



Limestone Calcination Kinetics in Microfluidized Bed Thermogravimetric Analysis (MFB-TGA) for Calcium Looping


Abstract
:1. Introduction
2. Results
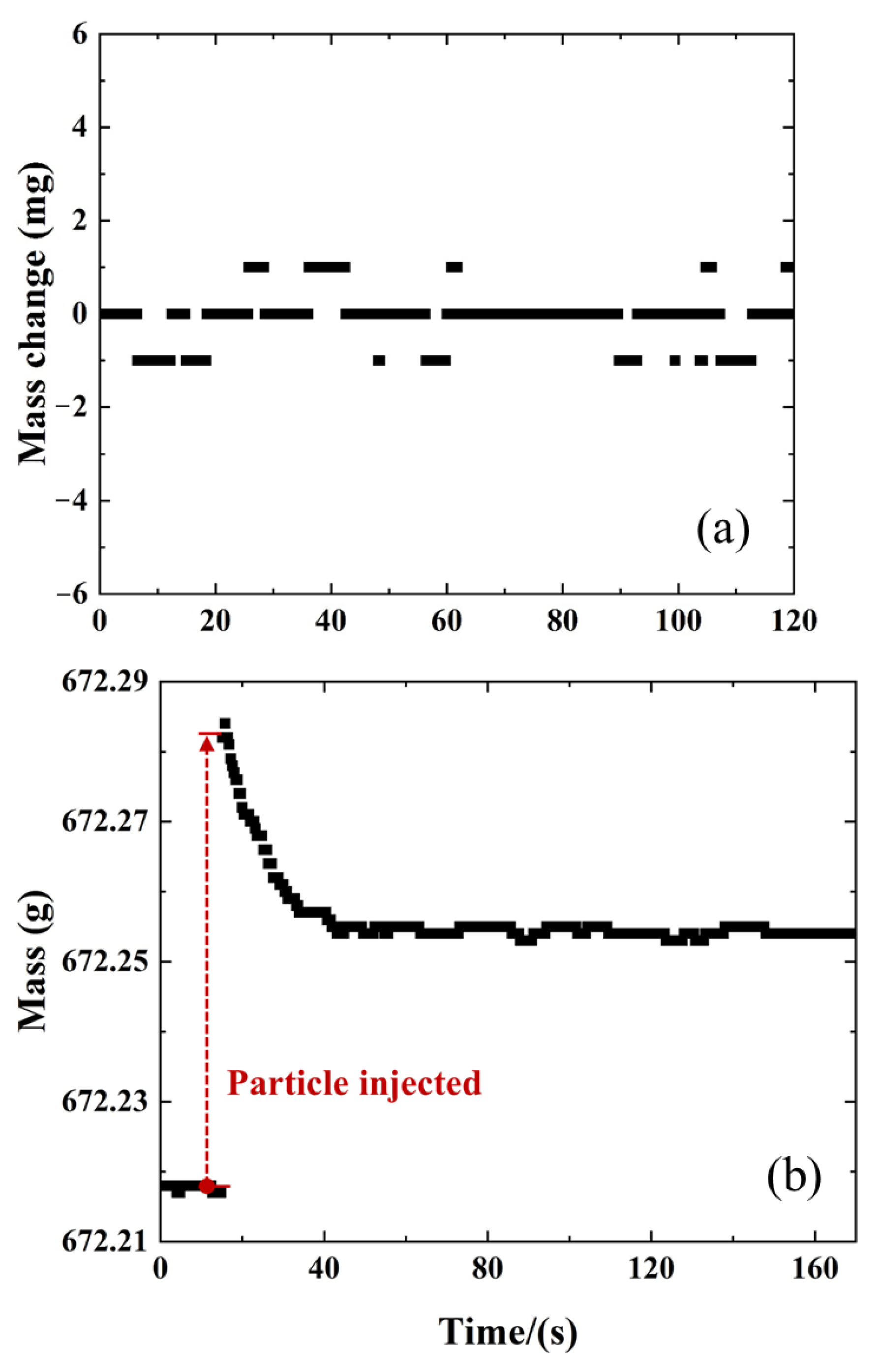
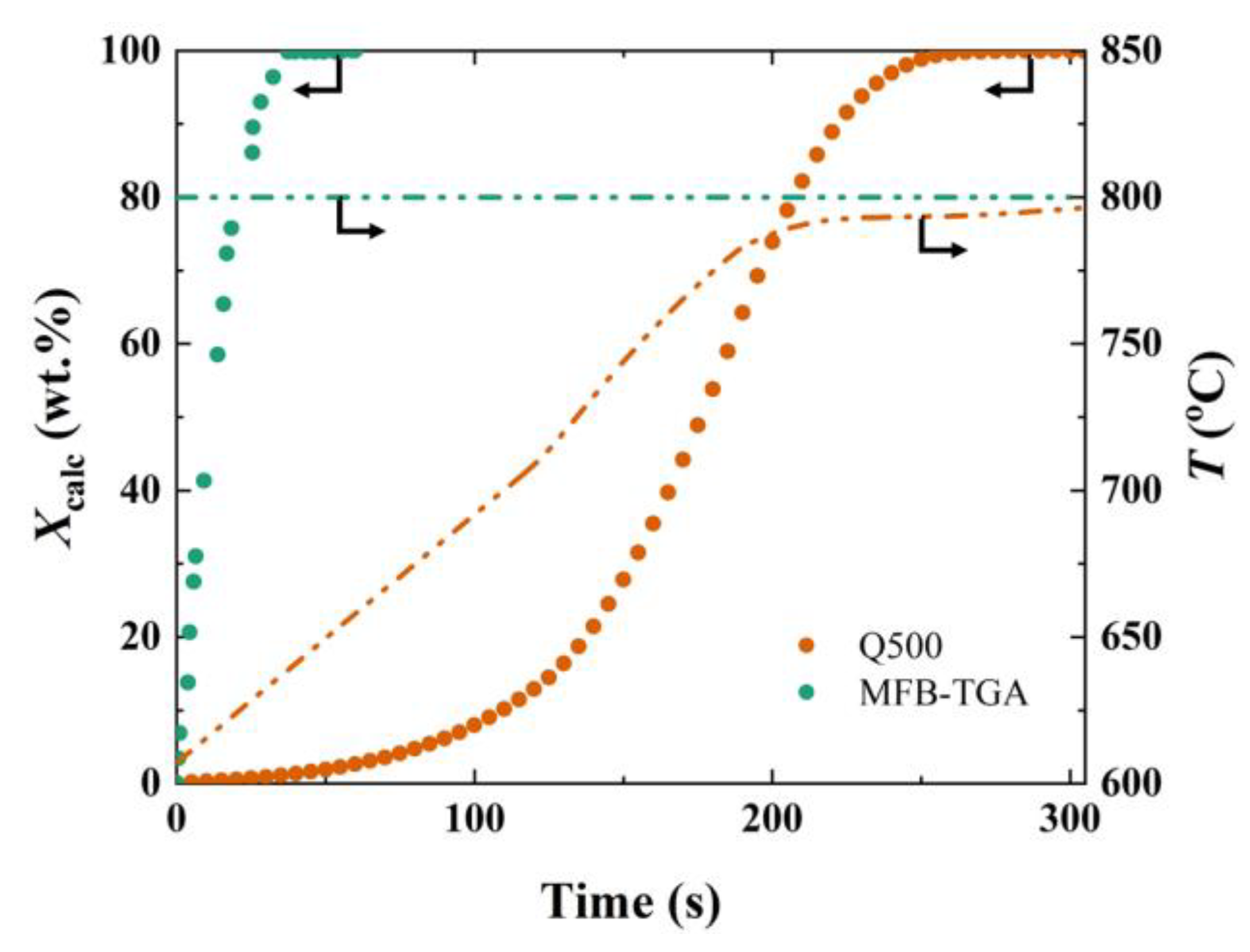
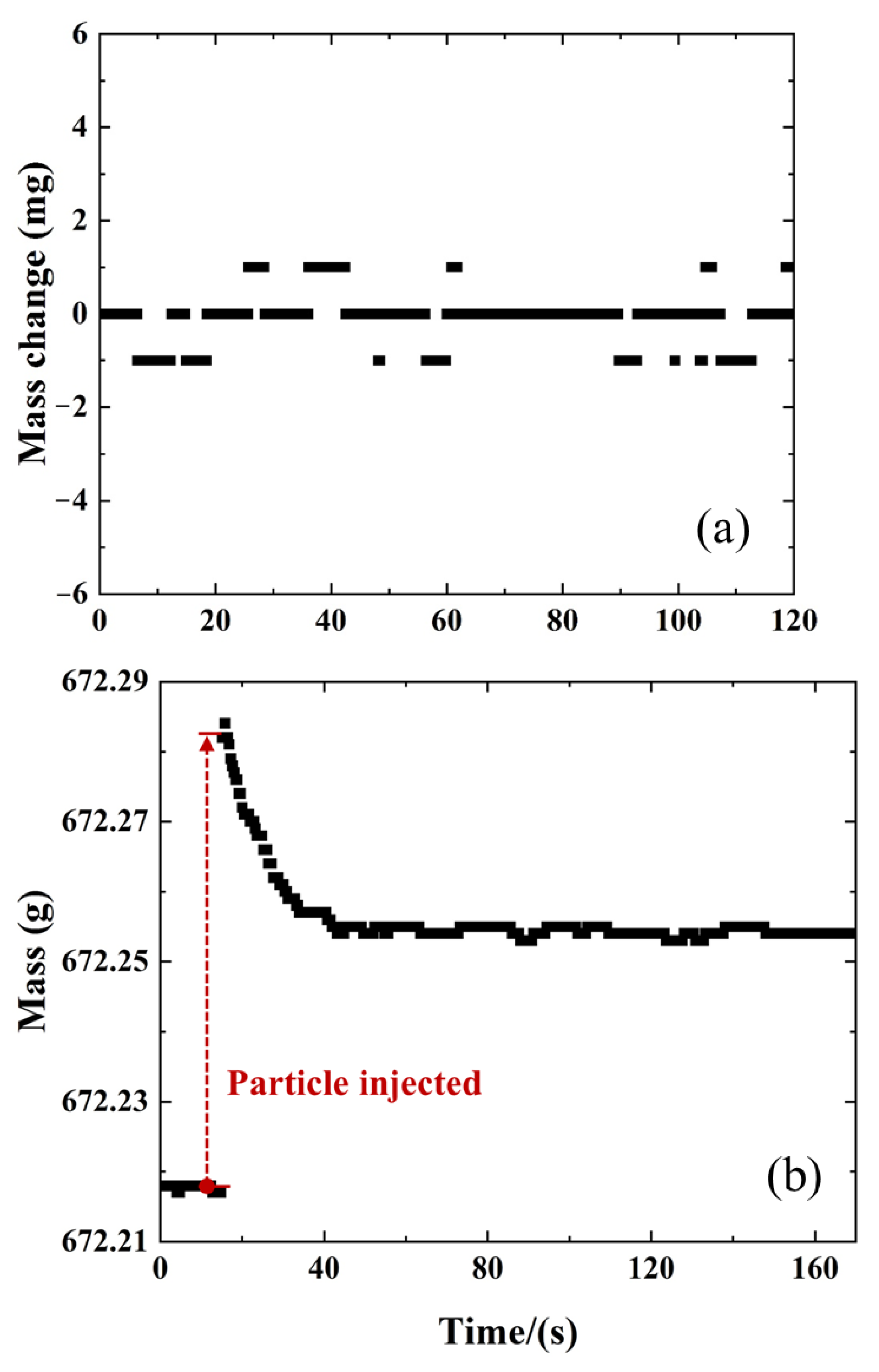
2.1. Comparison of MFB-TGA with Regular TGA
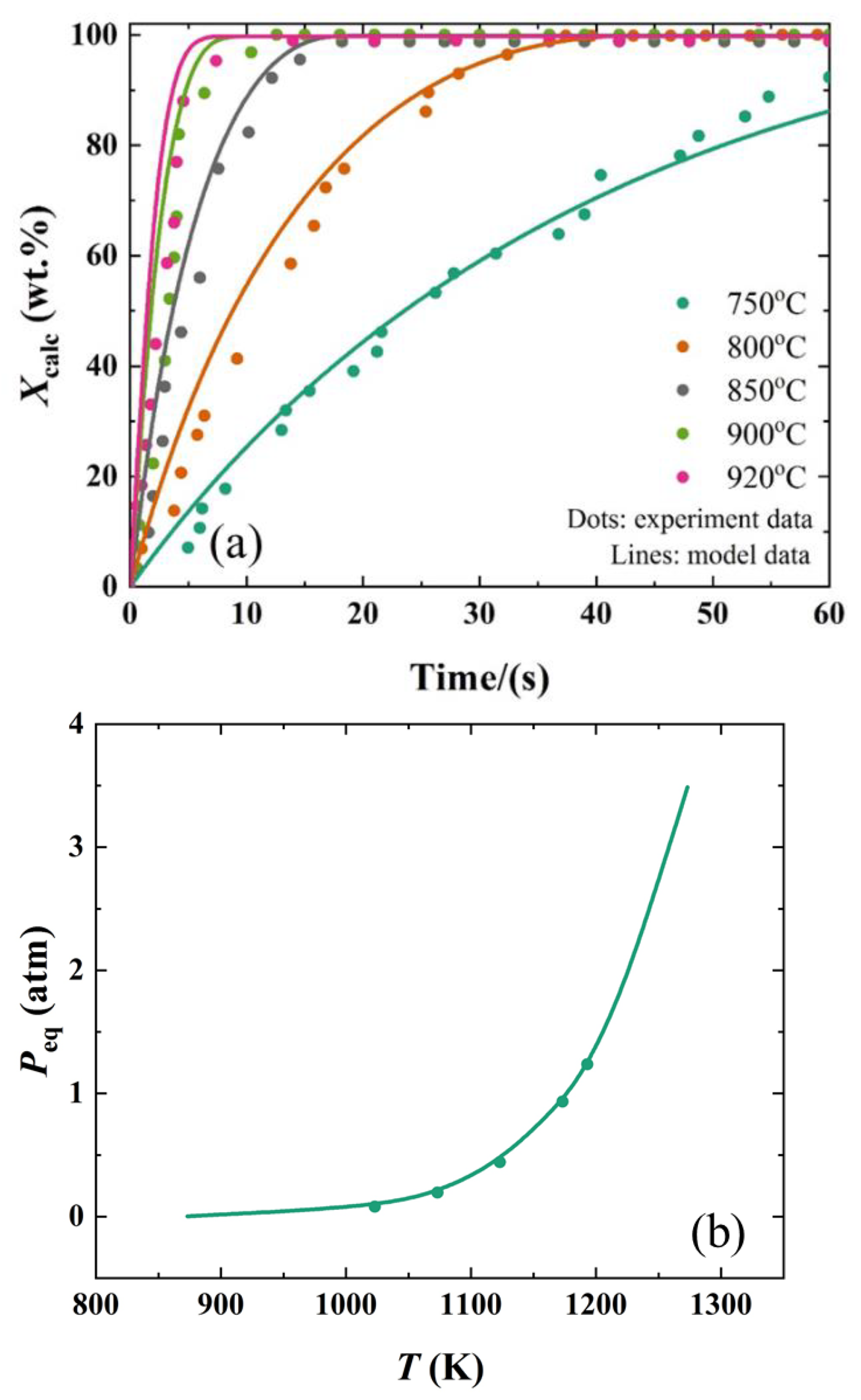
2.2. Effect of Temperature on the Calcination Kinetics
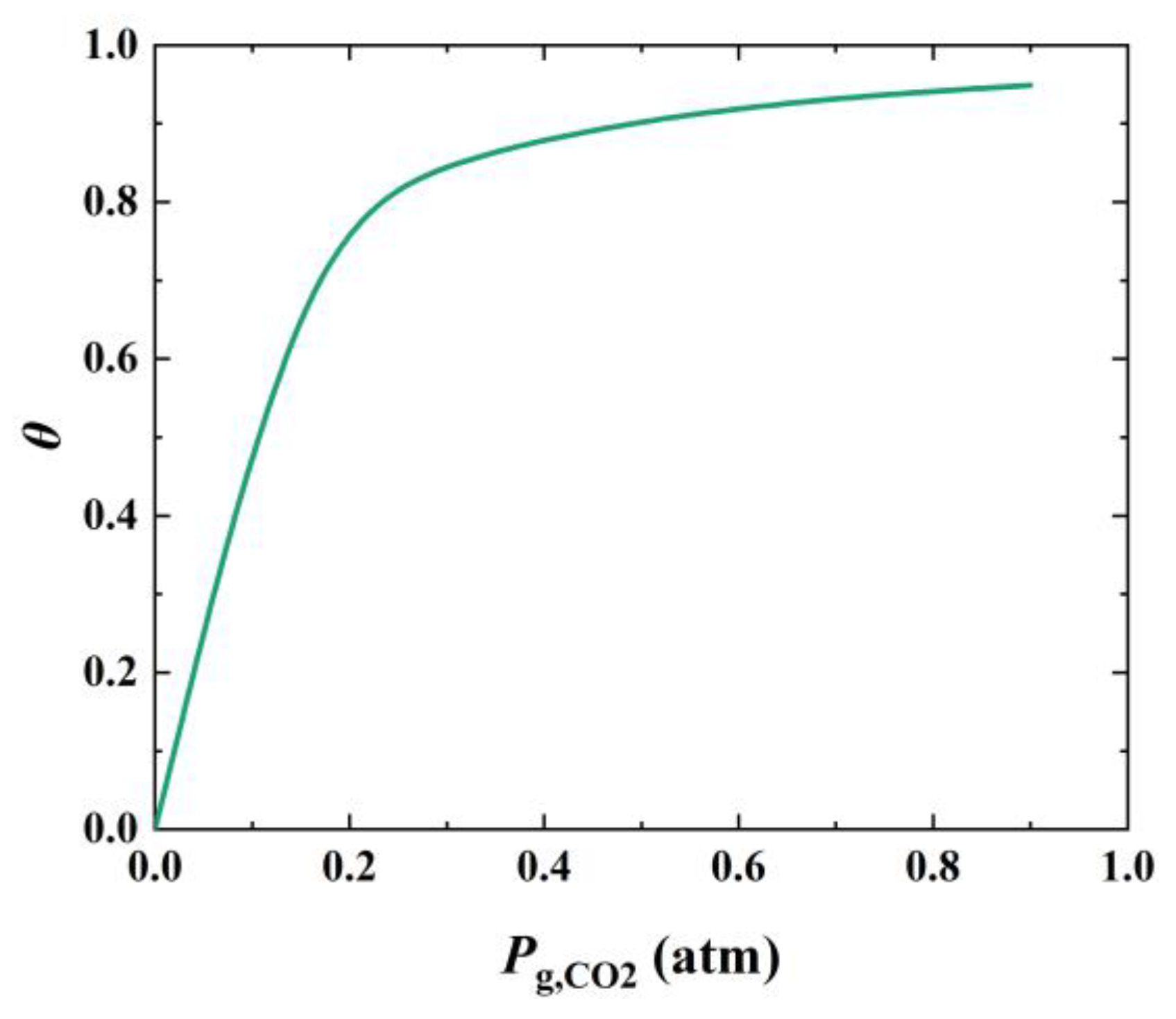
2.3. Effect of CO2 Concentration on the Calcination Kinetics
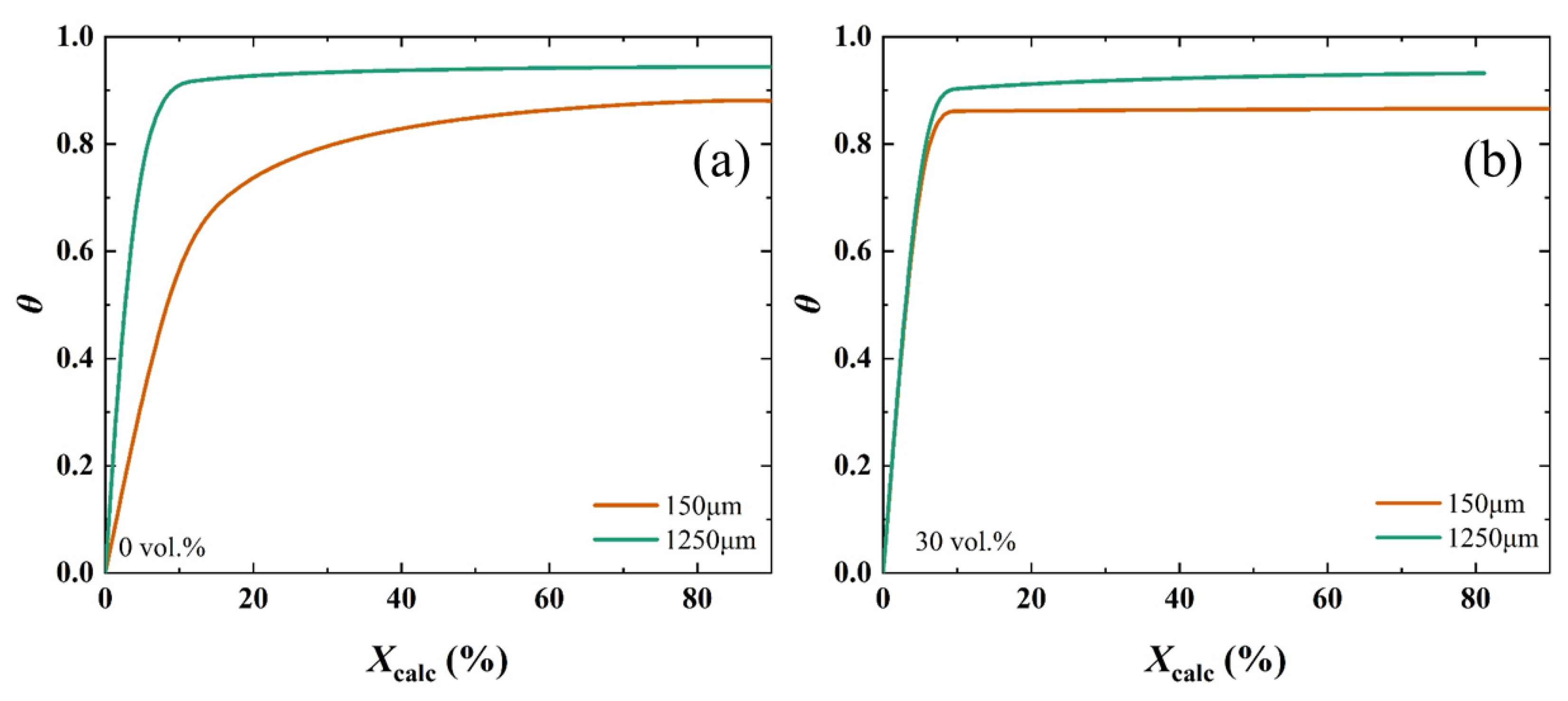
2.4. Effect of Particle Size on the Calcination Kinetics
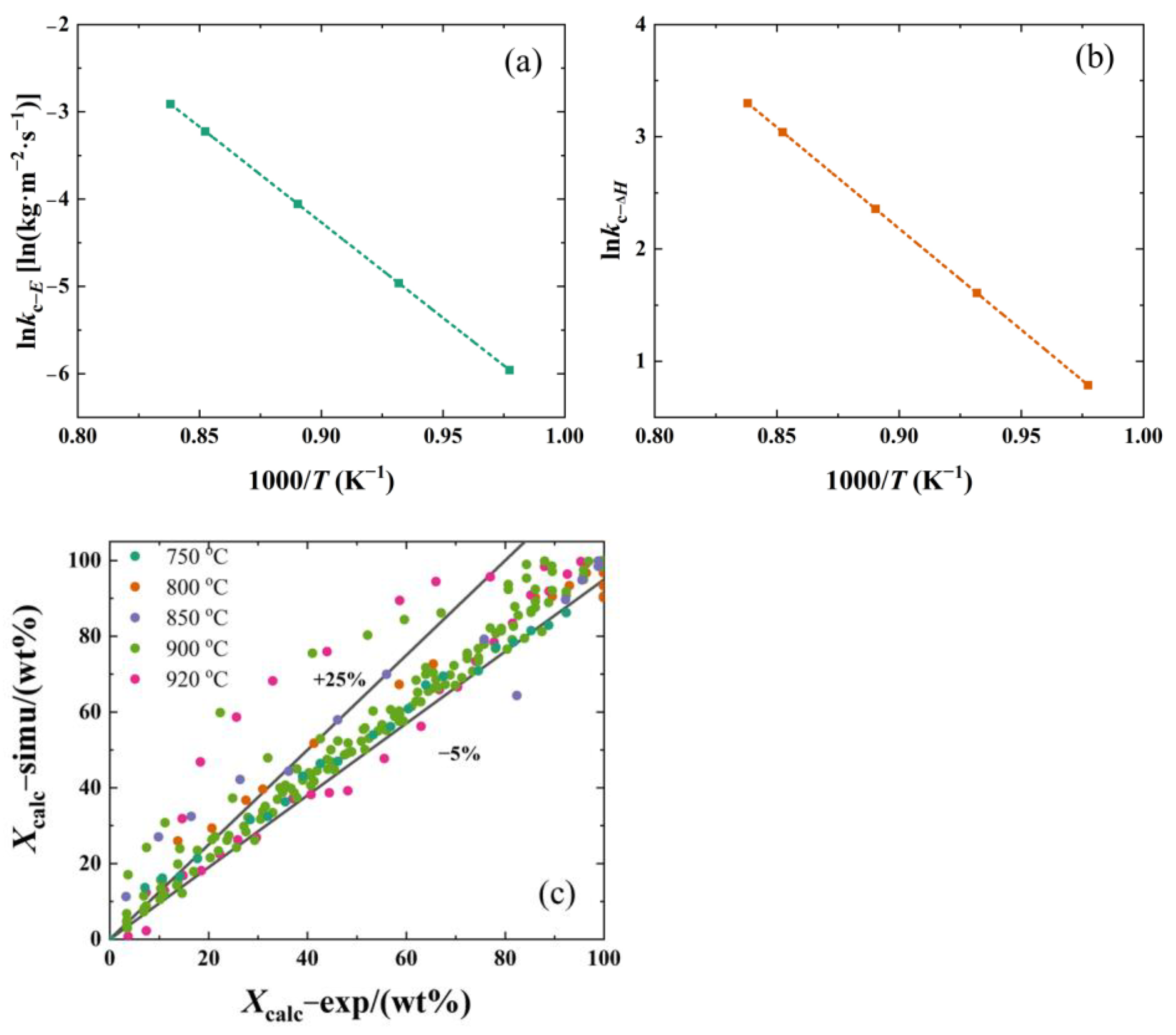
2.5. Determination of Kinetic Parameters
3. Discussion
4. Experiment
4.1. Microfluidized Bed Thermogravimetric Analysis (MFB-TGA)
4.2. Experimental Condition and Data Evaluation
5. Models
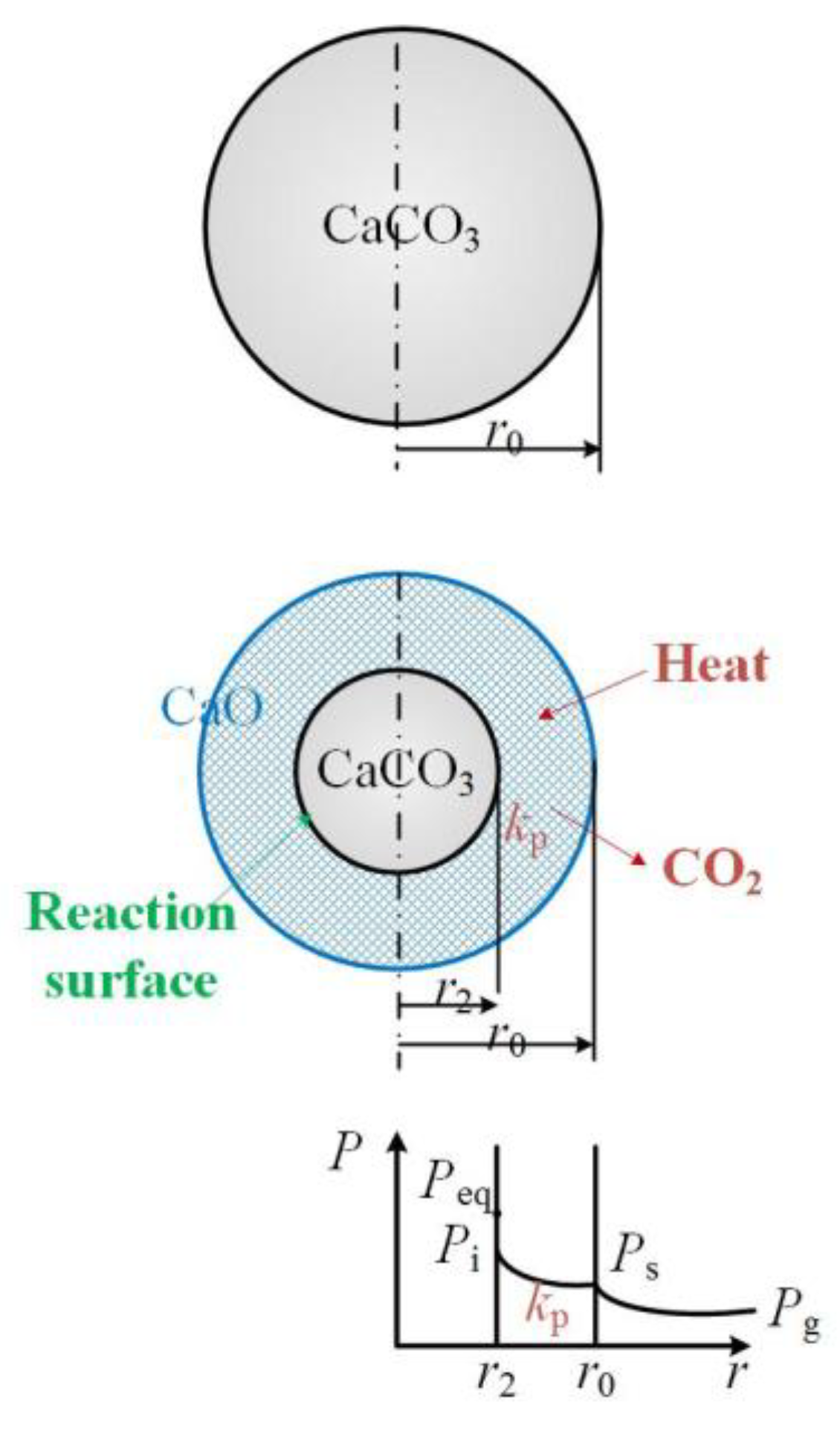
5.1. Calcination Kinetics
5.2. Intraparticle Diffusion
5.3. External Diffusion
5.4. Calcination Kinetic Model
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Han, R.; Wang, Y.; Xing, S.; Pang, C.; Hao, Y.; Song, C.; Liu, Q. Progress in reducing calcination reaction temperature of Calcium-Looping CO2 capture technology: A critical review. Chem. Eng. J. 2022, 450, 137952. [Google Scholar] [CrossRef]
- Perejón, A.; Romeo, L.M.; Lara, Y.; Lisbona, P.; Martínez, A.; Valverde, J.M. The Calcium-Looping technology for CO2 capture: On the important roles of energy integration and sorbent behavior. Appl. Energy 2016, 162, 787–807. [Google Scholar] [CrossRef] [Green Version]
