
Insight into the Effect of Anionic–Anionic Co-Doping on BaTiO3 for Visible Light Photocatalytic Water Splitting: A First-Principles Hybrid Computational Study


Abstract
1. Introduction
2. Computational Details
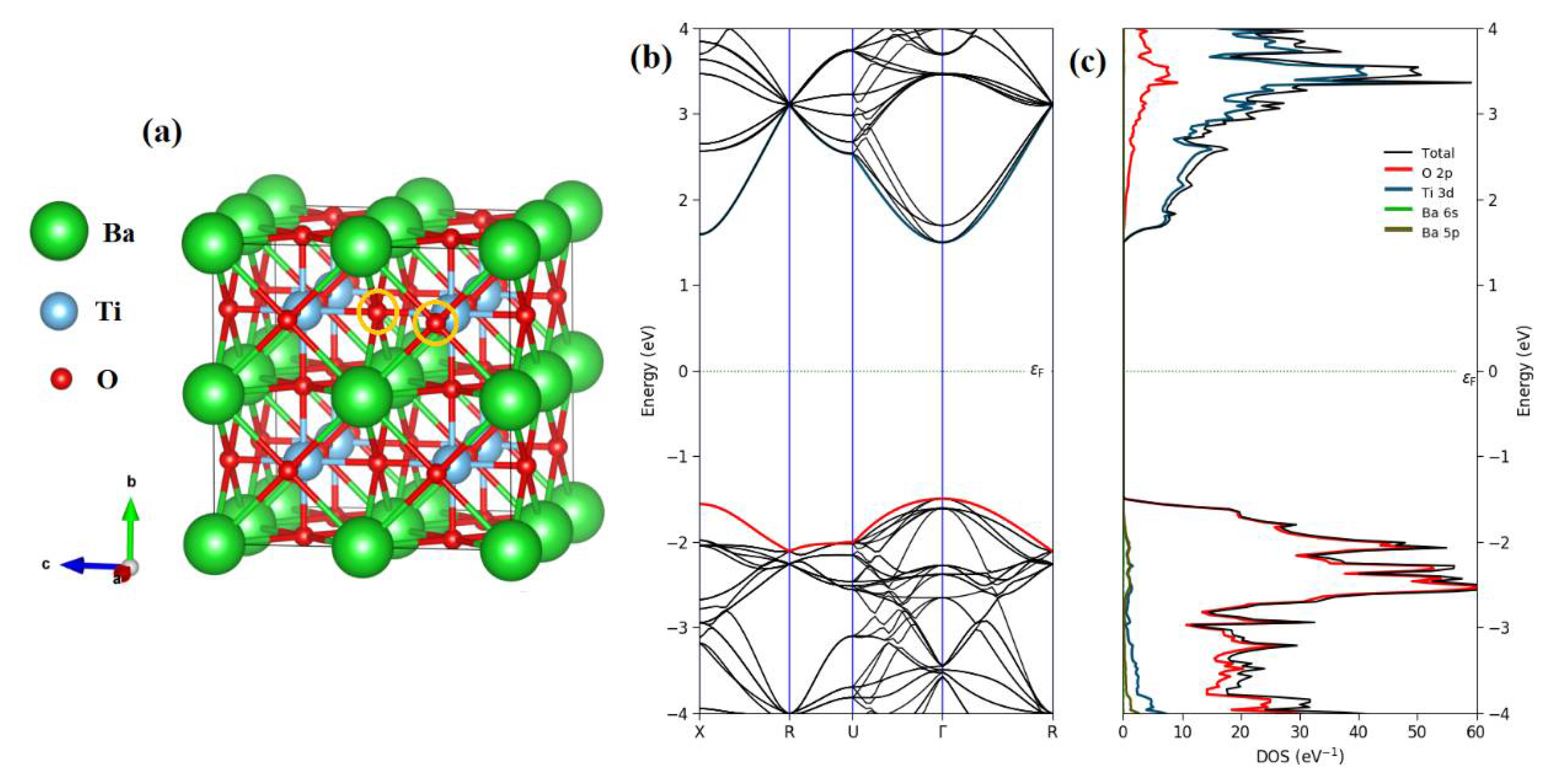
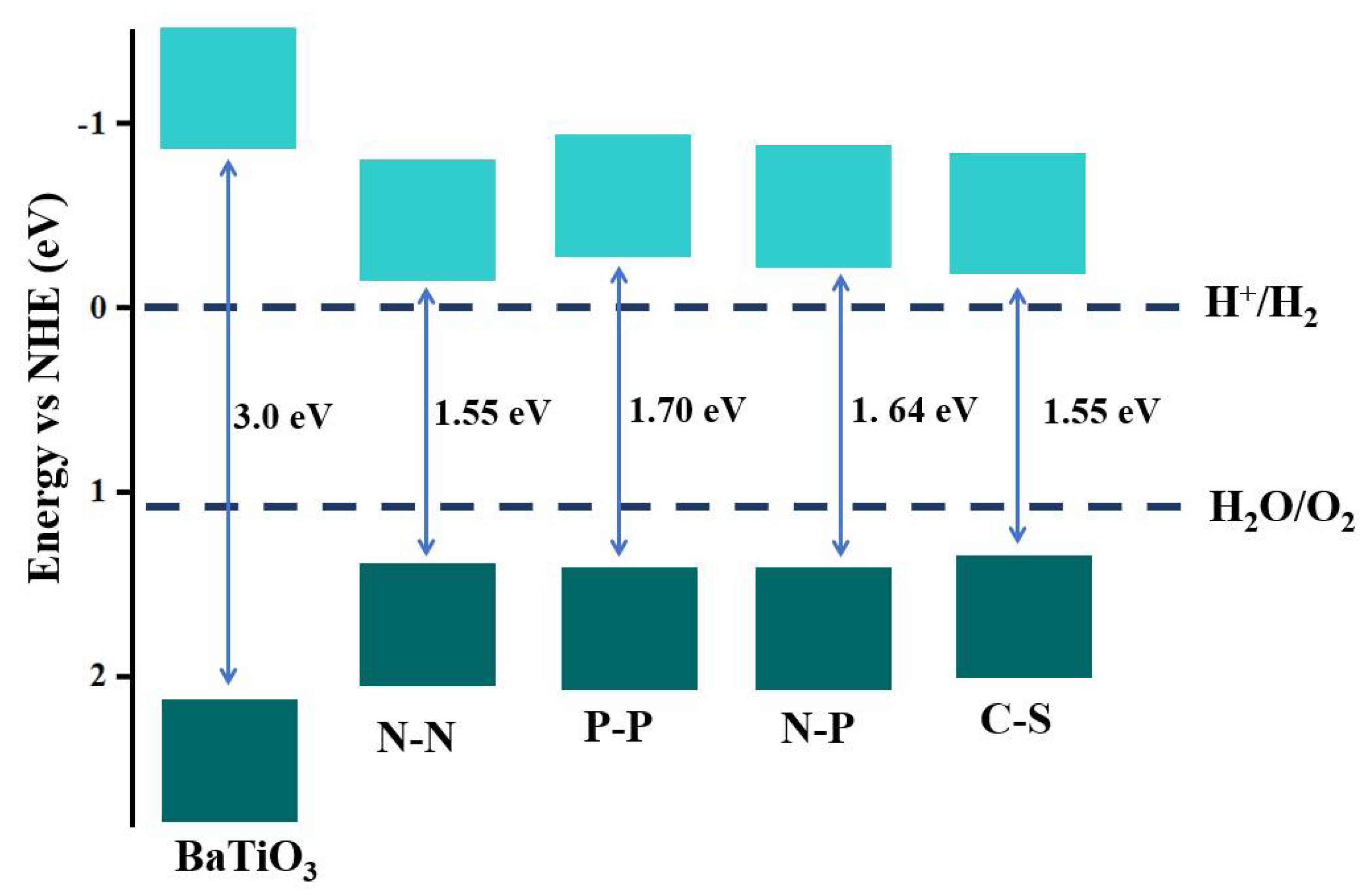
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.; Subramanian, G.; Pillai, S.C. Recent advances in photocatalysis for environmental applications. J. Environ. Chem. Eng. 2018, 6, 3531–3555. [Google Scholar] [CrossRef]
- Moniz, S.J.A.; Shevlin, S.A.; Martin, D.J.; Guo, Z.X.; Tang, J.W. Visible-light driven heterojunction photocatalysts for water splitting—A critical review. Energy Environ. Sci. 2015, 8, 731–759. [Google Scholar] [CrossRef]
- Wang, Q.; Domen, K. Particulate Photocatalysts for Light-Driven Water Splitting: Mechanisms, Challenges, and Design Strategies. Chem. Rev. 2020, 120, 919–985. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Schoonen, M.A. The absolute energy positions of conduction and valence bands of selected semiconducting minerals. Am. Miner. 2000, 85, 543–556. [Google Scholar] [CrossRef]
- Maeda, K. Rhodium-Doped Barium Titanate Perovskite as a Stable p-Type Semiconductor Photocatalyst for Hydrogen Evolution under Visible Light. ACS Appl. Mater. Interfaces 2014, 6, 2167–2173. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, D.; Liu, Z.; Ruan, M.; Guo, Z. Novel strategy for efficient water splitting through pyro-electric and pyro-photo-electric catalysis of BaTiO3 by using thermal resource and solar energy. Appl. Catal. B Environ. 2021, 284, 119686. [Google Scholar] [CrossRef]
- Gao, C.; Yu, H.; Zhang, L.; Zhao, Y.; Xie, J.; Li, C.; Cui, K.; Yu, J. Ultrasensitive Paper-Based Photoelectrochemical Sensing Platform Enabled by the Polar Charge Carriers-Created Electric Field. Anal. Chem. 2020, 92, 2902–2906. [Google Scholar] [CrossRef]
- Zhou, Z.; Tang, H.X.; Sodano, A.H. Vertically Aligned Arrays of BaTiO3 Nanowires. ACS Appl. Mater. Interfaces 2013, 5, 11894–11899. [Google Scholar] [CrossRef]
