1. Introduction
Despite the increasing role of processes related to “green chemistry”, oil refining remains one of the key industrial areas for energy supply. Large-scale refining processes include hydro-refining processes, among which the hydrotreatment of various oil distillates dominates.
Hydrotreatment is a catalytic process which is designed to remove heteroatomic compounds from oil fractions in order to bring them to meet environmental standards [
1,
2,
3]. Supported catalysts containing MoS
2/WS
2 promoted by Ni(Co) [
2,
4,
5] as an active compound and various porous oxides like gamma alumina [
3,
4,
6] as a support are usually used as catalysts in industry. These catalysts have sufficiently high activity in the conversion of straight-run oil fractions and oil fractions with the addition of fractions of secondary processes up to 15%.
The main recent trend is the increase of the depth of conversion of oil, which causes a deterioration in the composition of the oil fraction that undergoes refining. As a result, there is a necessity to develop new catalytic systems possessing high activity in the processing of heavier oil feedstocks.
There are a number of ways to increase the activity of a Ni(Co)—Mo(W)—S system, but the main principle includes a search for synthesis properties with the greatest amount of sulfide active sites. One of the methods is to develop catalytic systems without support, i.e., consisting entirely of active metal sulfides. The high-dispersed polymetallic (NiMo or NiMoW) bulk catalysts of NEBULA series synthesized by the coprecipitation of the corresponding salts of metals precursors have gained the greatest fame [
7,
8]. In these catalysts, the portion of active sites in a given volume of catalyst increases many times over. Another reason to study bulk catalysts is the possibility of applying them to the hydroprocessing of heavy oils and residues.
The hydrotreating of heavy fractions and residues using traditional supported catalysts in fixed-bed reactors leads to rapid catalyst deactivation caused by coke formation [
9,
10]. Bulk catalysts do not possess similar problems, and therefore they can be used as catalysts in flow reactors [
11]. They can be used individually or as one of the layers in a reactor [
7].
Various approaches to the synthesis of bulk catalysts are described in the literature. Each of them includes preparation of the active phase precursor [
12,
13]. Under certain conditions, the formation of precursors with different morphologies and porous structures is possible, which in turn makes it possible to vary the properties of bulk catalysts in a fairly wide range [
13,
14]. The structure and catalytic properties of these catalysts will greatly depend upon the preparation method. When a synthesis method has to be chosen, the ratio of active components plays a very important role. According to the patent [
15], high activity levels in HDS and HDN reactions can be achieved at definite Ni/Mo/W ratios in the precursor. The influence of nickel content given fixed Mo and W contents in supported NiMoW catalysts being prepared by mechanical mixing is studied in [
14]. It is established that Mo and W present in the catalyst as corresponding crystallized sulfides. The implementation of nickel into the Mo and W mixture results in the de-stacking of layers of sulfides and in changes to slab height. Investigation of catalysts with different nickel contents showed that the best activity in hydrodeoxygenation is achieved at a Ni/Mo/W molar ratio of 1/3/3. In [
12], controversial results are given for NiMoW bulk catalysts with different nickel contents indicating that the catalyst with a Ni/Mo/W molar ratio of 1/2/2 has the highest activity in HDS of dibenzothiophene. This difference may be caused by the fact that preparation of the active-phase precursor in the latter work was made by the decomposition of thiosalts followed by mechanical mixing of this salt with nickel nitrate. The change in Mo/W ratio in bulk catalysts prepared from mixed oxides is studied in [
16]. It is noted that the W in samples does not enter the ɸ
y layered structure of the oxide precursor. No synergetic effect of Mo—W pairing was observed, and the activity and selectivity of catalysts were influenced only by promoter atoms. Obviously, changing the preparation method may make it possible to change the structure features of the oxide precursor from the active phase. This would lead to the manifestation of differences in the optimal ratios between metals.
Considering catalyst development from the application point of view, it is necessary to choose a preparation method of bulk catalysts which allows for the preparation of granular catalysts. One of the perspective methods is the synthesis of the active component precursor in the solution by mixing all salts together followed by drying of the suspension, thermal treatment, and formation of granules. However, the available literature data almost do not contain any data on the influence of the ratio of active metals on the precursor and of the catalyst on the activity of bulk sulfide catalysts in the target hydrotreating reactions.
Bulk Ni/Mo/W granular catalysts with different molar ratios of active metals were synthesized and studied in the present work. The dependence of catalysts’ properties on the ratio of active metals was established. Sulfide bulk catalysts were tested for the hydrotreating of vacuum gasoil and compared with commercial NiMo—supported hydrotreating catalyst for vacuum gasoil hydrotreating.
2. Results and Discussion
2.1. Thermal Analysis Data
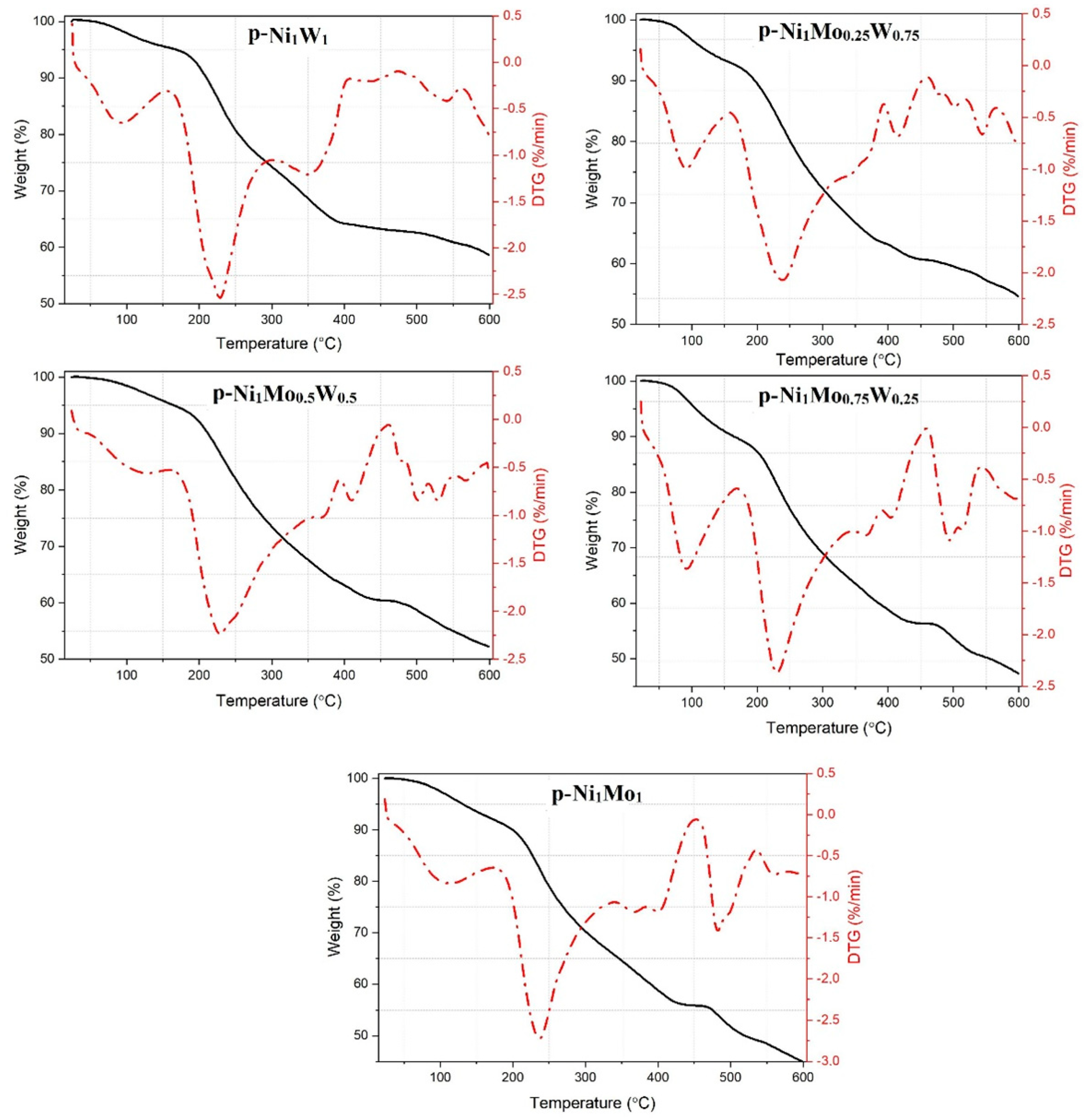
The TG and DTG curves of the powders of NiMoW precursors (designated as p—Ni
1Mo
xW
y, where x and y are the molar weight of Mo and W) of bulk catalysts (molar ratios 1/0/1, 1/0.25/0.75, 1/0.5/0.5, 1/0.75/0.25, and 1/1/0) are given in
Figure 1. During heating, there are three areas of weight loss observed for all precursors-temperature ranges of 50–140, 180–350, and 400–600 °C. Such types of DTG curves are characteristic for the decomposition of citrate complex compounds [
17,
18,
19]. The loss of samples’ weight at 50–150 °C is caused by the removal of adsorbed water and ammonium. Great weight loss (25% of total samples weight) is observed at 180–350 °C due to the decomposition of citrate ligands to acetondicarboxylate complexes or oxycarbonate compounds [
17,
20]. The specific widening of the DTG curve is observed for samples with an atomic ratio of Mo/W ≥ 1 (p—Ni
1Mo
1, p—Ni
1Mo
0.75W
0.25, and p—Ni
1Mo
0.5W
0.5), which indicates uneven decomposition of the powders. This can be accounted for by the partial decomposition of organic compounds or the formation of more stable molybdenum citrate complexes. The complete decomposition of the citrate complex compounds is carried out at 350 °C. When the atomic ratio Mo/W is ≤ 1 (p—Ni
1W
1, p—Ni
1Mo
0.25W
0.75), complete decomposition of citrate complexes is observed at lower temperatures—300–310 °C. This may reveal a higher thermal stability of Mo—citrate complexes in comparison with W—containing complexes. Weight loss in the temperature range of 400–600 °C is accompanied by a great exo-effect caused by the oxidation of the residual fragments of organic molecules and the formation of Ni—, Mo—, and W—oxide compounds and their phase transitions [
21,
22,
23].