- Rivero, M.A.; Rodrigues, D.; Pinheiro, C.I.; Cardoso, J.P.; Mendes, L.F. Solid-gas reactors driven by concentrated solar energy with potential application to calcium looping: A comparative review. Renew. Sust. Energ. Rev. 2022, 158, 112048. [Google Scholar] [CrossRef]
- Qin, C.; He, D.; Zhang, Z.; Tan, L.; Ran, J. The consecutive calcination/sulfation in calcium looping for CO2 capture: Particle modeling and behaviour investigation. Chem. Eng. J. 2018, 334, 2238–2249. [Google Scholar] [CrossRef]
- Di Giuliano, A.; Gallucci, K. Sorption enhanced steam methane reforming based on nickel and calcium looping: A review. Chem. Eng. Process. 2018, 130, 240–252. [Google Scholar] [CrossRef]
- Blamey, J.; Anthony, E.; Wang, J.; Fennell, P. The calcium looping cycle for large-scale CO2 capture. Prog. Energy Combust. Sci. 2010, 36, 260–279. [Google Scholar] [CrossRef]
- Hashemi, S.M.; Sedghkerdar, M.H.; Mahinpey, N. Calcium looping carbon capture: Progress and prospects. Can. J. Chem. Eng. 2022, 100, 2140–2171. [Google Scholar] [CrossRef]
- Hornberger, M.; Spörl, R.; Scheffknecht, G. Calcium looping for CO2 capture in cement plants-pilot scale test. Energy Procedia 2017, 114, 6171–6174. [Google Scholar] [CrossRef]
- Haaf, M.; Stroh, A.; Hilz, J.; Helbig, M.; Ströhle, J.; Epple, B. Process modelling of the calcium looping process and validation against 1 MWth pilot testing. Energy Procedia 2017, 114, 167–178. [Google Scholar] [CrossRef]