- Fang, T.; Hu, H.; Liu, J.; Jiang, M.; Zhou, S.; Fu, J.; Wang, W.; Yang, Y. Type-II Band Alignment Enhances Unassisted Photoelectrochemical Water-Splitting Performance of the BaTiO3/CdS Ferroelectric Heterostructure Photoanode under Solar Light Irradiation. J. Phys. Chem. C 2021, 125, 18734–18742. [Google Scholar] [CrossRef]
- Fernandes, E.; Gomes, J.; Martins, R.C. Semiconductors Application Forms and Doping Benefits to Wastewater Treatment: A Comparison of TiO2, WO3, and g-C3N4. Catalysts 2022, 12, 1218. [Google Scholar] [CrossRef]
- Upadhyay, S.; Shrivastava, J.; Solanki, A.; Choudhary, S.; Sharma, V.; Kumar, P.; Singh, N.; Satsangi, V.R.; Shrivastav, R.; Waghmare, U.V.; et al. Enhanced photoelectrochemical response of BaTiO3 with Fe doping: Experiments and first-principles analysis. J. Phys. Chem. C 2011, 115, 24373–24380. [Google Scholar] [CrossRef]
- Nageri, M.; Kumar, V. Manganese-doped BaTiO3 nanotube arrays for enhanced visible light photocatalytic applications. Mater. Chem. Phys. 2018, 213, 400–405. [Google Scholar] [CrossRef]
- Demircivi, P.; Simsek, E.B. Visible-light-enhanced photoactivity of perovskite-type W-doped BaTiO3 photocatalyst for photodegradation of tetracycline. J. Alloys Compd. 2019, 774, 795–802. [Google Scholar] [CrossRef]
- Cui, Y.; Sun, H.; Shen, G.; Jing, P.; Pu, Y. Effect of Dual-Cocatalyst Surface Modification on Photodegradation Activity, Pathway, and Mechanisms with Highly Efficient Ag/BaTiO3/MnOx. Langmuir 2020, 36, 498–509. [Google Scholar] [CrossRef]
- Xie, P.; Yang, F.; Li, R.; Ai, C.; Lin, C.; Lin, S. Improving hydrogen evolution activity of perovskite BaTiO3 with Mo doping: Experiments and first-principles analysis. Int. J. Hydrogen Energy 2019, 44, 11695–11704. [Google Scholar] [CrossRef]
- Cao, J.; Ji, Y.; Tian, C.; Yi, Z. Synthesis and enhancement of visible light activities of nitrogen-doped BaTiO3. J. Alloys Compd. 2014, 615, 243–248. [Google Scholar] [CrossRef]
- Sharma, V.; Pilania, G.; Rossetti, G.A.; Slenes, K.; Ramprasad, R. Comprehensive examination of dopants and defects in BaTiO3 from first principles. Phys. Rev. B Condens. Matter Mater. Phys. 2013, 87, 134109. [Google Scholar] [CrossRef]
