
Preparation of Extremely Active Ethylene Tetramerization Catalyst [iPrN(PAr2)2−CrCl2]+[B(C6F5)4]– (Ar = −C6H4-p-SiR3)

 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
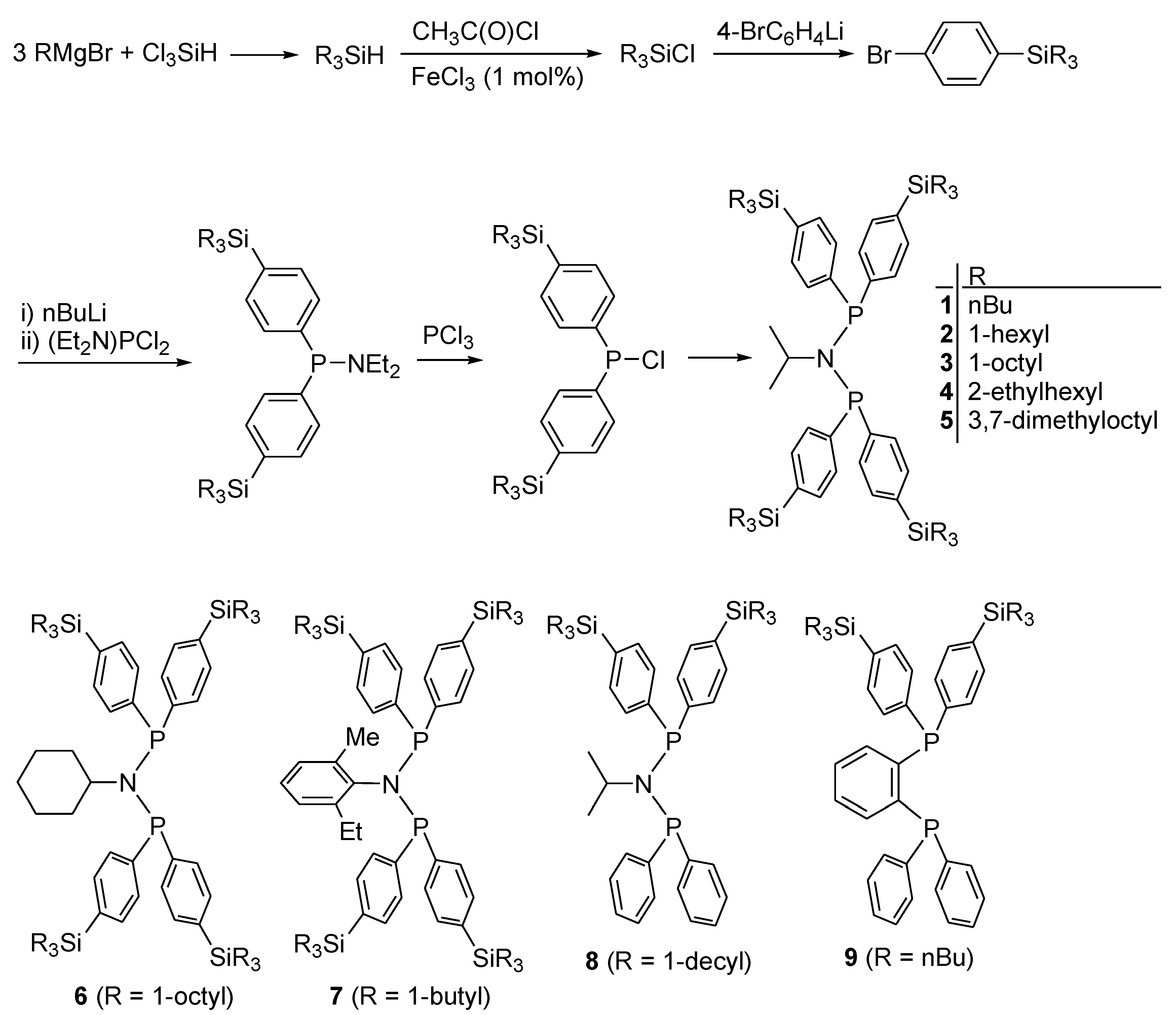
2.1. Preparation of Ligands
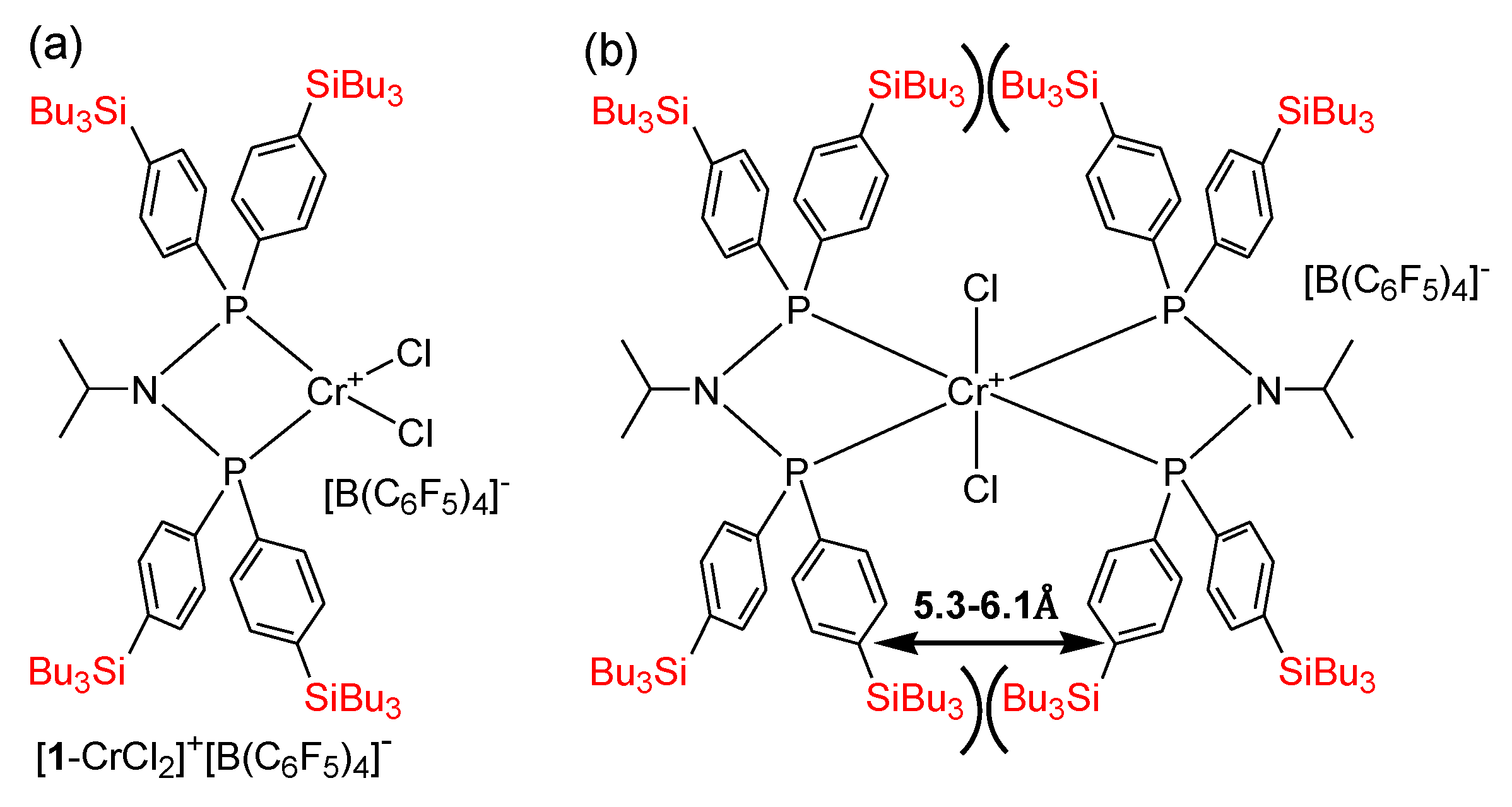
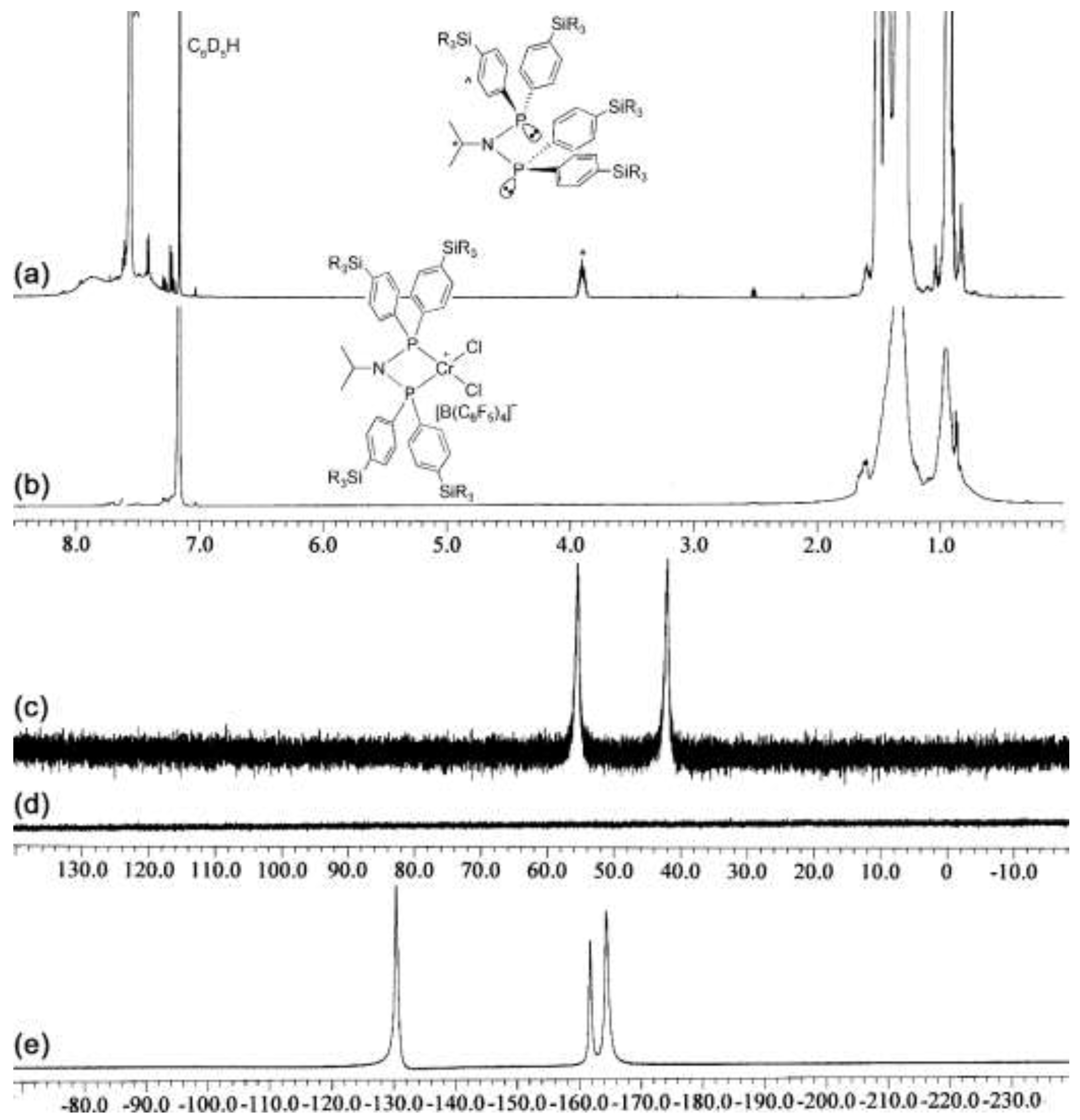
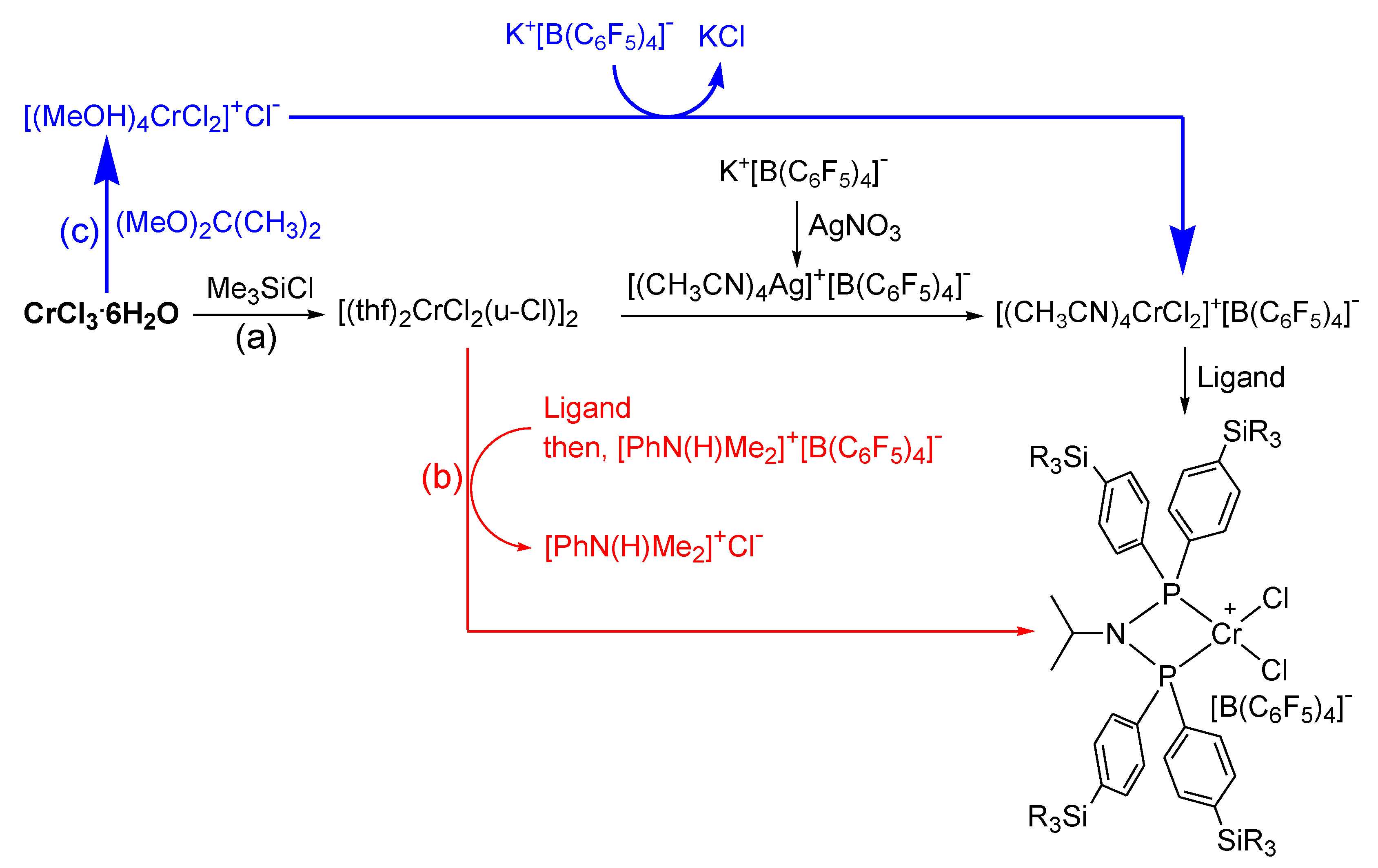
2.2. Preparation of Chromium Complexes
2.3. Oligomerization Studies
3. Experimental Section
3.1. General Remarks
3.2. (1-Octyl)3SiCl
3.3. BrC6H4-p-Si(1-octyl)3
3.4. Et2NP[C6H4-p-Si(1-octyl)3]2
3.5. ClP[C6H4-p-Si(1-octyl)3]2
3.6. iPrN(PAr2)2 (Ar = −C6H4-p-Si(1-octyl)3)
3.7. Preparation of [iPrN(PAr2)2−CrCl2]+[B(C6F5)4]– Using [CrCl2(NCCH3)4]+[B(C6F5)4]–
3.8. Attempt to Prepare [iPrN(PAr2)2−CrCl2]+[B(C6F5)4]– Using [PhN(H)Me2]+[B(C6F5)4]– and [CrCl2(μ-Cl)(thf)2]2
3.9. [CrCl2(NCCH3)4]+[B(C6F5)4]–
3.10. Ethylene Tetramerization
4. Conclusions
5. Patent
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- De Klerk, A. Contributions of Burtron H. Davis to fischer-tropsch refining catalysis: Dehydration as applied to processes for 1-octene production. Top. Catal. 2014, 57, 715–722. [Google Scholar] [CrossRef]
- Bohley, R.C.; Jacobsen, G.B.; Pelt, H.L.; Schaart, B.J.; Schenk, M.; van Oeffelen, D.A.G. Process for Producing 1-Octene. W.O. Patent 1992010450A1, 25 June 1992. [Google Scholar]
- Klinkenberg, J.L.; Lawry, K.P. Sterically Encumbered and Poorly Electron-Donating Oxaphosphaadamantane Ligands for the Pd-Catalyzed Telomerization of Butadiene with Methanol. Org. Process Res. Dev. 2019, 23, 1654–1658. [Google Scholar] [CrossRef]
- Keim, W. Oligomerization of Ethylene to α-Olefins: Discovery and Development of the Shell Higher Olefin Process (SHOP). Angew. Chem. Int. Ed. 2013, 52, 12492–12496. [Google Scholar] [CrossRef]
- Meiswinkel, A.; Wöhl, A.; Müller, W.; Bölt, H.V.; Mosa, F.M.; Al-Hazmi, M.H. Developing linear-alpha-olefins technology—From laboratory to a commercial plant. Oil Gas Eur. Mag. 2012, 38, 103–106. [Google Scholar]