It should be pointed out that the use of citric acid for the preparation of bulk catalysts has some advantages in comparison to traditional methods like co-precipitation/hydrothermal synthesis. Citric acid has proven itself as a complexing agent in the preparation of impregnating solutions for hydrotreating catalysts, which contain Ni(or Co), Mo, and W in a wide range of concentrations [
24,
25,
26]. Another point is that the simultaneous dissolution of Ni, Mo, W, and citric acid followed by spray drying allows us to form precursor with a uniform distribution of active metals in the volume of a catalyst. Spray drying compared to co-precipitation/hydrothermal synthesis provides rapid synthesis (~1–2 h) of the precursor in the solid state. However, the precursor obtained after spray drying is not suitable for plasticizing. Therefore, it was necessary to select the temperature for calcination. Based on these considerations, we chose a temperature of 300 °C to calcine all precursors, despite the fact that complete decomposition of citrate ligands occurs at temperatures above 300 °C.
2.2. XRD Data
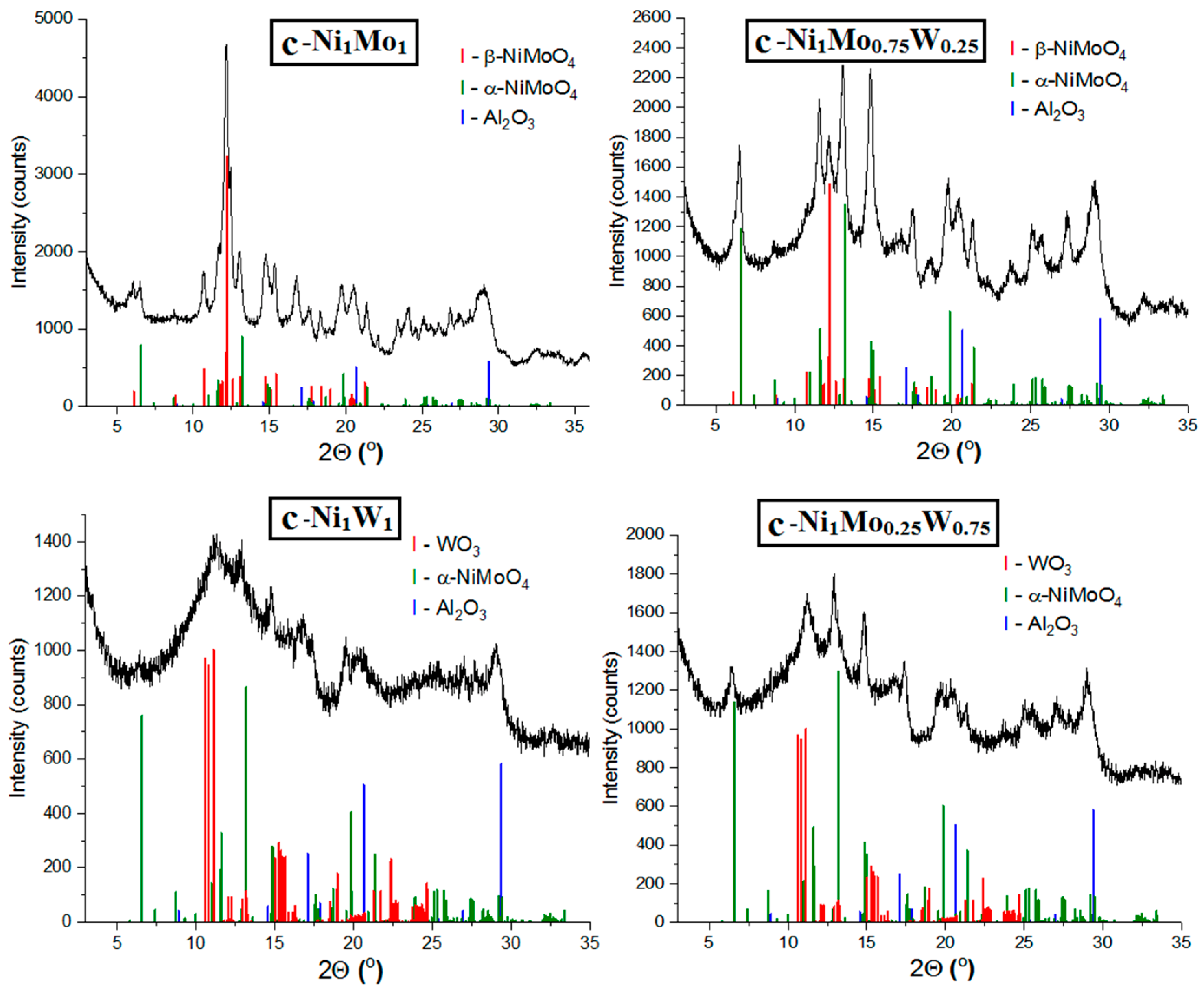
The catalysts (designated as c—Ni1MoxWy, where x and y are the molar weight of Mo and W) after drying at 300 °C have been studied using the XRD method. The resulting data revealed that the samples are X-ray amorphous. Therefore, the catalysts were calcined at 500 °C for 2 h in order to find the characteristic peaks in XRD spectra and to show the difference between samples.
XRD patterns of the catalysts (c—Ni
1Mo
1, c—Ni
1Mo
0.75W
0.25, c—Ni
1Mo
0.25W
0.75, and c—Ni
1W
1) calcined at 500 °C are demonstrated in
Figure 2. The catalysts, which contain Ni and Mo, are characterized by the formation of α—NiMoO
4 (PDF# 00—033—0948, a = 9.509Å, b = 8.759 Å, c = 7.667 Å, β = 113.13°) and/or β—NiMoO
4 (PDF# 00—012—0348, 00—045—0142, a = 10.18 Å, b = 9.241 Å, c = 7.018 Å, β = 107.09°). The main difference between these phases is the coordination of Mo. The α—phase contains Mo in an octahedral coordination, and β—phase is characterized by a tetrahedral Mo coordination [
27,
28]. Apart from that difference, the α—phase is considered more stable, while the β—phase responds to high—temperature modification [
29,
30].
Among the studied catalysts, the c—Ni
1Mo
1 and c—Ni
1Mo
0.75W
0.25 samples enriched with molybdenum can be marked out due to their very similar diffraction patterns. Both catalysts contain β—NiMoO
4 and α—NiMoO
4 phases. The main difference lies in the ratios of these phases in the samples. The polymorphs’ β—NiMoO
4 phase slightly prevails in the c—Ni
1Mo
1 catalyst, while the c—Ni
1Mo
0.75W
0.25 catalyst contains more of the α—NiMoO
4 phase (
Table 1). According to the obtained results, it can be suggested that W content in catalysts correlates with prevailing content of stable α—modification over β—NiMoO
4.
