Cerium d-Block Element (Co, Ni) Bimetallic Oxides as Catalysts for the Methanation of CO2: Effect of Pressure



Abstract
:
1. Introduction
2. Results and Discussion
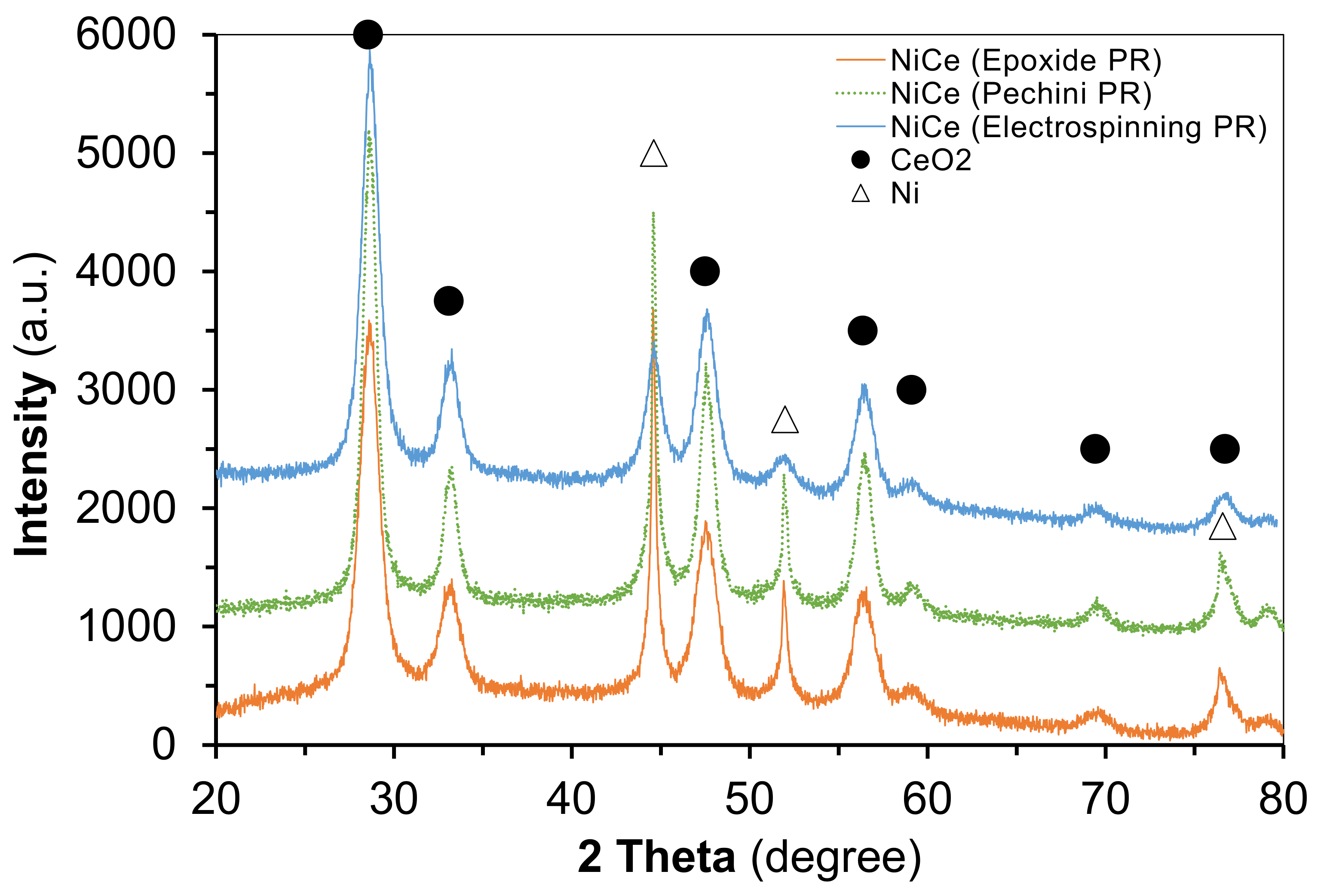
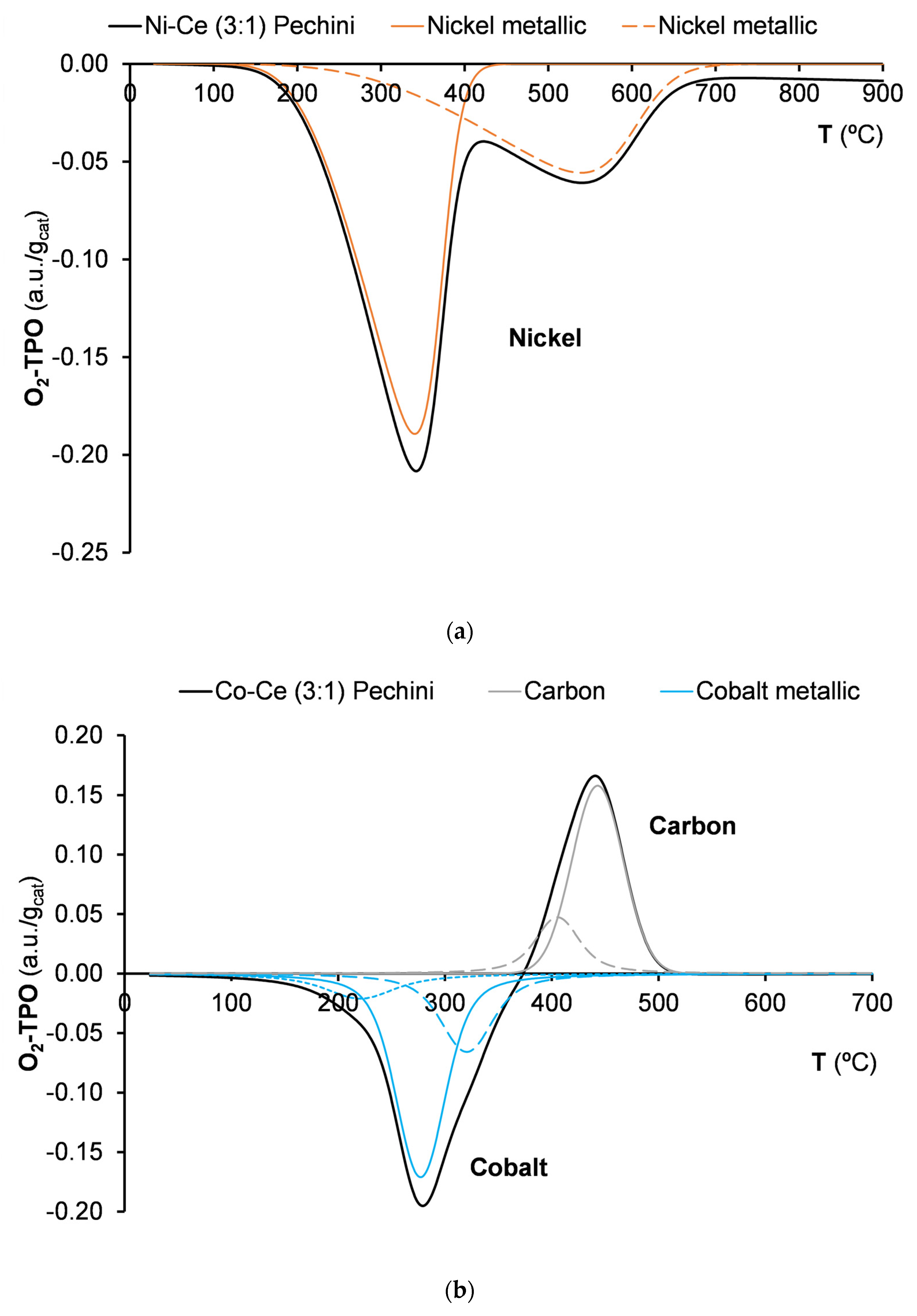
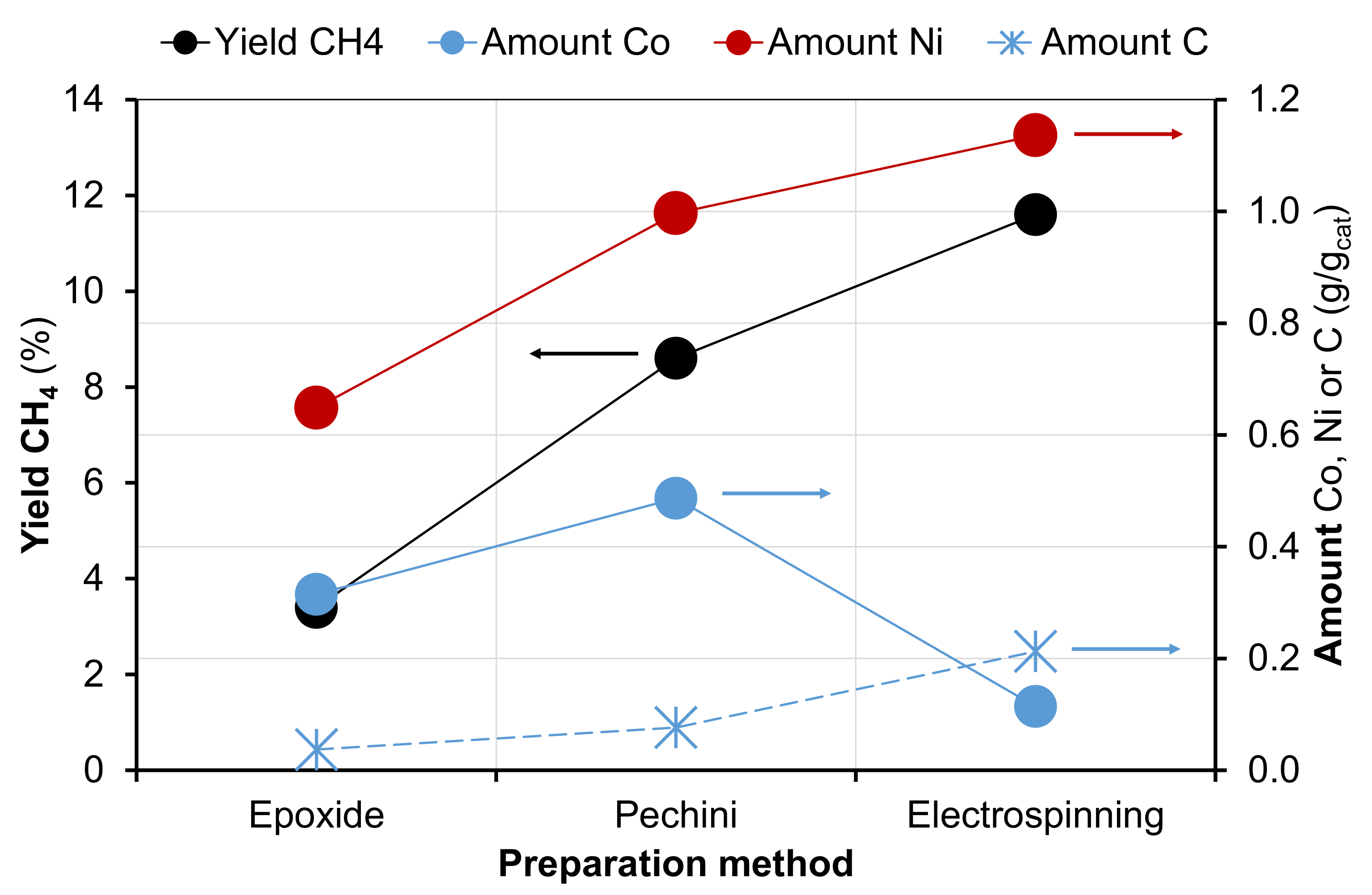
2.1. Characterization of Catalysts before Catalytic Reaction
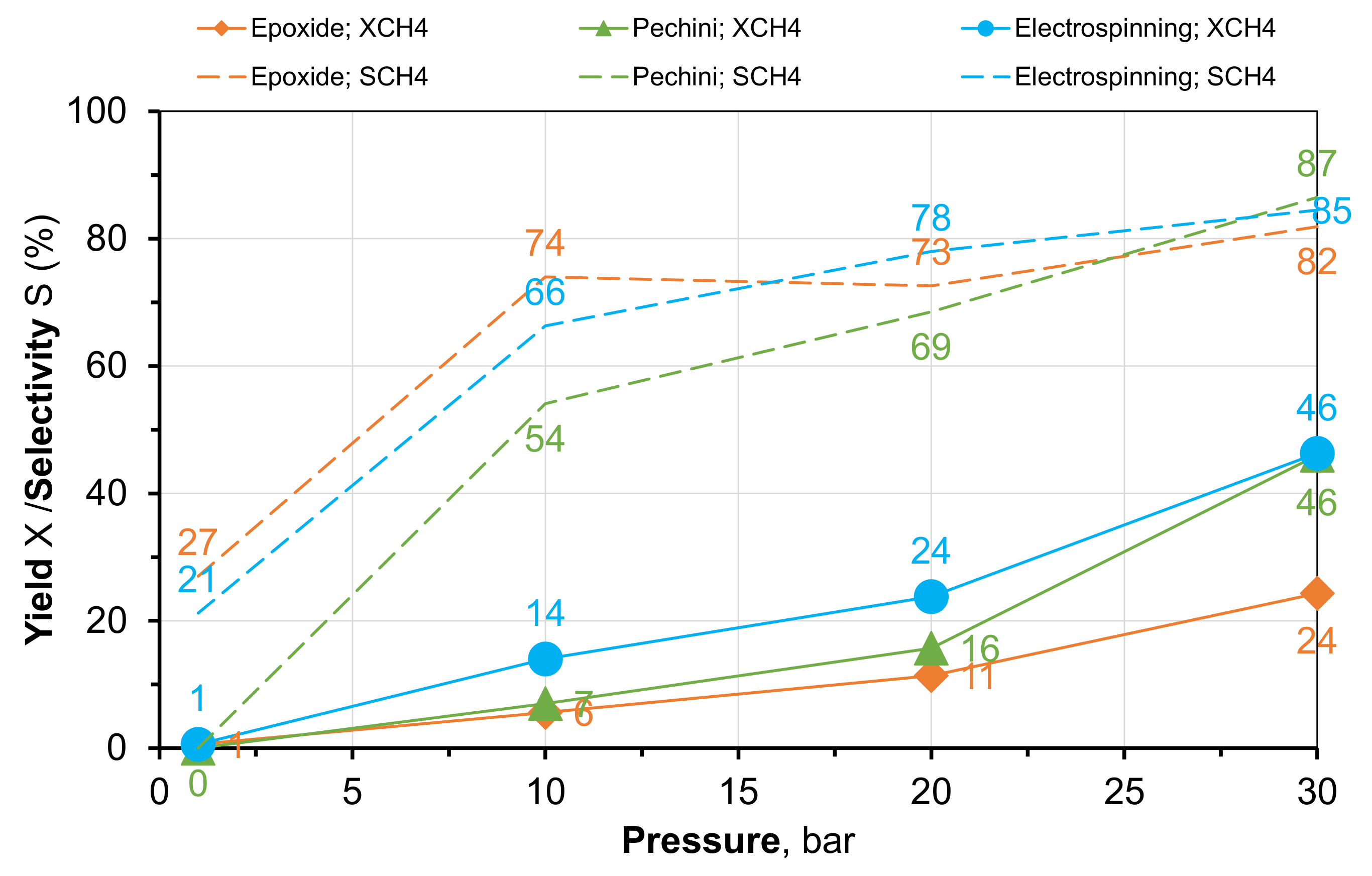
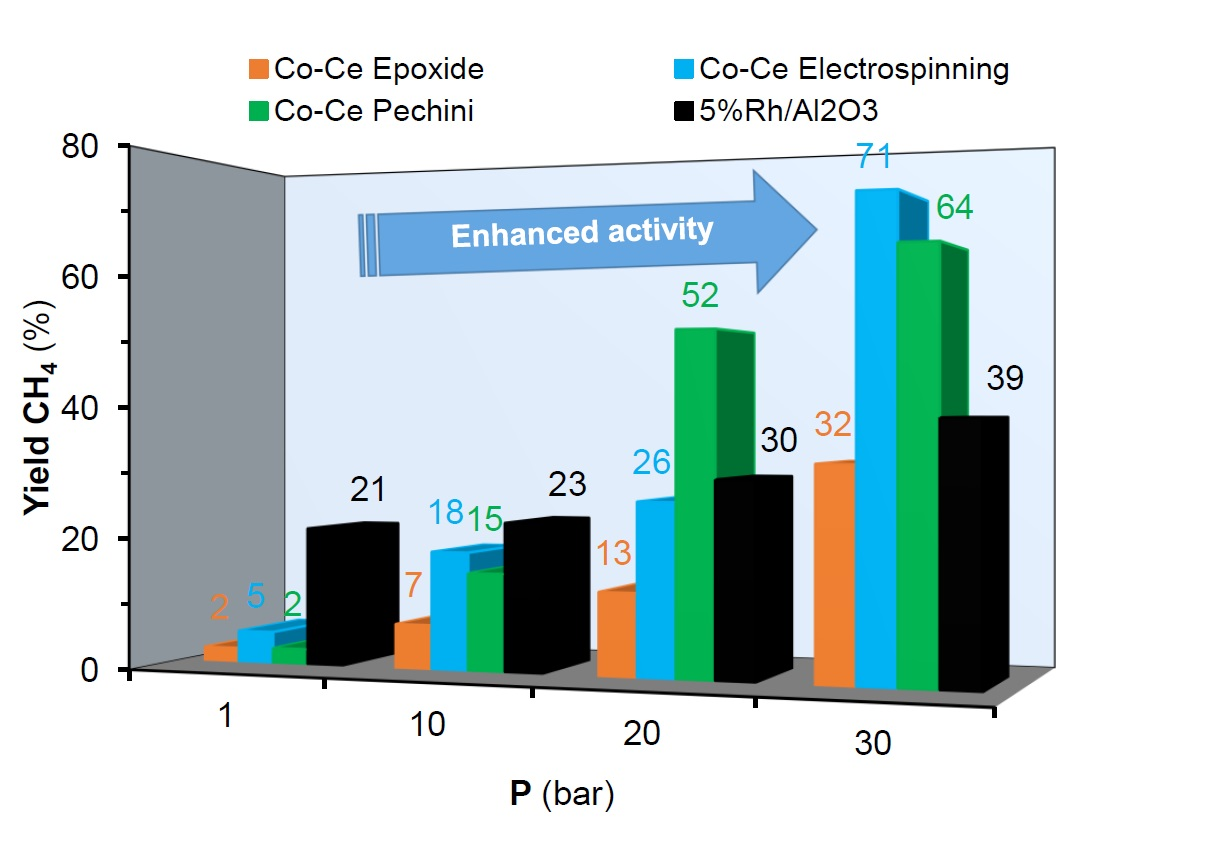
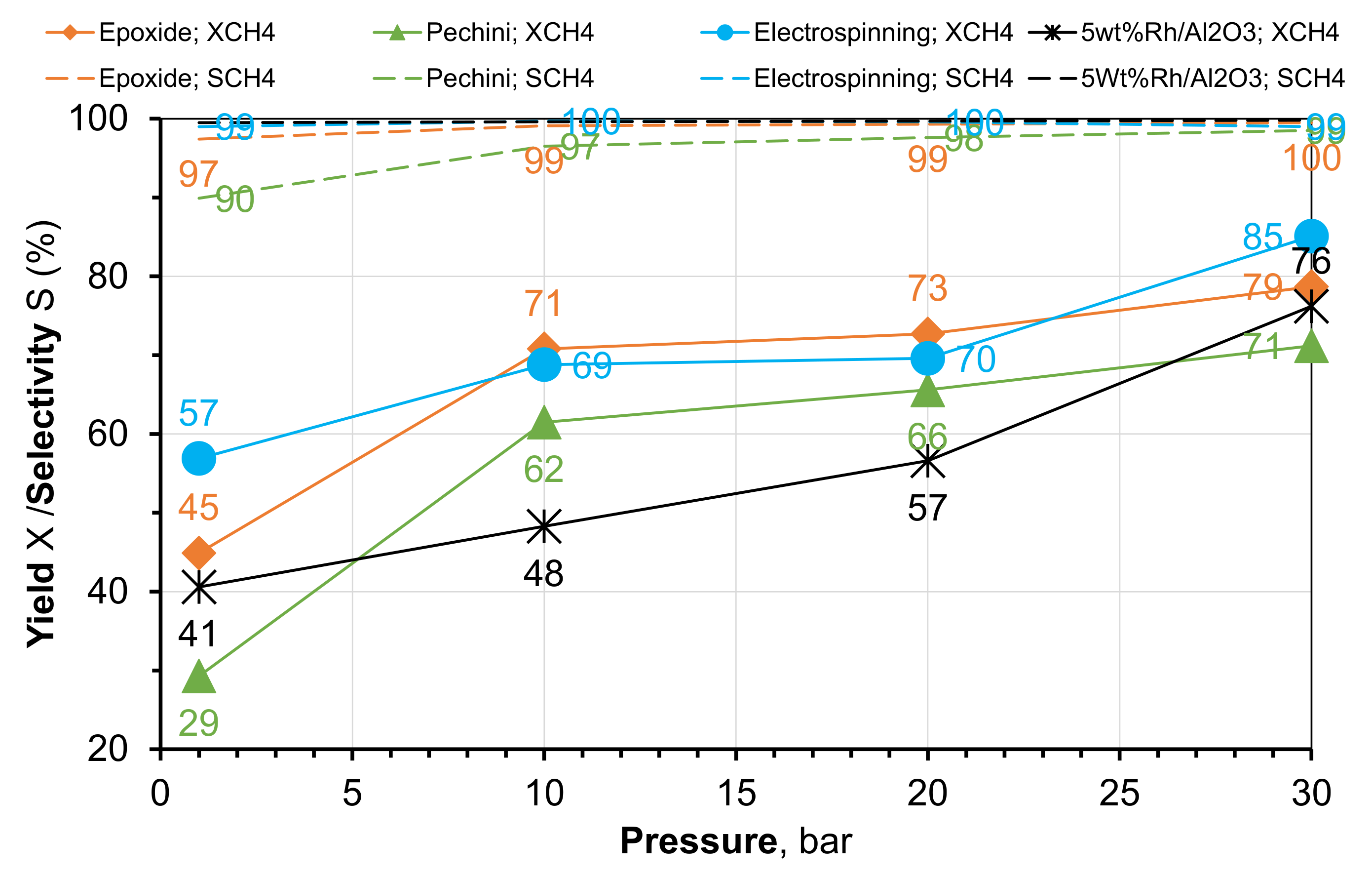
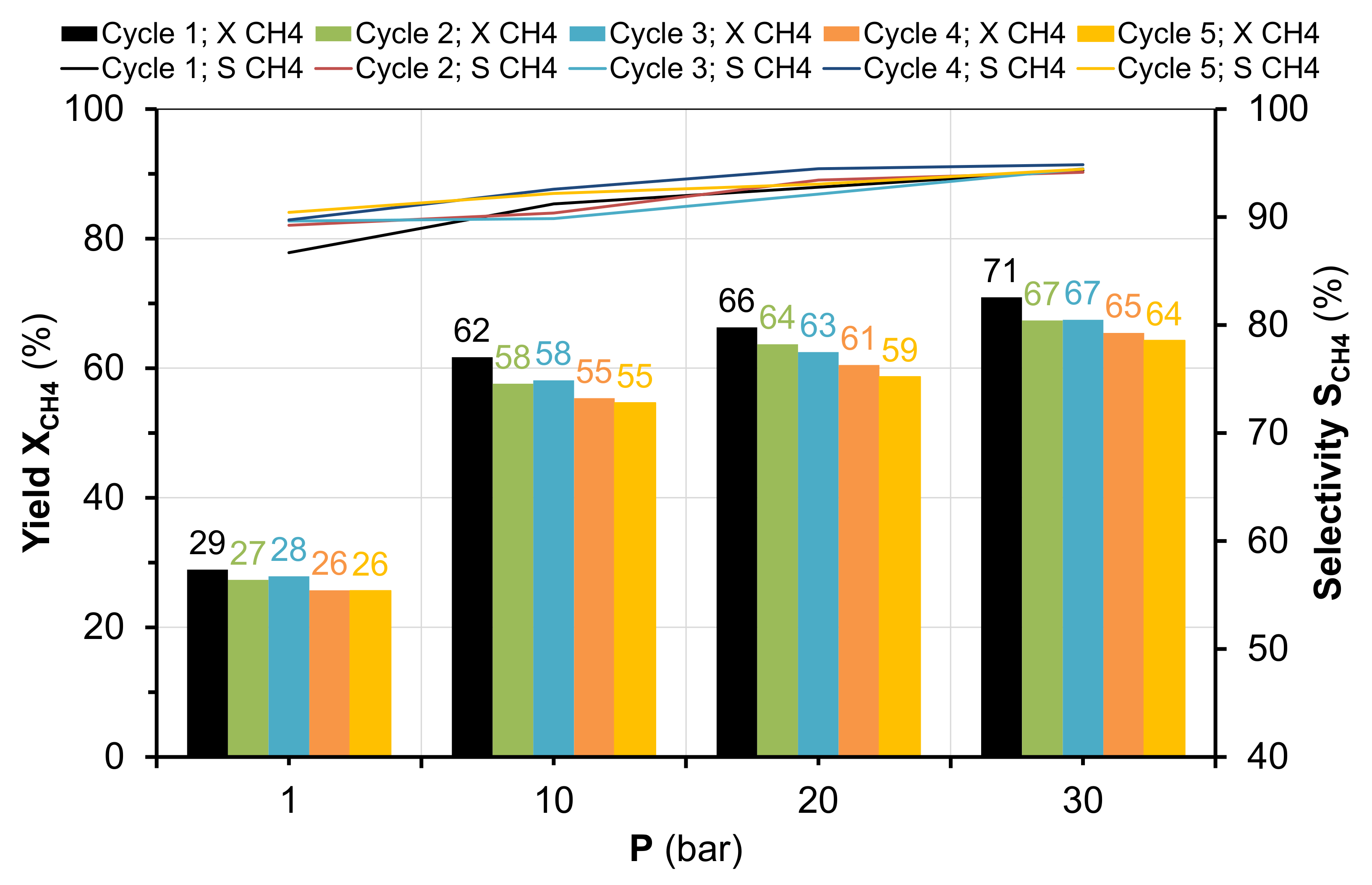
2.2. Methanation of CO2 under Pressure
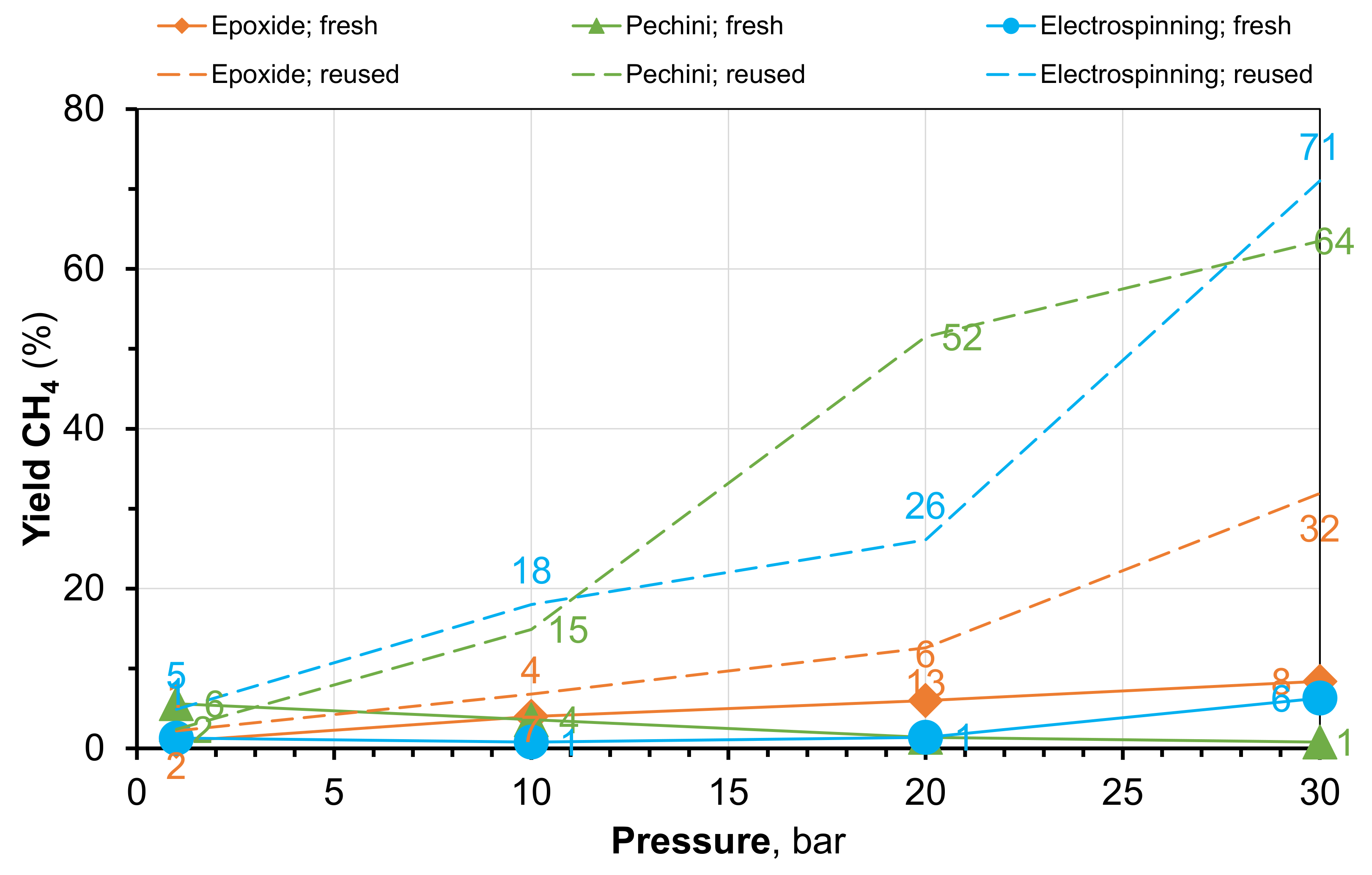
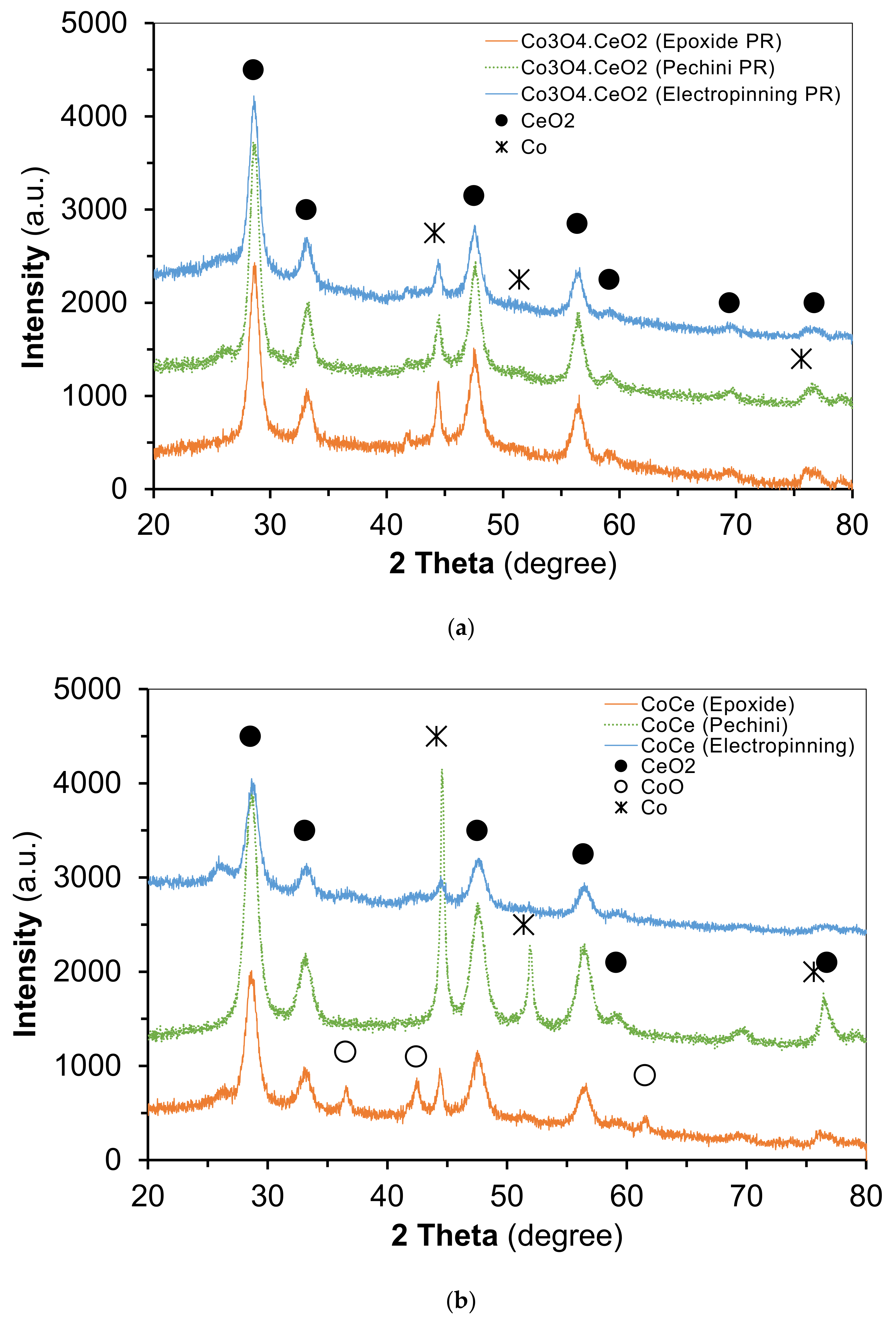
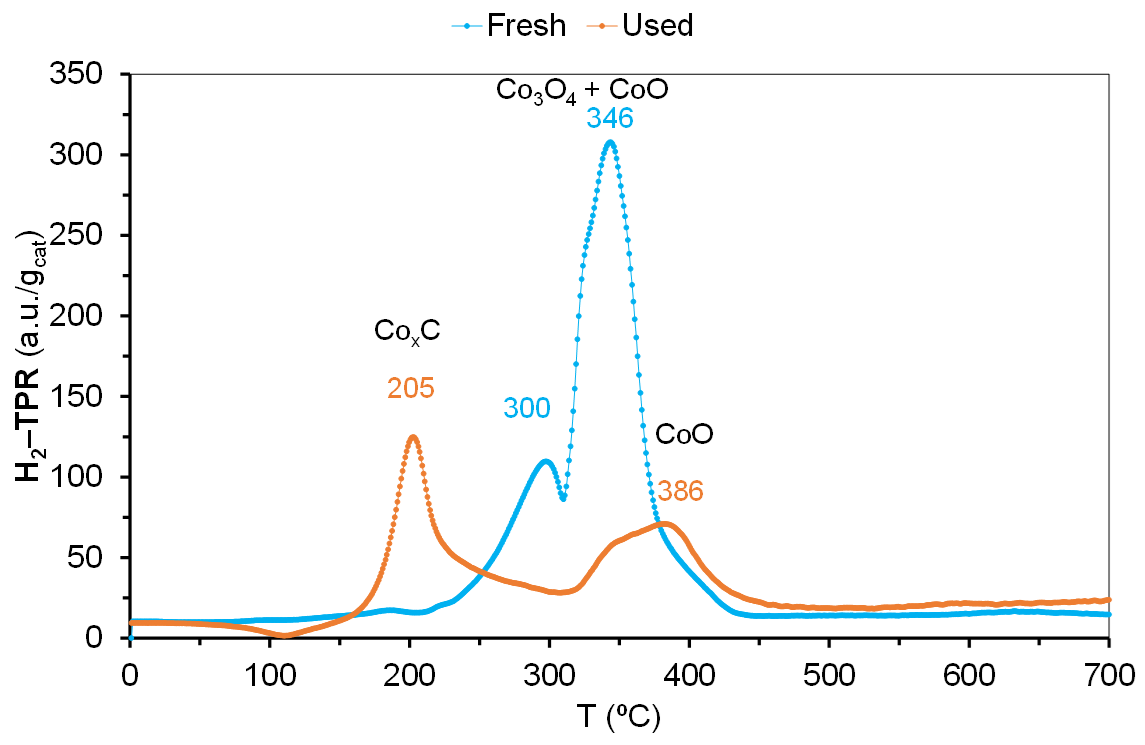
2.3. Characterization of Catalysts after Catalytic Reaction
2.4. Kinetic Considerations
3. Materials and Methods