- Young, C.T.; von Goetze, R.; Tomov, A.K.; Zaccaria, F.; Britovsek, G.J.P. The Mathematics of Ethylene Oligomerisation and Polymerisation. Top. Catal. 2020, 63, 294–318. [Google Scholar] [CrossRef] [Green Version]
- Overett, M.J.; Blann, K.; Bollmann, A.; Dixon, J.T.; Haasbroek, D.; Killian, E.; Maumela, H.; McGuinness, D.S.; Morgan, D.H. Mechanistic Investigations of the Ethylene Tetramerisation Reaction. J. Am. Chem. Soc. 2005, 127, 10723–10730. [Google Scholar] [CrossRef]
- Bollmann, A.; Blann, K.; Dixon, J.T.; Hess, F.M.; Killian, E.; Maumela, H.; McGuinness, D.S.; Morgan, D.H.; Neveling, A.; Otto, S.; et al. Ethylene Tetramerization: A New Route to Produce 1-Octene in Exceptionally High Selectivities. J. Am. Chem. Soc. 2004, 126, 14712–14713. [Google Scholar] [CrossRef]
- Kuhlmann, S.; Blann, K.; Bollmann, A.; Dixon, J.T.; Killian, E.; Maumela, M.C.; Maumela, H.; Morgan, D.H.; Prétorius, M.; Taccardi, N.; et al. N-substituted diphosphinoamines: Toward rational ligand design for the efficient tetramerization of ethylene. J. Catal. 2007, 245, 279–284. [Google Scholar] [CrossRef]
- Dixon, J.T.; Green, M.J.; Hess, F.M.; Morgan, D.H. Advances in selective ethylene trimerisation—A critical overview. J. Organomet. Chem. 2004, 689, 3641–3668. [Google Scholar] [CrossRef]
- Tang, S.; Liu, Z.; Yan, X.; Li, N.; Cheng, R.; He, X.; Liu, B. Kinetic studies on the pyrrole–Cr-based Chevron-Phillips ethylene trimerization catalyst system. Appl. Catal. A 2014, 481, 39–48. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Park, D.S.; Lee, D.H.; Eo, S.C.; Park, S.Y.; Jeong, M.S.; Kang, Y.Y.; Lee, J.; Lee, B.Y. A chromium precursor for the Phillips ethylene trimerization catalyst: (2-ethylhexanoate)2CrOH. Dalton Trans. 2015, 44, 11004–11012. [Google Scholar] [CrossRef]
