


Asymmetric Synthesis of Enantiomerically Pure Aliphatic and Aromatic D-Amino Acids Catalyzed by Transaminase from Haliscomenobacter hydrossis

Abstract
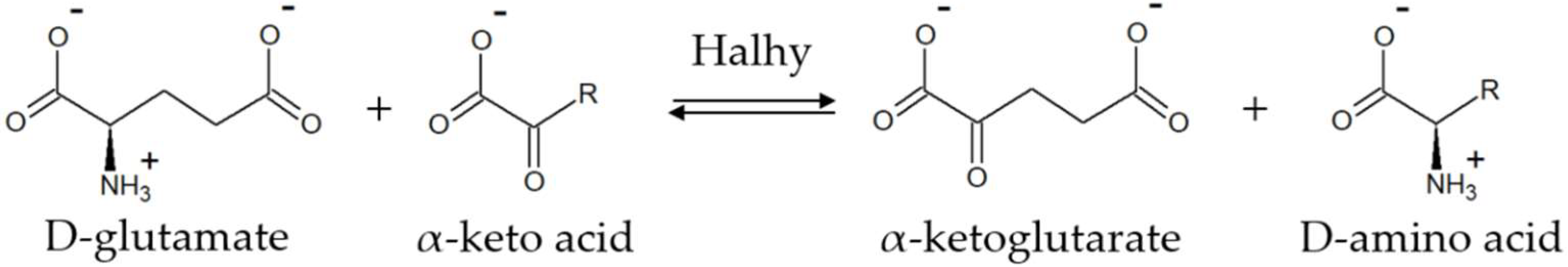
:1. Introduction
2. Results
2.1. α-Keto Acid Substrate Scope of Halhy
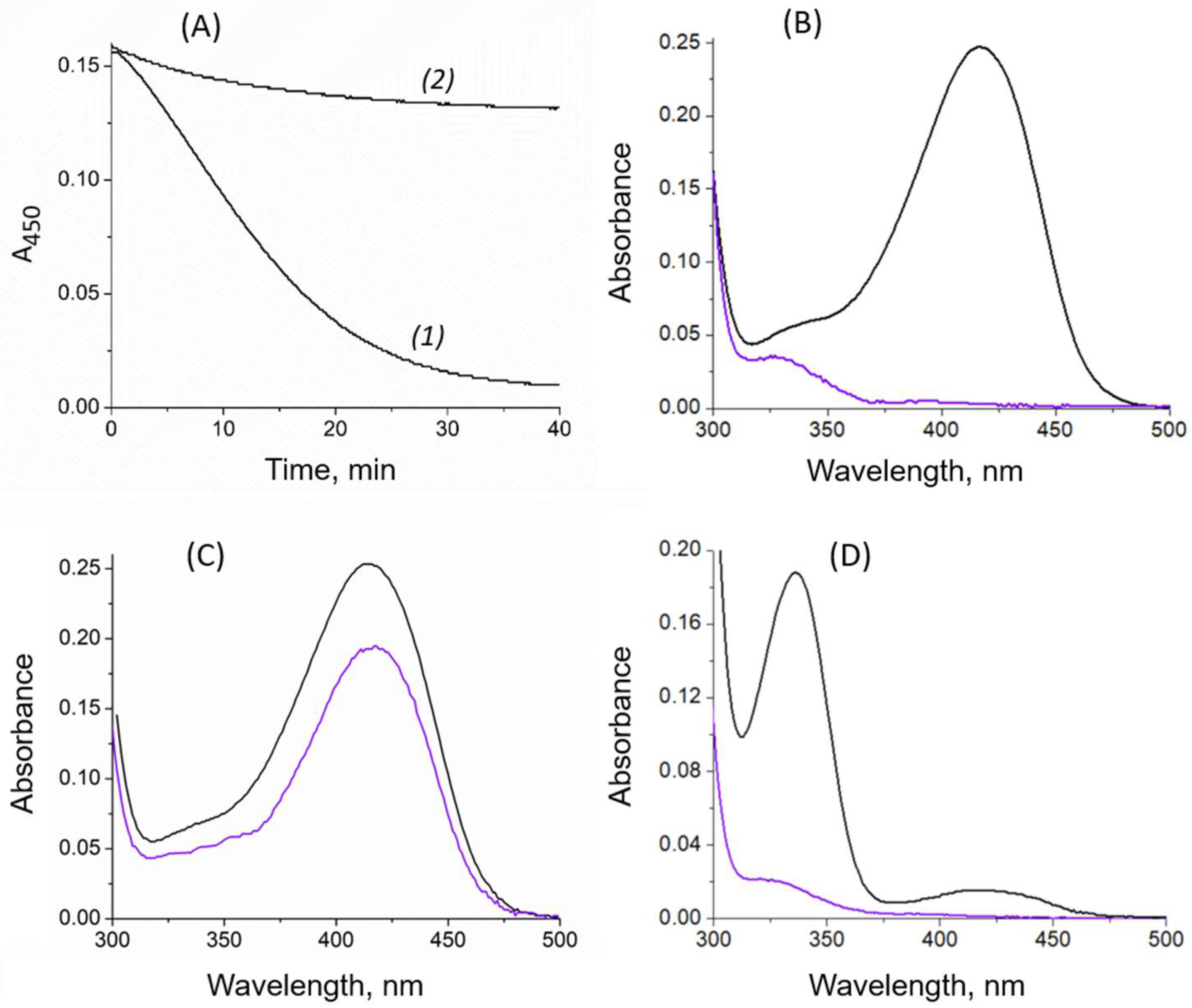
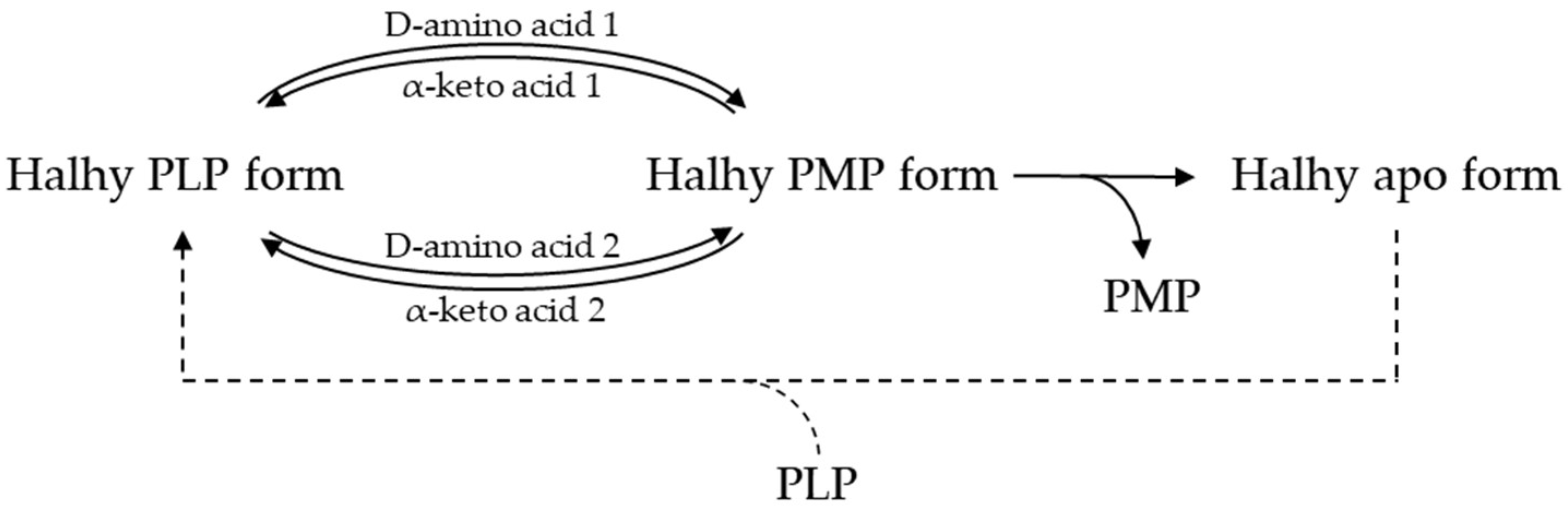
2.2. Cofactor Leakage under Reaction Conditions
2.3. Asymmetric Synthesis of D-Amino Acids
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of Recombinant Halhy
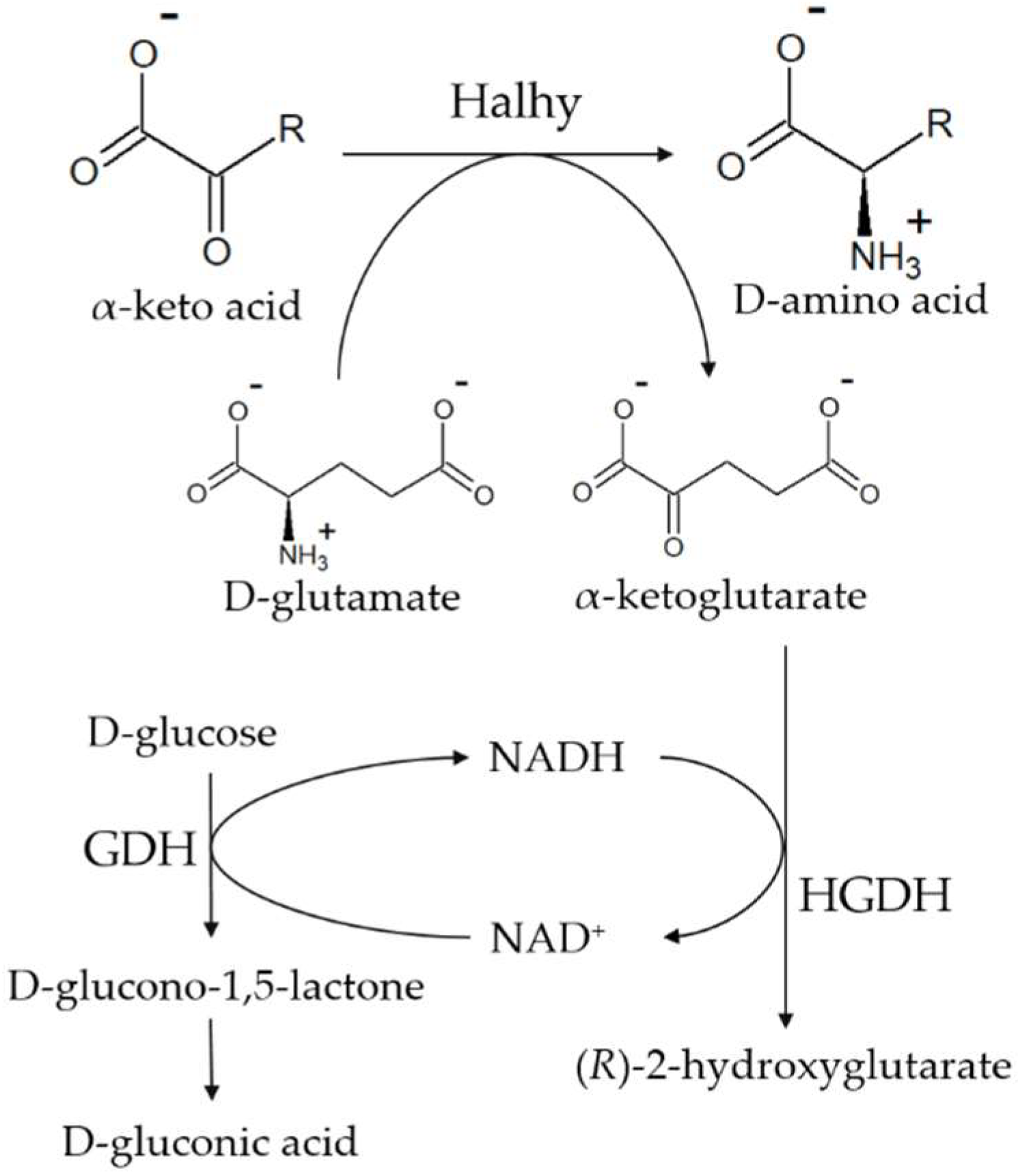
4.2. Enzyme Activity Assay
4.3. Cofactor Leakage Assay
4.4. Enzymatic Synthesis of D-Amino Acids
Analysis of the Product Yield and the Enantiomeric Excess of D-Amino Acids
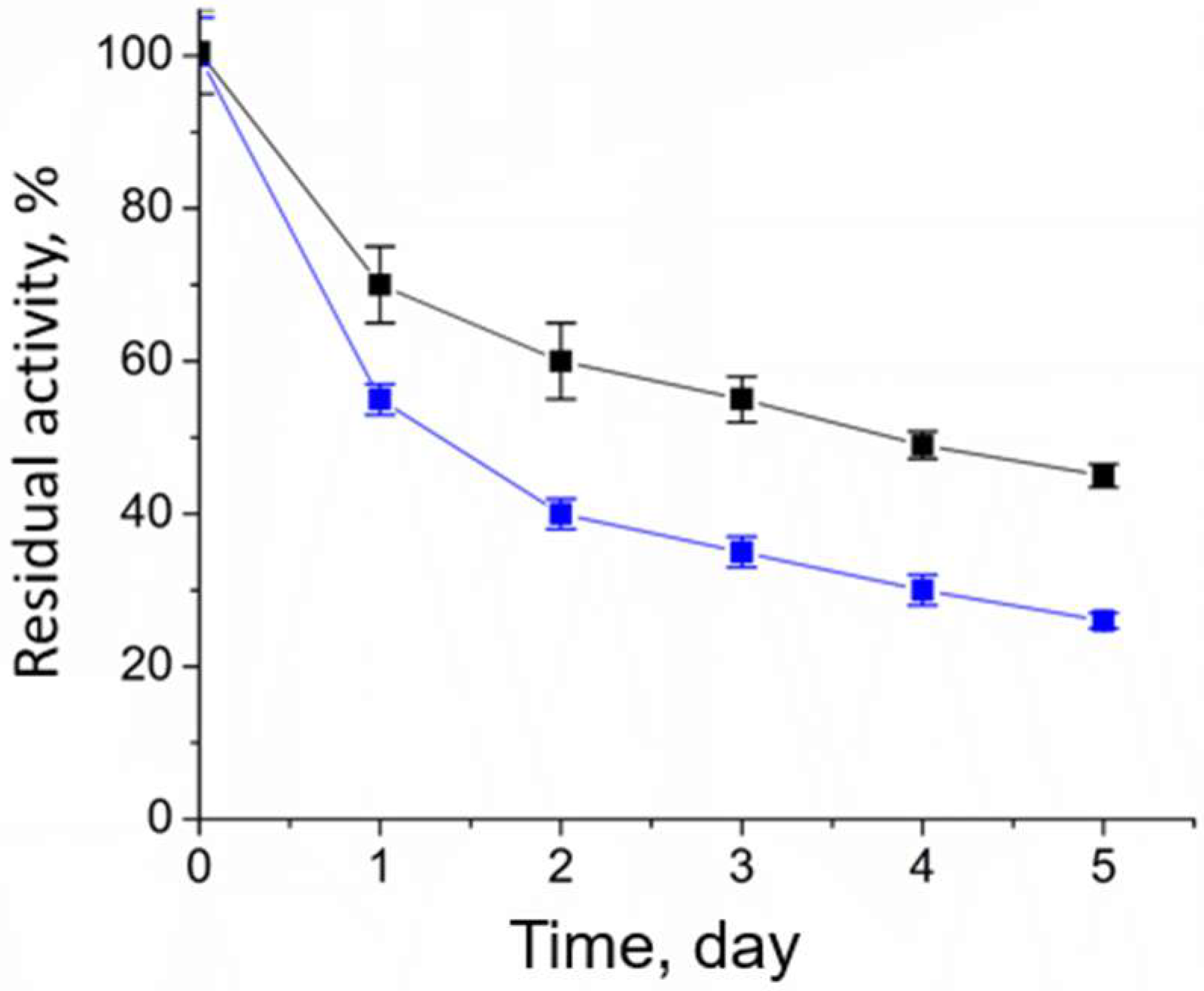
4.5. Analysis of the Operational Stability of Halhy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Eluent | UV Detection | Retention Time, min |
|---|---|---|---|
| 2-oxobutyrate | 20 mM NaH2PO4, pH 2.2, 5% methanol | 210 nm | 3.0 |
| 2-oxovalerate | 20 mM NaH2PO4, pH 2.2, 5% methanol | 210 nm | 7.4 |
| 3-methyl-2-oxobutyrate | 20 mM NaH2PO4, pH 2.2, 5% methanol | 210 nm | 6.1 |
| 2-oxohexanoate | 20 mM NaH2PO4, pH 3.0, 15% methanol | 210 nm | 5.8 |
| 3-methyl-2-oxovalerate | 20 mM NaH2PO4, pH 2.2, 5% methanol | 210 nm | 15.7 |
| D-tryptophan | 20 mM NaH2PO4, pH 3.0, 15% methanol | 280 nm | 6.2 |
| D-tyrosine | 20 mM NaH2PO4, pH 2.2, 5% methanol | 280 nm | 3.9 |
| 2-oxo-4-phenyl-butyric acid | 20 mM NaH2PO4, pH 3.0, 30% methanol | 210 nm | 9.5 |
Appendix B
| Instrument | ӒKTA Purifier, Cytiva, Marlborough, MA, USA |
|---|---|
| Column | Zorbax Eclipse XDB-C18, 5 µM, 4.6 mm × 150 mm, Agilent Technologies, Inc., Santa Clara, CA, USA |
| Buffer A | 0.1% trifluoroacetic acid in water |
| Buffer B | 0.1% trifluoroacetic acid in 100% methanol |
| Elution | linear gradient of Buffer B from 20 to 70% in 15 min |
| Flow rate | 1.0 mL/min |
