
Advances in Green Catalysis for the Synthesis of Medicinally Relevant N-Heterocycles


 and
and 
Abstract
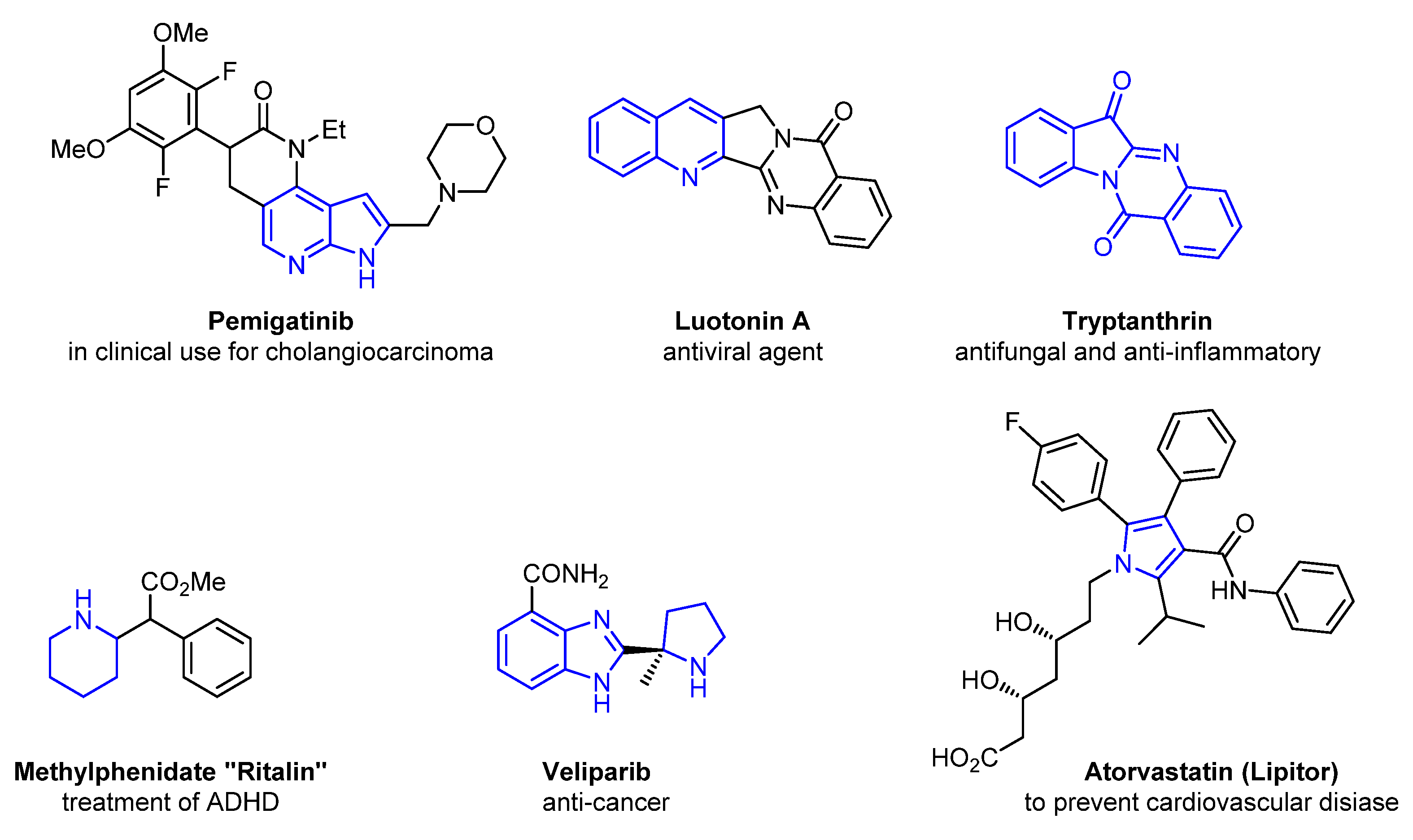
1. Introduction
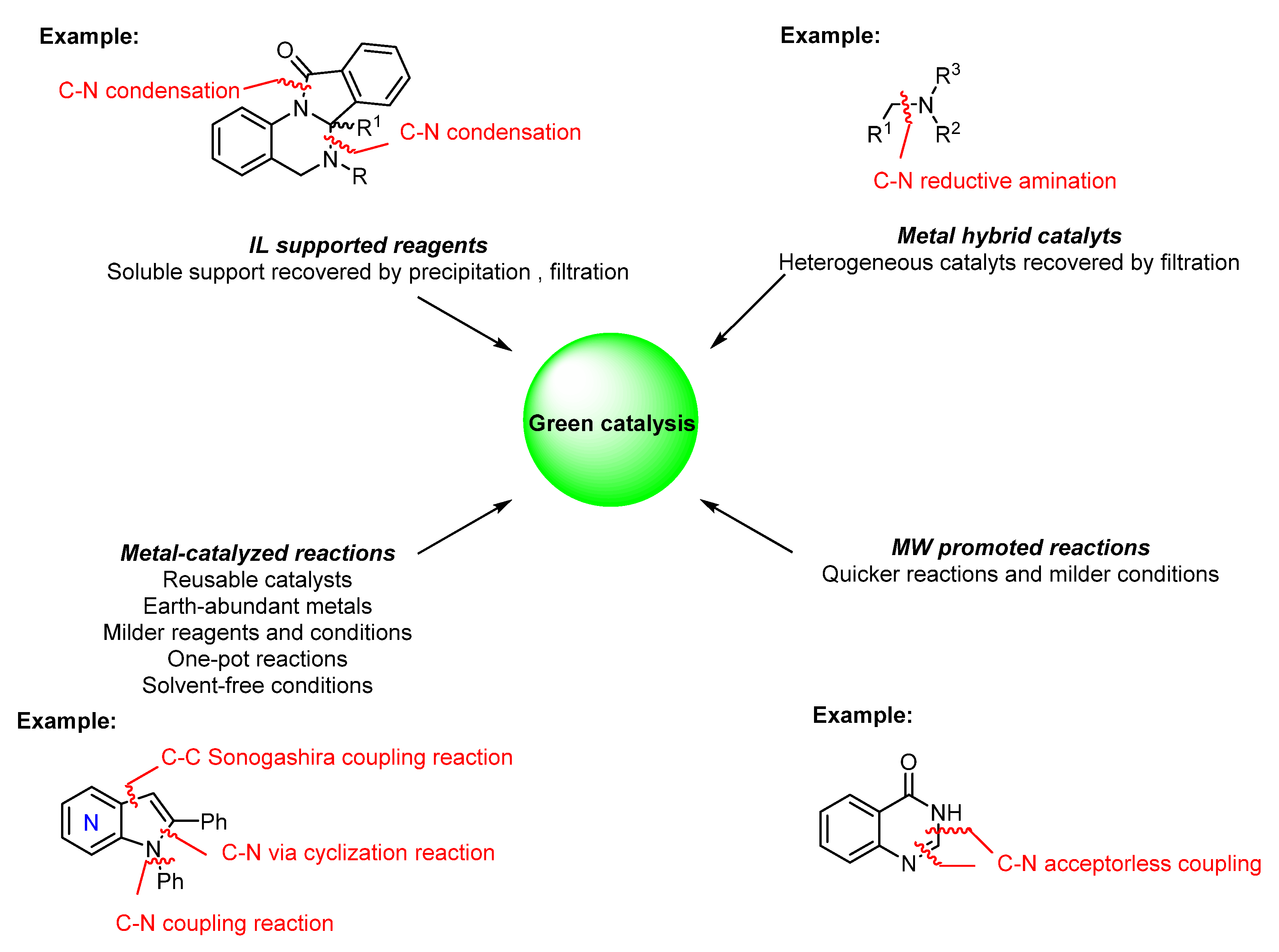
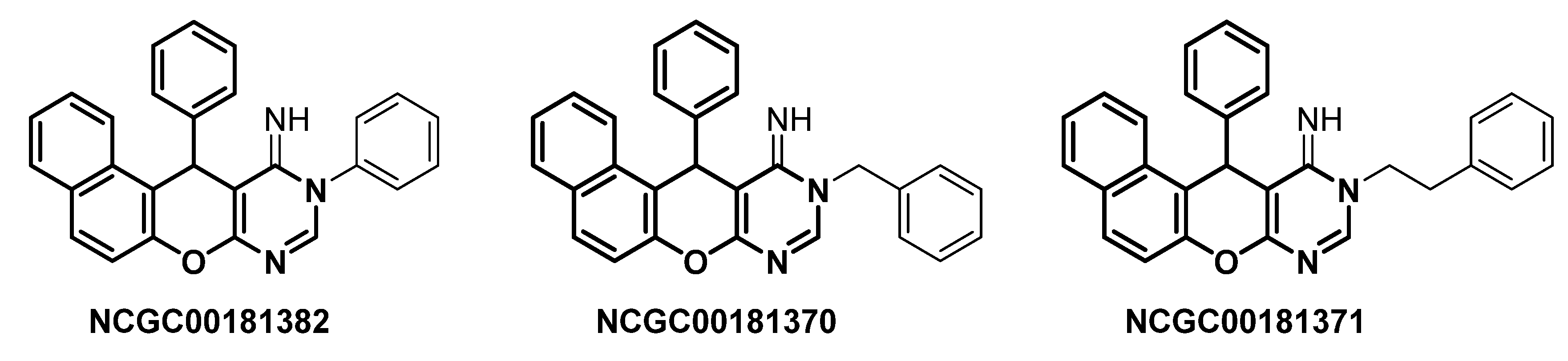
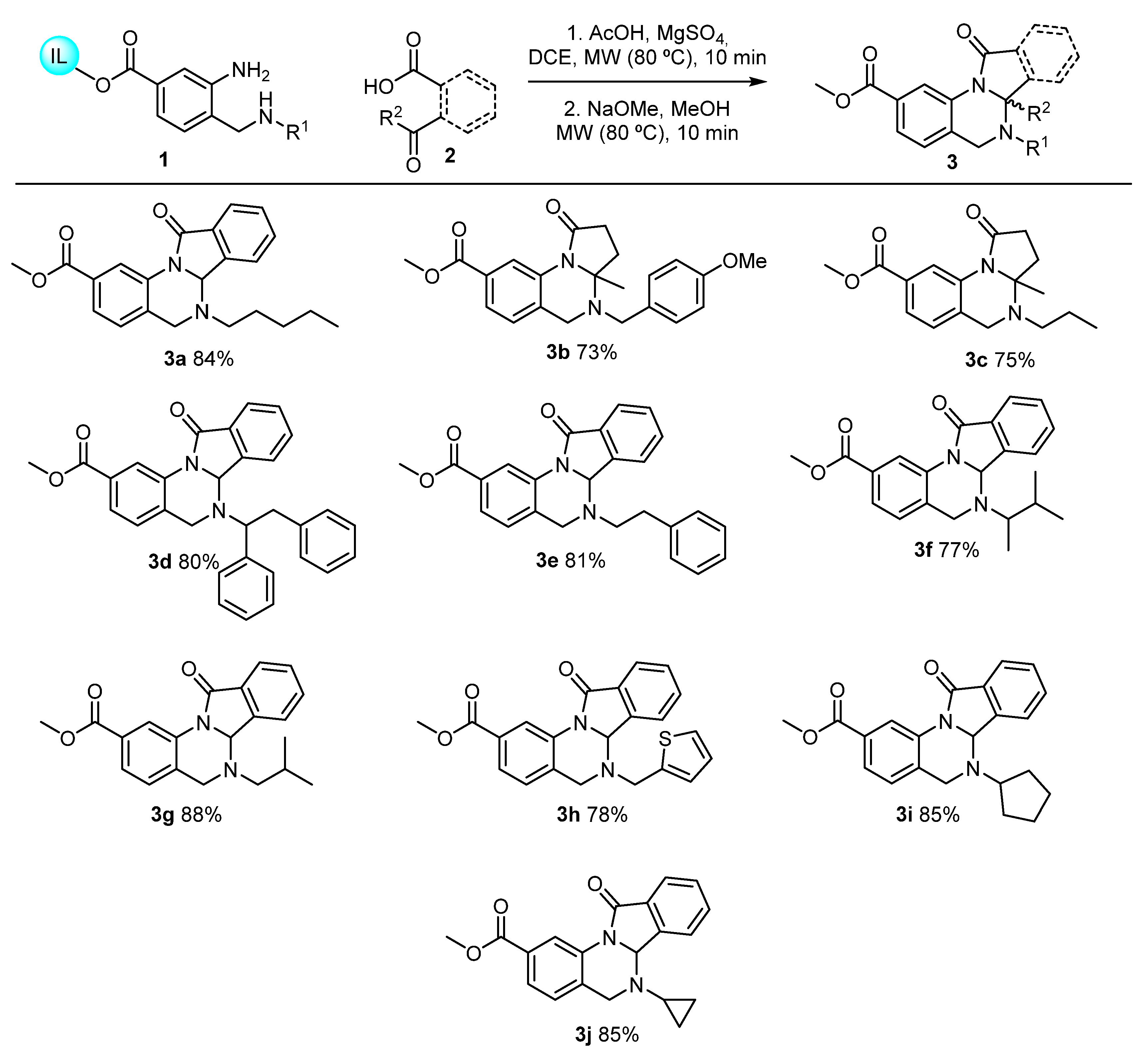
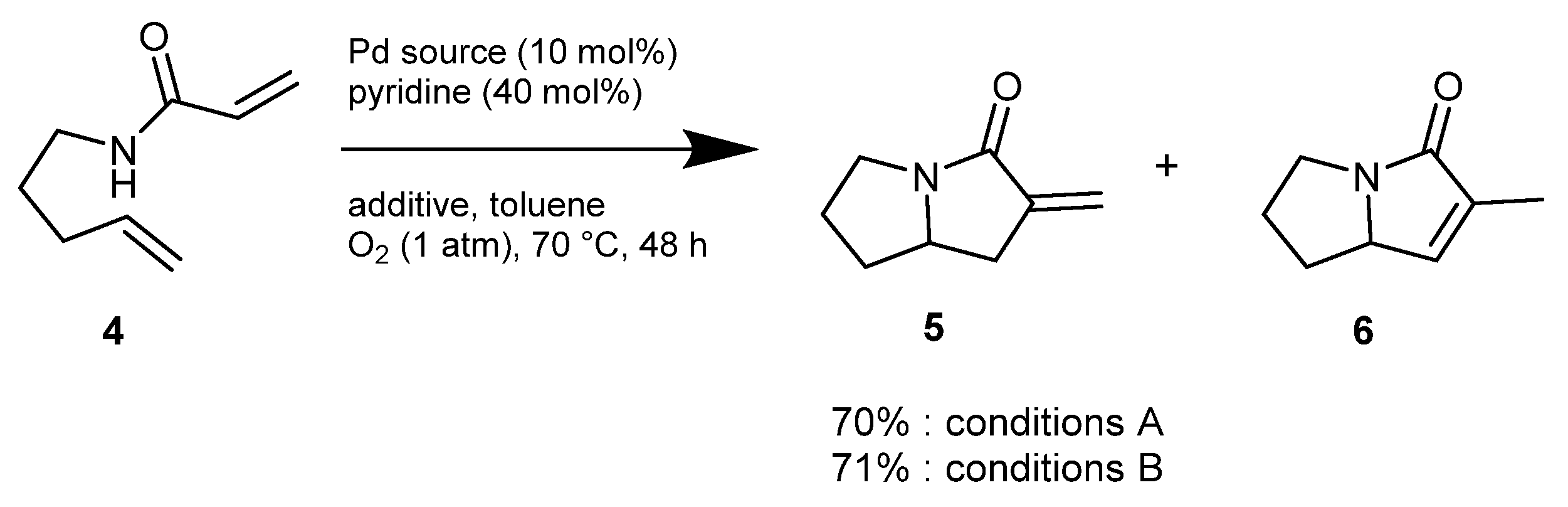
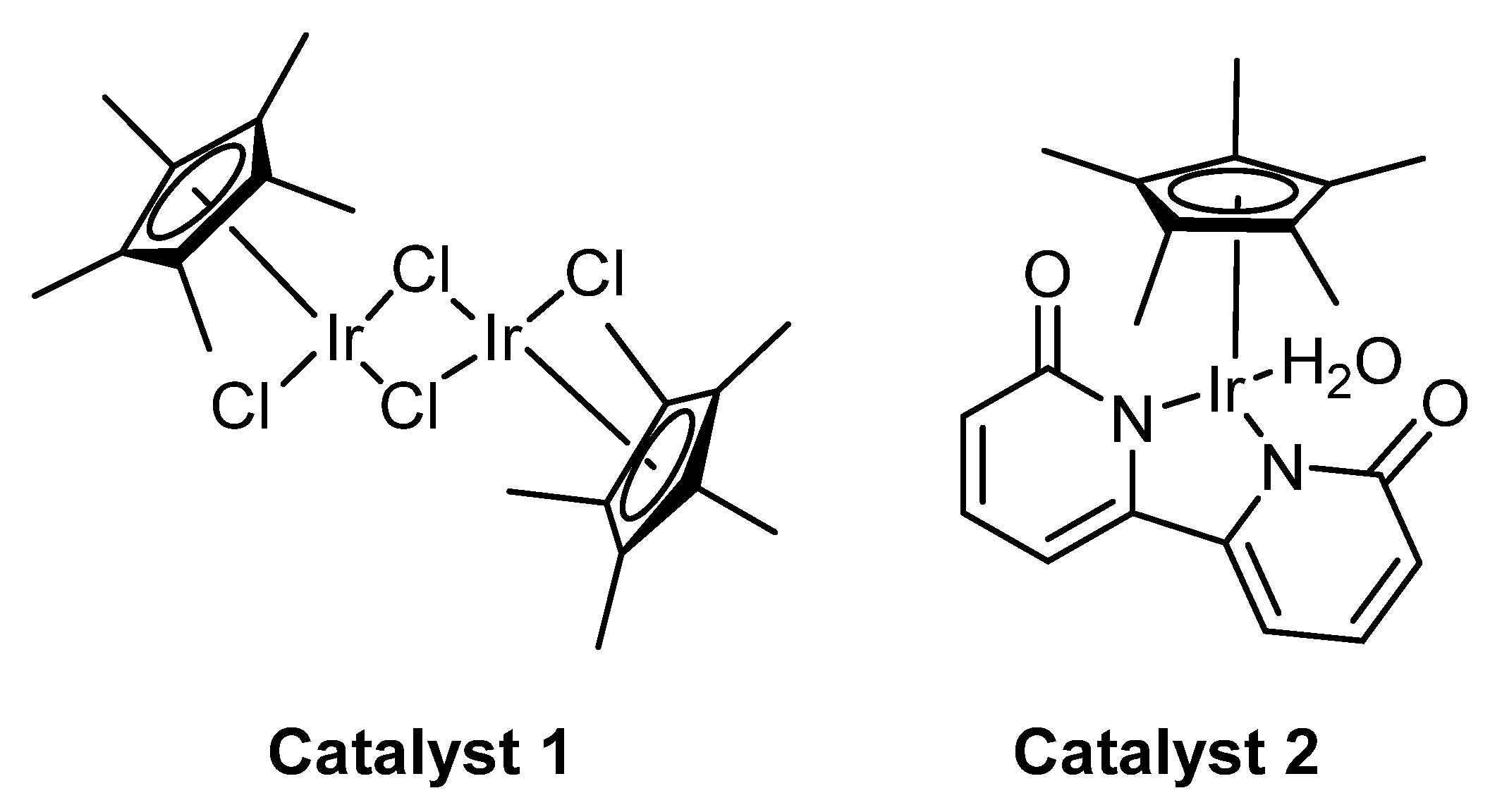
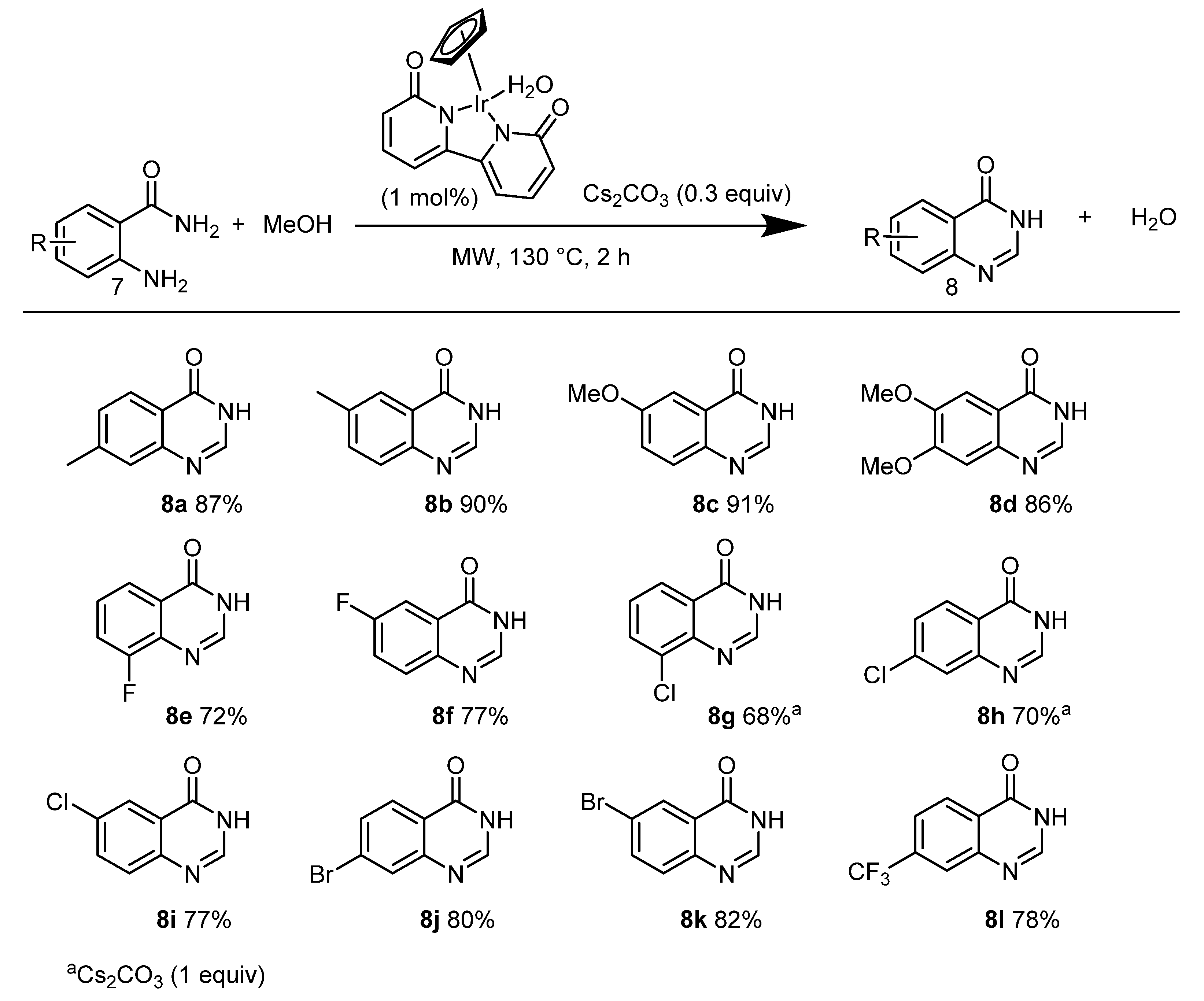
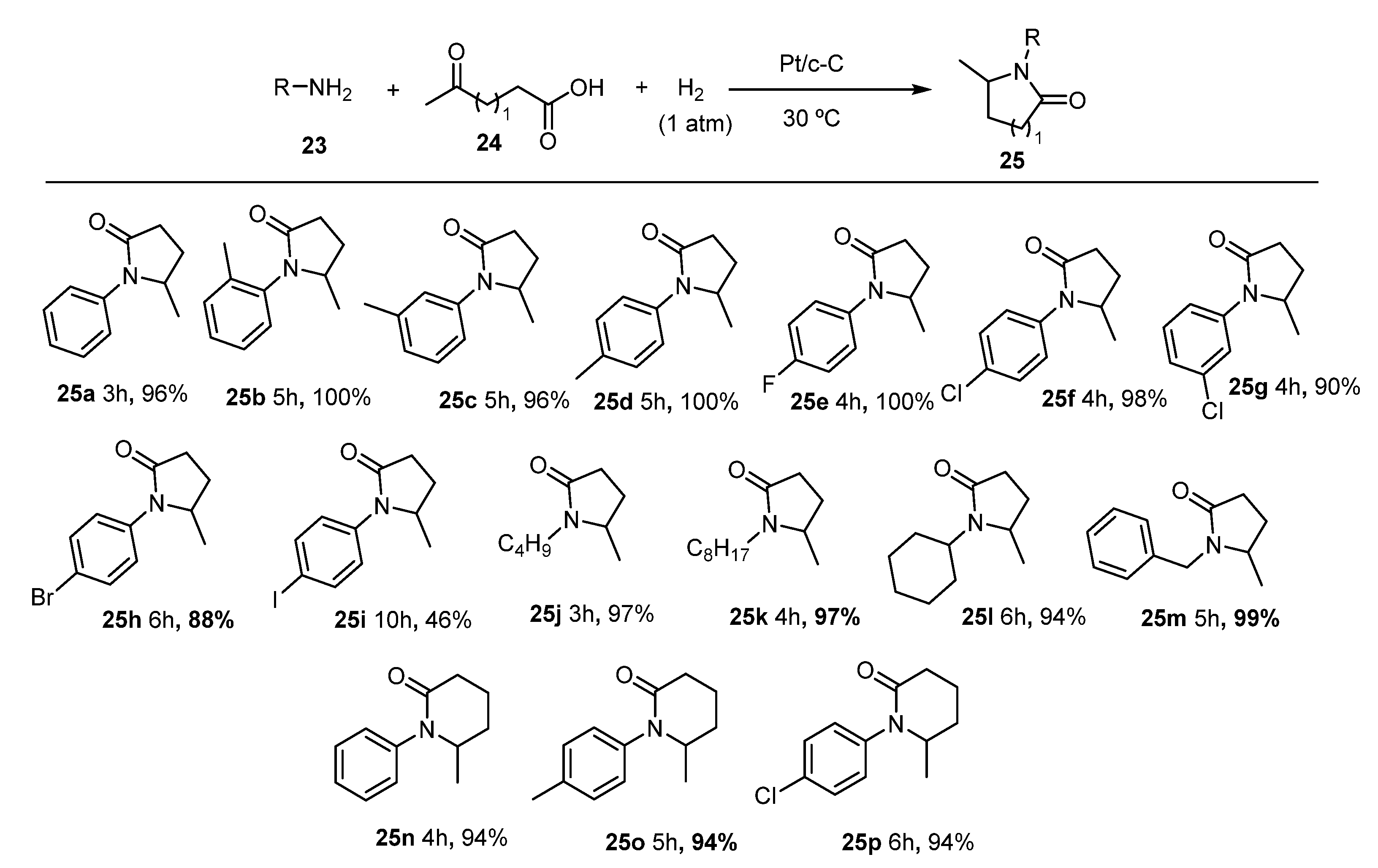
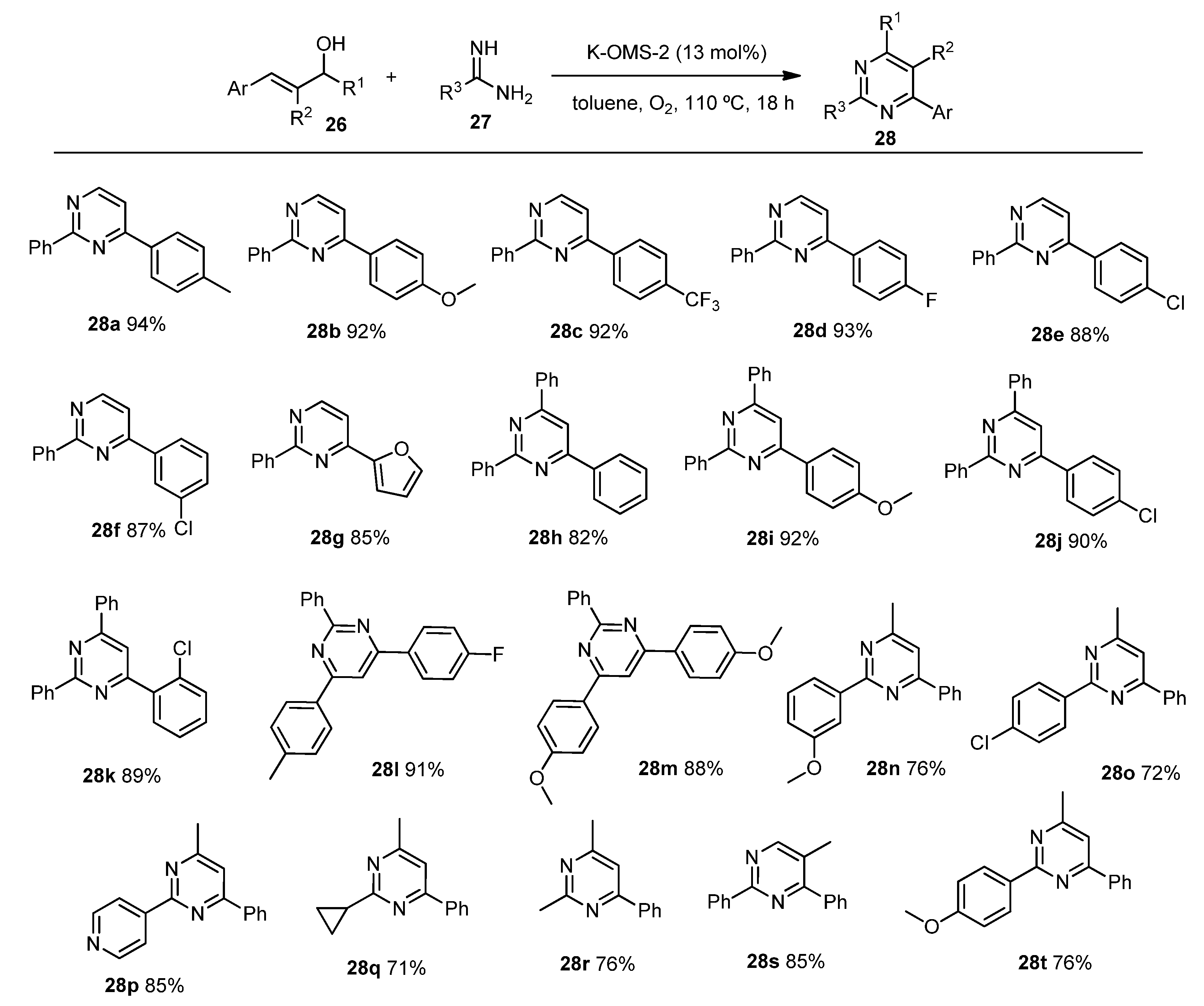
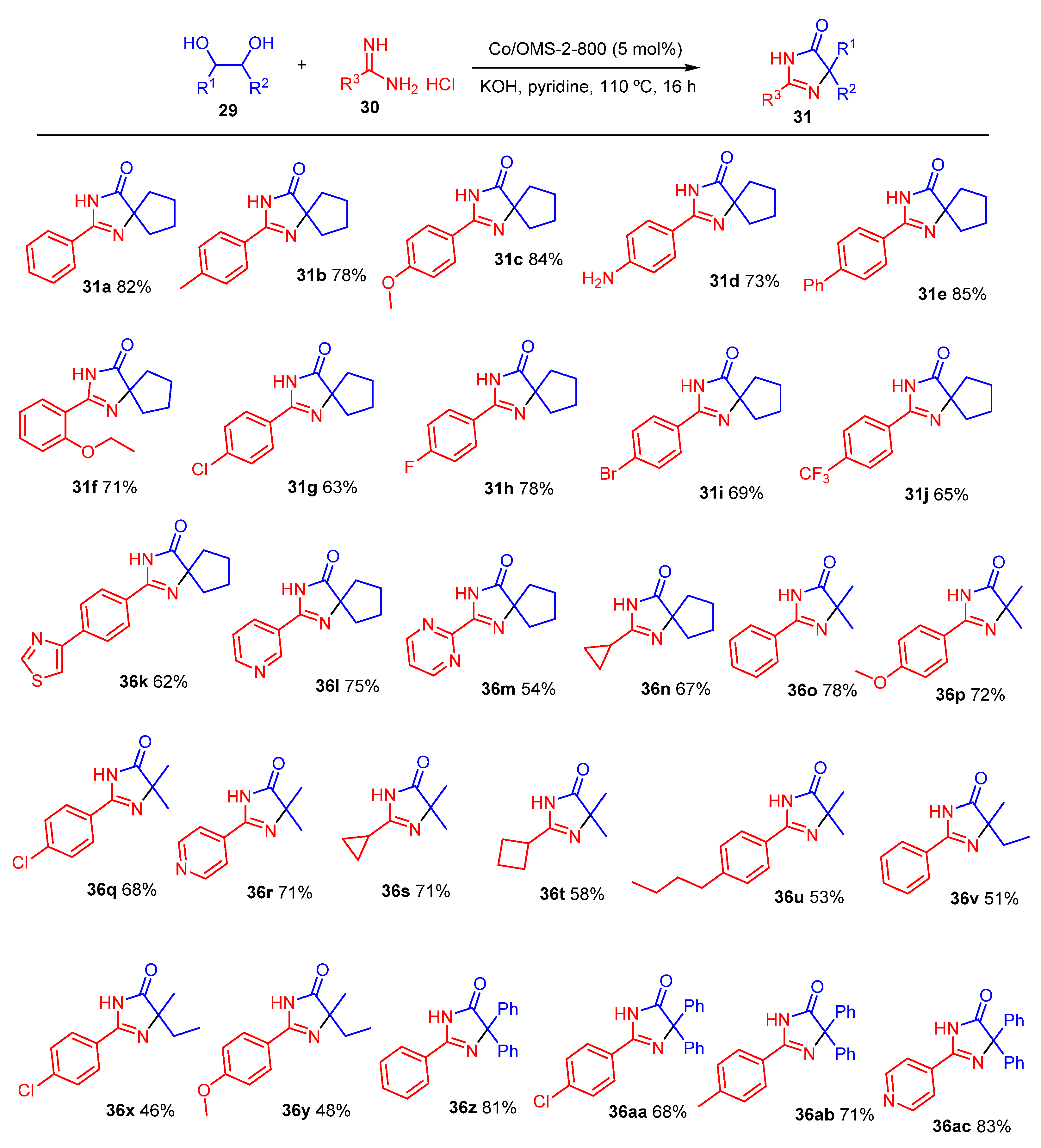
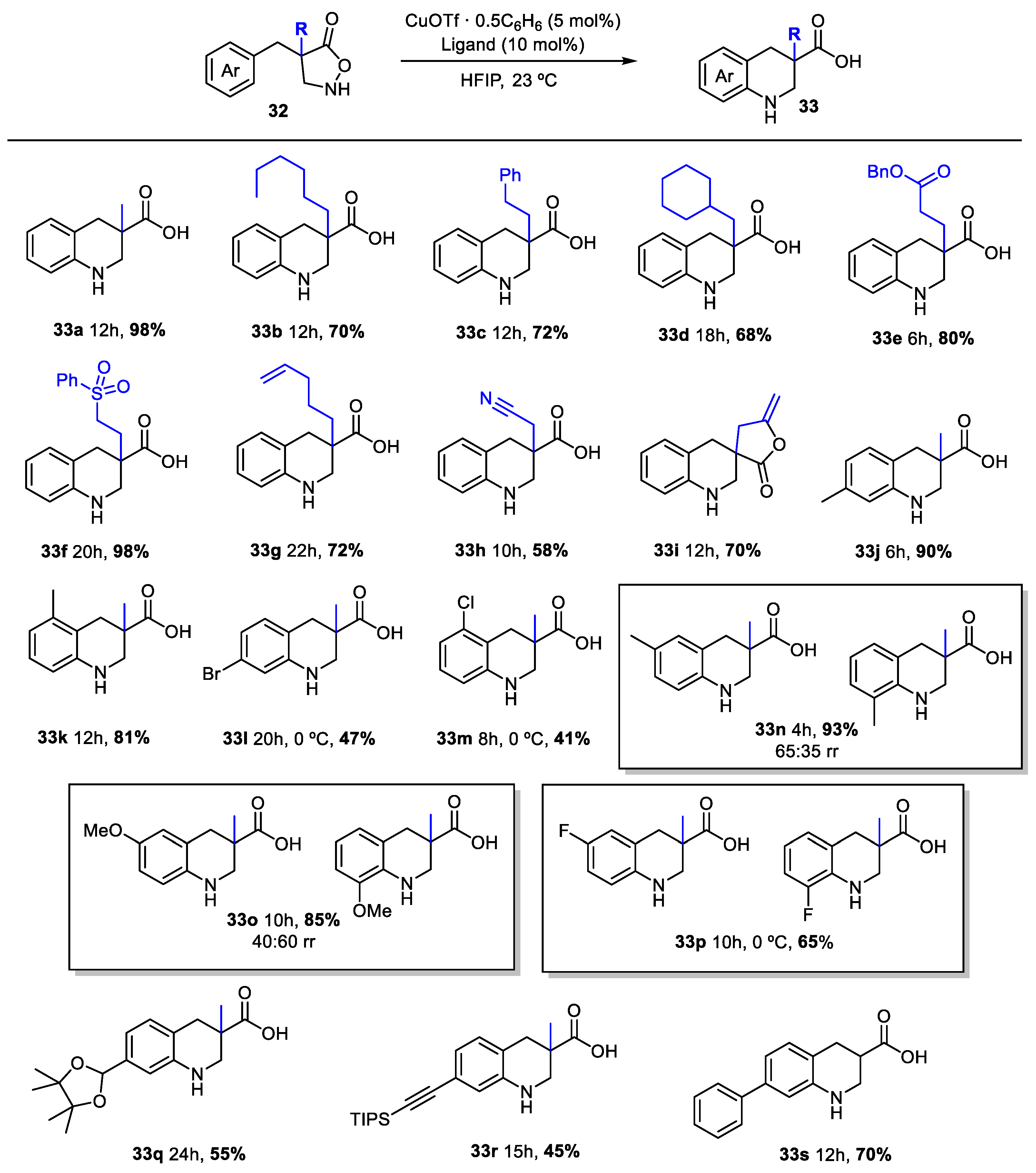
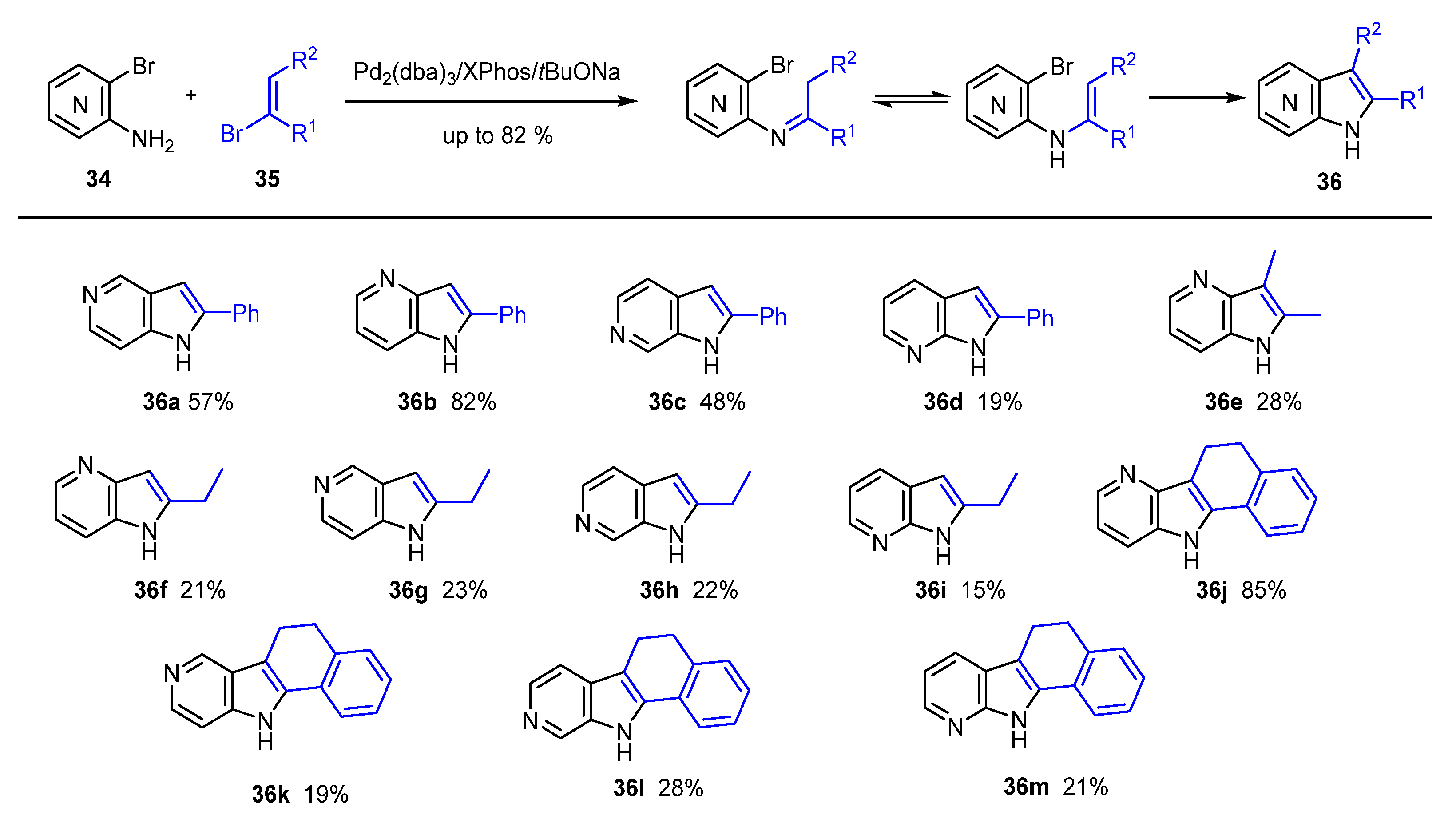
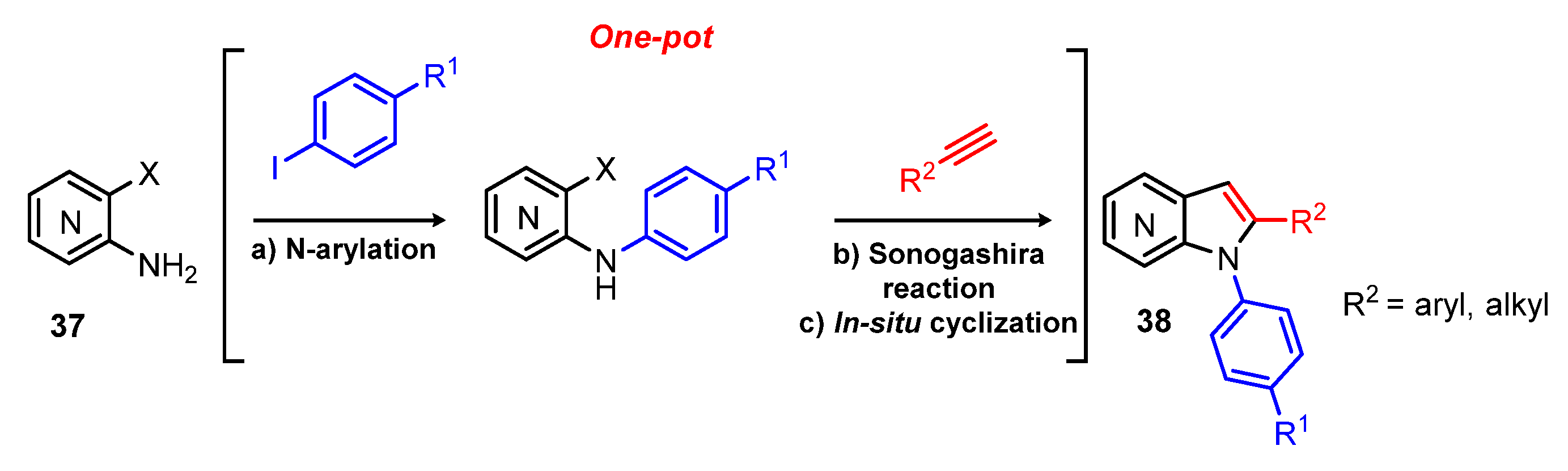
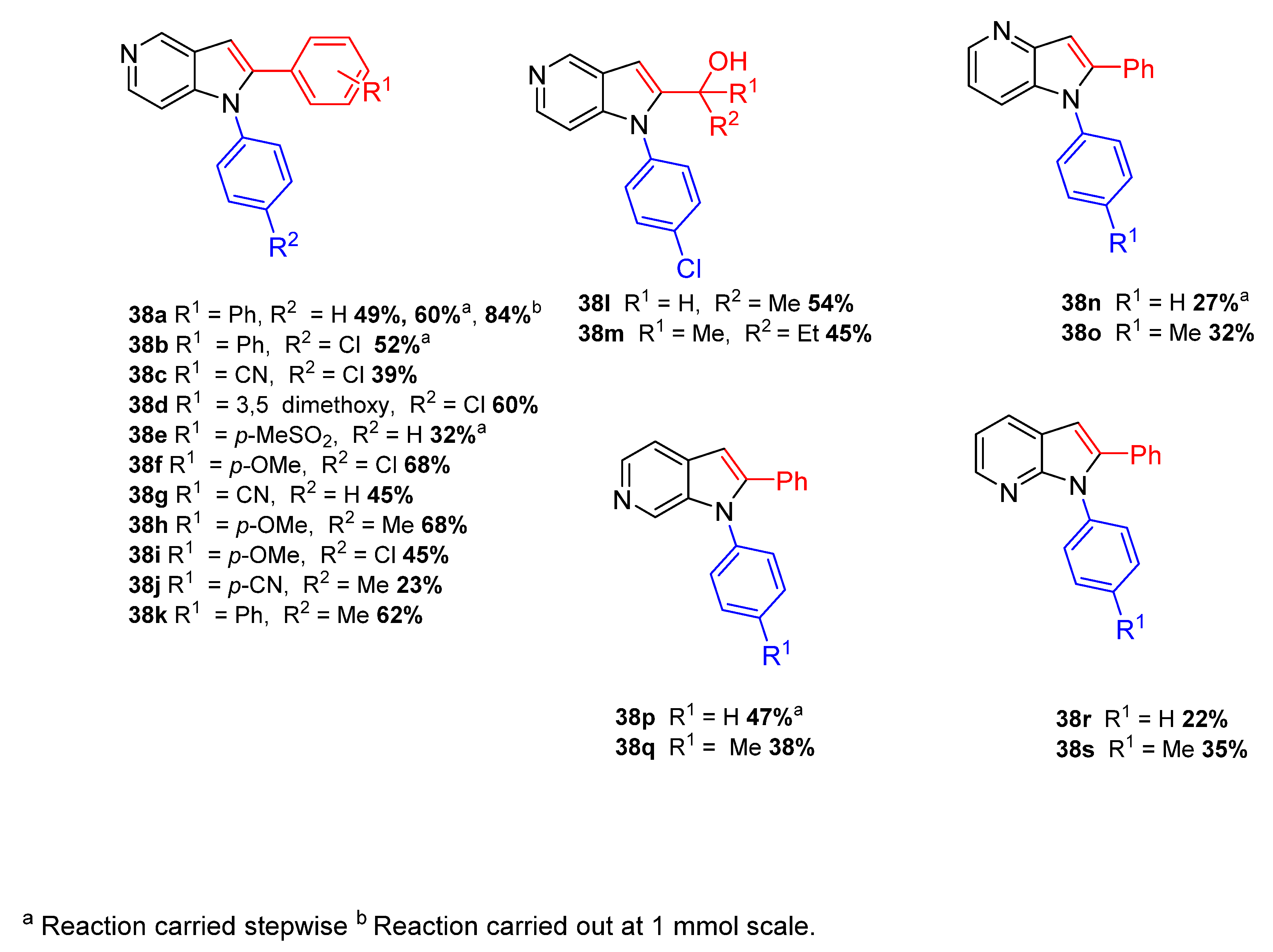
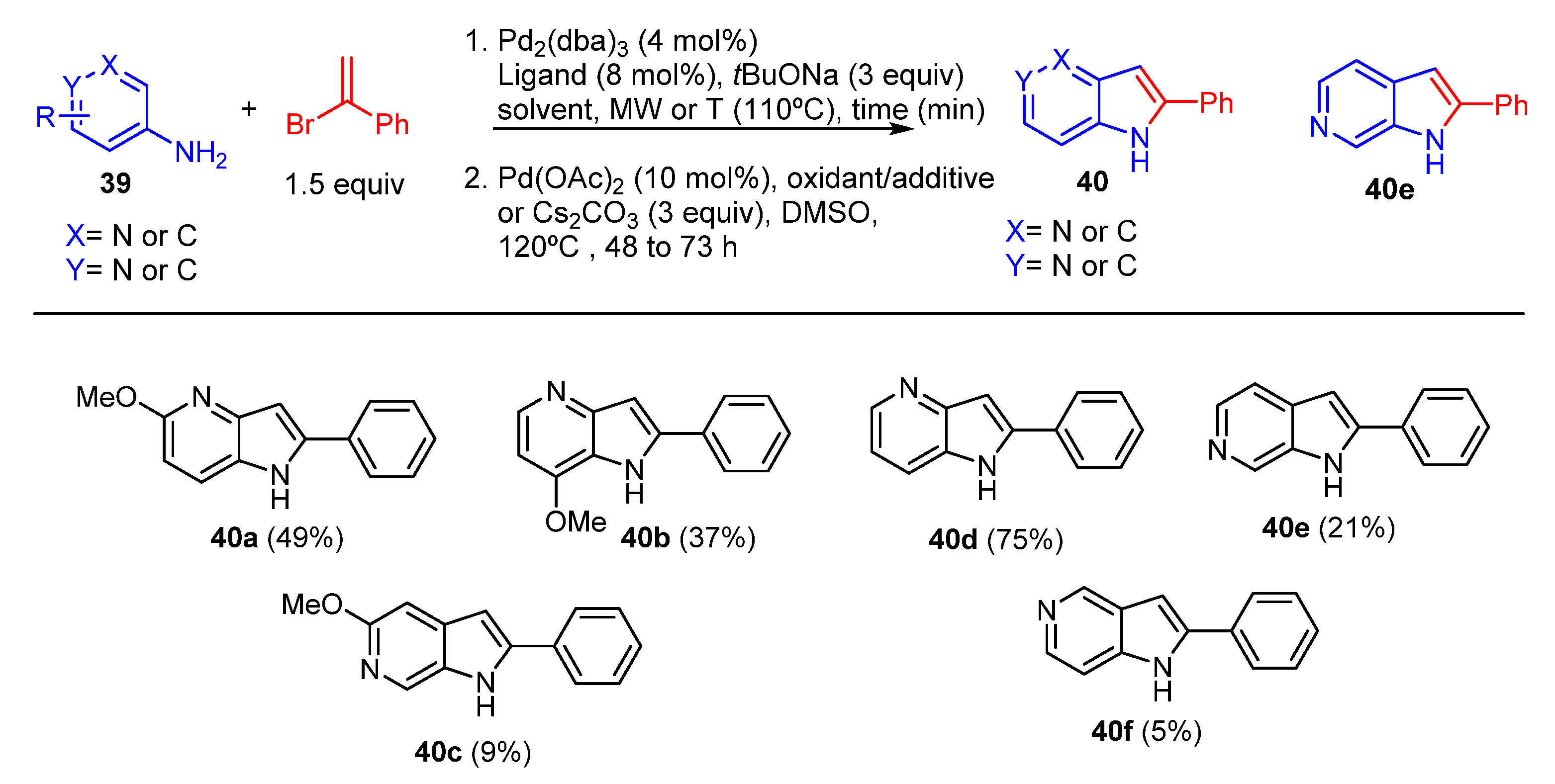
2. Green Catalytic Synthesis of N-Heterocycles
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Han, Y.; Dong, W.; Guo, Q.; Li, X.; Huang, L. The importance of indole and azaindole scaffold in the development of antitumor agents. Eur. J. Med. Chem. 2020, 203, 112506. [Google Scholar] [CrossRef]
- Cagir, A.; Jones, S.H.; Gao, R.; Eisenhauer, B.M.; Hecht, S.M.; Luotonin, A. A naturally occurring human DNA topoisomerase I poison. J. Am. Chem. Soc. 2003, 125, 13628–13629. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.-L.; Yip, H.-Y.; Mak, N.-K.; Leung, K.-N. Modulatory effects and action mechanisms of tryptanthrin on murine myeloid Leukemia cells. Cell. Mol. Immunol. 2009, 6, 335–342. [Google Scholar] [CrossRef]