- Yang, F.; Yang, L.; Ai, C.; Xie, P.; Lin, S.; Wang, C.; Lu, X. Tailoring Bandgap of Perovskite BaTiO3 by Transition Metals Co-Doping for Visible-Light Photoelectrical Applications: A First-Principles Study. Nanomaterials 2018, 8, 455. [Google Scholar] [CrossRef]
- Alshoaibi, A.; Kanoun, M.B.; Haq, B.U.; AlFaify, S.; Goumri-Said, S. Ytterbium doping effects into the Ba and Ti sites of perovskite barium titanate: Electronic structures and optical properties. Results Phys. 2020, 18, 103257. [Google Scholar] [CrossRef]
- Alshoaibi, A.; Kanoun, M.B.; Haq, B.U.; AlFaify, S.; Goumri-Said, S. Insights into the Impact of Yttrium Doping at the Ba and Ti Sites of BaTiO3 on the Electronic Structures and Optical Properties: A First-Principles Study. ACS Omega 2020, 5, 15502–15509. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Yang, C.-L.; Wang, M.-S.; Ma, X.-G. Chalcogens doped BaTiO3 for visible light photocatalytic hydrogen production from water splitting. Spectrochim. Acta Part A 2019, 208, 65–72. [Google Scholar] [CrossRef]
- Ohno, T.; Tsubota, T.; Nakamura, Y.; Sayama, K. Preparation of S, C cation-codoped SrTiO3 and its photocatalytic activity under visible light. Appl. Catal. A Gen. 2005, 288, 74–79. [Google Scholar] [CrossRef]
- Niu, M.; Cheng, D.; Cao, D. Enhanced photoelectrochemical performance of anatase TiO2 by metal-assisted SeO coupling for water splitting. Int. J. Hydrogen Energy 2013, 38, 1251–1257. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B. Review of First-Principles Studies of TiO2: Nanocluster, Bulk, and Material Interface. Catalysts 2020, 10, 972. [Google Scholar] [CrossRef]
- Zhang, J.; Tse, K.; Wong, M.; Zhang, Y.; Zhu, J. A brief review of co-doping. Front. Phys. 2016, 11, 117405. [Google Scholar] [CrossRef]
- Gai, Y.; Li, J.; Li, S.-S.; Xia, J.-B.; Wei, S.-H. Design of Narrow-Gap TiO2: A Passivated Codoping Approach for Enhanced Photoelectrochemical Activity. Phys. Rev. Lett. 2009, 102, 036402. [Google Scholar] [CrossRef]
- Kanoun, M.B.; Alshoaibi, A.; Goumri-Said, S. Hybrid Density Functional Investigation of Cu Doping Impact on the Electronic Structures and Optical Characteristics of TiO2 for Improved Visible Light Absorption. Materials 2022, 15, 5645. [Google Scholar] [CrossRef]