- Alferov, K.A.; Belov, G.P.; Meng, Y. Chromium catalysts for selective ethylene oligomerization to 1-hexene and 1-octene: Recent results. Appl. Catal. A Gen. 2017, 542, 71–124. [Google Scholar] [CrossRef]
- Sydora, O.L. Selective Ethylene Oligomerization. Organometallics 2019, 38, 997–1010. [Google Scholar] [CrossRef]
- Bariashir, C.; Huang, C.; Solan, G.A.; Sun, W.H. Recent advances in homogeneous chromium catalyst design for ethylene tri-, tetra-, oligo- and polymerization. Coordin. Chem. Rev. 2019, 385, 208–229. [Google Scholar] [CrossRef] [Green Version]
- McGuinness, D.S.; Overett, M.; Tooze, R.P.; Blann, K.; Dixon, J.T.; Slawin, A.M.Z. Ethylene Tri- and Tetramerization with Borate Cocatalysts: Effects on Activity, Selectivity, and Catalyst Degradation Pathways. Organometallics 2007, 26, 1108–1111. [Google Scholar] [CrossRef]
- McGuinness, D.S.; Rucklidge, A.J.; Tooze, R.P.; Slawin, A.M.Z. Cocatalyst Influence in Selective Oligomerization: Effect on Activity, Catalyst Stability, and 1-Hexene/1-Octene Selectivity in the Ethylene Trimerization and Tetramerization Reaction. Organometallics 2007, 26, 2561–2569. [Google Scholar] [CrossRef]
- Rucklidge, A.J.; McGuinness, D.S.; Tooze, R.P.; Slawin, A.M.Z.; Pelletier, J.D.A.; Hanton, M.J.; Webb, P.B. Ethylene Tetramerization with Cationic Chromium(I) Complexes. Organometallics 2007, 26, 2782–2787. [Google Scholar] [CrossRef]
- Hanton, M.J.; Tenza, K. Bis(imino)pyridine Complexes of the First-Row Transition Metals: Alternative Methods of Activation. Organometallics 2008, 27, 5712–5716. [Google Scholar] [CrossRef]
- Stennett, T.E.; Haddow, M.F.; Wass, D.F. Avoiding MAO: Alternative Activation Methods in Selective Ethylene Oligomerization. Organometallics 2012, 31, 6960–6965. [Google Scholar] [CrossRef]
- Hirscher, N.A.; Agapie, T. Stoichiometrically Activated Catalysts for Ethylene Tetramerization using Diphosphinoamine-Ligated Cr Tris(hydrocarbyl) Complexes. Organometallics 2017, 36, 4107–4110. [Google Scholar] [CrossRef] [Green Version]