- Tsou, H.-R.; Mamuya, N.; Johnson, B.D.; Reich, M.F.; Gruber, B.C.; Ye, F.; Nilakantan, R.; Shen, R.; Discafani, C.; DeBlanc, R.; et al. 6-Substituted-4-(3-bromophenylamino) quinazolines as putative irreversible inhibitors of the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor (HER-2) tyrosine kinases with enhanced antitumor activity. J. Med. Chem. 2001, 44, 2719–2734. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, A.E.; Barker, A.J.; Davies, D.H.; Brown, D.S.; Green, L.R.; Cartlidge, S.A.; Woodburn, J.R. Specific inhibition of epidermal growth factor receptor tyrosine kinase by 4-anilinoquinazolines. Breast Cancer Res. Treat. 1996, 38, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kung, P.-P.; Casper, M.D.; Cook, K.L.; Wilson-Lingardo, L.; Risen, L.M.; Vickers, T.A.; Ranken, R.; Blyn, L.B.; Wyatt, J.R.; Cook, P.D.; et al. Structure—activity relationships of novel 2-substituted quinazoline antibacterial agents. J. Med. Chem. 1999, 42, 4705–4713. [Google Scholar] [CrossRef]
- Jung, I.-P.; Ha, N.-R.; Lee, S.-C.; Ryoo, S.-W.; Yoon, M.-Y. Development of potent chemical antituberculosis agents targeting Mycobacterium tuberculosis acetohydroxy acid synthase. Int. J. Antimicrob. Agents 2016, 48, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.L.; Cha, H.C.; Jahng, Y. Recent advances in the studies on luotonins. Molecules 2011, 16, 4861–4883. [Google Scholar] [CrossRef]