- Bui, M.; Adjiman, C.S.; Bardow, A.; Anthony, E.J.; Boston, A.; Brown, S.; Fennell, P.S.; Fuss, S.; Galindo, A.; Hackett, L.A. Carbon capture and storage (CCS): The way forward. Energy Environ. Sci. 2018, 11, 1062–1176. [Google Scholar] [CrossRef]
- Li, Z.; Cai, N.; Huang, Y.; Han, H. Synthesis, experimental studies, and analysis of a new calcium-based carbon dioxide absorbent. Energy Fuels 2005, 19, 1447–1452. [Google Scholar] [CrossRef]
- Rodriguez, N.; Alonso, M.; Grasa, G.; Abanades, J.C. Heat requirements in a calciner of CaCO3 integrated in a CO2 capture system using CaO. Chem. Eng. J. 2008, 138, 148–154. [Google Scholar] [CrossRef] [Green Version]
- Grasa, G.S.; Abanades, J.C. CO2 capture capacity of CaO in long series of carbonation/calcination cycles. Ind. Eng. Chem. Res. 2006, 45, 8846–8851. [Google Scholar] [CrossRef]
- Hyatt, E.P.; Cutler, I.B.; Wadsworth, M.E. Calcium carbonate decomposition in carbon dioxide atmosphere. J. Am. Ceram. Soc. 1958, 41, 70–74. [Google Scholar] [CrossRef]
- Khinast, J.; Krammer, G.; Brunner, C.; Staudinger, G. Decomposition of limestone: The influence of CO2 and particle size on the reaction rate. Chem. Eng. Sci. 1996, 51, 623–634. [Google Scholar] [CrossRef]
- Hills, A. The mechanism of the thermal decomposition of calcium carbonate. Chem. Eng. Sci. 1968, 23, 297–320. [Google Scholar] [CrossRef]