- Hirscher, N.A.; Perez Sierra, D.; Agapie, T. Robust Chromium Precursors for Catalysis: Isolation and Structure of a Single-Component Ethylene Tetramerization Precatalyst. J. Am. Chem. Soc. 2019, 141, 6022–6029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirscher, N.A.; Arnett, C.H.; Oyala, P.H.; Agapie, T. Characterization of Cr-Hydrocarbyl Species via Pulse EPR in the Study of Ethylene Tetramerization Catalysis. Organometallics 2020, 39, 4420–4429. [Google Scholar] [CrossRef]
- Lee, H.-J.; Baek, J.-W.; Seo, Y.-H.; Lee, H.-C.; Jeong, S.-M.; Lee, J.; Lee, C.-G.; Lee, B.-Y. Preparation of High-Purity Ammonium Tetrakis(pentafluorophenyl)borate for the Activation of Olefin Polymerization Catalysts. Molecules 2021, 26, 2827. [Google Scholar] [CrossRef] [PubMed]
- Ewart, S.W.; Kolthammer, B.W.S.; Smith, D.M.; Hanton, M.J.; Dixon, J.T.; Morgan, D.H.; Debod, H.; Gabrielli, W.F.; Evans, S.J. Oligomerization of Olefinic Compounds in the Presence of an Activated Oligomerization Catalyst. W.O. Patent 2010092554A1, 19 August 2010. [Google Scholar]
- Kim, E.H.; Lee, H.M.; Jeong, M.S.; Ryu, J.Y.; Lee, J.; Lee, B.Y. Methylaluminoxane-Free Chromium Catalytic System for Ethylene Tetramerization. ACS Omega 2017, 2, 765–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.H.; Lee, H.M.; Park, H.S.; Kim, S.D.; Kwon, S.J.; Tahara, A.; Nagashima, H.; Lee, B.Y. MAO-free and extremely active catalytic system for ethylene tetramerization. Appl. Organomet. Chem. 2019, 33, e4829. [Google Scholar] [CrossRef]
- Park, H.S.; Kim, T.H.; Baek, J.W.; Lee, H.J.; Kim, T.J.; Ryu, J.Y.; Lee, J.; Lee, B.Y. Extremely Active Ethylene Tetramerization Catalyst Avoiding the Use of Methylaluminoxane: [iPrN{P(C6H4-p-SiR3)2}2CrCl2]+[B(C6F5)4]–. ChemCatChem 2019, 11, 4351–4359. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, L.; Ma, X.; Liu, Y.; Mi, P.; Liu, Z.; Zhang, J. Effect of an additional donor on decene formation in ethylene oligomerization catalyzed by a Cr/PCCP system: A combined experimental and DFT study. Catal. Sci. Technol. 2021, 11, 4596–4604. [Google Scholar] [CrossRef]
