

Origin of the Unexpected Enantioselectivity in the Enzymatic Reductions of 5-Membered-Ring Heterocyclic Ketones Catalyzed by Candida parapsilosis Carbonyl Reductases

 , and
, and
Abstract
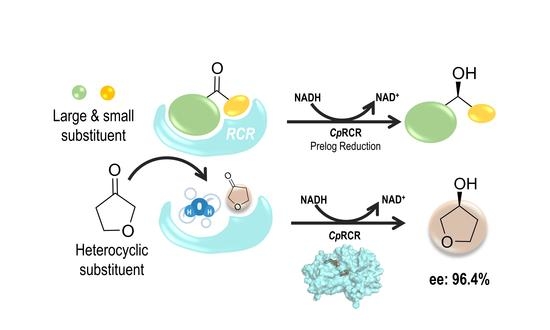
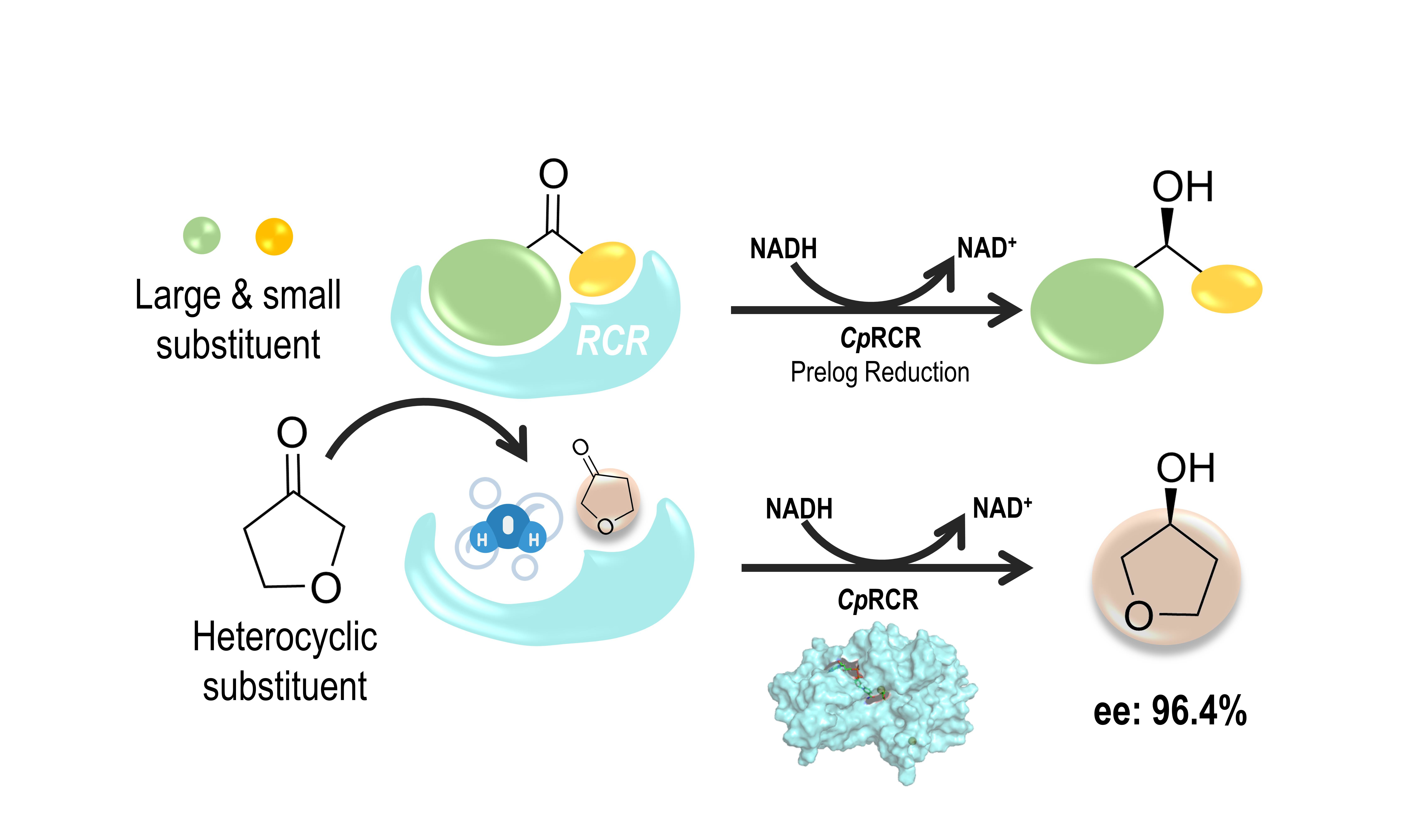
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
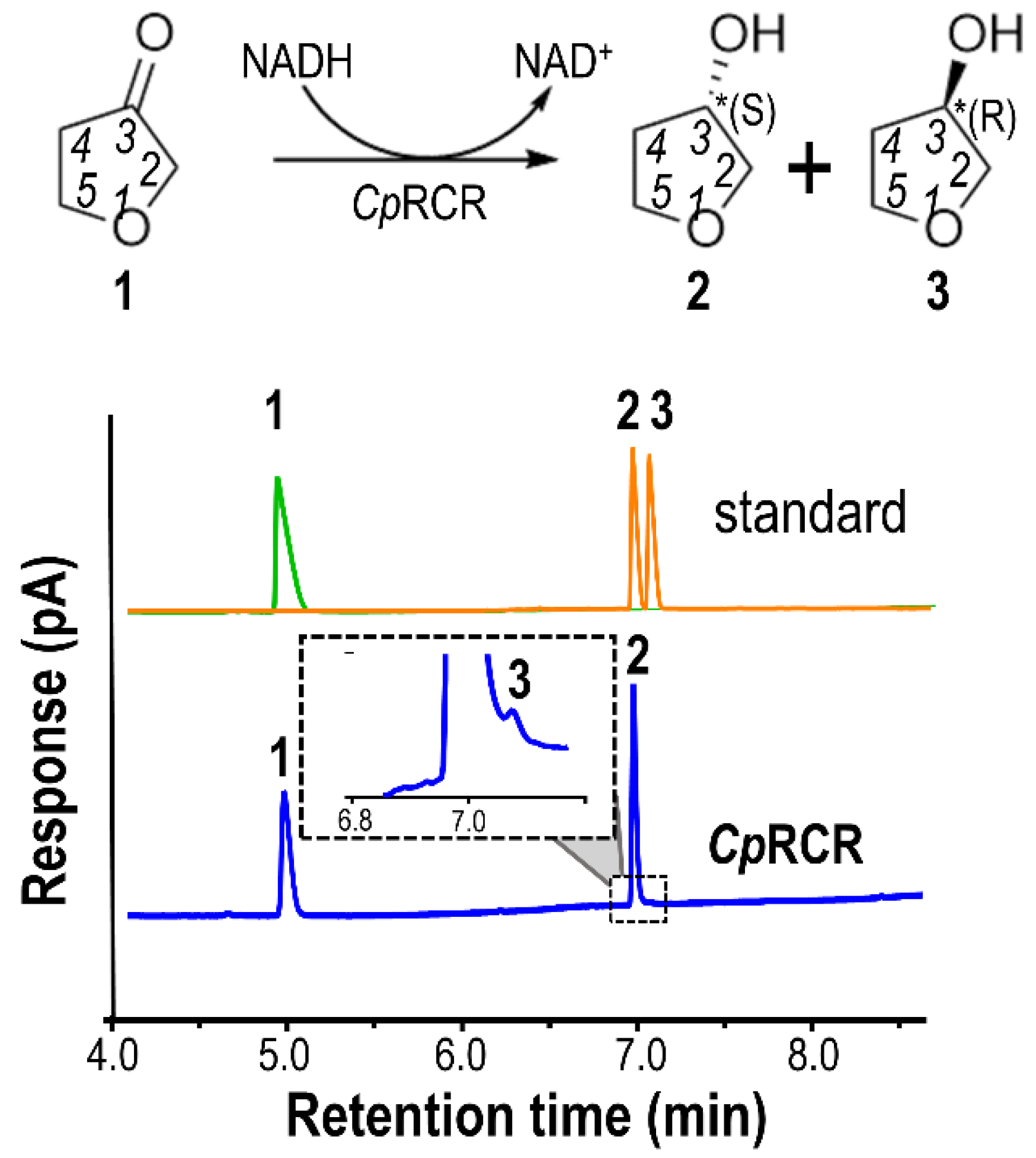
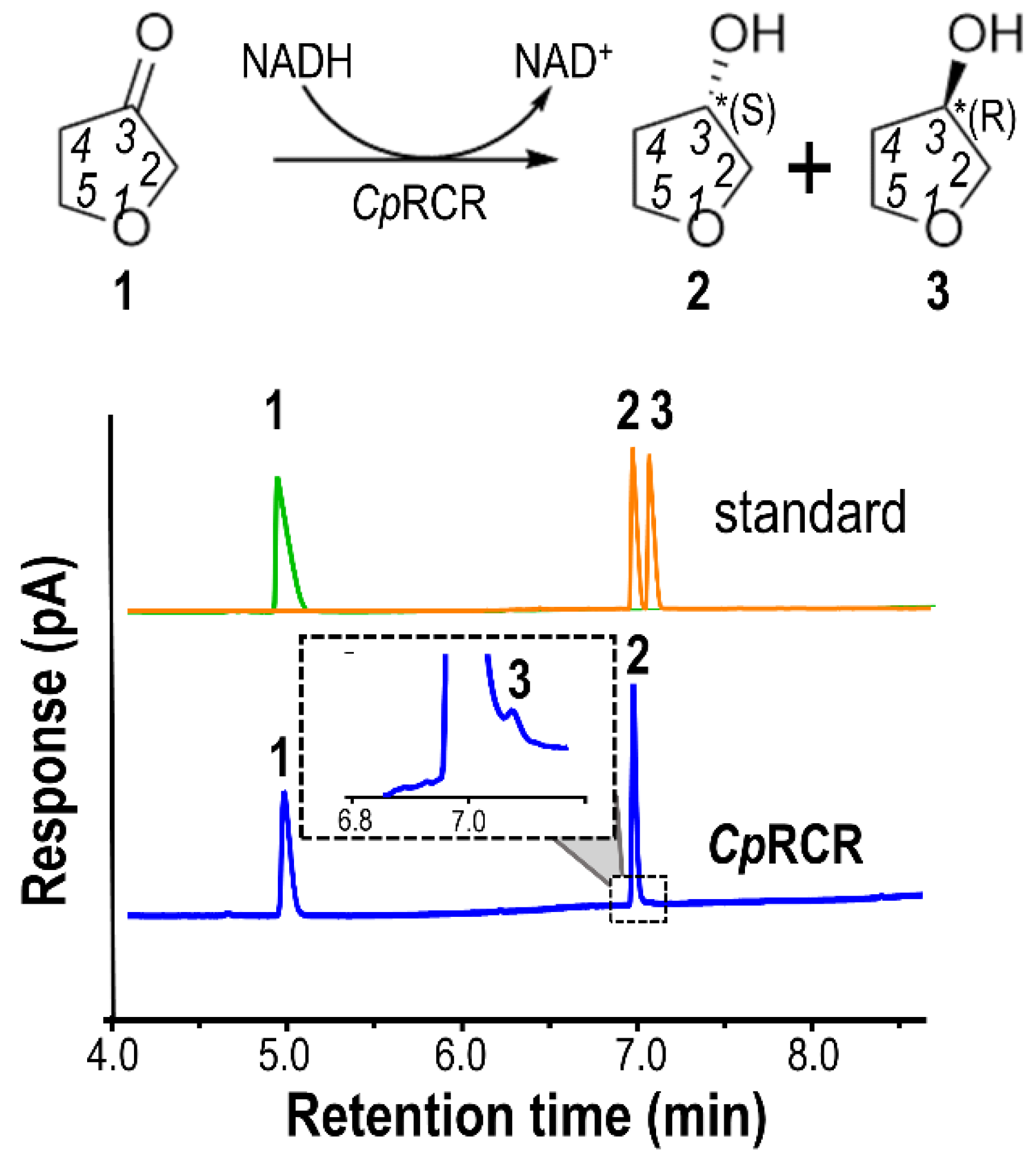
2.1. Kinetics and Stereoselectivity of DHF and DHT with CpRCR
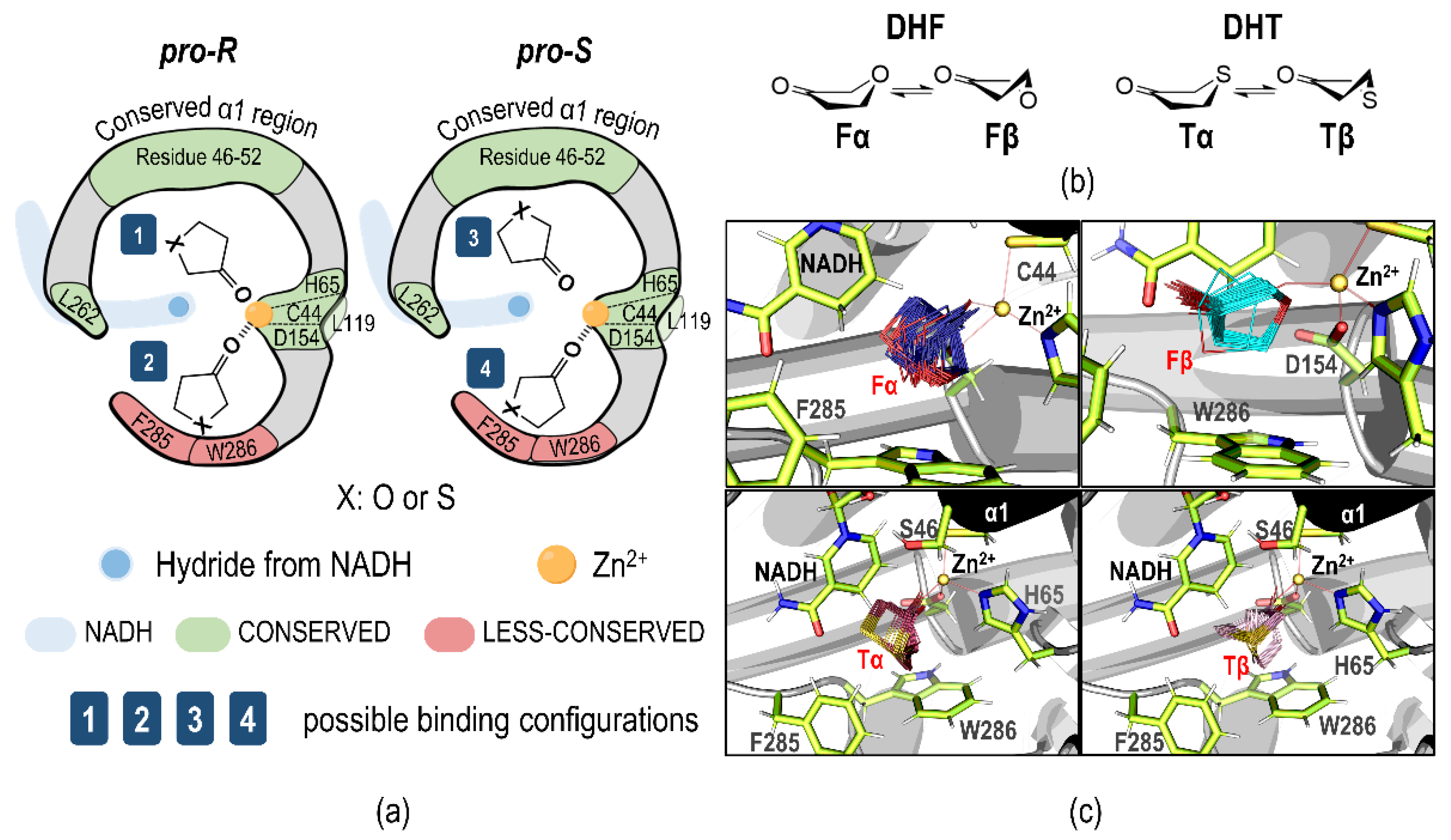
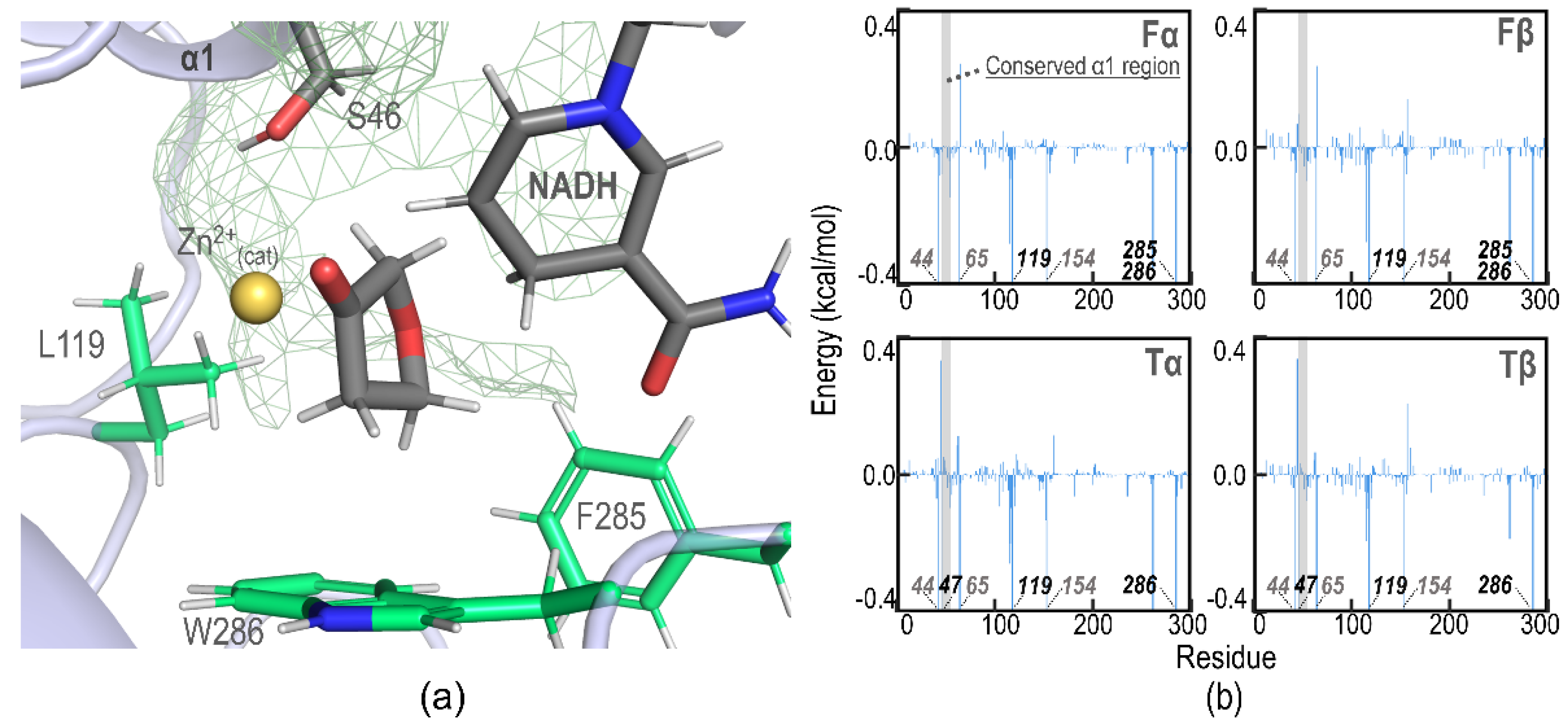
2.2. Plausible Binding Orientations
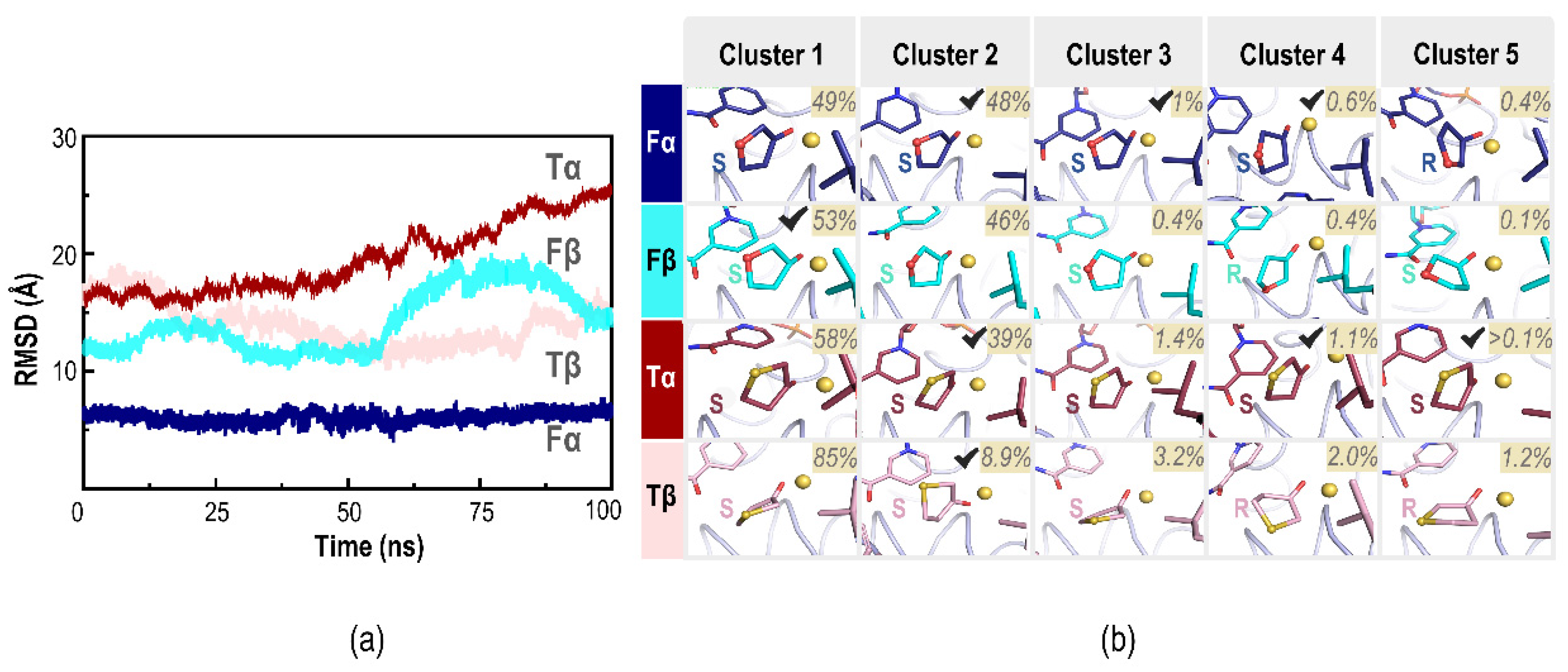
2.3. Molecular Dynamics Simulations
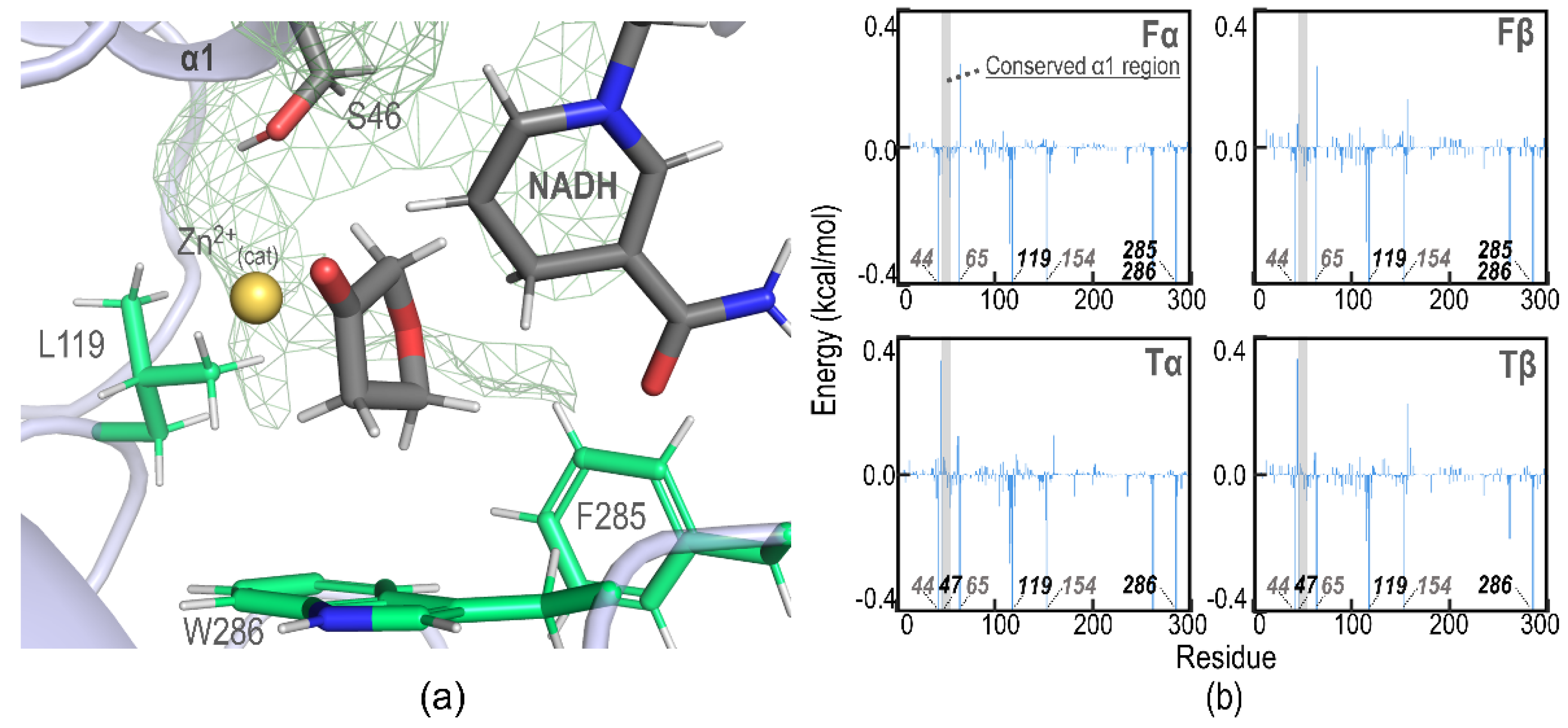
2.4. Binding Affinity Analysis
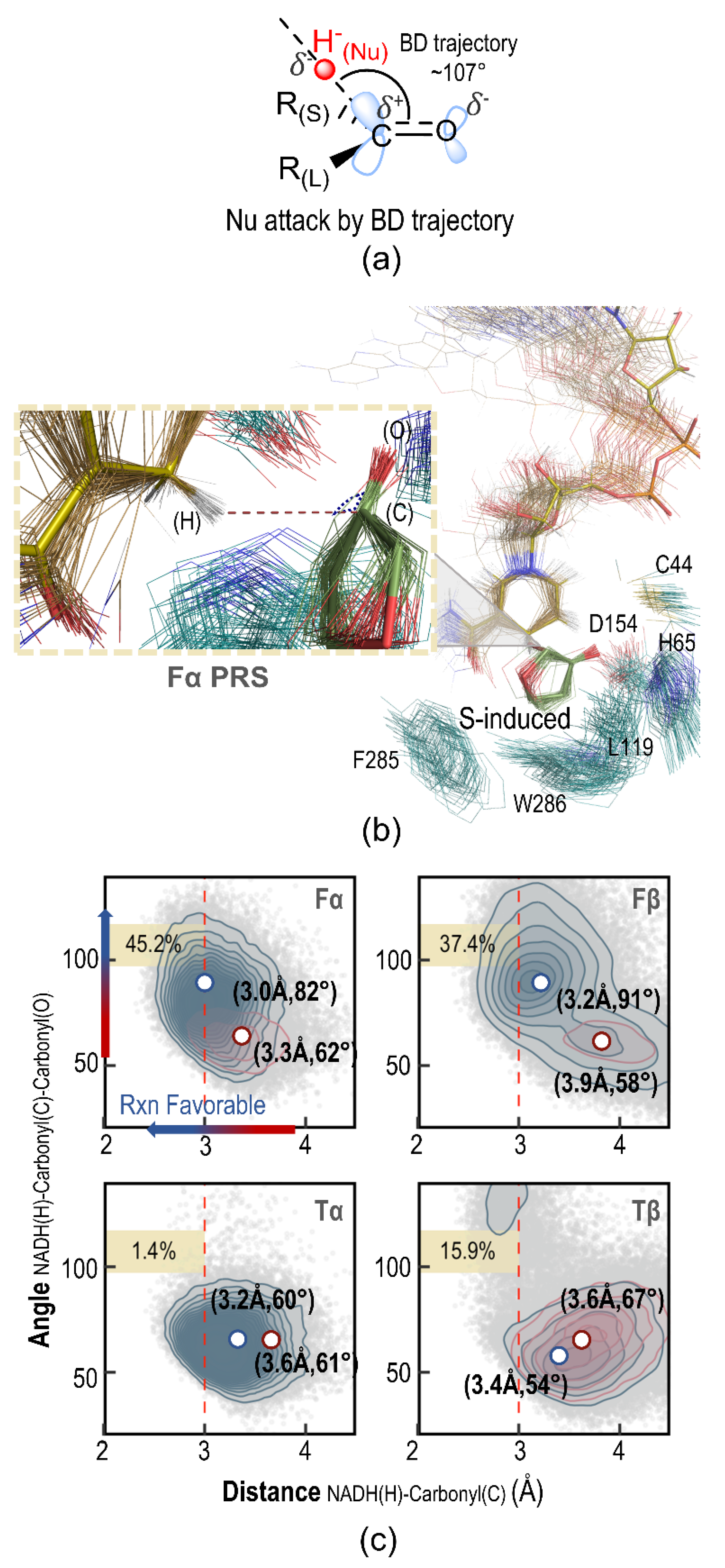
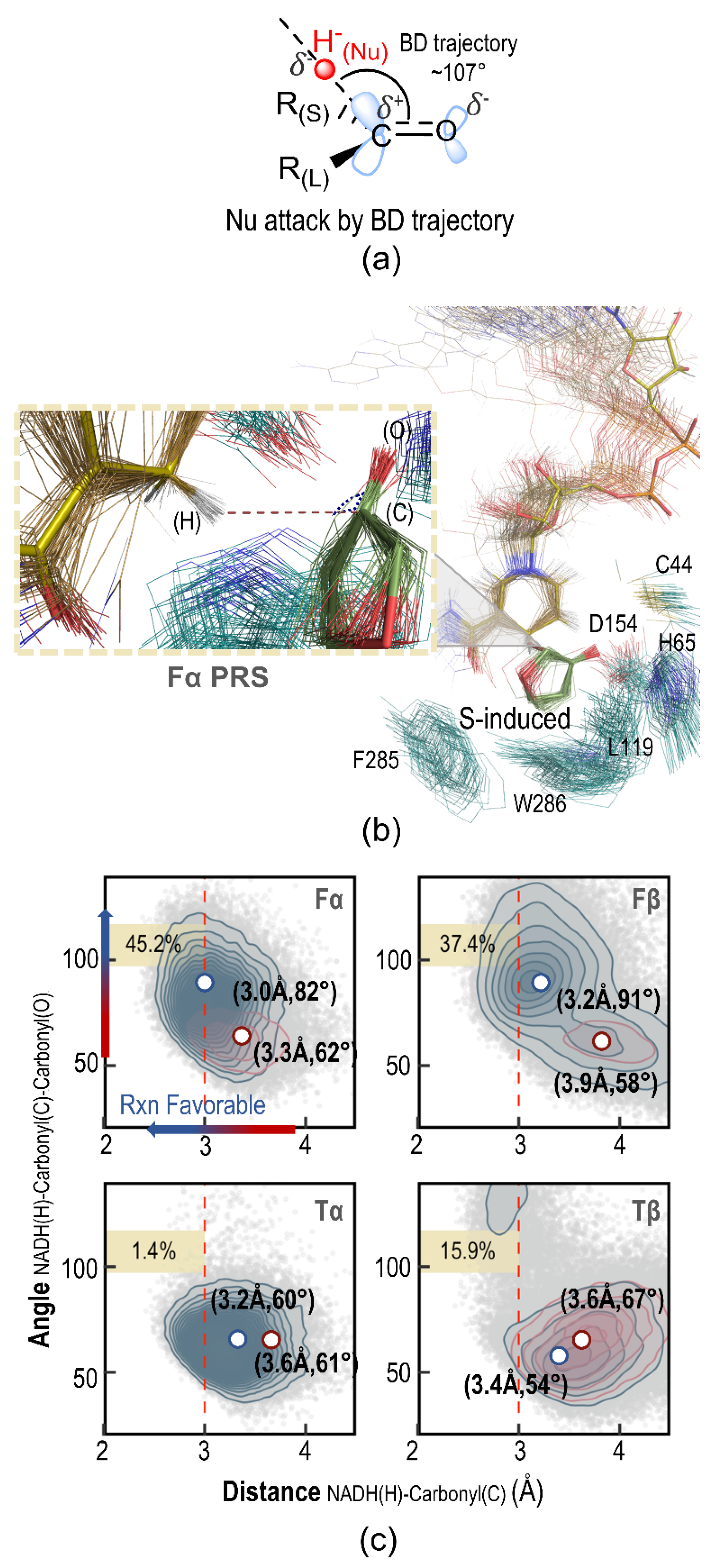