- Thompson, R.B. Foundations for blockbuster drugs in federally sponsored research. FASEB J. 2001, 15, 1671–1676. [Google Scholar] [CrossRef]
- Pantaine, L.R.E.; Milligan, J.A.; Matsui, J.K.; Kelly, C.B.; Molander, G.A. Photoredox radical/polar crossover enables construction of saturated nitrogen heterocycles. Org. Lett. 2019, 21, 2317–2321. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Reddy, K.R. Isolation and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2002, 102, 4303–4428. [Google Scholar] [CrossRef] [PubMed]
- Daştan, A.; Kulkarni, A.; Török, B. Environmentally benign synthesis of heterocyclic compounds by combined microwave-assisted heterogeneous catalytic approaches. Green Chem. 2012, 14, 17–37. [Google Scholar] [CrossRef]
- Jedhe, R.; Paike, V.; Sun, C.-M. Rapid synthesis of novel isoindolo [1,2-a] quinazoline on ionic liquid support under microwave irradiation. Green Process. Synth. 2013, 2, 251–258. [Google Scholar] [CrossRef]
- Yang, D.; Lo, K.-Y.; Ye, L. Palladium-catalyzed aerobic oxidative cyclization of aliphatic alkenyl amides for the construction of pyrrolizidine and indolizidine derivatives. Synlett 2017, 28, 1570–1575. [Google Scholar] [CrossRef]