- Ogawa, T.; Lindeperg, F.; Stradiotto, M.; Turculet, L.; Sydora, O.L. Chromium N-phosphinoamidine ethylene tri-/tetramerization catalysts: Designing a step change in 1-octene selectivity. J. Catal. 2021, 394, 444–450. [Google Scholar] [CrossRef]
- Makume, B.F.; Holzapfel, C.W.; Maumela, M.C.; Willemse, J.A.; van den Berg, J.A. Ethylene Tetramerisation: A Structure-Selectivity Correlation. ChemPlusChem 2020, 85, 2308–2315. [Google Scholar] [CrossRef]
- Rosenthal, U. PNPN-H in Comparison to other PNP, PNPN and NPNPN Ligands for the Chromium Catalyzed Selective Ethylene Oligomerization. ChemCatChem 2020, 12, 41–52. [Google Scholar] [CrossRef]
- Boelter, S.D.; Davies, D.R.; Margl, P.; Milbrandt, K.A.; Mort, D.; Vanchura, B.A.; Wilson, D.R.; Wiltzius, M.; Rosen, M.S.; Klosin, J. Phospholane-Based Ligands for Chromium-Catalyzed Ethylene Tri- and Tetramerization. Organometallics 2020, 39, 976–987. [Google Scholar] [CrossRef]
- Boelter, S.D.; Davies, D.R.; Milbrandt, K.A.; Wilson, D.R.; Wiltzius, M.; Rosen, M.S.; Klosin, J. Evaluation of Bis(phosphine) Ligands for Ethylene Oligomerization: Discovery of Alkyl Phosphines as Effective Ligands for Ethylene Tri- and Tetramerization. Organometallics 2020, 39, 967–975. [Google Scholar] [CrossRef]
- Maley, S.M.; Kwon, D.-H.; Rollins, N.; Stanley, J.C.; Sydora, O.L.; Bischof, S.M.; Ess, D.H. Quantum-mechanical transition-state model combined with machine learning provides catalyst design features for selective Cr olefin oligomerization. Chem. Sci. 2020, 11, 9665–9674. [Google Scholar] [CrossRef]
- Chabbra, S.; Smith, D.M.; Bell, N.L.; Watson, A.J.B.; Bühl, M.; Cole-Hamilton, D.J.; Bode, B.E. First experimental evidence for a bis-ethene chromium(I) complex forming from an activated ethene oligomerization catalyst. Sci. Adv. 2020, 6, eabd7057. [Google Scholar] [CrossRef]