- Borgwardt, R.H. Calcination kinetics and surface-area of dispersed limestone particles. AlChE J. 1985, 31, 103–111. [Google Scholar] [CrossRef]
- Rajeswara, R. Kinetics of calcium carbonate decomposition. Chem. Eng. Res. Des. 1989, 67, 38–47. [Google Scholar]
- Garcıa-Labiano, F.; Abad, A.; De Diego, L.; Gayán, P.; Adánez, J. Calcination of calcium-based sorbents at pressure in a broad range of CO2 concentrations. Chem. Eng. Sci. 2002, 57, 2381–2393. [Google Scholar] [CrossRef] [Green Version]
- Ylätalo, J.; Parkkinen, J.; Ritvanen, J.; Tynjälä, T.; Hyppänen, T. Modeling of the oxy-combustion calciner in the post-combustion calcium looping process. Fuel 2013, 113, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Szekely, J.; Evans, J. A structural model for gas-solid reactions with a moving boundary. Chem. Eng. Sci. 1970, 25, 1091–1107. [Google Scholar] [CrossRef]
- Ar, I.; Dogu, G. Calcination kinetics of high purity limestones. Chem. Eng. J. 2001, 83, 131–137. [Google Scholar] [CrossRef]
- Escardino, A.; Garcia-Ten, J.; Feliu, C.; Saburit, A.; Cantavella, V. Kinetic study of the thermal decomposition process of calcite particles in air and CO2 atmosphere. Ind. Eng. Chem. Res. 2013, 19, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Galwey, A.K.; Brown, M.E. Application of the Arrhenius equation to solid state kinetics: Can this be justified? Thermochim. Acta 2002, 386, 91–98. [Google Scholar] [CrossRef]