The c—Ni1W1 sample contains a large amount of poorly crystallized phase, which is indicated by the presence of a strongly widened intensive peak at 9–12° (2ϴ). The intense reflexes at these angles are characteristic for WO3 (PDF# 00—020—1323). Therefore, it is possible that the poorly crystallized phase corresponds to WO3. Despite the enrichment of c—Ni1W1 catalyst by W, there are no reflections, which are characteristic for the NiWO4 phase (PDF# 00—015—0755) in the spectrum.
The XRD pattern of the c—Ni1Mo0.25W0.75 sample contains peaks of which the location and relative intensities are features of the α—NiMoO4 phase (PDF# 00—033—0948). A strongly broadened peak related to the WO3 phase is also observed.
In addition to the above, the peaks characteristic of the metastable phase of γ—Al
2O
3 (PDF# 00—029—0063) with a spinel structure are detected in the spectra of all studied catalysts. However, the definite crystal lattice parameter (a = 8.022(3) Å) is much greater than the typical value (7.830–7.940 Å). An increase in the lattice parameter may indicate cationic modification. In particular, the NiAl
2O
4 spinel is characterized by a larger lattice parameter (PDF# 00—010—0339, a = 8.048Å). The average D
XRD of alumina in all catalysts was similar—4.5 nm (
Table 1).
2.3. Nitrogen Adsorption—Desorption Data
Basically, the open literature provides data on the textural properties of bulk catalysts with low surface area and pore volume [
16,
31,
32]. The reason for this is that NiMoW compounds do not form porous structure by themselves. Moreover, the bulk catalysts described in the literature [
31,
32] are mainly used in the form of powders rather than granular catalysts. On the contrary, granular catalysts have been used and studied in the present paper. The feature of the synthesis was the use of a binding component—aluminum hydroxide. Such an approach should yield the formation of a porous structure. Indeed, according to the data of
Table 2, all of the catalysts have a relatively high specific surface area (72–116 m
2/g), but their total pore volumes are still small (0.16–0.22 cm
3/g). The γ—Al
2O
3 in the catalysts performs three important functions: it increases the strength of the catalyst, provides the formation of granules, and increases the surface area and pore volume of the catalysts.
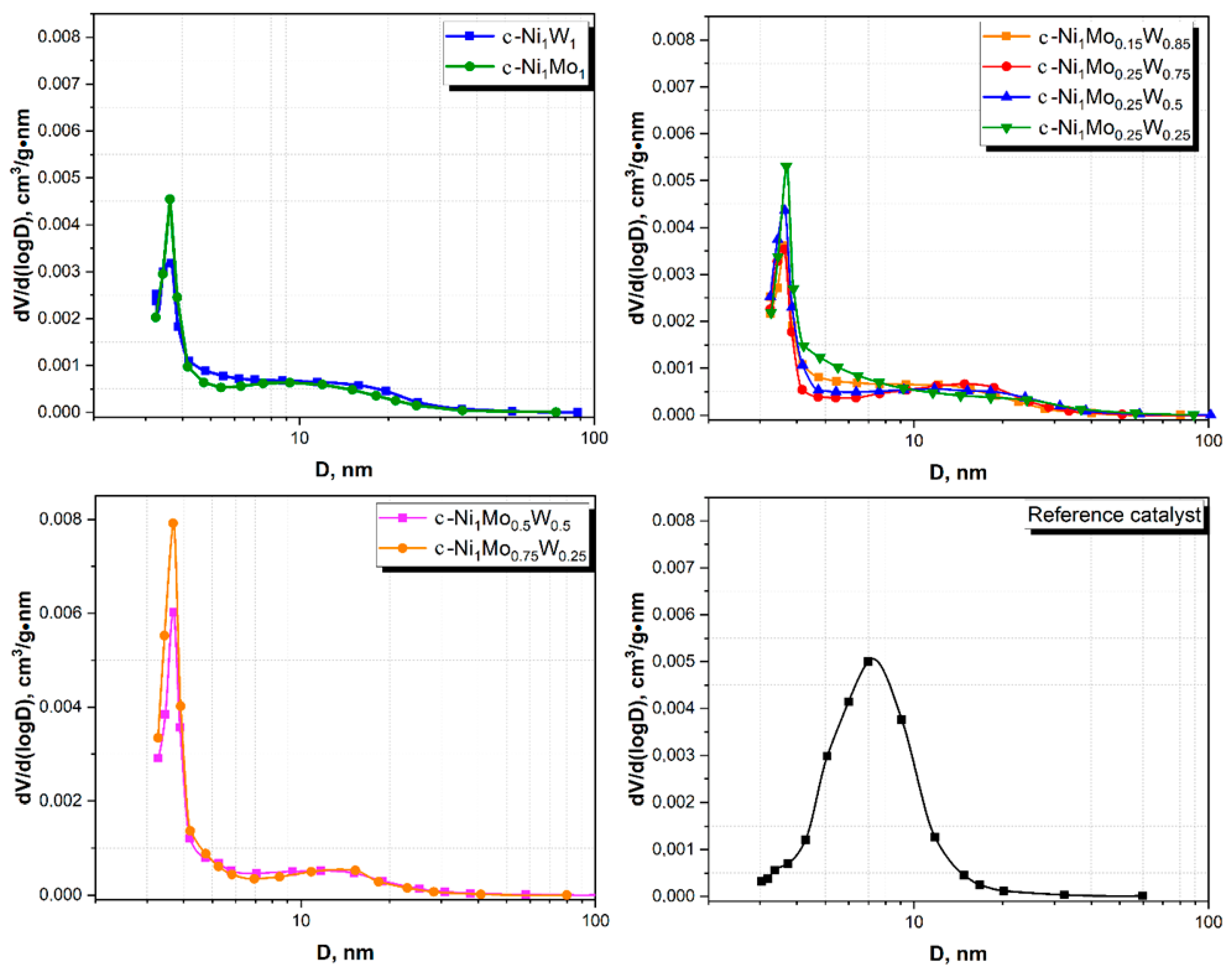
The comparison of c—Ni1Mo1 and c—Ni1W1 catalysts shows a higher specific surface area for the sample with W, while their pore volumes are similar. However, another tendency is observed for the mixed c—Ni1MoxWy catalysts: the higher the W content, the lower the specific surface area. In addition, bulk samples are inferior in terms of specific surface area and pore volume to the reference catalyst.
Figure 3 shows pore size distribution curves of the catalysts. c—Ni
1W
1 possesses a more-developed porous structure in comparison with the c—Ni
1Mo
1 catalyst. The c—Ni
1W
1 sample has a sufficiently wide pore size distribution—from 5 to 35 nm. The c—Ni
1Mo
1 catalyst also has a wide pore size distribution. However, the volume of wide mesopores with the diameters of 7–25 nm decreases. The mixed NiMoW catalysts, such as c—Ni
1Mo
0.15W
0.85, c—Ni
1Mo
0.25W
0.75, c—Ni
1Mo
0.25W
0.5, and c—Ni
1Mo
0.5W
0.25, are also characterized by wide pore size distribution curves with diameters of 5–35 nm. The main difference lies in the range of 5–10 nm and in the amount of small mesopores (<4 nm). It is noted that the lower the W content, the greater the amount of small mesopores. The presence of wider mesopores in the bulk catalysts as compared to the reference samples is observed. This is not the disadvantage of bulk catalysts, because it provides feedstock molecules access to active sites of catalysts.