| Temperature | 25 °C |
| Injection volume | 10 µL |
| Detection | UV, 340 nm |
| Compound | RT, min | |
|---|---|---|
| L-isomer | D-isomer | |
| norvaline | 16.0 | 18.6 |
| valine | 16.5 | 19.4 |
| norleucine | 17.8 | 22.0 |
| isoleucine | 17.6 | 21.6 |
| leucine | 17.4 | 20.8 |
| phenylalanine | 17.1 | 20.0 |
| tryptophan | 16.0 | 17.7 |
| tyrosine | 20.0 | 29.0 |
| homophenylalanine | 20.1 | 26.3 |
| homoalanine * | 19.2 | 21.7 |
References
- Bell, E.L.; Finnigan, W.; France, S.P.; Green, A.P.; Hayes, M.A.; Hepworth, L.J.; Lovelock, S.L.; Niikura, H.; Osuna, S.; Romero, E.; et al. Biocatalysis. Nat. Rev. Methods Prim. 2021, 1, 46. [Google Scholar] [CrossRef]
- Winkler, C.K.; Schrittwieser, J.H.; Kroutil, W. Power of Biocatalysis for Organic Synthesis. ACS Cent. Sci. 2021, 7, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. 2021, 60, 88–119. [Google Scholar] [CrossRef]
- Bornscheuer, U.T. The Fourth Wave of Biocatalysis Is Approaching. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2018, 376, 20170063. [Google Scholar] [CrossRef] [PubMed]
- Patel, R. Biocatalytic Synthesis of Chiral Alcohols and Amino Acids for Development of Pharmaceuticals. Biomolecules 2013, 3, 741–777. [Google Scholar] [CrossRef] [Green Version]
- Brundiek, H.; Höhne, M. Transaminases—A Biosynthetic Route for Chiral Amines. In Applied Biocatalysis: From Fundamental Science to Industrial Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; pp. 199–218. ISBN 9783527677122. [Google Scholar]
- Zawodny, W.; Montgomery, S.L. Evolving New Chemistry: Biocatalysis for the Synthesis of Amine-Containing Pharmaceuticals. Catalysts 2022, 12, 595. [Google Scholar] [CrossRef]
- Grishin, D.V.; Zhdanov, D.D.; Pokrovskaya, M.V.; Sokolov, N.N. D-Amino Acids in Nature, Agriculture and Biomedicine. Front. Life Sci. 2020, 13, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Ma, Q.; Zhu, H. Distribution, Industrial Applications, and Enzymatic Synthesis of d-Amino Acids. Appl. Microbiol. Biotechnol. 2015, 99, 3341–3349. [Google Scholar] [CrossRef]