- Yip, K.-T.; Yang, D. Pd(II)-catalyzed intramolecular amidoarylation of alkenes with molecular oxygen as sole oxidant. Org. Lett. 2011, 13, 2134–2137. [Google Scholar] [CrossRef]
- Xing, D.; Yang, D. Pd(II)-catalyzed intramolecular 1,2-aminoalkylation of conjugated 1,3-dienes for the synthesis of pyrrolizidines. Org. Lett. 2013, 15, 4370–4373. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Lo, K.-Y.; Gu, Q.; Yang, D. Pd-catalyzed intramolecular aminoalkylation of unactivated alkenes: Access to diverse N-heterocycles. Org. Lett. 2017, 19, 308–311. [Google Scholar] [CrossRef]
- Du, W.; Gu, Q.; Li, Z.; Yang, D. Palladium(II)-catalyzed intramolecular tandem aminoalkylation via divergent C(sp3)–H functionalization. J. Am. Chem. Soc. 2015, 137, 1130–1135. [Google Scholar] [CrossRef]
- Khan, I.; Zaib, S.; Batool, S.; Abbas, N.; Ashraf, Z.; Iqbal, J.; Saeed, A. Quinazolines and quinazolinones as ubiquitous structural fragments in medicinal chemistry: An update on the development of synthetic methods and pharmacological diversification. Bioorg. Med. Chem. 2016, 24, 2361–2381. [Google Scholar] [CrossRef]
- Li, F.; Lu, L.; Liu, P. Acceptorless dehydrogenative coupling of o-aminobenzamides with the activation of methanol as a C1 source for the construction of quinazolinones. Org. Lett. 2016, 18, 2580–2583. [Google Scholar] [CrossRef]