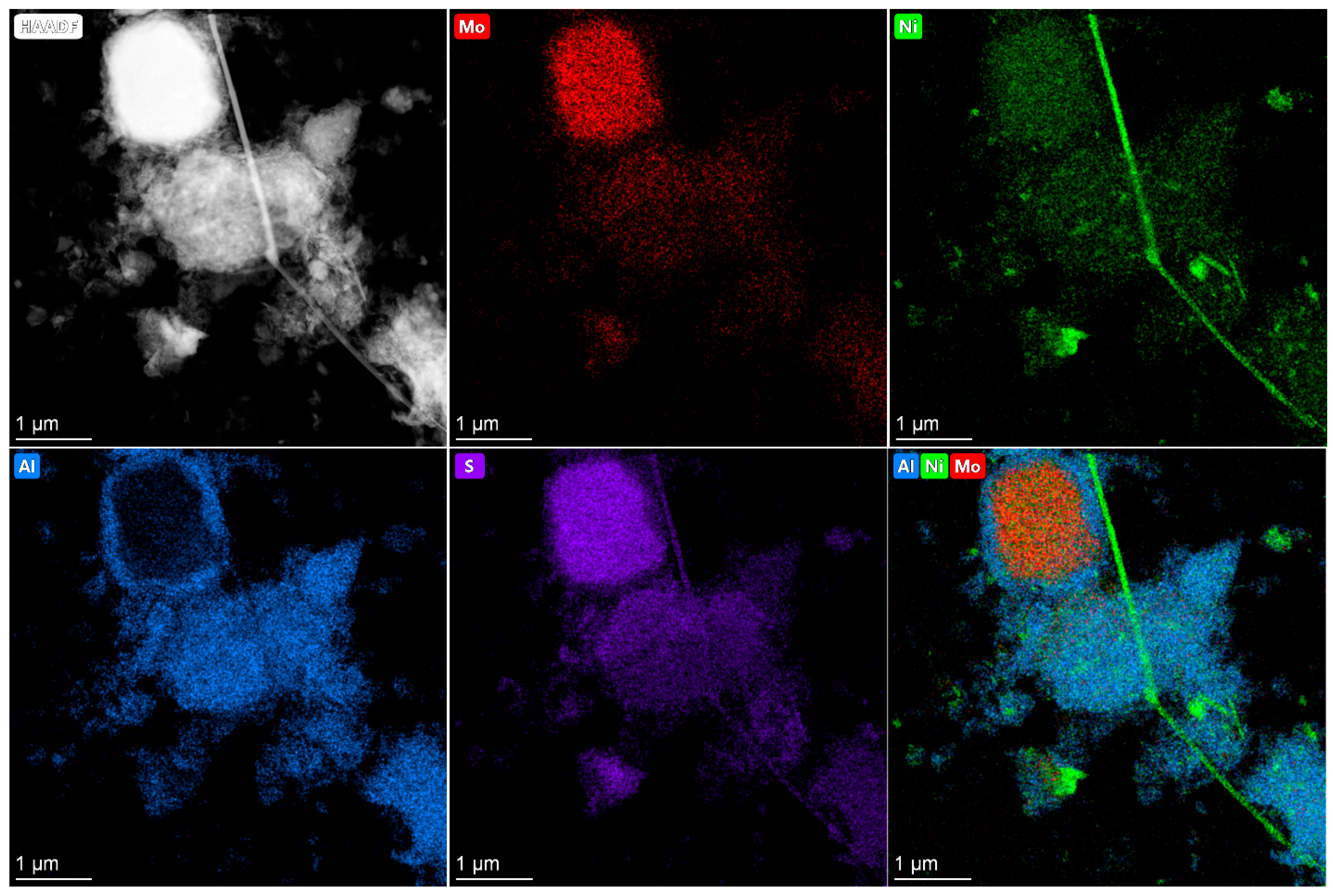
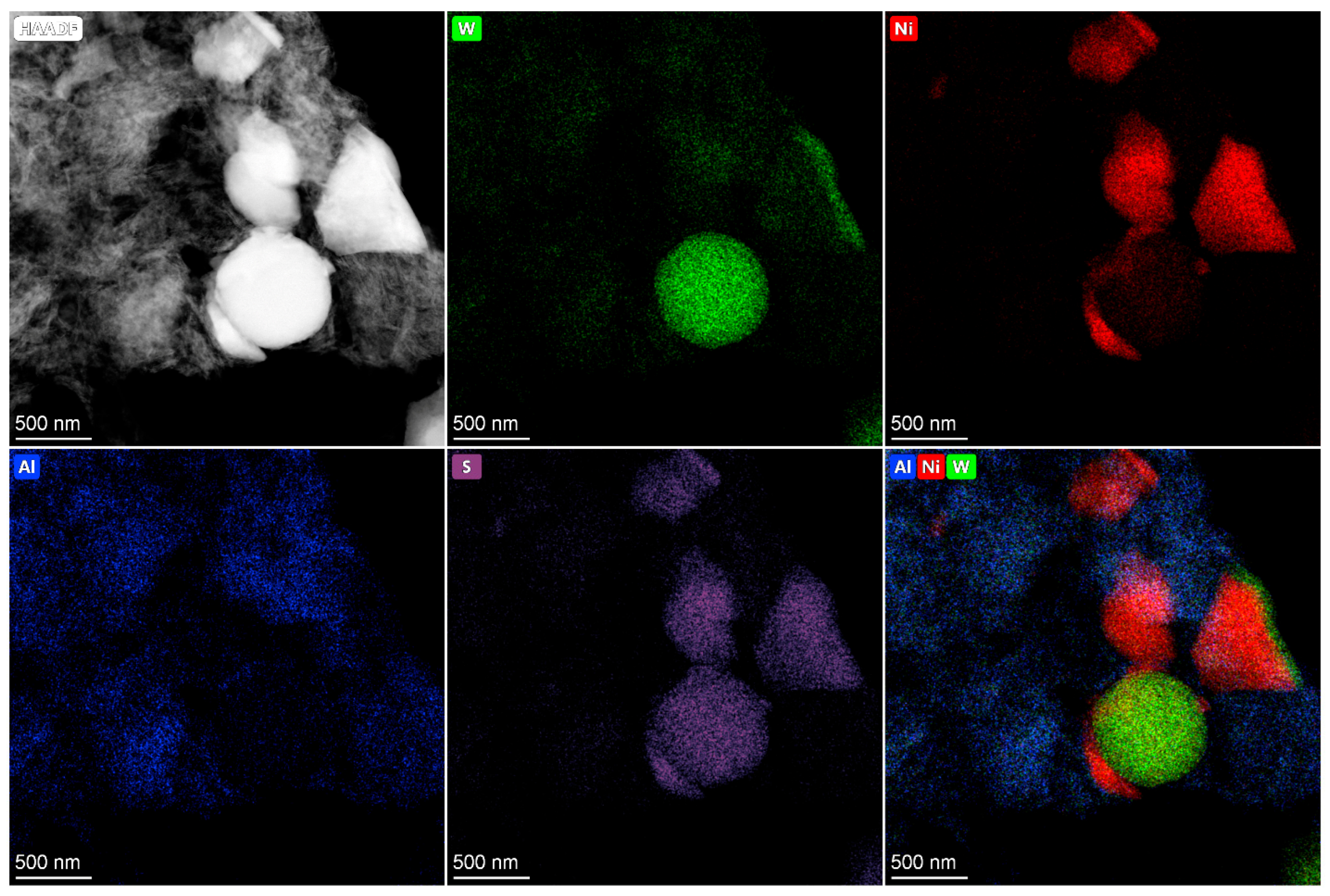
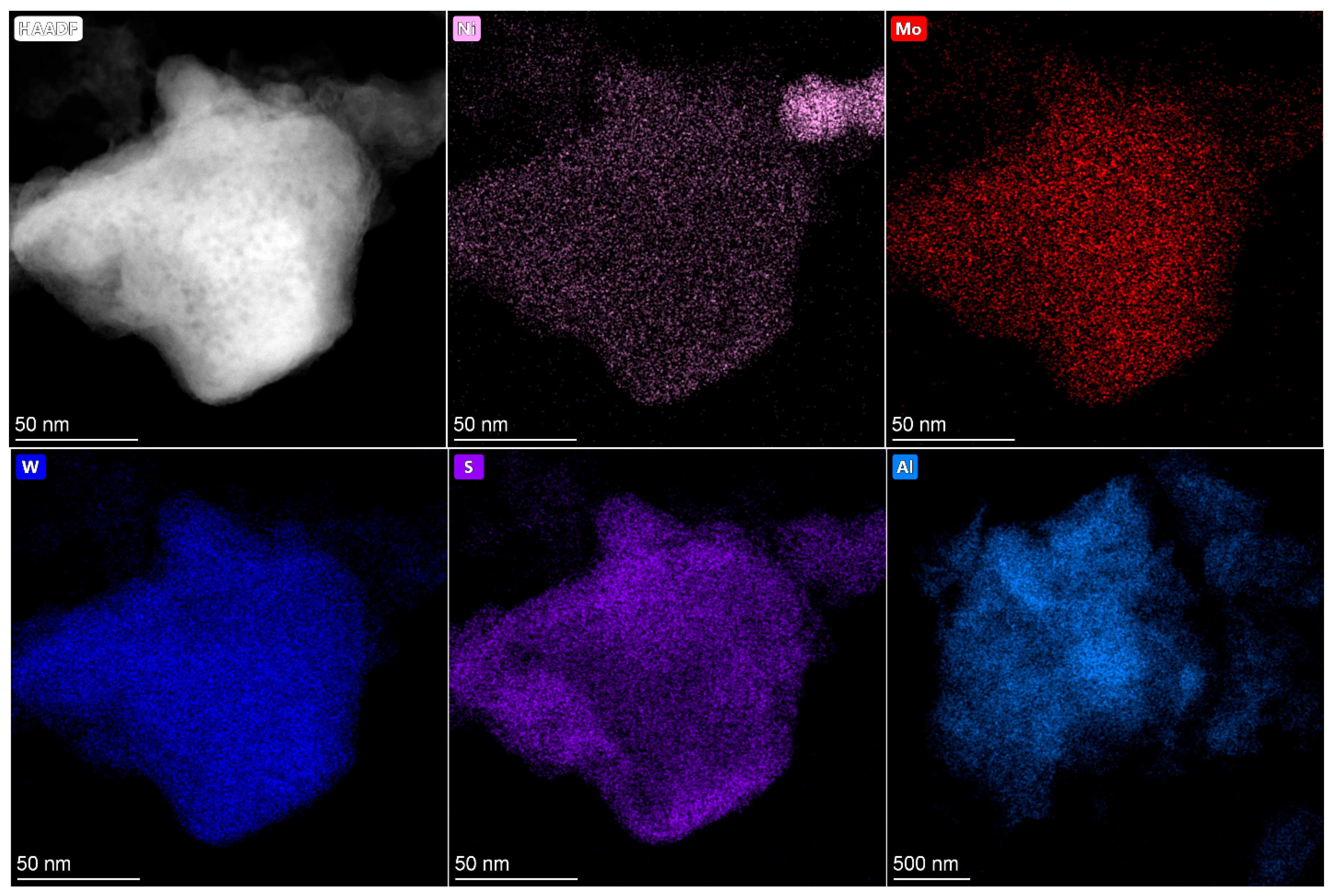
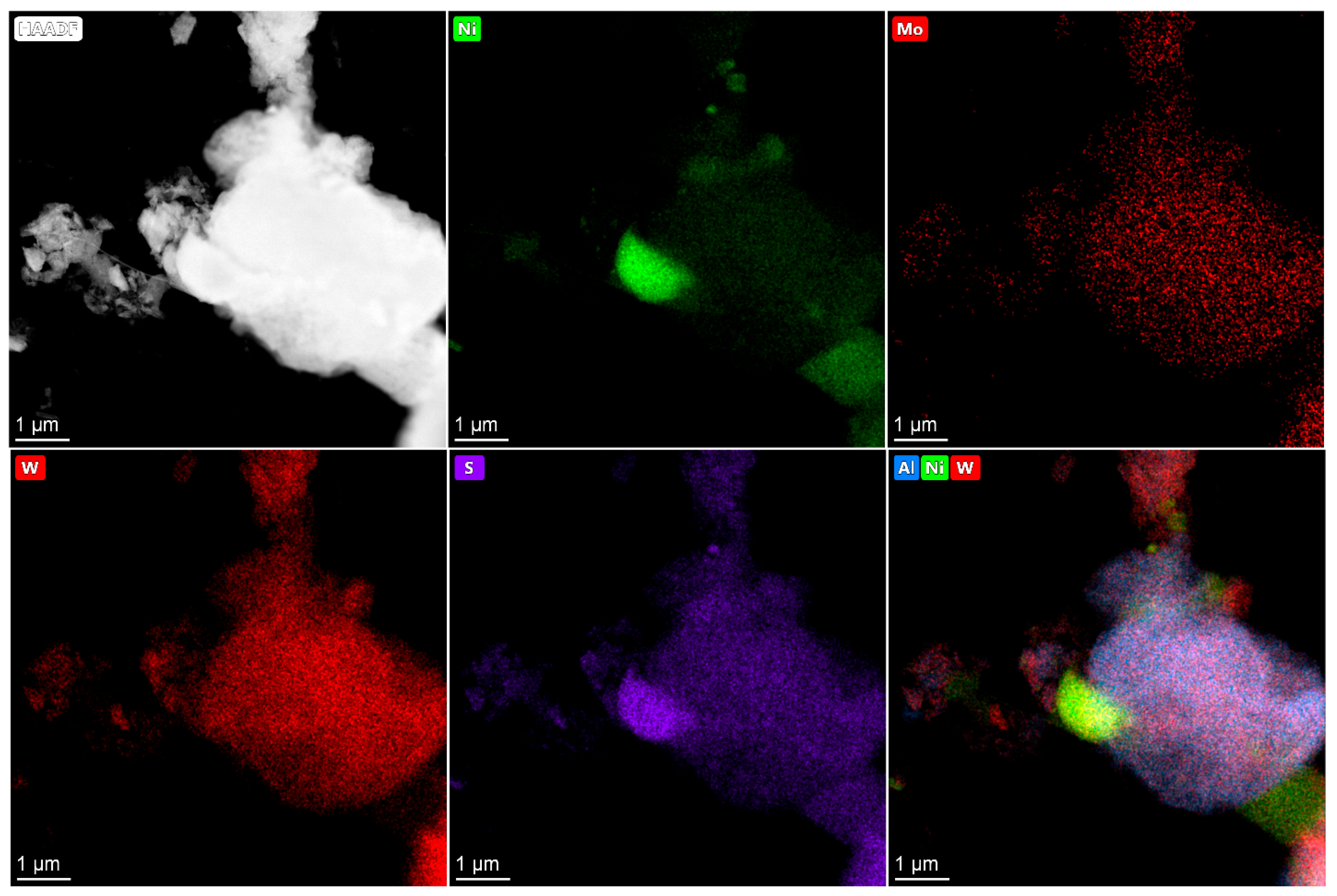
2.4. HRTEM and HAADF Data
The active component in supported Co(Ni)Mo(W) hydrotreating catalysts consists of Mo or W sulfides, the edges of which are decorated by promotor atoms [
33,
34]. The high dispersity of active component particles and their uniform distribution on the surface provide high catalytic activity. Conversely, bulk catalysts have a high concentration of active metals per volume and a large amount of the so-called Co(Ni)Mo(W)—S phase. As was mentioned above, the bulk catalysts used in the present work are also characterized by the texture developed due to the presence of binding agent, which provides sulfur and nitrogen compounds access to active metals [
35]. The main question is what active compounds are formed in the bulk catalysts?
In present work, after testing, the bulk catalysts were washed with hexane to remove residual hydrocarbons and then studied using HRTEM and HAADF—imaging (high-angle annular dark field) (
Figure 4,
Figure 5,
Figure 6,
Figure 7 and
Figure 8). The bimetallic c—Ni
1Mo
1 and c—Ni
1W
1 bulk catalysts consist of two types of components: 1. sulfide NiMo or NiW particles, which are uniformly dispersed on an alumina binding agent (
Figure 4); 2. bulk NiMo and NiW sulfide particles (
Figure 5 and
Figure 6). Layers of particles located on alumina binding agent are similar to the particles, which are usually present in supported NiMo/Al
2O
3 or NiW/Al
2O
3 hydrotreating catalysts [
34,
36]. Bulk sulfide particles are presented as large Mo or W sulfides, in which some Ni is uniformly distributed in the volume (
Figure 5 and
Figure 6). The other portion of Ni is located nearby these sulfide particles and forms areas enriched by Ni.