- Ikram, M.; Ul-Haq, M.A.; Haider, A.; Haider, J.; Ul-Hamid, A.; Shahzadi, I.; Bari, M.A.; Ali, S.; Goumri-Said, S.; Kanoun, M.B. The enhanced photocatalytic performance and first-principles computational insights of Ba doping-dependent TiO2 quantum dots. Nanoscale Adv. 2022, 4, 3996–4008. [Google Scholar] [CrossRef]
- Niishiro, R.; Kato, H.; Kudo, A. Nickel and either tantalum or niobium-codoped TiO2 and SrTiO3 photocatalysts with visible-light response for H2 or O2 evolution from aqueous solutions. Phys. Chem. Chem. Phys. 2005, 7, 2241–2245. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Wang, Y.; Zhang, X.; Fan, Y.; Liu, Y.; Yi, Z. Role of P in improving V-doped SrTiO3 visible light photocatalytic activity for water splitting: A first—Principles study. Int. J. Hydrogen Energy 2021, 46, 20492–20502. [Google Scholar] [CrossRef]
- Wang, B.; Kanhere, P.D.; Chen, Z.; Nisar, J.; Pathak, B.; Ahuja, R. Anion-doped NaTaO3 for visible light photocatalysis. J. Phys. Chem. C 2013, 44, 22518–22524. [Google Scholar] [CrossRef]
- Wang, G.-Z.; Chen, H.; Wu, G.; Kuang, A.-L.; Yuan, H.-K. Hybrid Density Functional Study on Mono- and Codoped NaNbO3 for Visible-Light Photocatalysis. ChemPhysChem 2016, 17, 489. [Google Scholar] [CrossRef]
- Maarouf, A.A.; Gogova, D.; Fadlallah, M.M. Metal-doped KNbO3 for visible light photocatalytic water splitting: A first principles investigation. Appl. Phys. Lett. 2021, 119, 063901. [Google Scholar] [CrossRef]
- Liu, P.; Nisar, J.; Pathak, B.; Ahuja, R. Cationic–anionic mediated charge compensation on La2Ti2O7 for visible light photocatalysis. Phys. Chem. Chem. Phys. 2013, 15, 17150–17157. [Google Scholar] [CrossRef]
- Fo, Y.; Ma, Y.; Dong, H.; Zhou, X. Tuning the electronic structure of BaTiO3 for an enhanced photocatalytic performance using cation–anion codoping: A first-principles study. New J. Chem. 2021, 45, 8228. [Google Scholar] [CrossRef]
- Fo, Y.; Zhou, X. A theoretical study on tetragonal BaTiO3 modified by surface co-doping for photocatalytic overall water splitting. Int. J. Hydrogen Energy 2022, 47, 19073–19085. [Google Scholar] [CrossRef]
