO2. J. CO2 Util. 2019, 33, 242–252. [Google Scholar] [CrossRef]
- Kim, A.; Debecker, D.P.; Devred, F.; Dubois, V.; Sanchez, C.; Sassoye, C. CO2 methanation on Ru/TiO2 catalysts: On the effect of mixing anatase and rutile TiO2 supports. Appl. Catal. B Environ. 2018, 220, 615–625. [Google Scholar] [CrossRef]
- Liu, J.; Li, C.; Wang, F.; He, S.; Chen, H.; Zhao, Y.; Wei, M.; Evans, D.G.; Duan, X. Enhanced low-temperature activity of CO2 methanation over highly-dispersed Ni/TiO2 catalyst. Catal. Sci. Technol. 2013, 3, 2627–2633. [Google Scholar] [CrossRef]
- Song, H.; Yang, J.; Zhao, J.; Chou, L. Methanation of carbon dioxide over a highly dispersed Ni/La2O3 catalyst. Chin. J. Catal. 2010, 31, 21–23. [Google Scholar] [CrossRef]
- Takami, K.; Takahash, T. Kinetics of the methanation of carbon dioxide over a supported La2O3. Can. J. Eng. 1988, 66, 343–347. [Google Scholar]
- Li, S.; Tang, H.; Gong, D.; Ma, Z.; Liu, Y. Loading Ni/La2O3 on SiO2 for CO methanation from syngas. Catal. Today 2017, 297, 298–307. [Google Scholar] [CrossRef]
- Kock, E.M.; Kogler, M.; Bielz, T.; Klotzer, B.; Penner, S. In situ FT-IR spectroscopic study of CO2 and CO adsorption on Y2O3, ZrO2, and yttria-stabilized ZrO2. J. Phy.s Chem. C Nanomater. Interfaces 2013, 117, 17666–17673. [Google Scholar] [CrossRef]
- Ayub, N.A.; Bahruji, H.; Mahadi, A.H. Barium promoted Ni/Sm2O3 catalysts for enhanced CO2 methanation. RSC Adv. 2021, 11, 31807–31816. [Google Scholar] [CrossRef]
- Ilsemann, J.; Sonström, A.; Gesing, T.M.; Anwander, R.; Bäumer, M. Highly active Sm2O3-Ni xerogel catalysts for CO2 methanation. ChemCatChem 2019, 11, 1732–1741. [Google Scholar] [CrossRef]
- Bian, Z.; Chan, Y.M.; Yu, Y.; Kawi, S. Morphology dependence of catalytic properties of Ni/CeO2 for CO2 methanation: A kinetic and mechanism study. Catal. Today 2020, 347, 31–38. [Google Scholar] [CrossRef]
- Guo, Y.; Mei, S.; Yuan, K.; Wang, D.-J.; Liu, H.-C.; Yan, C.-H.; Zhang, Y.-W. Low-temperature CO2 methanation over CeO2-supported Ru single atoms, nanoclusters, and nanoparticles competitively tuned by strong metal–support interactions and H-spillover effect. ACS Catal. 2018, 8, 6203–6215. [Google Scholar] [CrossRef]
- Ye, R.-P.; Li, Q.; Gong, W.; Wang, T.; Razink, J.J.; Lin, L.; Qin, Y.-Y.; Zhou, Z.; Adidharma, H.; Tang, J.; et al. High-performance of nanostructured Ni/CeO2 catalyst on CO2 methanation. Appl. Catal. B Environ. 2020, 268, 118474. [Google Scholar] [CrossRef]
- Sakpal, T.; Lefferts, L. Structure-dependent activity of CeO2 supported Ru catalysts for CO2 methanation. J. Catal. 2018, 367, 171–180. [Google Scholar] [CrossRef]
- Jiang, M.; Wang, B.; Yao, Y.; Li, Z.; Ma, X.; Qin, S.; Sun, Q. A comparative study of CeO2-Al2O3 support prepared with different methods and its application on MoO3/CeO2-Al2O3 catalyst for sulfur-resistant methanation. Appl. Surf. Sci. 2013, 285, 267–277. [Google Scholar] [CrossRef]
- Ho, P.H.; de Luna, G.S.; Angelucci, S.; Canciani, A.; Jones, W.; Decarolis, D.; Ospitali, F.; Aguado, E.R.; Rodríguez-Castellón, E.; Fornasari, G.; et al. Understanding structure-activity relationships in highly active La promoted Ni catalysts for CO2 methanation. Appl. Catal. B Environ. 2020, 278, 119256. [Google Scholar] [CrossRef]