2.5. Pre-Reaction State Analysis
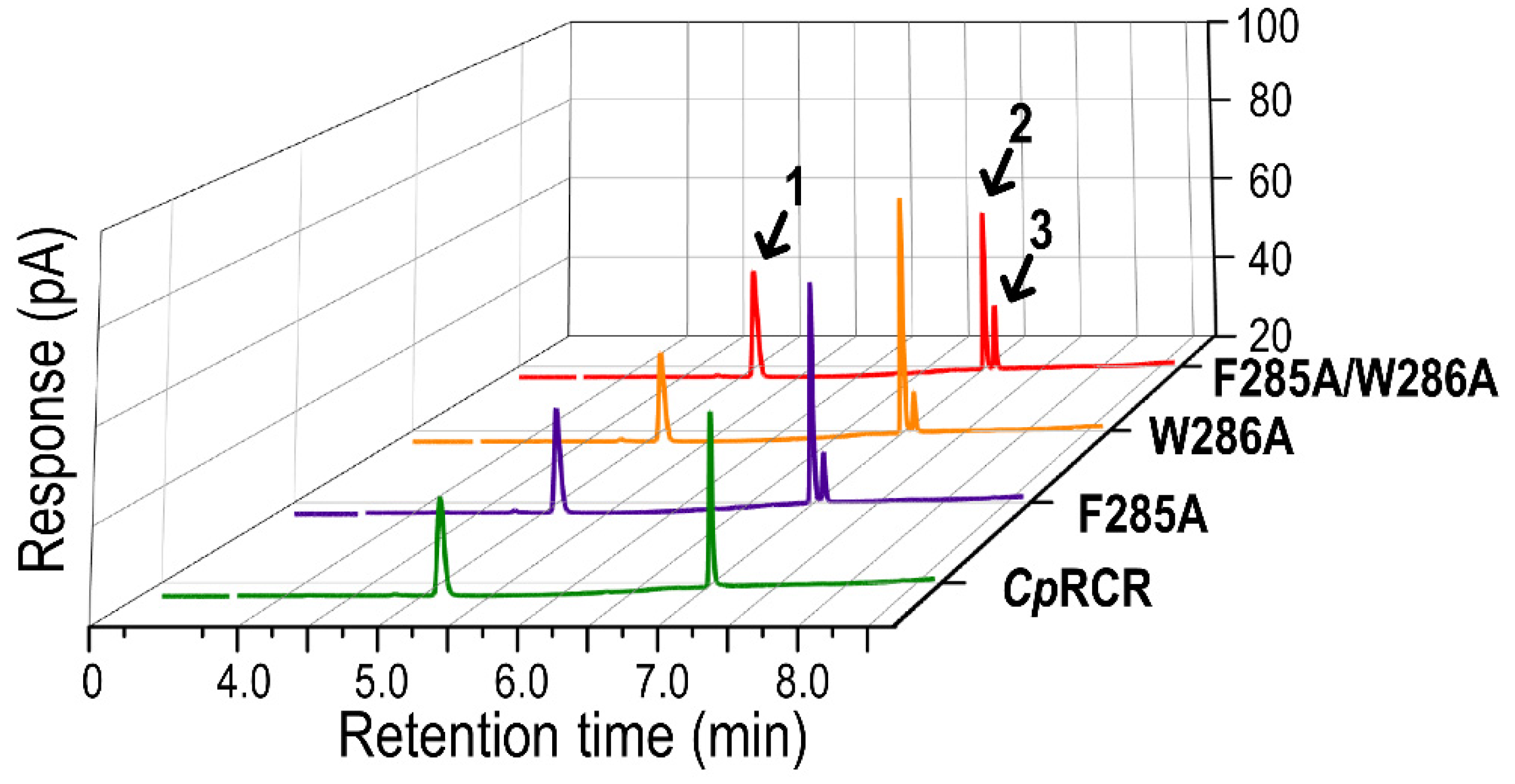
2.6. Experimental Validation
2.7. Prespective on Small Cavities in Alcohol Dehydrogenases
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Gene Expression and Enzyme Purification
4.3. Enzyme Kinetic Assay
4.4. Stereoselectivity Assay
4.5. Computational System Preparation
4.6. Molecular Dynamics Simulations
4.7. Molcular Mechanics-Poisson-Boltzmann Surface Area (MM/PBSA)
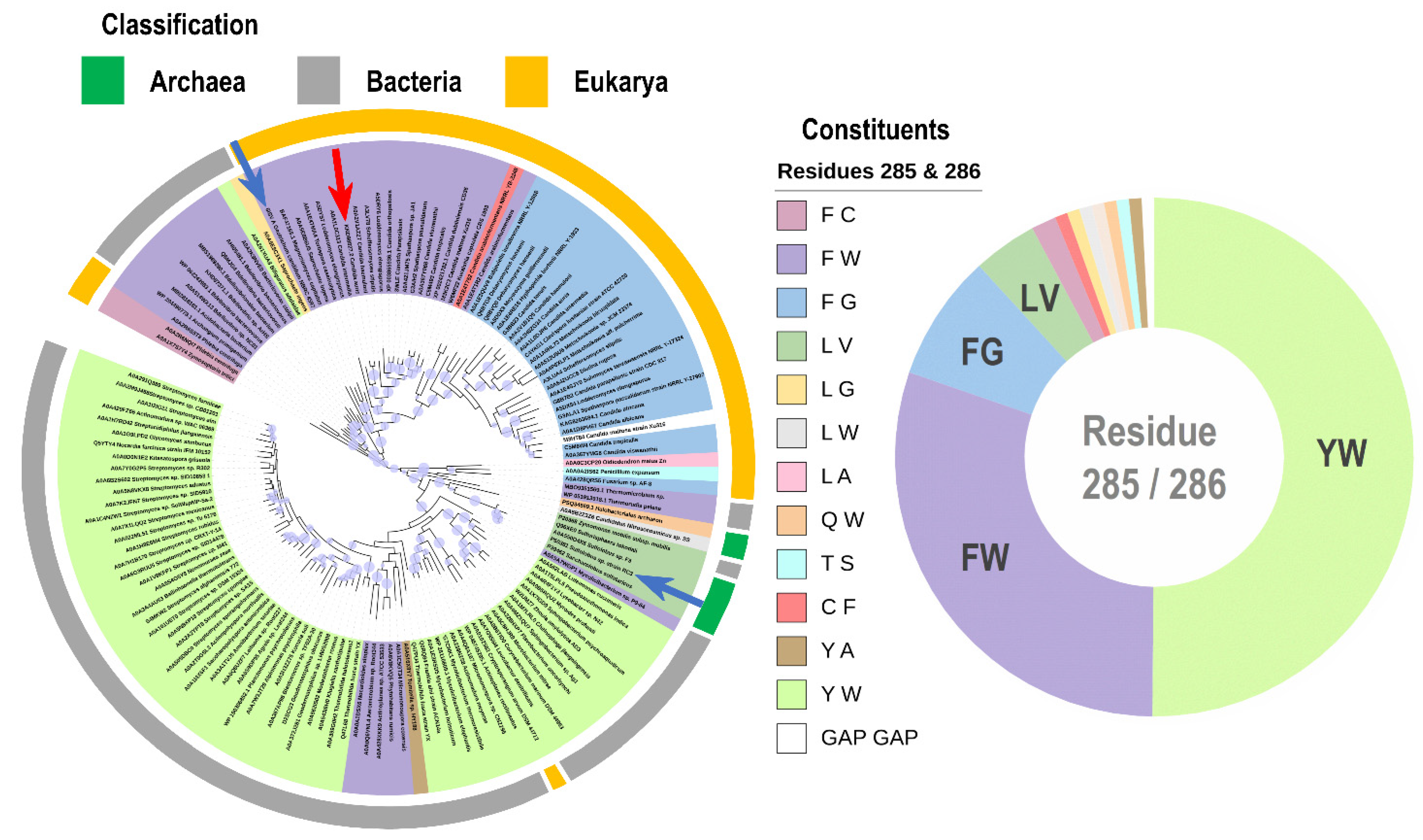
4.8. Homology Sequence Analysis and Phyolgenetic Tree
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gladiali, S.; Alberico, E. Asymmetric transfer hydrogenation: Chiral ligands and applications. Chem. Soc. Rev. 2006, 35, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wu, L.; Nie, Y.; Xu, Y. Biosynthesis of chiral cyclic and heterocyclic alcohols via CO/C–H/C–O asymmetric reactions. Catal. Sci. Technol. 2021, 11, 2637–2651. [Google Scholar] [CrossRef]