- Lu, L.; Ma, J.; Qu, P.; Li, F. Effective recognition of different types of amino groups: From aminobenzenesulfonamides to amino-(N-alkyl) benzenesulfonamides via. iridium-catalyzed N-alkylation with alcohols. Org. Lett. 2015, 17, 2350–2353. [Google Scholar] [CrossRef]
- Li, F.; Wang, N.; Lu, L.; Zhu, G. Regioselective hydration of terminal alkynes catalyzed by a neutral gold(I) complex [(IPr)AuCl] and one-pot synthesis of optically active secondary alcohols from terminal alkynes by the combination of [(IPr)AuCl] and Cp*RhCl[(R,R)-TsDPEN]. J. Org. Chem. 2015, 80, 3538–3546. [Google Scholar] [CrossRef] [PubMed]
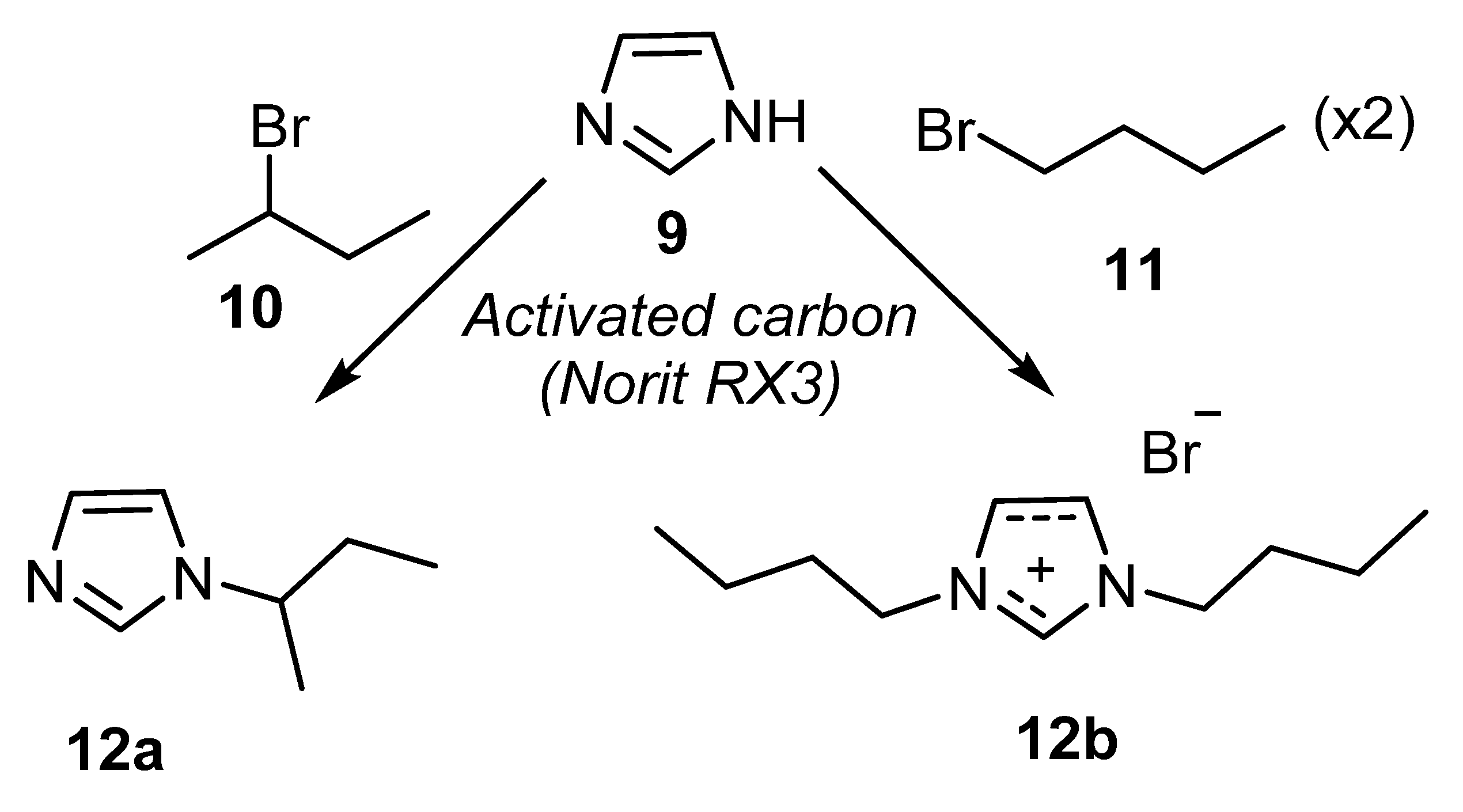
- Durán-Valle, C.J.; Madrigal-Martínez, M.; Martínez-Gallego, M.; Fonseca, I.M.F.L.; Matos, I.A.; Rego, A. Activated carbon as a catalyst for the synthesis of N-alkylimidazoles and imidazolium ionic liquids. Catal. Today 2012, 187, 108–114. [Google Scholar] [CrossRef]
- Bedair, A.; El-Hady, N.A.; El-Latif, M.; Fakery, A.; El-Agrody, A. 4-Hydroxycoumarin in heterocyclic synthesis: Part III. Synthesis of some new pyrano [2,3-d]pyrimidine, 2-substituted [1,2,4] triazolo [1,5-c] pyrimidine and pyrimido [1,6-b] [1,2,4] triazine derivatives. Il Farm. 2000, 55, 708–714. [Google Scholar] [CrossRef]