- Liang, C.; Tian, H.; Gao, G.; Zhang, S.; Liu, Q.; Dong, D.; Hu, X. Methanation of CO2 over alumina supported nickel or cobalt catalysts: Effects of the coordination between metal and support on formation of the reaction intermediates. Int. J. Hydrog. Energy 2020, 45, 531–543. [Google Scholar] [CrossRef]
- Mihet, M.; Lazar, M.D. Methanation of CO2 on Ni/γ-Al2O3: Influence of Pt, Pd or Rh promotion. Catal. Today 2018, 306, 294–299. [Google Scholar] [CrossRef]
- Hongmanorom, P.; Ashok, J.; Zhang, G.; Bian, Z.; Wai, M.H.; Zeng, Y.; Xi, S.; Borgna, A.; Kawi, S. Enhanced performance and selectivity of CO2 methanation over phyllosilicate structure derived Ni-Mg/SBA-15 catalysts. Appl. Catal. B Environ. 2021, 282, 19564. [Google Scholar] [CrossRef]
- Li, T.; Wang, S.; Gao, D.-N.; Wang, S.-D. Effect of support calcination temperature on the catalytic properties of Ru/Ce0.8Zr0.2O2 for methanation of carbon dioxide. J. Fuel Chem. Technol. 2014, 42, 1440–1446. [Google Scholar] [CrossRef]
- Wu, H.; Zou, M.; Guo, L.; Ma, F.; Mo, W.; Yu, Y.; Mian, I.; Liu, J.; Yin, S.; Tsubaki, N. Effects of calcination temperatures on the structure–activity relationship of Ni–La/Al2O3 catalysts for syngas methanation. RSC Adv. 2020, 10, 4166–4174. [Google Scholar] [CrossRef] [Green Version]
- Sexton, B.A.; Hughes, A.E.; Turney, T.W. An XPS and TPR study of the reduction of promoted cobalt-kieselguhr Fischer-Tropsch catalysts. J. Catal. 1986, 97, 390–406. [Google Scholar] [CrossRef]
- Castillo, J.; Arteaga-Pérez, L.E.; Karelovic, A.; Jiménez, R. The consequences of surface heterogeneity of cobalt nanoparticles on the kinetics of CO methanation. Catal. Sci. Technol. 2019, 9, 6415–6427. [Google Scholar] [CrossRef]
- Le, T.A.; Kim, M.S.; Lee, S.H.; Kim, T.W.; Park, E.D. CO and CO2 methanation over supported Ni catalysts. Catal. Today 2017, 293–294, 89–96. [Google Scholar] [CrossRef]
- Liu, K.; Xu, X.; Xu, J.; Fang, X.; Liu, L.; Wang, X. The distributions of alkaline earth metal oxides and their promotional effects on Ni/CeO2 for CO2 methanation. J. CO2 Util. 2020, 38, 113–124. [Google Scholar] [CrossRef]
- Le, T.A.; Kim, J.; Kang, J.K.; Park, E.D. CO and CO methanation over Ni/Al@Al2O3 core–shell catalyst. Catal. Today 2020, 356, 622–630. [Google Scholar] [CrossRef]
- Xu, L.; Cui, Y.; Chen, M.; Wen, X.; Lv, C.; Wu, X.; Wu, C.-E.; Miao, Z.; Hu, X. Screening transition metals (Mn, Fe, Co, and Cu) promoted Ni-based CO2 methanation bimetal catalysts with advanced low-temperature activities. Ind. Eng. Chem. Res. 2021, 60, 8056–8072. [Google Scholar] [CrossRef]
- Yu, Y.; Mottaghi-Tabar, S.; Iqbal, M.W.; Yu, A.; Simakov, D.S.A. CO2 methanation over alumina-supported cobalt oxide and carbide synthesized by reverse microemulsion method. Catal. Today 2021, 379, 250–261. [Google Scholar] [CrossRef]
- Hadjiivanov, K.; Ivanova, E.; Dimitrov, L.; Knözinger, H. FTIR spectroscopic study of CO adsorption on Rh–ZSM-5: Detection of Rh+ −CO species. J. Mol. Struct. 2003, 661–662, 459–463. [Google Scholar] [CrossRef]