- Leuchtenberger, W.; Huthmacher, K.; Drauz, K. Biotechnological Production of Amino Acids and Derivatives: Current Status and Prospects. Appl. Microbiol. Biotechnol. 2005, 69, 1–8. [Google Scholar] [CrossRef]
- Ivanov, K.; Stoimenova, A.; Obreshkova, D.; Saso, L. Biotechnology in the Production of Pharmaceutical Industry Ingredients: Amino Acids. Biotechnol. Biotechnol. Equip. 2013, 27, 3620–3626. [Google Scholar] [CrossRef]
- Radkov, A.D.; Moe, L.A. Bacterial Synthesis of D-Amino Acids. Appl. Microbiol. Biotechnol. 2014, 98, 5363–5374. [Google Scholar] [CrossRef] [PubMed]
- Pollegioni, L.; Rosini, E.; Molla, G. Advances in Enzymatic Synthesis of D-Amino Acids. Int. J. Mol. Sci. 2020, 21, 3206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Jing, X.; Zhang, W.; Nie, Y.; Xu, Y. Highly Selective Synthesis of d -Amino Acids from Readily Available l -Amino Acids by a One-Pot Biocatalytic Stereoinversion Cascade. RSC Adv. 2019, 9, 29927–29935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, A.; Li, J.; Yu, Y.; Zhang, D.; Nie, Y.; Xu, Y. Enzymatic Cascade Systems for D-Amino Acid Synthesis: Progress and Perspectives. Syst. Microbiol. Biomanuf. 2021, 1, 397–410. [Google Scholar] [CrossRef]
- Park, E.-S.; Dong, J.-Y.; Shin, J.-S. Biocatalytic Asymmetric Synthesis of Unnatural Amino Acids through the Cascade Transfer of Amino Groups from Primary Amines onto Keto Acids. ChemCatChem 2013, 5, 3538–3542. [Google Scholar] [CrossRef]
- Zhou, H.; Meng, L.; Yin, X.; Liu, Y.; Xu, G.; Wu, J.; Wu, M.; Yang, L. Artificial Biocatalytic Cascade with Three Enzymes in One Pot for Asymmetric Synthesis of Chiral Unnatural Amino Acids. Eur. J. Org. Chem. 2019, 2019, 6470–6477. [Google Scholar] [CrossRef]