- Müller, T.; Dixon, J.T.; Haumann, M.; Wasserscheid, P. Trimerization and tetramerization of ethylene in continuous gas-phase reaction using a Cr-based supported liquid phase catalyst. React. Chem. Eng. 2019, 4, 131–140. [Google Scholar] [CrossRef]
- Hirscher, N.A.; Labinger, J.A.; Agapie, T. Isotopic labelling in ethylene oligomerization: Addressing the issue of 1-octene vs. 1-hexene selectivity. Dalton Trans. 2019, 48, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Baek, J.W.; Hyun, Y.B.; Lee, H.J.; Lee, J.C.; Bae, S.M.; Seo, Y.H.; Lee, D.G.; Lee, B.Y. Selective Trimerization of α-Olefins with Immobilized Chromium Catalyst for Lubricant Base Oils. Catalysts 2020, 10, 990. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; McGuinness, D.S.; Wierenga, T.S.; Young, C.T. Single- and Double-Coordination Mechanism in Ethylene Tri- and Tetramerization with Cr/PNP Catalysts. ACS Catal. 2015, 5, 4152–4166. [Google Scholar] [CrossRef]
- Do, L.H.; Labinger, J.A.; Bercaw, J.E. Spectral Studies of a Cr(PNP)–MAO System for Selective Ethylene Trimerization Catalysis: Searching for the Active Species. ACS Catal. 2013, 3, 2582–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabeah, J.; Bauer, M.; Baumann, W.; McConnell, A.E.C.; Gabrielli, W.F.; Webb, P.B.; Selent, D.; Brückner, A. Formation, Operation and Deactivation of Cr Catalysts in Ethylene Tetramerization Directly Assessed by Operando EPR and XAS. ACS Catal. 2013, 3, 95–102. [Google Scholar] [CrossRef]
- Savela, R.; Zawartka, W.; Leino, R. Iron-Catalyzed Chlorination of Silanes. Organometallics 2012, 31, 3199–3206. [Google Scholar] [CrossRef]
- He, J.; Waggoner, N.W.; Dunning, S.G.; Steiner, A.; Lynch, V.M.; Humphrey, S.M. A PCP Pincer Ligand for Coordination Polymers with Versatile Chemical Reactivity: Selective Activation of CO2 Gas over CO Gas in the Solid State. Angew. Chem. Int. Ed. 2016, 55, 12351–12355. [Google Scholar] [CrossRef] [Green Version]