The HAADF images of c—Ni
1Mo
xW
y catalysts also demonstrate areas with sulfide active metals distributed on alumina. This behavior is similar to the bimetallic c—Ni
1Mo
1 and c—Ni
1W
1 catalysts (
Figure 7 and
Figure 8). The presence of metals’ distribution on the binding agent was also observed in [
37,
38] for bulk catalysts. In addition, bulk components of nickel sulfide and mixed NiMoW, NiMo, and NiW sulfide particles are detected. The distribution of Mo and W in the bulk particles relative to each other is quite uniform and similar to the catalysts with different Ni/Mo/W ratios. It is shown in [
39] that Mo and W atoms in bulk NiMoW sulfide catalysts are localized near each other at a distance of 0.6 nm. This corresponds to the distance in the metal–metal second coordination sphere in hexagonal lattice of metal sulfides (5.48 Å). Thus, it can be assumed that Mo and W atoms form mixed bimetallic sulfide particles of active components, the corners and edges of which are decorated by nickel atoms. The model of such particles is given in [
39].
2.5. Catalytic Activity of the Catalysts
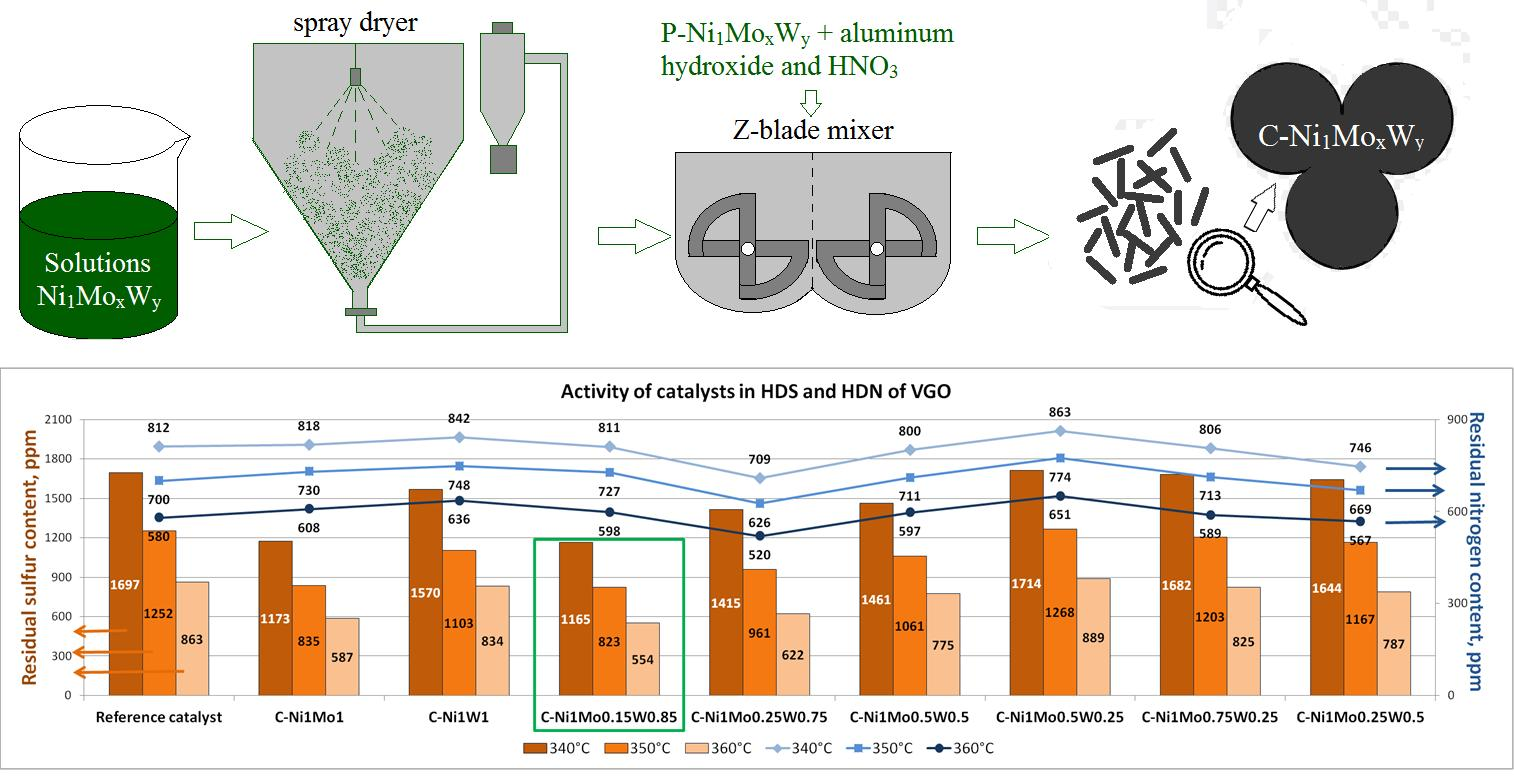
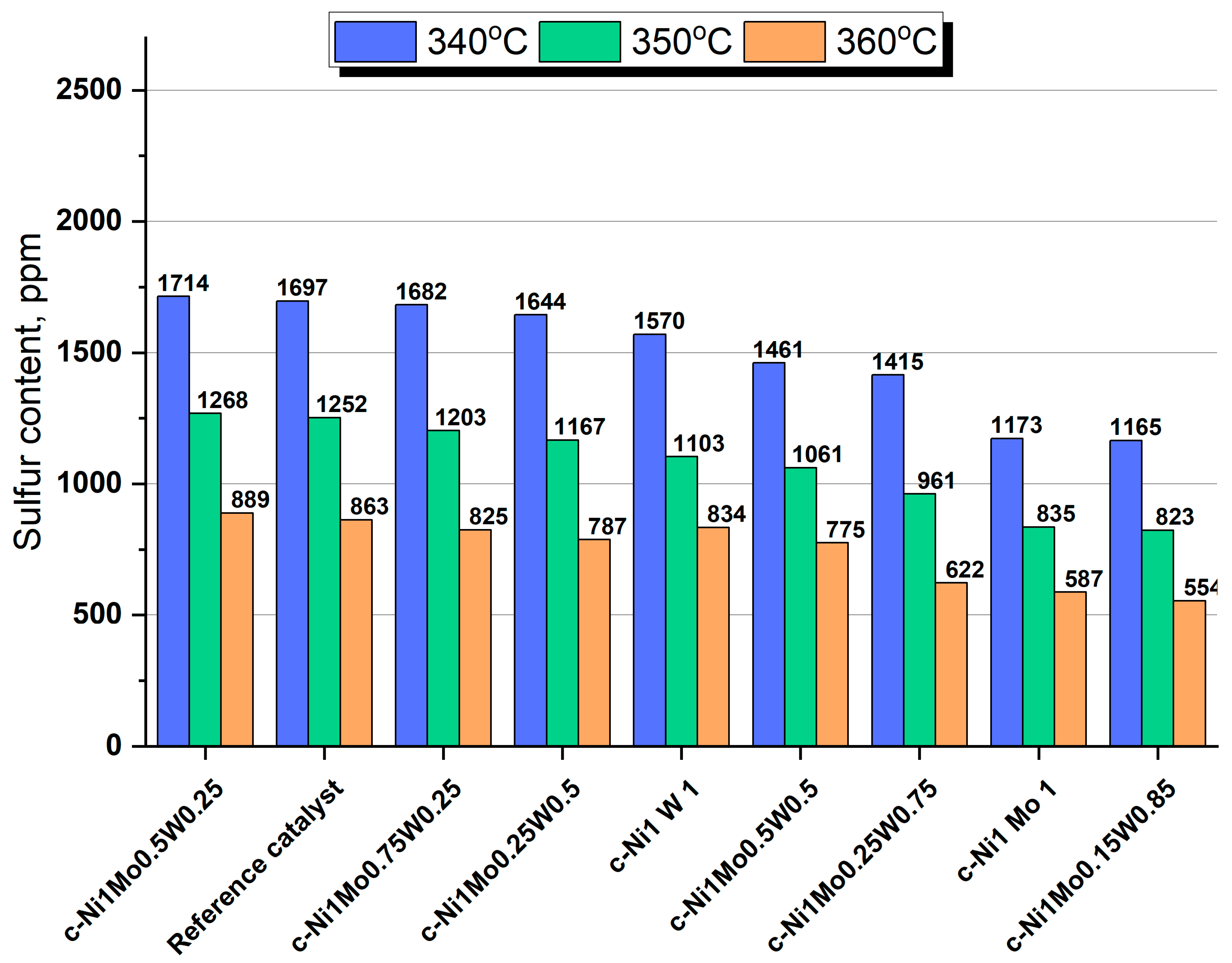
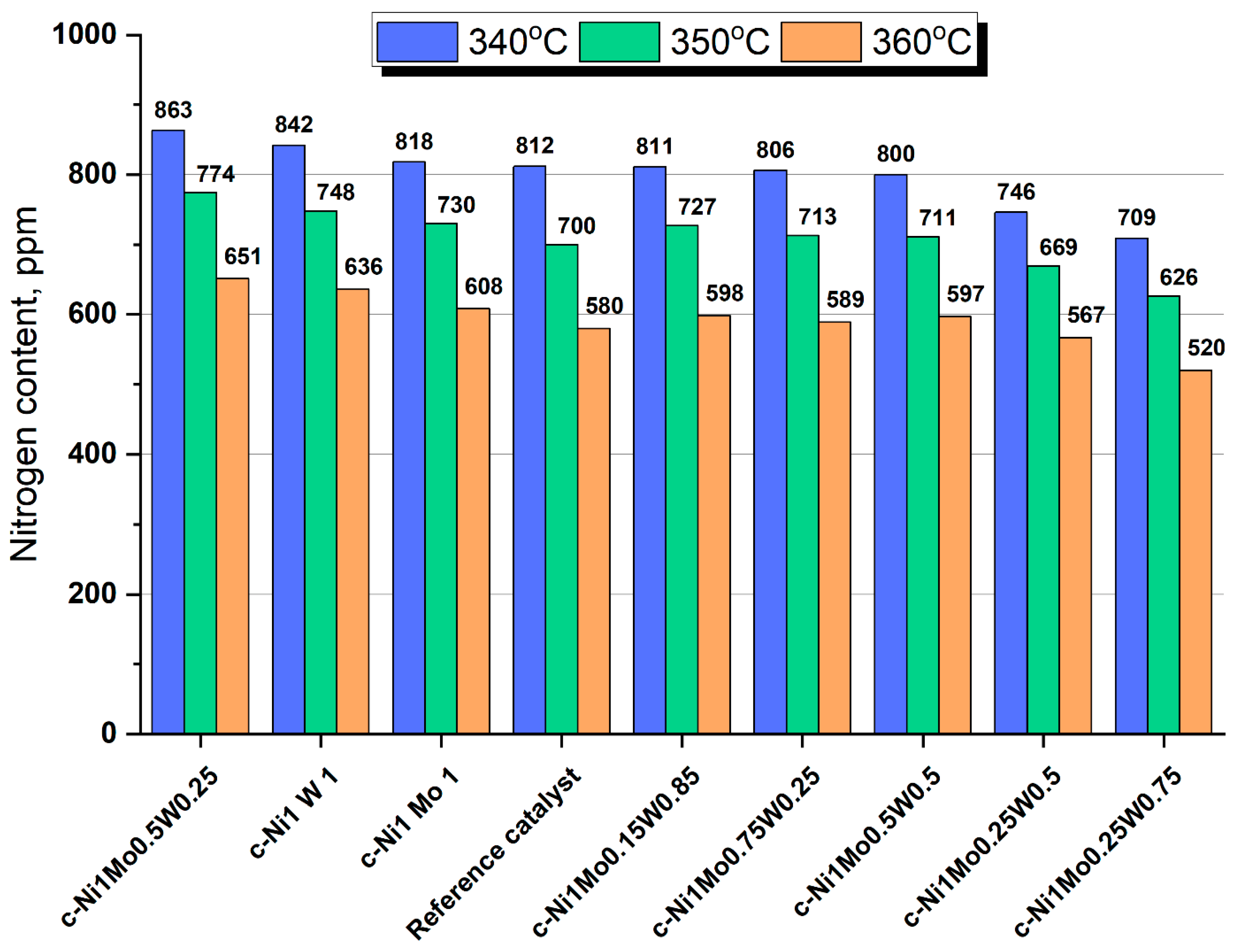
Bulk catalysts and the reference supported catalyst were tested in the hydrotreating of straight—run vacuum gasoil (SR VGO). According to the obtained results, the ratio of active metals in the considered catalysts influences the catalytic activity in HDS and HDN reactions (
Figure 9 and
Figure 10). It should be noted that the bulk density and textural properties of the catalysts were similar. Thus, changes in catalytic activity were caused by the difference in the properties of the active phase of the catalysts.
Bimetallic c—Ni1Mo1 and c—Ni1W1 catalysts are superior in terms of HDS activity to the reference sample. The HDS activity of the c—Ni1Mo1 catalyst is 1.5 times higher than that of the reference sample, while the c—Ni1W1 catalyst is just slightly more active than the reference sample. Almost all of the mixed catalysts are more active than the supported catalyst. The exception is the c—Ni1Mo0.5W0.25 catalyst, which has the same HDS activity.
The best results in HDS of SR VGO among mixed catalysts were achieved for the c—Ni
1Mo
0.15W
0.85 catalyst, for which the smallest amounts of Mo and the largest content of W led to the most significant improvement of HDS activity. However, its activity is similar to bimetallic c—Ni
1Mo
1 catalyst. It should be emphasized that there is a tendency in the mixed series for a worsening of HDS catalytic activity when the Mo content is becoming larger, and the W content is becoming smaller. Due to the similar textural properties of bulk catalysts, we can conclude that such changes in catalytic behavior should be explained by the electronic influence of active metals in this system. We believe that the observed dependencies could be like the ones detected in our previous work for the mixed CoNiMo/Al
2O
3 catalysts [
40], when Co—Ni interaction resulted in changes in the formation of active species. In our case, the addition of the third metal (W) may change the properties of nickel. This influences the interaction between Mo and W in an oxide precursor (which is confirmed by the XRD data) and then in the sulfide catalyst. According to the HAADF data, the mixed series contains many NiMoW compounds with various structures. It is noted in [