- Parmeggiani, F.; Rué Casamajo, A.; Walton, C.J.W.; Galman, J.L.; Turner, N.J.; Chica, R.A. One-Pot Biocatalytic Synthesis of Substituted D-Tryptophans from Indoles Enabled by an Engineered Aminotransferase. ACS Catal. 2019, 9, 3482–3486. [Google Scholar] [CrossRef]
- Silva, M.V.d.M.; Costa, I.C.R.; de Souza, R.O.M.A.; Bornscheuer, U.T. Biocatalytic Cascade Reaction for the Asymmetric Synthesis of L- and D-homoalanine. ChemCatChem 2019, 11, 407–411. [Google Scholar] [CrossRef]
- Walton, C.J.W.; Parmeggiani, F.; Barber, J.E.B.; McCann, J.L.; Turner, N.J.; Chica, R.A. Engineered Aminotransferase for the Production of D-Phenylalanine Derivatives Using Biocatalytic Cascades. ChemCatChem 2018, 10, 470–474. [Google Scholar] [CrossRef]
- Bae, H.-S.; Lee, S.-G.; Hong, S.-P.; Kwak, M.-S.; Esaki, N.; Soda, K.; Sung, M.-H. Production of Aromatic D-Amino Acids from α-Keto Acids and Ammonia by Coupling of Four Enzyme Reactions. J. Mol. Catal. B Enzym. 1999, 6, 241–247. [Google Scholar] [CrossRef]
- Eliot, A.C.; Kirsch, J.F. Pyridoxal Phosphate Enzymes: Mechanistic, Structural, and Evolutionary Considerations. Annu. Rev. Biochem. 2004, 73, 383–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunstein, A.E. 10 Amino Group Transfer. In Enzymes; Karger Publishers: Basel, Switzerland, 1973; pp. 379–481. [Google Scholar]
- Bezsudnova, E.Y.; Popov, V.O.; Boyko, K.M. Structural Insight into the Substrate Specificity of PLP Fold Type IV Transaminases. Appl. Microbiol. Biotechnol. 2020, 104, 2343–2357. [Google Scholar] [CrossRef] [PubMed]
- Tanizawa, K.; Masus, Y.; Asano, S.; Tanaka, H.; Sodas, K. Thermostable D-Amino Acid Aminotransferase from a Thermophilic Bacillus Species. Purification, Characterization, and Active Site Sequence Determination. J. Biol. Chem. 1989, 264, 2445–2449. [Google Scholar] [CrossRef] [PubMed]
- Yonaha, K.; Misono, H.; Yamamoto, T.; Soda, K. D-Amino Acid Aminotransferase of Bacillus sphaericus. Enzymologic and Spectrometric Properties. J. Biol. Chem. 1975, 250, 6983–6989. [Google Scholar] [CrossRef]