- Sa, S.P.; Lee, Y.H. Ligand compound, organic chromium compound, catalyst system for oligomerization of olefins and method for oligomerization of olefins using the catalyst system. KR Patent 101757835B1, 7 July 2017. [Google Scholar]
- Overett, M.J.; Blann, K.; Bollmann, A.; de Villiers, R.; Dixon, J.T.; Killian, E.; Maumela, M.C.; Maumela, H.; McGuinness, D.S.; Morgan, D.H.; et al. Carbon-bridged diphosphine ligands for chromium-catalysed ethylene tetramerisation and trimerisation reactions. J. Mol. Catal. A Chem. 2008, 283, 114–119. [Google Scholar] [CrossRef]
- Baker, B.A.; Bošković, Ž.V.; Lipshutz, B.H. (BDP)CuH: A “Hot” Stryker’s Reagent for Use in Achiral Conjugate Reductions. Org. Lett. 2008, 10, 289–292. [Google Scholar] [CrossRef]
- Lee, D.G.; Baek, J.W.; Lee, J.H.; Lee, H.J.; Seo, Y.H.; Lee, J.; Lee, C.G.; Lee, B.Y. Replacement of the Common Chromium Source CrCl3(thf)3 with Well-Defined [CrCl2(μ-Cl)(thf)2]2. Molecules 2021, 26, 1167. [Google Scholar] [CrossRef]
- Hardcastle, K.I.; Skovlin, D.O.; Eidawad, A.-H. Synthesis and X-ray molecular structure of trans-dichlorotetramethanolchromium(III) chloride, [Cr(MeOH)4Cl2]Cl. J. Chem. Soc. Chem. Commun. 1975, 190. [Google Scholar] [CrossRef]
- Klosin, J.; Milbrandt, K.A.; Boelter, S.D.; Wilson, D.R.; Rosen, M.S.; Welsh, D.M.; Margl, P.M.; Koh, K.M.; Pearson, D.M.; Huacuja, R. Chromium Complex and Catalyst Therefrom. W.O. Patent 2016149024A2, 22 September 2016. [Google Scholar]
- Mogorosi, M.M.; Maumela, M.C.; Overett, M.J. Oligomerisation of Ethylene to Mixtures of 1-hexene and 1-octene. W.O. Patent 2014181248A1, 13 November 2014. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst (Substituent) | Activity (Kg/g-Cr/h) | 1-C8 (wt%) | 1-C6 (wt%) | cy-C6 (wt%) b | >C10 (wt%) | PE (wt%) |