- Grunberg, E.; Titsworth, E.H. Chemotherapeutic Properties of Heterocyclic Compounds: Monocyclic Compounds with Five-Membered Rings. Annu. Rev. Microbiol. 1973, 27, 317–346. [Google Scholar] [CrossRef]
- Albarrán-Velo, J.; González-Martínez, D.; Gotor-Fernández, V. Stereoselective biocatalysis: A mature technology for the asymmetric synthesis of pharmaceutical building blocks. Biocatal. Biotransform. 2018, 36, 102–130. [Google Scholar] [CrossRef]
- Ager, D.J.; Prakash, I.; Schaad, D.R. 1,2-Amino Alcohols and Their Heterocyclic Derivatives as Chiral Auxiliaries in Asymmetric Synthesis. Chem. Rev. 1996, 96, 835–876. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Marini, F.; Sternativo, S.; Santi, C.; Bagnoli, L.; Temperini, A. Intramolecular addition of carbon radicals to aldehydes: Synthesis of enantiopure tetrahydrofuran-3-ols. Tetrahedron 2007, 63, 5482–5489. [Google Scholar] [CrossRef]
- Gadakh, S.K.; Santhosh Reddy, R.; Sudalai, A. Enantioselective synthesis of HIV protease inhibitor amprenavir via Co-catalyzed HKR of 2-(1-azido-2-phenylethyl)oxirane. Tetrahedron Asymmetry 2012, 23, 898–903. [Google Scholar] [CrossRef]
- Nie, Y.; Wang, S.; Xu, Y.; Luo, S.; Zhao, Y.-L.; Xiao, R.; Montelione, G.T.; Hunt, J.F.; Szyperski, T. Enzyme Engineering Based on X-ray Structures and Kinetic Profiling of Substrate Libraries: Alcohol Dehydrogenases for Stereospecific Synthesis of a Broad Range of Chiral Alcohols. ACS Catal. 2018, 8, 5145–5152. [Google Scholar] [CrossRef]