- Kobayashi, J.; Shimizu, Y.; Mutaguchi, Y.; Doi, K.; Ohshima, T. Characterization of D-Amino Acid Aminotransferase from Lactobacillus salivarius. J. Mol. Catal. B Enzym. 2013, 94, 15–22. [Google Scholar] [CrossRef]
- Lee, S.-G.; Hong, S.-P.; Song, J.J.; Kim, S.-J.; Kwak, M.-S.; Sung, M.-H. Functional and Structural Characterization of Thermostable D-Amino Acid Aminotransferases from Geobacillus spp. Appl. Environ. Microbiol. 2006, 72, 1588–1594. [Google Scholar] [CrossRef] [Green Version]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Zhao, J.; Chen, L.; Li, Y.; Yu, B.; Tian, X.; Min, Z.; Xu, S.; Gu, H.; Sun, J.; et al. Application of Transition-Metal Catalysis, Biocatalysis, and Flow Chemistry as State-of-the-Art Technologies in the Synthesis of LCZ696. J. Org. Chem. 2020, 85, 6844–6853. [Google Scholar] [CrossRef]
- Slabu, I.; Galman, J.L.; Lloyd, R.C.; Turner, N.J. Discovery, Engineering, and Synthetic Application of Transaminase Biocatalysts. ACS Catal. 2017, 7, 8263–8284. [Google Scholar] [CrossRef]
- Bakunova, A.K.; Nikolaeva, A.Y.; Rakitina, T.V.; Isaikina, T.Y.; Khrenova, M.G.; Boyko, K.M.; Popov, V.O.; Bezsudnova, E.Y. The Uncommon Active Site of D-Amino Acid Transaminase from Haliscomenobacter Hydrossis: Biochemical and Structural Insights into the New Enzyme. Molecules 2021, 26, 5053. [Google Scholar] [CrossRef]
- Yu, X.; Bresser, J.; Schall, I.; Djurdjevic, I.; Buckel, W.; Wang, X.; Engel, P.C. Development of a Satisfactory and General Continuous Assay for Aminotransferases by Coupling with (R)-2-Hydroxyglutarate Dehydrogenase. Anal. Biochem. 2012, 431, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, G.; Dostalek, M.; Hsu, E.L.; Nguyen, L.P.; Stec, D.F.; Bradfield, C.A.; Guengerich, F.P. Structural Identification of Diindole Agonists of the Aryl Hydrocarbon Receptor Derived from Degradation of Indole-3-Pyruvic Acid. Chem. Res. Toxicol. 