- Choi, J.G.; Rhee, H.K.; Moon, S.H. IR and TPD study of fresh and carbon-deposited Co/Al2O3 catalysts. Appl. Catal. A Gen. 1985, 13, 269–280. [Google Scholar] [CrossRef]
- Watson, C.D.; Martinelli, M.; Cronauer, D.C.; Kropf, A.J.; Jacobs, G. Low temperature water-gas shift: Enhancing stability through optimizing Rb loading on Pt/ZrO2. Catalysts 2021, 11, 210. [Google Scholar] [CrossRef]
- Das, T.; Deo, G. Synthesis, characterization and in situ DRIFTS during the CO2 hydrogenation reaction over supported cobalt catalysts. J. Mol. Catal. A Chem. 2011, 350, 75–82. [Google Scholar] [CrossRef]
- Baltrusaitis, J.; Schuttlefield, J.; Zeitler, E.; Grassian, V.H. Carbon dioxide adsorption on oxide nanoparticle surfaces. Chem. Eng. J. 2011, 170, 471–481. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | BET Surface Area b (m2/g) | Pore Volume b (cm3/g) | Average Pore Diameter b (nm) | Cobalt Dispersion c (%) | CASA c (m2/gcat) | CO2 Uptake d (μmol/gcat) |
|---|---|---|---|---|---|---|
| Co0.1Ce0.9Ox(300) | 90 | 0.23 | 10.2 | 4.65 | 0.74 | 124 |
| Co0.1Ce0.9Ox(500) | 72 | 0.28 | 18.9 | 5.98 | 0.95 | 146 |
| Co0.1Ce0.9Ox(700) | 34 | 0.22 | 26.3 | 3.27 | 0.52 | 59 |
| Co/CeO2(300) | 130 | 0.12 | 3.9 | 3.87 | 0.94 | 196 |
| Co/CeO2(500) | 128 | 0.14 | 4.3 | 4.65 | 1.13 | 186 |
| Co/CeO2(700) | 60 | 0.13 | 8.6 | 3.72 | 0.90 | 122 |
| Vibrational Mode Assignments | IR Peak Position (cm−1) | References | ||||||
|---|---|---|---|---|---|---|---|---|
| Co0.1Ce0.9Ox(300) | Ce/CeO2(500) | Literature | ||||||
| Adsorption | Hydrogenation | Adsorption | Hydrogenation | |||||
| 40 °C | 300 °C | 40 °C | 300 °C | |||||
| Absorbed CO* species on metallic cobalt | 2184 2142 2045 | 2115 2075 | 2119 | 2115 | 2181, 2168, 2118, 2084, 2048 | [70,71] | ||
| Absorbed CO2 | 2200–2400 | [69] | ||||||
| Bicarbonate (HCO3−) | [33,72,73,74] | |||||||
| ν(OH) νasy(OCO) νs(OCO) ν(COH) | 1602 1408 1217 | 1217 | 1216 | 1654 1434 | 3636, 3667 1216 | 3600–3627, 3620 1655, 1652, 1650 1440, 1424, 1435 1225, 1220, 1228 | ||
| Carbonate (CO3−) | 1550 and 1380 | [33,69,74] | ||||||
| Monodentate νasy(OCO) νs(OCO) ν(CO) | 1450 1385 | 1394 | 1475 1380 | 1457 1354 | 1452 1349, 1396 1075 | 1446–1590 1380–1395 1040 | ||
| Bidentate νasy(OCO) νs(OCO) | 1293 | 1288 | 1575 1286 | 1545 1265 1331 | 1577 1279 | 1284 | 1535–1670 1243–1355 | |
| Formate ν(CH) ν(OCO) | 2850 | 2713, 2849 2944 | 2848 1523 | 2838, 2719 2940 | 2756–2866, 2905 1540, 1510, 1450 | [69,72,73] | ||
| Methane | 3014 | 1305, 3014 | 1305 and 3015 | [73] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.H.; Kim, H.B.; Park, E.D. CO and CO2 Methanation over CeO2-Supported Cobalt Catalysts. Catalysts 2022, 12, 212. https://doi.org/10.3390/catal12020212
Nguyen TH, Kim HB, Park ED. CO and CO2 Methanation over CeO2-Supported Cobalt Catalysts. Catalysts. 2022; 12(2):212. https://doi.org/10.3390/catal12020212
Chicago/Turabian StyleNguyen, Thuy Ha, Han Bom Kim, and Eun Duck Park. 2022. "CO and CO2 Methanation over CeO2-Supported Cobalt Catalysts" Catalysts 12, no. 2: 212. https://doi.org/10.3390/catal12020212
APA StyleNguyen, T. H., Kim, H. B., & Park, E. D. (2022). CO and CO2 Methanation over CeO2-Supported Cobalt Catalysts. Catalysts, 12(2), 212. https://doi.org/10.3390/catal12020212