3.1. Synthesis and Characterization
3.2. Catalytic Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ashok, J.; Pati, S.; Hongmanorom, P.; Tianxi, Z.; Junmei, C.; Kawi, S. A review of recent catalyst advances in CO2 methanation processes. Catal. Today 2020, 356, 471–489. [Google Scholar] [CrossRef]
- Thema, M.; Bauer, F.; Sterner, M. Power-to-Gas: Electrolysis and methanation status review. Renew. Sustain. Energ. Rev. 2019, 112, 775–787. [Google Scholar] [CrossRef]
- Gotz, M.; Lefebvre, J.; Mors, F.; Koch, A.M.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable Power-to-Gas: A technological and economic review. Renew. Energ. 2016, 85, 1371–1390. [Google Scholar] [CrossRef] [Green Version]
- Melaet, G.; Ralston, W.T.; Li, C.S.; Alayoglu, S.; An, K.; Musselwhite, N.; Kalkan, B.; Somorjai, G.A. Evidence of Highly Active Cobalt Oxide Catalyst for the Fischer-Tropsch Synthesis and CO2 Hydrogenation. J. Am. Chem. Soc. 2014, 136, 2260–2263. [Google Scholar] [CrossRef]
- Pan, Q.S.; Peng, J.X.; Sun, T.J.; Wang, S.; Wang, S.D. Insight into the reaction route of CO2 methanation: Promotion effect of medium basic sites. Catal. Commun. 2014, 45, 74–78. [Google Scholar] [CrossRef]
- Dauenhauer, P.J.; Dreyer, B.J.; Degenstein, N.J.; Schmidt, L.D. Millisecond reforming of solid biomass for sustainable fuels. Angew. Chem. Int. Edit. 2007, 46, 5864–5867. [Google Scholar] [CrossRef] [Green Version]
- Bacariza, M.C.; Spataru, D.; Karam, L.; Lopes, J.M.; Henriques, C. Promising Catalytic Systems for CO2 Hydrogenation into CH4: A Review of Recent Studies. Processes 2020, 8, 1646. [Google Scholar] [CrossRef]