2009, 22, 1905–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchikami, Y.; Yoshimura, T.; Gutierrez, A.; Soda, K.; Esaki, N. Construction and Properties of a Fragmentary D-Amino Acid Aminotransferase. J. Biochem. 1998, 124, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Sugio, S.; Petsko, G.A.; Manning, J.M.; Soda, K.; Ringe, D. Crystal Structure of a D-Amino Acid Aminotransferase: How the Protein Controls Stereoselectivity. Biochemistry 1995, 34, 9661–9669. [Google Scholar] [CrossRef] [PubMed]
- Roura Padrosa, D.; Alaux, R.; Smith, P.; Dreveny, I.; López-Gallego, F.; Paradisi, F. Enhancing PLP-Binding Capacity of Class-III ω-Transaminase by Single Residue Substitution. Front. Bioeng. Biotechnol. 2019, 7, 282. [Google Scholar] [CrossRef] [Green Version]
- Börner, T.; Rämisch, S.; Reddem, E.R.; Bartsch, S.; Vogel, A.; Thunnissen, A.M.W.H.; Adlercreutz, P.; Grey, C. Explaining Operational Instability of Amine Transaminases: Substrate-Induced Inactivation Mechanism and Influence of Quaternary Structure on Enzyme-Cofactor Intermediate Stability. ACS Catal. 2017, 7, 1259–1269. [Google Scholar] [CrossRef]
- Pavkov-Keller, T.; Strohmeier, G.A.; Diepold, M.; Peeters, W.; Smeets, N.; Schürmann, M.; Gruber, K.; Schwab, H.; Steiner, K. Discovery and Structural Characterisation of New Fold Type IV-Transaminases Exemplify the Diversity of This Enzyme Fold. Sci. Rep. 2016, 6, 38183. [Google Scholar] [CrossRef]


| Substrate | Structure | Corresponding Amino Acid | Vmax, U/mg | kcat, s−1 | Km, mM | kcat/Km, s−1 M−1 |
|---|---|---|---|---|---|---|
| pyruvate | 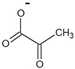 | D-alanine | 380 ± 10 | 215 ± 6 * | 2.1 ± 0.1 * | 103,000 ± 8000 * |
| 2-oxobutyrate |  | D-homoalanine | 71 ± 1 | 40.0 ± 0.6 | 1.6 ± 0.1 | 25,000 ± 2000 |
| 2-oxovalerate | 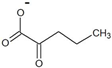 | D-norvaline | 48 ± 4 | 27 ± 2 | 13.5 ± 0.3 | 2000 ± 200 |