- Regnier, G.L.; Canevari, R.J.; Le Douarec, J.C.; Holstorp, S.; Daussy, J. Triphenylpropylpiperazine derivatives as new potent analgetic substances. J. Med. Chem. 1972, 15, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.; Schmid, F.A.; Schmid, M.M.; Brown, G.F. Effect of compounds on a spectrum of rat tumors. Cancer Chemother. Rep. Part 2 1972, 3, 231–308. [Google Scholar]
- Mohr, S.J.; Chirigos, M.A.; Fuhrman, F.S.; Pryor, J.W. Pyran copolymer as an effective adjuvant to chemotherapy against a murine leukemia and solid tumor. Cancer Res. 1975, 35, 3750–3754. [Google Scholar]
- Hackler, R.E.; Jourdan, G.P. Condensed Pyrimidine Derivatives and Their Use as Fungicides, Insecticides, and Miticides. CA Patent 2,131,545,A1, 16 March 1999. [Google Scholar]
- Patnaik, S.; Marugan, J.; Liu, K.; Zheng, W.; Thorsell, A.; Eskay, R.; Southall, N.; Heilig, M.; Inglese, J.; Austin, C. Identification of Small Molecule Antagonists of the Neuropeptide-S Receptor; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010.
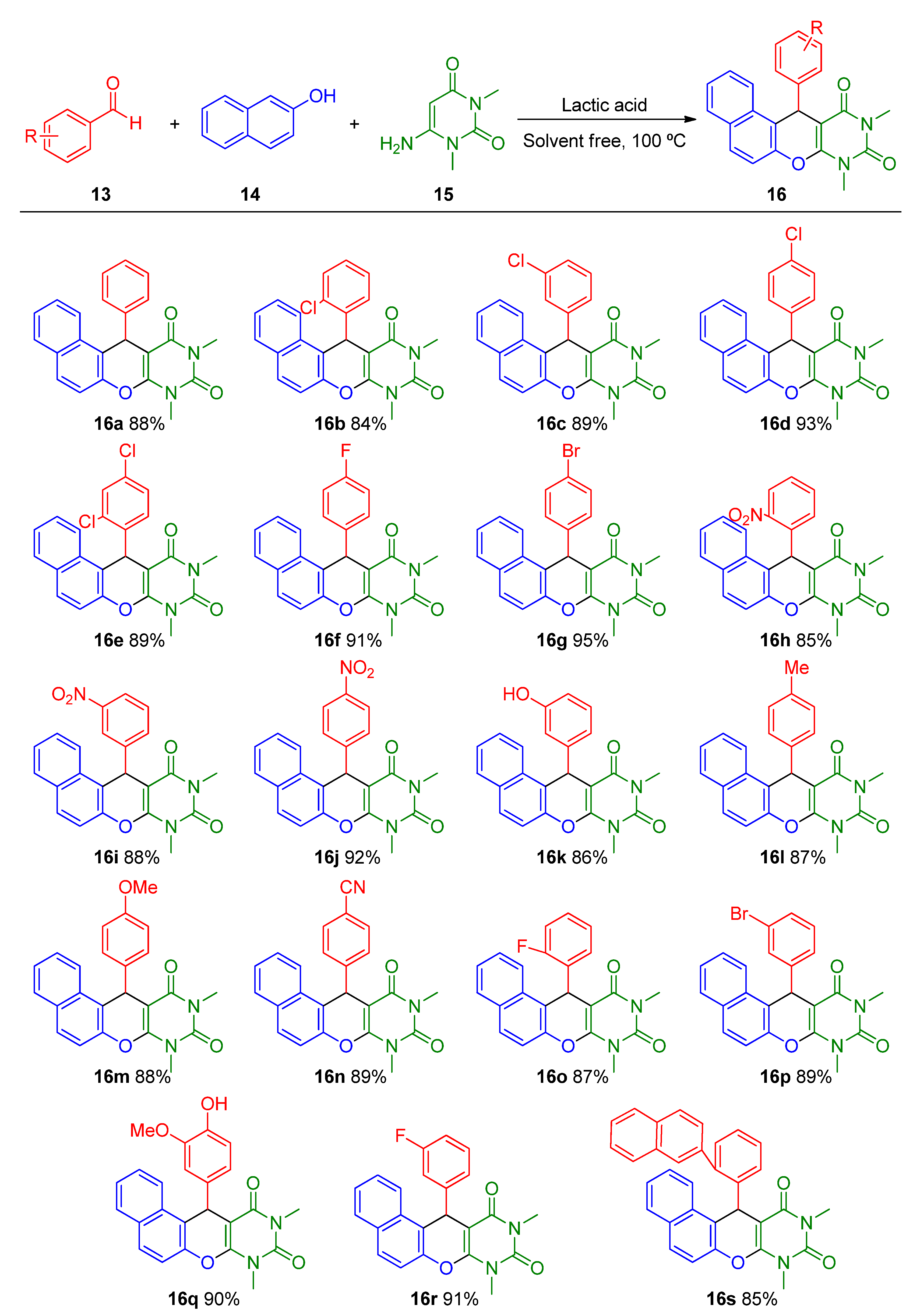
- Fatahpour, M.; Hazeri, N.; Maghsoodlou, M.T.; Lashkari, M. One-pot condensation approach for synthesis of diverse naphthopyranopyrimidines utilizing lactic acid as efficient and eco-friendly catalyst. Polycycl. Aromat. Compd. 2017, 39, 311–317. [Google Scholar] [CrossRef]