- Sreedhar, I.; Varun, Y.; Singh, S.A.; Venugopal, A.; Reddy, B.M. Developmental trends in CO2 methanation using various catalysts. Catal. Sci. Technol. 2019, 9, 4478–4504. [Google Scholar] [CrossRef]
- Janlamool, J.; Praserthdam, P.; Jongsomjit, B. Ti-Si composite oxide-supported cobalt catalysts for CO2 hydrogenation. J. Nat. Gas Chem. 2011, 20, 558–564. [Google Scholar] [CrossRef]
- Branco, J.B.; da Silva, R.P.; Ferreira, A.C. Methanation of CO2 over Cobalt-Lanthanide Aerogels: Effect of Calcination Temperature. Catalysts 2020, 10, 704. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. CO2 Reduction on Supported Ru/Al2O3 Catalysts: Cluster Size Dependence of Product Selectivity. ACS Catal. 2013, 3, 2449–2455. [Google Scholar] [CrossRef]
- Beuls, A.; Swalus, C.; Jacquemin, M.; Heyen, G.; Karelovic, A.; Ruiz, P. Methanation of CO2: Further insight into the mechanism over Rh/gamma-Al2O3 catalyst. Appl. Catal. B-Environ. 2012, 113, 2–10. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. Heterogeneous Catalysis on Atomically Dispersed Supported Metals: CO2 Reduction on Multifunctional Pd Catalysts. ACS Catal. 2013, 3, 2094–2100. [Google Scholar] [CrossRef]
- Ocampo, F.; Louis, B.; Kiwi-Minsker, L.; Roger, A.C. Effect of Ce/Zr composition and noble metal promotion on nickel based CexZr1-xO2 catalysts for carbon dioxide methanation. Appl. Catal. A-Gen. 2011, 392, 36–44. [Google Scholar] [CrossRef]
- Massa, F.; Coppola, A.; Scala, F. A thermodynamic study of sorption-enhanced CO2 methanation at low pressure. J. CO2 Util. 2020, 35, 176–184. [Google Scholar] [CrossRef]
- Gao, J.J.; Wang, Y.L.; Ping, Y.; Hu, D.C.; Xu, G.W.; Gu, F.N.; Su, F.B. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Skrzypek, J.; Lachowska, M.; Grzesik, M.; Sloczynski, J.; Nowak, P. Thermodynamics and Kinetics of Low-Pressure Methanol Synthesis. Chem. Eng. J. Biochem. Eng. 1995, 58, 101–108. [Google Scholar] [CrossRef]
- Gaikwad, R.; Bansode, A.; Urakawa, A. High-pressure advantages in stoichiometric hydrogenation of carbon dioxide to methanol. J. Catal. 2016, 343, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.B.; Højlund Nielsen, P.E. Methanol Synthesis. In Handbook of Heterogeneous Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- BASF, German Patent n° 415 686, 441 433 and 462 837, 1923. Available online: https://patents.google.com/patent/WO2017140800A1/en (accessed on 27 December 2021).