| 3-methyl-2-oxobutyrate | 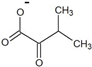 | D-valine | 46 ± 2 | 26 ± 1 | 18 ± 2 | 1400 ± 200 |
| 2-oxohexanoate | 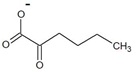 | D-norleucine | 4.6 ± 0.4 | 2.6 ± 0.2 | 43 ± 5 | 60 ± 10 |
| 3-methyl-2-oxovalerate |  | D-isoleucine | 17.6 ± 0.4 | 10 ± 0.2 | 200 ± 50 | 50 ± 20 |
| 4-methyl-2-oxovalerate | 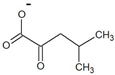 | D-leucine | 35 ± 2 | 20 ± 1 | 110 ± 6 | 200 ± 10 |
| trimethylpyruvate | 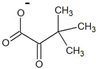 | D-tert-leucine | ND | |||
| phenylpyruvate | 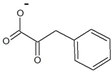 | D-phenylalanine | 35 ± 2 | 20 ± 1 | 36 ± 3 | 560 ± 70 |
| indol-3-pyruvate |  | D-tryptophan | 3.7 ± 0.2 | 2.1 ± 0.1 | 5.0 ± 0.2 | 420 ± 40 |
| 4-hydroxyphenylpyruvate | 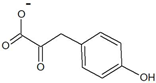 | D-tyrosine | 34 ± 2 | 19 ± 1 | 8 ± 1 | 2400 ± 400 |
| 2-oxo-4-phenylbutyrate | 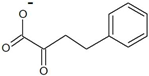 | D-homophenylalanine | 6.7 ± 0.4 | 3.8 ± 0.2 | 16.0 ± 0.8 | 240 ± 20 |
| Reaction Conditions | min−1 | Half-Life, min | |||
|---|---|---|---|---|---|
| Amino Donor | Amino Acceptor | Temperature, °C | [PLP], µM | ||
| 100 mM D-alanine | 10 mM α-ketoglutarate | 40 | 0 | 0.048 ± 0.006 | 10 ± 1 |
| 100 mM D-alanine | 10 mM α-ketoglutarate | 40 | 100 | 0.030 ± 0.004 | 17 ± 2 |
| 100 mM D-alanine | 10 mM α-ketoglutarate | 30 | 0 | 0.010 ± 0.001 | 50 ± 5 |
| 100 mM D-alanine | 10 mM α-ketoglutarate | 30 | 100 | 0.008 ± 0.002 | 62 ± 15 |
| 100 mM D-alanine | 50 mM α-ketoglutarate | 30 | 0 | 0.005 ± 0.001 | 100 ± 20 |
| 50 mM D-alanine | 50 mM α-ketoglutarate | 30 | 0 | 0.0030 ± 0.0003 | 170 ± 20 |
| 50 mM D-glutamate | 50 mM 2-oxovalerate | 30 | 0 | 0.0021 ± 0.0003 | 240 ± 30 |
| Substrate | Product | Product Yield, % | ee, % | |
|---|---|---|---|---|
| 2-oxobutyrate | D-homoalanine |  | 99 | >99.5 |
| 2-oxovalerate | D-norvaline | 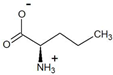 | 95.7 | >99.3 |