- Larsen, T.O.; Svendsen, A.; Smedsgaard, J. Biochemical characterization of ochratoxin a-producing strains of the genus penicillium. Appl. Environ. Microbiol. 2001, 67, 3630–3635. [Google Scholar] [CrossRef]
- Ariza, M.R.; Larsen, T.O.; Petersen, B.O.; Duus, J.Ø.; Christophersen, C.; Barrero, A.F. A Novel alkaloid serantrypinone and the spiro azaphilone daldinin D frompenicillium thymicola. J. Nat. Prod. 2001, 64, 1590–1592. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Na, Y.H.; Hong, S.H.; Lee, J.H.; Park, W.-K.; Baek, D.-J.; Koh, H.Y.; Cho, Y.S.; Choo, H.; Pae, A.N. Novel quinazolinone derivatives as 5-HT7 receptor ligands. Bioorganic Med. Chem. 2008, 16, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Kurogi, Y.; Inoue, Y.; Tsutsumi, K.; Nakamura, S.; Nagao, K.; Yoshitsugu, H.; Tsuda, Y. Synthesis and hypolipidemic activities of novel 2-[4-[(diethoxyphosphoryl) methyl] phenyl] quinazolines and 4(3 h)-quinazolinones. J. Med. Chem. 1996, 39, 1433–1437. [Google Scholar] [CrossRef]
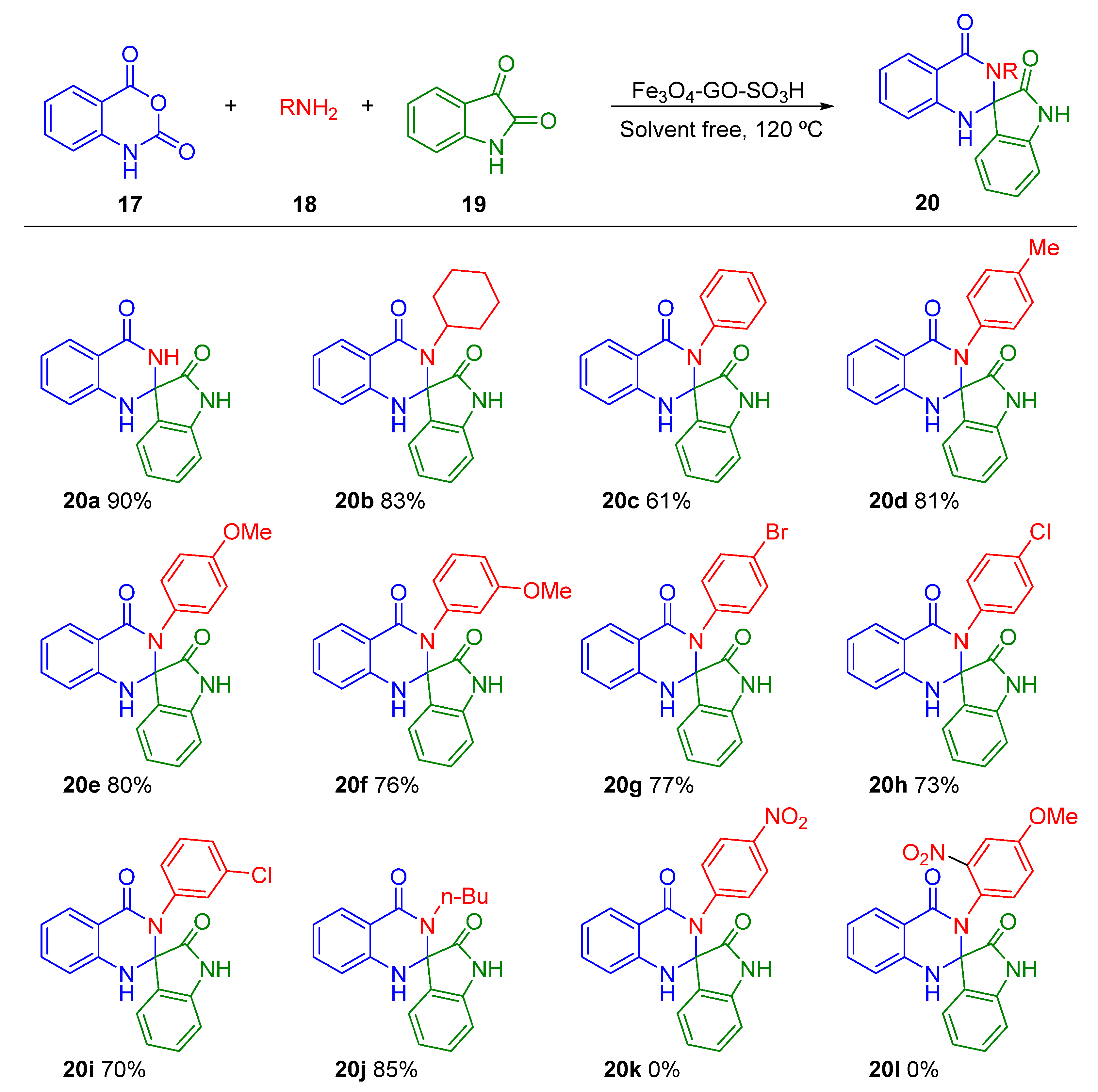
- Abdi, M.; Rostamizadeh, S.; Zekri, N. An efficient and green synthesis of 1′hspiro [isoindoline-1,2′-quinazoline]-3,4′(3′h)-dione derivatives in the presence of nano Fe3O4-GO-SO3H. Polycycl. Aromat. Compd. 2019, 39, 413–424. [Google Scholar] [CrossRef]
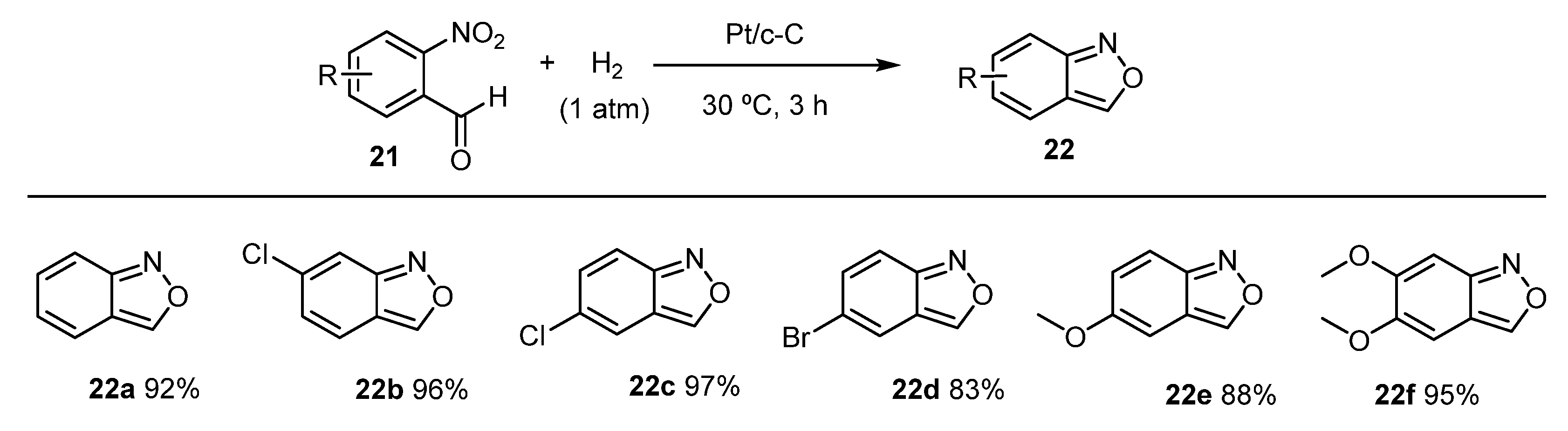