- Liu, P.; Nisar, J.; Pathak, B.; Ahuja, R. Hybrid density functional study on SrTiO3 for visible light photocatalysis. Int. J. Hydrogen Energy 2012, 16, 11611–11617. [Google Scholar] [CrossRef]
- Wang, J.; Teng, J.; Pu, L.; Huang, J.; Wang, Y.; Li, Q. Double-hole-mediated coupling of anionic dopants in perovskite NaNbO3 for efficient solar water splitting. Int. J. Quantum Chem. 2019, 119, e25930. [Google Scholar] [CrossRef]
- Wang, G.; Huang, Y.; Kuang, A.; Yuan, H.; Li, Y.; Chen, H. Double-Hole-Mediated Codoping on KNbO3 for Visible Light Photocatalysis. Inorg. Chem. 2016, 55, 9620–9631. [Google Scholar] [CrossRef]
- Wang, J.; Huang, J.; Meng, J.; Li, Q.; Yang, J. Single-and few-layer BiOI as promising photocatalysts for solar water splitting. Phys. Chem. Chem. Phys. 2016, 18, 17517. [Google Scholar] [CrossRef]
- Smidstrup, S.; Markussen, T.; Vancraeyveld, P.; Wellendorff, J.; Schneider, J.; Gunst, T.; Verstichel, B.; Stradi, D.; Khomyakov, P.A.; Vej-Hansen, U.G.; et al. QuantumATK: An integrated platform of electronic and atomic-scale modelling tools. J. Phys. Condens. Matter 2020, 32, 015901. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Van Setten, M.; Giantomassi, M.; Bousquet, E.; Verstraete, M.; Hamann, D.; Gonze, X.; Rignanese, G.-M. The PseudoDojo: Training and grading a 85 element optimized norm-conserving pseudopotential table. Comput. Phys. Commun. 2018, 226, 39–54. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Marques, M.; Teles, L.K. Approximation to density functional theory for the calculation of band gaps of semiconductors. Phys. Rev. B 2008, 78, 125116. [Google Scholar] [CrossRef]
- Kanoun, M.B.; Goumri-Said, S.; Schwingenschlögl, U.; Manchon, A. Magnetism in Sc-doped ZnO with zinc vacancies: A hybrid density functional and GGA+U approaches. Chem. Phys. Lett. 2012, 532, 96–99. [Google Scholar] [CrossRef]
- Ravel, B.; Stern, E.A.; Vedrinskii, R.I.; Kraizman, V. Local structure and the phase transitions of BaTiO3. Ferroelectrics 1998, 206, 407–430. [Google Scholar] [CrossRef]
- Chakraborty, A.; Liton, M.N.H.; Sarker, M.S.I.; Rahman, M.M.; Khan, M.K.R. A comprehensive DFT evaluation of catalytic and optoelectronic properties of BaTiO3 polymorphs. Phys. B Condens. Matter 2023, 648, 414418. [Google Scholar] [CrossRef]