- Wang, S.; Nie, Y.; Xu, Y.; Zhang, R.; Ko, T.-P.; Huang, C.-H.; Chan, H.-C.; Guo, R.-T.; Xiao, R. Unconserved substrate-binding sites direct the stereoselectivity of medium-chain alcohol dehydrogenase. Chem. Commun. 2014, 50, 7770–7772. [Google Scholar] [CrossRef]
- Gu, J.; Sim, B.R.; Li, J.; Yu, Y.; Qin, L.; Wu, L.; Shen, Y.; Nie, Y.; Zhao, Y.-L.; Xu, Y. Evolutionary coupling-inspired engineering of alcohol dehydrogenase reveals the influence of distant sites on its catalytic efficiency for stereospecific synthesis of chiral alcohols. Comput. Struct. Biotechnol. J. 2021, 19, 5864–5873. [Google Scholar] [CrossRef]
- Dhoke, G.V.; Davari, M.D.; Schwaneberg, U.; Bocola, M. QM/MM calculations revealing the resting and catalytic states in zinc-dependent medium-chain dehydrogenases/reductases. ACS Catal. 2015, 5, 3207–3215. [Google Scholar] [CrossRef]
- Byuri, S.; Yilei, Z. Assessment on the pre-reaction state of enzyme: Could we understand catalytic activity with near transition-state molecular dynamic simulation?—A review. Synth. Biol. J. 2022, 3, 567. [Google Scholar]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein. Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Kruse, H.; Goerigk, L.; Grimme, S. Why the Standard B3LYP/6-31G* Model Chemistry Should Not Be Used in DFT Calculations of Molecular Thermochemistry: Understanding and Correcting the Problem. J. Org. Chem. 2012, 77, 10824–10834. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Li, P.; Merz, K.M. MCPB.py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Modeling 2016, 56, 599–604. [Google Scholar] [CrossRef]