- Ramezani, M.; Tremain, P.; Doroodchi, E.; Moghtaderi, B. Determination of carbonation/calcination reaction kinetics of a limestone sorbent in low CO2 partial pressures using TGA experiments. Energy Procedia 2017, 114, 259–270. [Google Scholar] [CrossRef]
- Guler, C.; Dollimore, D.; Heal, G. The investigation of the decomposition kinetics of calcium carbonate alone and in the presence of some clays using the rising temperature technique. Thermochim. Acta 1982, 54, 187–199. [Google Scholar] [CrossRef]
- Dennis, J.; Hayhurst, A. The effect of CO2 on the kinetics and extent of calcination of limestone and dolomite particles in fluidised beds. Chem. Eng. Sci. 1987, 42, 2361–2372. [Google Scholar] [CrossRef]
- Silcox, G.D.; Kramlich, J.C.; Pershing, D.W. A mathematical model for the flash calcination of dispersed calcium carbonate and calcium hydroxide particles. Ind. Eng. Chem. Res. 1989, 28, 155–160. [Google Scholar] [CrossRef]
- Rao, T.R. Kinetics of calcium carbonate decomposition. Chem. Eng. Technol. 1996, 19, 373–377. [Google Scholar] [CrossRef]
- Fuertes, A.; Marban, G.; Rubiera, F. Kinetics of thermal decomposition of limestone particles in a fluidized bed reactor. Chem. Eng. Re.s Des. 1993, 71, 421–428. [Google Scholar]
- Wang, Y.; Lin, S.; Suzuki, Y. Limestone calcination with CO2 capture (II): Decomposition in CO2/steam and CO2/N2 atmospheres. Energy fuels 2008, 22, 2326–2331. [Google Scholar] [CrossRef]
- Valverde, J.M.; Sanchez-Jimenez, P.E.; Perez-Maqueda, L.A. Limestone calcination nearby equilibrium: Kinetics, CaO crystal structure, sintering and reactivity. J. Phys. Chem. C 2015, 119, 1623–1641. [Google Scholar] [CrossRef]