39] that the addition of W in the catalyst influences nickel sulfidation temperature. When the Ni/Mo/W ratios were 2/1/1 and 4/2/1, the addition of W to NiMo resulted in the decrease of the sulfidation temperature of 50 wt.% of Ni in the catalyst from 250 to 185 and to 150 °C, respectively. When Ni/Mo/W was 2/1/1, the addition of W resulted in the increase in the end point of nickel sulfidation from 300 to 350 °C. When the Ni/Mo/W ratio was 4/2/1, the end point of nickel sulfidation decreased to 180 °C. Narrowing the temperature range of Ni sulfidation probably results in the increase in the portion of individual Ni sulfide particles, which are not built—in in the structure of individual and mixed Ni and/or W sulfides. Moreover, it can be supposed that W addition leads to stronger interaction between molybdenum and nickel, as evidenced by the formation of a more stable α—NiMoO
4 phase in calcined catalysts. This can provide a close localization of nickel and molybdenum during the sulfiding stage which prevents the formation of individual nickel sulfide particles and then promotes the formation of a mixed bulk sulfide phase. This leads to the increased portion of more active sites being formed in the mixed sulfides. Based on the results in [
40] and the tendency of increasing HDS activity with the increase of W content and the decrease of Mo content, we can suggest the following: particles of W sulfide decorated with small amounts of Mo and large amounts of Ni atoms are responsible for the higher activity of the addressed reaction in the mixed system.
The dependence of HDN activity on catalysts’ composition significantly differs from that of HDS activity. The c—Ni1Mo0.5W0.25, c—Ni1Mo0.15W0.85, c—Ni1Mo0.5W0.5, c—Ni1Mo1, and c—Ni1W1 catalysts are inferior in HDN activity to the reference catalyst. The c—Ni1Mo0.75W0.25 catalyst has similar activity to the reference catalyst. Only two catalysts—c—Ni1Mo0.25W0.5 and c—Ni1Mo0.25W0.75—are superior in terms of HDN activity to the reference sample. However, it should be noted that the difference in HDN activity between the bulk and supported catalysts is not very great for SR VGO hydrotreating.
Summarizing the results, the choice of the unsupported catalyst for the hydrotreating process, especially for SR VGO, is absolutely justified because of the high activity in HDS reactions. When the Ni/Mo/W ratio is 1/0.15/0.85, the activity of the bulk catalyst is almost 1.5 times higher than that of the supported catalyst. The difference in catalysts’ compositions is very hard to establish correctly. However, it can be supposed that the proportion of active sites increases due to the increase in the portion of mixed—bulk NiMoW sulfide particles in such W contents. Since the addition of W leads to the increase in the interaction between molybdenum and nickel ions, as evidenced by an increase in α—NiMoO4 in calcined catalysts according XRD, it probably leads to a decrease in the portion of individual nickel sulfides at the sulfided stage.