| 3-methyl-2-oxobutyrate | D-valine | 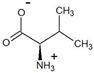 | 99 | >99 |
| 2-oxohexanoate | D-norleucine | 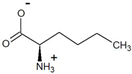 | 99 | >99 |
| 3-methyl-2-oxovalerate | D-isoleucine | 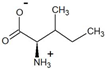 | 90 | >99 |
| 4-methyl-2-oxovalerate | D-leucine | 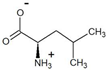 | 98.5 * | >99.4 * |
| phenylpyruvate | D-phenylalanine | 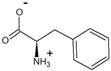 | 95.6 * | >99.3 * |
| indol-3-pyruvate | D-tryptophan |  | 75 ** | >99.6 |
| 4-hydroxyphenylpyruvate | D-tyrosine | 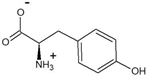 | 85 | >99.7 |
| 2-oxo-4-phenylbutyrate | D-homophenylalanine | 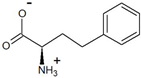 | 99 | >99.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakunova, A.K.; Isaikina, T.Y.; Popov, V.O.; Bezsudnova, E.Y. Asymmetric Synthesis of Enantiomerically Pure Aliphatic and Aromatic D-Amino Acids Catalyzed by Transaminase from Haliscomenobacter hydrossis. Catalysts 2022, 12, 1551. https://doi.org/10.3390/catal12121551
Bakunova AK, Isaikina TY, Popov VO, Bezsudnova EY. Asymmetric Synthesis of Enantiomerically Pure Aliphatic and Aromatic D-Amino Acids Catalyzed by Transaminase from Haliscomenobacter hydrossis. Catalysts. 2022; 12(12):1551. https://doi.org/10.3390/catal12121551
Chicago/Turabian StyleBakunova, Alina K., Tatiana Y. Isaikina, Vladimir O. Popov, and Ekaterina Yu. Bezsudnova. 2022. "Asymmetric Synthesis of Enantiomerically Pure Aliphatic and Aromatic D-Amino Acids Catalyzed by Transaminase from Haliscomenobacter hydrossis" Catalysts 12, no. 12: 1551. https://doi.org/10.3390/catal12121551
APA StyleBakunova, A. K., Isaikina, T. Y., Popov, V. O., & Bezsudnova, E. Y. (2022). Asymmetric Synthesis of Enantiomerically Pure Aliphatic and Aromatic D-Amino Acids Catalyzed by Transaminase from Haliscomenobacter hydrossis. Catalysts, 12(12), 1551. https://doi.org/10.3390/catal12121551