|---|---|---|---|---|---|---|---|
| 1 | [1−CrCl2]+[B(C6F5)4]– (−Si(nBu)3) | 6900 | 75.0 | 8.1 | 4.0 | 12.6 | 0.3 |
| 2 | [2−CrCl2]+[B(C6F5)4]– (−Si(1-hexyl)3) | 6400 | 75.2 | 8.2 | 4.4 | 12.0 | 0.2 |
| 3 | [3−CrCl2]+[B(C6F5)4]– (−Si(1-octyl)3) | 11,100 | 75.0 | 9.2 | 4.9 | 10.7 | 0.2 |
| 4 c | [3−CrCl2]+[B(C6F5)4]– (−Si(1-octyl)3) | 12,500 | 56.1 | 25.2 | 4.2 | 13.6 | 1.0 |
| 5 | [4−CrCl2]+[B(C6F5)4]– (−Si(2-ethylhexyl)3) | 7000 | 64.7 | 11.4 | 8.9 | 14.7 | 0.2 |
| 6 | [5−CrCl2]+[B(C6F5)4]– (−Si(3,7-dimethyloctyl)3) | 8900 | 70.1 | 9.4 | 5.0 | 15.2 | 0.3 |
| 7 | [6−CrCl2]+[B(C6F5)4]– (C6H11N−; −Si(1-octyl)3) | 9200 | 74.0 | 9.1 | 4.7 | 12.0 | 0.1 |
| 8 | [8−CrCl2]+[B(C6F5)4]– (asymmetric) | 7200 | 69.5 | 12.2 | 5.1 | 11.3 | 1.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Baek, J.W.; Lee, D.G.; Ko, J.H.; Lee, D.G.; Cho, K.S.; Lee, J.W.; Lee, B.Y. Preparation of Extremely Active Ethylene Tetramerization Catalyst [iPrN(PAr2)2−CrCl2]+[B(C6F5)4]– (Ar = −C6H4-p-SiR3). Catalysts 2021, 11, 1122. https://doi.org/10.3390/catal11091122
Lee JH, Baek JW, Lee DG, Ko JH, Lee DG, Cho KS, Lee JW, Lee BY. Preparation of Extremely Active Ethylene Tetramerization Catalyst [iPrN(PAr2)2−CrCl2]+[B(C6F5)4]– (Ar = −C6H4-p-SiR3). Catalysts. 2021; 11(9):1122. https://doi.org/10.3390/catal11091122
Chicago/Turabian StyleLee, Jung Hyun, Jun Won Baek, Dong Geun Lee, Ji Hyeong Ko, Dong Geun Lee, Kye Sung Cho, Jin Woo Lee, and Bun Yeoul Lee. 2021. "Preparation of Extremely Active Ethylene Tetramerization Catalyst [iPrN(PAr2)2−CrCl2]+[B(C6F5)4]– (Ar = −C6H4-p-SiR3)" Catalysts 11, no. 9: 1122. https://doi.org/10.3390/catal11091122
APA StyleLee, J. H., Baek, J. W., Lee, D. G., Ko, J. H., Lee, D. G., Cho, K. S., Lee, J. W., & Lee, B. Y. (2021). Preparation of Extremely Active Ethylene Tetramerization Catalyst [iPrN(PAr2)2−CrCl2]+[B(C6F5)4]– (Ar = −C6H4-p-SiR3). Catalysts, 11(9), 1122. https://doi.org/10.3390/catal11091122