- Bahmanpour, A.M.; Signorile, M.; Krocher, O. Recent progress in syngas production via catalytic CO2 hydrogenation reaction. Appl. Catal. B-Environ. 2021, 295, 120319. [Google Scholar] [CrossRef]
- Ciesielski, R.; Shtyka, O.; Zakrzewski, M.; Kubicki, J.; Maniukiewicz, W.; Kedziora, A.; Maniecki, T.P. Mechanistic Studies of Methanol Synthesis Reaction over Cu and Pd-Cu Catalysts. Kinet. Catal. 2020, 61, 623–630. [Google Scholar] [CrossRef]
- Ipatieff, V.N.; Monroe, G.S. Synthesis of Methanol from Carbon Dioxide and Hydrogen over Copper Alumina Catalysts—Mechanism of Reaction. J. Am. Chem. Soc. 1945, 67, 2168–2171. [Google Scholar] [CrossRef]
- Luna, M.L.; Timoshenko, J.; Kordus, D.; Rettenmaier, C.; Chee, S.W.; Hoffman, A.S.; Bare, S.R.; Shaikhutdinov, S.; Cuenya, B.R. Role of the Oxide Support on the Structural and Chemical Evolution of Fe Catalysts during the Hydrogenation of CO2. ACS Catal. 2021, 11, 6175–6185. [Google Scholar] [CrossRef]
- Franken, T.; Heel, A. Are Fe based catalysts an upcoming alternative to Ni in CO2 methanation at elevated pressure? J. CO2 Util. 2020, 39, 101175. [Google Scholar] [CrossRef]
- Li, W.H.; Liu, Y.; Mu, M.C.; Ding, F.S.; Liu, Z.M.; Guo, X.W.; Song, C.S. Organic acid-assisted preparation of highly dispersed Co/ZrO2 catalysts with superior activity for CO2 methanation. Appl. Catal. B-Environ. 2019, 254, 531–540. [Google Scholar] [CrossRef]
- Zhou, Y.W.; Jiang, Y.X.; Qin, Z.Z.; Xie, Q.R.; Ji, H.B. Influence of Zr, Ce, and La on Co3O4 catalyst for CO2 methanation at low temperature. Chin. J. Chem. Eng. 2018, 26, 768–774. [Google Scholar] [CrossRef]
- Jimenez, J.D.; Wen, C.; Lauterbach, J. Design of highly active cobalt catalysts for CO2 hydrogenation via the tailoring of surface orientation of nanostructures. Catal. Sci. Technol. 2019, 9, 1970–1978. [Google Scholar] [CrossRef]
- Li, W.H.; Nie, X.W.; Jiang, X.; Zhang, A.F.; Ding, F.S.; Liu, M.; Liu, Z.M.; Guo, X.W.; Song, C.S. ZrO2 support imparts superior activity and stability of Co catalysts for CO2 methanation. Appl. Catal. B-Environ. 2018, 220, 397–408. [Google Scholar] [CrossRef]
- Jampaiah, D.; Damma, D.; Chalkidis, A.; Venkataswamy, P.; Bhargava, S.K.; Reddy, B.M. MOF-derived ceria-zirconia supported Co3O4 catalysts with enhanced activity in CO2 methanation. Catal. Today 2020, 356, 519–526. [Google Scholar] [CrossRef]
- Branco, J.B.; Ferreira, A.C.; Vieira, F.; Martinho, J.F. Cerium-Based Bimetallic Oxides as Catalysts for the Methanation of CO2: Influence of the Preparation Method. Energy Fuels 2021, 35, 6725–6737. [Google Scholar] [CrossRef]
- Branco, J.B.; Ferreira, A.C.; Goncalves, A.P.; Soares, C.O.; Gasche, T.A. Synthesis of methanol using copper-f block element bimetallic oxides as catalysts and greenhouse gases (CO2, CH4) as feedstock. J. Catal. 2016, 341, 24–32. [Google Scholar] [CrossRef]
- Li, Z.H.; Si, M.Y.; Xin, L.; Liu, R.J.; Liu, R.X.; Lu, J. Cobalt catalysts for Fischer-Tropsch synthesis: The effect of support, precipitant and pH value. Chin. J. Chem. Eng. 2018, 26, 747–752. [Google Scholar] [CrossRef]
- Zhao, K.; Calizzi, M.; Moioli, E.; Li, M.; Borsay, A.; Lombardo, L.; Mutschler, R.; Luo, W.; Zuttel, A. Unraveling and optimizing the metal-metal oxide synergistic effect in a highly active CoxCoO1-x catalyst for CO2 hydrogenation. J. Energy Chem. 2021, 53, 241–250. [Google Scholar] [CrossRef]
- Puga, A.V. On the nature of active phases and sites in CO and CO2 hydrogenation catalysts. Catal. Sci. Technol. 2018, 8, 5681–5707. [Google Scholar] [CrossRef]
- Manukyan, K.V.; Avetisyan, A.G.; Shuck, C.E.; Chatilyan, H.A.; Rouvimov, S.; Kharatyan, S.L.; Mukasyan, A.S. Nickel Oxide Reduction by Hydrogen: Kinetics and Structural Transformations. J. Phys. Chem. C 2015, 119, 16131–16138. [Google Scholar] [CrossRef]
- Tomic-Tucakovic, B.; Majstorovic, D.; Jelic, D.; Mentus, S. Thermogravimetric study of the kinetics of Co3O4 reduction by hydrogen. Thermochim. Acta 2012, 541, 15–24. [Google Scholar] [CrossRef]
- Lin, Q.; Liu, B.; Jiang, F.; Fang, X.J.; Xu, Y.B.; Liu, X.H. Assessing the formation of cobalt carbide and its catalytic performance under realistic reaction conditions and tuning product selectivity in a cobalt-based FTS reaction. Catal. Sci. Technol. 2019, 9, 3238–3258. [Google Scholar] [CrossRef]
- Alotaibi, N.; Hammud, H.H.; Karnati, R.K.; Hussain, S.G.; Mazher, J.; Prakasam, T. Cobalt-carbon/silica nanocomposites prepared by pyrolysis of a cobalt 2,2′-bipyridine terephthalate complex for remediation of cationic dyes. RSC Adv. 2020, 10, 17660–17672. [Google Scholar] [CrossRef]
- Kwak, G.; Kim, D.E.; Kim, Y.T.; Park, H.G.; Kang, S.C.; Ha, K.S.; Jun, K.W.; Lee, Y.J. Enhanced catalytic activity of cobalt catalysts for Fischer-Tropsch synthesis via carburization and hydrogenation and its application to regeneration. Catal. Sci. Technol. 2016, 6, 4594–4600. [Google Scholar] [CrossRef]
- Choi, Y.I.; Yang, J.H.; Park, S.J.; Sohn, Y. Energy Storage and CO2 Reduction Performances of Co/Co2C/C Prepared by an Anaerobic Ethanol Oxidation Reaction Using Sacrificial SnO2. Catalysts 2020, 10, 1116. [Google Scholar] [CrossRef]
- Dai, E.G.; Xu, J.; Qiu, J.J.; Liu, S.C.; Chen, P.; Liu, Y. Co@Carbon and Co3O4@Carbon nanocomposites derived from a single MOF for supercapacitors. Sci. Rep. 2017, 7, 12588. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kawashima, K.; Wygant, B.R.; Mabayoje, O.; Liu, Y.; Wang, J.H.; Mullins, C.B. Transformation of a Cobalt Carbide (Co3C) Oxygen Evolution Precatalyst. Acs Appl Energ. Mater 2018, 1, 5145–5150. [Google Scholar] [CrossRef]