- Janotti, A.; van de Walle, C.G. Native point defects in ZnO. Phys. Rev. B 2007, 76, 165202. [Google Scholar] [CrossRef]
- Kanoun, M.B. Vacancy defects- and strain-tunable electronic structures and magnetism in two-dimensional MoTe2: Insight from first-principles calculations Surf. Interfaces 2021, 27, 101442. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a (Å) | b (Å) | c (Å) | Eb (eV) | Ef (eV) | ||
|---|---|---|---|---|---|---|
| Ti-Rich | O-Rich | |||||
| Pure | 8.005 | 8.005 | 8.005 | - | - | - |
| N | 8.014 | 8.014 | 8.014 | - | −10.10 | −2.75 |
| C | 8.008 | 8.008 | 8.073 | - | −5.33 | −0.08 |
| S | 8.070 | 8.064 | 8.423 | - | −4.31 | 0.94 |
| P | 8.012 | 8.012 | 8.232 | - | −2.08 | 3.18 |
| N–N | 8.088 | 8.015 | 8.088 | 1.317 | −17.53 | −6.23 |
| C–S | 8.267 | 8.017 | 8.107 | 2.179 | −11.89 | −1.32 |
| N–P | 8.297 | 8.024 | 8.086 | 3.038 | −13.12 | −2.62 |
| P–P | 8.321 | 7.980 | 8.321 | 4.637 | −8.79 | 1.72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goumri-Said, S.; Kanoun, M.B. Insight into the Effect of Anionic–Anionic Co-Doping on BaTiO3 for Visible Light Photocatalytic Water Splitting: A First-Principles Hybrid Computational Study. Catalysts 2022, 12, 1672. https://doi.org/10.3390/catal12121672
Goumri-Said S, Kanoun MB. Insight into the Effect of Anionic–Anionic Co-Doping on BaTiO3 for Visible Light Photocatalytic Water Splitting: A First-Principles Hybrid Computational Study. Catalysts. 2022; 12(12):1672. https://doi.org/10.3390/catal12121672
Chicago/Turabian StyleGoumri-Said, Souraya, and Mohammed Benali Kanoun. 2022. "Insight into the Effect of Anionic–Anionic Co-Doping on BaTiO3 for Visible Light Photocatalytic Water Splitting: A First-Principles Hybrid Computational Study" Catalysts 12, no. 12: 1672. https://doi.org/10.3390/catal12121672
APA StyleGoumri-Said, S., & Kanoun, M. B. (2022). Insight into the Effect of Anionic–Anionic Co-Doping on BaTiO3 for Visible Light Photocatalytic Water Splitting: A First-Principles Hybrid Computational Study. Catalysts, 12(12), 1672. https://doi.org/10.3390/catal12121672