- Ramon Fernandez, J.; Turrado, S.; Carlos Abanades, J. Calcination kinetics of cement raw meals under various CO2 concentrations. React. Chem. Eng. 2019, 4, 2129–2140. [Google Scholar] [CrossRef] [Green Version]
- Barker, R. The reversibility of the reaction CaCO3⇄ CaO+ CO2. J. Chem. Technol. Biotechnol. 1973, 23, 733–742. [Google Scholar] [CrossRef]
- Calvo, E.G.; Arranz, M.; Leton, P. Effects of impurities in the kinetics of calcite decomposition. Thermochim. Acta 1990, 170, 7–11. [Google Scholar] [CrossRef]
- Valverde, J.M. On the negative activation energy for limestone calcination at high temperatures nearby equilibrium. Chem. Eng. Sci. 2015, 132, 169–177. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, S.; Suzuki, Y. Study of limestone calcination with CO2 capture: Decomposition behavior in a CO2 atmosphere. Energy Fuels 2007, 21, 3317–3321. [Google Scholar] [CrossRef]
- Scaltsoyiannes, A.; Lemonidou, A. CaCO3 decomposition for calcium-looping applications: Kinetic modeling in a fixed-bed reactor. Chem. Eng. Sci. X 2020, 8, 100071. [Google Scholar] [CrossRef]
- Lv, G.; Lu, J.; Cai, L.; Xie, X.; Liu, Z. Experimental study on the dynamic process of NO reduction in a precalciner. Ind. Eng. Chem. Res. 2011, 50, 4366–4372. [Google Scholar]