- Kim, M.; Nam, D.H.; Park, H.Y.; Kwon, C.; Eom, K.; Yoo, S.; Jang, J.; Kim, H.J.; Cho, E.; Kwon, H. Cobalt-carbon nanofibers as an efficient support-free catalyst for oxygen reduction reaction with a systematic study of active site formation. J. Mater Chem. A 2015, 3, 14284–14290. [Google Scholar] [CrossRef]
- Schild, C.; Wokaun, A.; Baiker, A. On the Mechanism of CO and CO2 Hydrogenation Reactions on Zirconia-Supported Catalysts—A Diffuse Reflectance FTIR Study.1. Identification of Surface Species and Methanation Reactions on Palladium Zirconia Catalysts. J. Mol. Catal. 1990, 63, 223–242. [Google Scholar] [CrossRef]
- Bui, M.; Adjiman, C.S.; Bardow, A.; Anthony, E.J.; Boston, A.; Brown, S.; Fennell, P.S.; Fuss, S.; Galindo, A.; Hackett, L.A.; et al. Carbon capture and storage (CCS): The way forward. Energ. Environ. Sci. 2018, 11, 1062–1176. [Google Scholar] [CrossRef] [Green Version]
- Lapidus, A.L.; Gaidai, N.A.; Nekrasov, N.V.; Tishkova, L.A.; Agafonov, Y.A.; Myshenkova, T.N. The mechanism of carbon dioxide hydrogenation on copper and nickel catalysts. Pet. Chem. 2007, 47, 75–82. [Google Scholar] [CrossRef]
- Miguel, V.; Mendes, A.; Madeira, L.M. Intrinsic kinetics of CO2 methanation over an industrial nickel-based catalyst. J. CO2 Util. 2018, 25, 128–136. [Google Scholar] [CrossRef]
- Koytsoumpa, E.I.; Karellas, S. Equilibrium and kinetic aspects for catalytic methanation focusing on CO2 derived Substitute Natural Gas (SNG). Renew. Sustain. Energ. Rev. 2018, 94, 536–550. [Google Scholar] [CrossRef]
- Dew, J.N.; White, R.R.; Sliepcevich, C.M. Hydrogenation of Carbon Dioxide on Nickel-Kieselguhr Catalyst. Ind. Eng. Chem. 1955, 47, 140–146. [Google Scholar] [CrossRef]
- Chiang, J.H.; Hopper, J.R. Kinetics of the Hydrogenation of Carbon-Dioxide over Supported Nickel. Ind. Eng. Chem. Prod. Res. Dev. 1983, 22, 225–228. [Google Scholar] [CrossRef]
- Herwijnen, T.V.; Doesburg, H.V.; Dejong, W.A. Kinetics of Methanation of CO and CO2 on a Nickel-Catalyst. J. Catal. 1973, 28, 391–402. [Google Scholar] [CrossRef]
- JCPDS. The Powder Diffraction File; JCPDS: Swarthmore, PA, USA, 1981. [Google Scholar]
- Oyama, S.T.; Zhang, X.M.; Lu, J.Q.; Gu, Y.F.; Fujitani, T. Epoxidation of propylene with H2 and O2 in the explosive regime in a packed-bed catalytic membrane reactor. J. Catal. 2008, 257, 1–4. [Google Scholar] [CrossRef]
- Froment, G.F.; Bischoff, K.B. Chemical reactor Anaylsis and Design; John Wiley & Sons: Hoboken, NJ, USA, 1979. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preparation Method | Surface Area a | H2-TPR | Basicity | EDS (Atomic Ratio | ||
|---|---|---|---|---|---|---|
| m2/g | m2/gNi or Co | Tm (°C) | molH2/molcat b | υA/υP c | Ni or Co/Ce) d | |
| 3NiO.CeO2 | ||||||
| Epoxide addition | 42.5 | 0.57 | 330 | 3.0 | 36.6 | 2.8 |
| Pechini | 46.8 | 3.25 | 179, 374, 733 | 2.9 | 146.4 | 2.7 |
| Electrospinning | 73.6 | 1.17 | 336, 697 | 3.0 | 252.9 | 2.9 |
| Co3O4.CeO2 | ||||||
| Epoxide addition | 29.5 | 0.84 | 341, 382, 655 | 3.9 | 244.0 | 2.8 |
| Pechini | 44.2 | 0.78 | 286, 434, 714 | 3.8 | 370.0 | 2.8 |
| Electrospinning | 58.2 | 1.15 | 300, 356, 683 | 4.1 | 717.0 | 3.0 |
| Catalyst | Conditions (T °C, P Bar, GHSV mL/g.h) | Yield CH4 (%) a | Sel CH4 (%) a | Sel CO (%) a | Ref. |
|---|---|---|---|---|---|
| 3NiO.CeO2 Epoxide | 300, 30, 15,000 | 78.7 (76.5) | 99.5 (99.8) | 0.5 (0.2) | This work |
| 3NiO.CeO2 Pechini | 300, 30, 15,000 | 71.2 (74.9) | 98.5 (99.8) | 0.9 (0.2) | This work |
| 3NiO.CeO2 Electrospinning | 300, 30, 15,000 | 85.1 (90.0) | 99 (98.4) | 1 (1.6) | This work |
| Co3O4.CeO2 Epoxide | 300, 30, 15,000 | 24.3 (31.9) | 81.9 (85.9) | 7.7 (14.1) | This work |
| Co3O4.CeO2 Pechini | 300, 30, 15,000 | 46.0 (63.5) | 86.5 (96.3) | 5.6 83.7) | This work |
| Co3O4.CeO2 Electrospinning | 300, 30, 15,000 | 46.3 (71.0) | 84.5 (97.4) | 5.3 (2.6) | This work |
| 5wt.% Rh/Al2O3 | 300, 30, 15,000 | 86.2 | 99.8 | 0.2 | This work |
| Co/Ce0.8Zr0.2O2 | 320, 15, 15,000 | 80.4 | 99 | 1 | [30] |
| Co/ZrO2 | 400, 30, 7200 | 84.2 | 99 | 1 | [26] |
| Co/ZrO2 | 400, 30, 3600 | 92.4 | 99.9 | 0.1 | [29] |
| Co nanorods | 300, 10, 18,000 | 78.4 | 98 | 2 | [28] |
| Zr-Co3O4 | 200, 5, 18,000 | 58.2 | 100 | 0 | [27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Branco, J.M.B.; Ferreira, A.C.; Martinho, J.F. Cerium d-Block Element (Co, Ni) Bimetallic Oxides as Catalysts for the Methanation of CO2: Effect of Pressure. Catalysts 2022, 12, 44. https://doi.org/10.3390/catal12010044
Branco JMB, Ferreira AC, Martinho JF. Cerium d-Block Element (Co, Ni) Bimetallic Oxides as Catalysts for the Methanation of CO2: Effect of Pressure. Catalysts. 2022; 12(1):44. https://doi.org/10.3390/catal12010044
Chicago/Turabian StyleBranco, Joaquim Miguel Badalo, Ana Cristina Ferreira, and Joana Filipa Martinho. 2022. "Cerium d-Block Element (Co, Ni) Bimetallic Oxides as Catalysts for the Methanation of CO2: Effect of Pressure" Catalysts 12, no. 1: 44. https://doi.org/10.3390/catal12010044
APA StyleBranco, J. M. B., Ferreira, A. C., & Martinho, J. F. (2022). Cerium d-Block Element (Co, Ni) Bimetallic Oxides as Catalysts for the Methanation of CO2: Effect of Pressure. Catalysts, 12(1), 44. https://doi.org/10.3390/catal12010044