3. Materials and Methods
3.1. Catalysts Preparation
Catalysts were prepared as follows. Citric acid (CA) (produced by Base of Chemreactives No.1, Russia) was dissolved in distilled water under intense stirring at 70 °C. Next, nickel hydroxide (produced by Acros Organics, Geel, Belgium) was added to the solution and stirred until complete preparation of a transparent green solution. Ammonium heptamolybdate (produced by Baltic Enterprise Ltd., Saint Petersburg, Russia) and ammonium metatungstane (produced by Sigma-Aldrich, St. Louis, MO, USA) were added to the solution and stirred until complete dissolution. The Ni/CA molar ratio was 1.5 in all solutions. The Ni/Mo/W molar ratio was varied. The resulting solutions were spray dried in order to obtain precursor of the active phase of bulk catalyst. The dried powder of the precursor was calcined at 325 °C. Thus, NiMoW precursor powders with the following Ni/Mo/W molar ratios were prepared: 1/1/0; 1/0.25/0.75; 1/0.5/0.5; 1/0.25/0.5, 1/0.75/0.25; 1/0.5/0.25; 1/0/1. The precursors were named as p—Ni1MoxWy, where x and y are the molar weights of Mo and W.
Granular NiMoW bulk catalysts were prepared as follows. The powders of NiMoW precursors were mixed with aluminum hydroxide (produced by IC SB RAS [
41]) and plasticizing agent in the Z—blade mixer. The ratio of aluminum hydroxide to powders of NiMoW precursors was 0.6 in all cases. Nitric acid was used as a plasticizing agent (n(HNO
3)/n(Al
2O
3) = 0.1). Then, the kneading paste was granulated by the VINCHI extruder using the spinneret with a trilobe shape (the size of 1.3 mm). The resulting extrudates were dried at 120 °C over the course of 8 h and calcined at 300 °C in air flow for 2 h. The designation of catalysts is given in
Table 3. The bulk density of calcined catalysts varied from 0.9 to 1.1 g/cm
3. 3.2. Sulfiding and Testing of Catalysts
Catalysts were tested in a fixed-bed pilot unit in a hydrotreatment of SR VGO containing 7080 ppm S and 958 ppm N. In all cases, catalysts were used in a granular shape. A total of 30 cm3 of a catalyst sample was diluted with silicon carbide (0.1–0.2 mm) in a volume ratio of 1:2 and loaded in the isothermal zone of the reactor. Next, catalysts were heated in hydrogen flow and sulfided by a mixture of straight-run gasoil and dimethyl disulfide (20 g of DMDS per 1 l of straight-run gasoil). Sulfidation of catalysts was carried out at 3.8 MPa, sulfidation mixture flow—2 h−1, and volume ratio H2/sulfidation mixture—300 Nm3/m3, at 240 °C for 16 h and at 340 °C for 16 h.
Next, catalysts were tested in a hydrotreatment of SR VGO. The process parameters were as follows: liquid hourly space velocity (LHSV) = 1.5 h−1; H2/feed = 600; P = 8.0 MPa; T = 340, 350, and 360 °C. Catalysts were tested for 48 h at each temperature. There were no liquid samples during first 24 h at each temperature. That period of time was marked as having no steady state. Next, every 4 h, 4 liquid product samples were selected for analysis. The content of total nitrogen (total N content) and total sulfur (total S content) in the feedstock and hydrotreating products was determined on an Xplorer—NS Analyzer (TE Instruments) using oxidative combustion with chemiluminescence and ultraviolet fluorescence detection according to ASTM D5762 and ASTM D4629 (for total N content) and ASTM D5453 (for total S content). The total inaccuracy of analysis was ±8 ppm.
3.3. Characterization Techniques
The XRD patterns of the powders were recorded by means of a STOE STADI MP powder diffractometer (STOE, Germany) equipped with a MYTHEN2 1K detector using MoKα radiation (λ = 0.7093 Å). The scanning interval varied from 2–69°, and the scanning pitch was 0.015° at 2ϴ. The resulting data were analyzed in order to obtain an average crystallite size. The dimensions of the coherent scattering region (CSR—DXRD) were determined from the broadening of the diffraction peaks. Phase analysis was conducted with the use of diffraction database ICDD PDF—2.
Thermal analysis of samples was carried out using a device for simultaneous thermal analysis: STA 449 C Jupiter of NETZSCH. A sample was placed in a corundum crucible. The air flow in the chamber with a sample was 30 mL/min. The sample was heated in a programmable temperature regime—heating rate of 10 °C/min from 30 to 600 °C. Experimental data were analyzed using the analysis software package NETZSCH Proteus Thermal Analysis.
Nitrogen adsorption–desorption data were obtained for catalysts calcined at 300 °C. The textural properties of the catalysts were determined by nitrogen physisorption using an ASAP 2400 instrument (Micrometrics, Norcross, GA, USA). Prior to analysis, samples were subjected to a N2 flow at 150 °C for 2 h. The BET surface areas were calculated from the nitrogen uptakes at relative pressures ranging from 0.05 to 0.30. The total pore volume was derived from the amount of nitrogen adsorbed at a relative pressure close to unity (in practice, P/P0 = 0.995) by assuming that all accessible pores have been filled with condensed nitrogen in the normal liquid state. The pore size distribution was calculated by the BJH method using the desorption branch of the isotherm. The uncertainty of the obtained results was 10%.
The structure of the sulfided catalysts was studied by HRTEM. For shooting in TEM and STEM modes, a ThemisZ two-corrector transmission electron microscope (Thermo Fisher Scientific, Waltham, MA, USA) with an accelerating voltage of 200 kV and a limiting resolution of 0.07 nm (TEM) and 0.06 nm (SEM) was used. Micrographs were recorded using a Ceta 16 CCD matrix (Thermo Fisher Scientific, USA). HAADF: Atomically resolved HAADF images were acquired in the STEM mode. Also called the STEM–HAADF technique, these images are generated by collecting electrons scattered at high angles on passing through the samples, using a Fischione HAADF detector. The acquired image intensity is roughly proportional to Z1.7, with Z as the atomic number of scattering atoms.
4. Conclusions
The present work demonstrates the possibility of the synthesis of granular unsupported catalysts. The influence of the Ni/Mo/W atomic ratio in the catalyst precursor on the properties of unsupported Ni—Mo—W catalysts was studied. Catalysts have been synthesized by plasticizing the Ni—Mo—W precursor with alumina hydroxide (binding agent) and nitric acid followed by extrusion and drying.
The TG and DTG data of the powders of the NiMoW precursors showed characteristic curves of the CA used as a chelating agent during synthesis. In addition, the higher thermal stability of the Mo citrate complexes in comparison with the W ones was detected. The XRD data of catalysts calcined at 550 °C show that the composition of the NiMoW precursor influences the formation of the compounds, respective of the formation of active component. Mo—containing catalysts comprise of the mixture of β—NiMoO4 and α—NiMoO4 phases. The main difference between catalysts is in the ratio of the phases. It is noted that W content in catalysts correlates with the prevailing content of stable α—modification over β—NiMoO4. No NiWO4 phase was detected by the XRD data. However, some portion of WO3 was observed in W—containing catalysts.
All unsupported catalysts have relatively high specific surface areas (72–116 m2/g) and small total pore volumes (0.16–0.22 cm3/g). The bulk catalysts contain large amounts of wide mesopores (5–30 nm) and small mesopores (<4 nm). The presence of wider mesopores in the bulk catalysts as compared to the reference samples was detected. HRTEM and HAADF data show that unsupported sulfide catalysts contain two types of phases: 1. sulfide NiMo or NiW particles, which are uniformly dispersed on alumina binding agent; 2. bulk nickel sulfide, mixed NiMoW, NiMo, and NiW sulfide particles. Depending on the composition of the catalyst, the ratio of the bulk components also changes.
The catalysts were tested in the hydrotreating of SR VGO. It was shown that unsupported catalysts were superior relative to the activity of the reference supported catalyst. The highest desulfurization activity in the hydrotreatment of SR VGO (LHSV = 1.5 h−1, H2/feed = 600, P = 8.0 MPa) was achieved with the c—Ni1Mo0.15W0.85 catalyst. It was also noted that the difference in HDN activity between the bulk and supported catalysts was not very great for SR VGO hydrotreating. Therefore, the crucial factor is HDS activity.



 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}