- Khraisha, Y.; Dugwell, D. Coal combustion and limestone calcination in a suspension reactor. Chem. Eng. Sci. 1992, 47, 993–1006. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Wang, H.; Cai, N. CaO carbonation kinetics determined using micro-fluidized bed thermogravimetric analysis. Fuel 2020, 264, 116823. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Z.; Cai, N. Redox reaction kinetics of a Fe-Cu-based oxygen carrier measured with microfluidized bed thermogravimetric analysis. Energy Fuels 2022, 36, 9672–9686. [Google Scholar] [CrossRef]
- Wang, H.; Li, Z.; Li, Y.; Cai, N. Reduced-order model for CaO carbonation kinetics measured using micro-fluidized bed thermogravimetric analysis. Chem. Eng. Sci. 2021, 229, 116039. [Google Scholar] [CrossRef]
- Giammaria, G.; Lefferts, L. Catalytic effect of water on calcium carbonate decomposition. J. CO2 Util. 2019, 33, 341–356. [Google Scholar] [CrossRef]
- Escardino, A.; Garcia-Ten, J.; Feliu, C. Kinetic study of calcite particle (powder) thermal decomposition: Part I. J. Eur. Ceram. Soc. 2008, 28, 3011–3020. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Liu, L.; Cai, N. Measuring the fast oxidation kinetics of a manganese oxygen carrier using microfluidized bed thermogravimetric analysis. Chem. Eng. J. 2020, 385, 123970. [Google Scholar] [CrossRef]
- Hu, N.; Scaroni, A.W. Calcination of pulverized limestone particles under furnace injection conditions. Fuel 1996, 75, 177–186. [Google Scholar] [CrossRef]
- Field, M.A. Rate of combustion of size-graded fractions of char from a low-rank coal between 1200 K and 2000 K. Combust. Flame 1969, 13, 237–252. [Google Scholar] [CrossRef]