- Walker, R.C.; De Souza, M.M.; Mercer, I.P.; Gould, I.R.; Klug, D.R. Large and fast relaxations inside a protein: Calculation and measurement of reorganization energies in alcohol dehydrogenase. J. Phys. Chem. B 2002, 106, 11658–11665. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate solvent-accessible surface areas from tetrahedrally directed neighbor densities. Biopolym. Orig. Res. Biomol. 1999, 50, 373–380. [Google Scholar] [CrossRef]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sim, B.R.; Gu, J.; Ley, Y.; Luo, S.; Liu, Y.; Chen, Q.; Nie, Y.; Zhao, Y.-L. Origin of the Unexpected Enantioselectivity in the Enzymatic Reductions of 5-Membered-Ring Heterocyclic Ketones Catalyzed by Candida parapsilosis Carbonyl Reductases. Catalysts 2022, 12, 1086. https://doi.org/10.3390/catal12101086
Sim BR, Gu J, Ley Y, Luo S, Liu Y, Chen Q, Nie Y, Zhao Y-L. Origin of the Unexpected Enantioselectivity in the Enzymatic Reductions of 5-Membered-Ring Heterocyclic Ketones Catalyzed by Candida parapsilosis Carbonyl Reductases. Catalysts. 2022; 12(10):1086. https://doi.org/10.3390/catal12101086
Chicago/Turabian StyleSim, Byu Ri, Jie Gu, Yvette Ley, Shenggan Luo, Yihan Liu, Qin Chen, Yao Nie, and Yi-Lei Zhao. 2022. "Origin of the Unexpected Enantioselectivity in the Enzymatic Reductions of 5-Membered-Ring Heterocyclic Ketones Catalyzed by Candida parapsilosis Carbonyl Reductases" Catalysts 12, no. 10: 1086. https://doi.org/10.3390/catal12101086
APA StyleSim, B. R., Gu, J., Ley, Y., Luo, S., Liu, Y., Chen, Q., Nie, Y., & Zhao, Y.-L. (2022). Origin of the Unexpected Enantioselectivity in the Enzymatic Reductions of 5-Membered-Ring Heterocyclic Ketones Catalyzed by Candida parapsilosis Carbonyl Reductases. Catalysts, 12(10), 1086. https://doi.org/10.3390/catal12101086