- Graham, T. On the law of the diffusion of gases. J. Membr. Sci. 1995, 100, 17–21.sss. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A (kg/m2/s) | E (kJ/mol) | a (/) | ΔH (kJ/mol) | C1 (s/K0.75) | S0 (m2/kg) | ε0 (/) | |
|---|---|---|---|---|---|---|---|
| 5.051 × 106 | 182 | 9.99 × 107 | 150 | 5 × 10−12 | 22.80 | 0.48 | 330 |
| Sample Mass/mg | Particle Size/μm | Temperature/°C | CO2/% | Superficial Velocity |
|---|---|---|---|---|
| 60–70 | 150/450/800/1250 | 750–920 | 0/30 | ~3 Umf |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Wang, Y.; Li, Z. Limestone Calcination Kinetics in Microfluidized Bed Thermogravimetric Analysis (MFB-TGA) for Calcium Looping. Catalysts 2022, 12, 1661. https://doi.org/10.3390/catal12121661
Li D, Wang Y, Li Z. Limestone Calcination Kinetics in Microfluidized Bed Thermogravimetric Analysis (MFB-TGA) for Calcium Looping. Catalysts. 2022; 12(12):1661. https://doi.org/10.3390/catal12121661
Chicago/Turabian StyleLi, Dan, Yang Wang, and Zhenshan Li. 2022. "Limestone Calcination Kinetics in Microfluidized Bed Thermogravimetric Analysis (MFB-TGA) for Calcium Looping" Catalysts 12, no. 12: 1661. https://doi.org/10.3390/catal12121661
APA StyleLi, D., Wang, Y., & Li, Z. (2022). Limestone Calcination Kinetics in Microfluidized Bed Thermogravimetric Analysis (MFB-TGA) for Calcium Looping. Catalysts, 12(12), 1661. https://doi.org/10.3390/catal12121661