- Wu, Y.; Zhao, Y.; Wang, H.; Zhang, F.; Li, R.; Xiang, J.; Wang, Z.; Han, B.; Liu, Z. Ambient reductive synthesis of N-heterocyclic compounds over cellulose-derived carbon supported Pt nanocatalyst under H2 atmosphere. Green Chem. 2020, 22, 3820–3826. [Google Scholar] [CrossRef]
- Gandhe, A.R.; Rebello, J.S.; Figueiredo, J.; Fernandes, J. Manganese oxide OMS-2 as an effective catalyst for total oxidation of ethyl acetate. Appl. Catal. B Environ. 2007, 72, 129–135. [Google Scholar] [CrossRef]
- Shen, J.; Meng, X. Selective synthesis of pyrimidines from cinnamyl alcohols and amidines using the heterogeneous OMS-2 catalyst. Catal. Commun. 2019, 138, 105846. [Google Scholar] [CrossRef]
- Xie, F.; Chen, X.; Zhang, X.; Luo, C.; Lin, S.; Chen, X.; Li, B.; Li, Y.; Zhang, M. OMS-2 nanorod-supported cobalt catalyst for aerobic dehydrocyclization of vicinal diols and amidines: Access to functionalized imidazolones. J. Catal. 2021, 398, 192–197. [Google Scholar] [CrossRef]
- Desai, N.C.; Wadekar, K.R.; Pandit, U.P.; Mehta, H.K.; Jadeja, D.J.; Pandya, M. Design, synthesis, biological evaluation and in-silico docking studies of some novel imidazolone derivatives as potent antimicrobial containing fluorine agents. Anal. Chem. Lett. 2021, 11, 469–496. [Google Scholar] [CrossRef]
- Keel, K.L.; Tepe, J.J. The preparation of (4 H)-imidazol-4-ones and their application in the total synthesis of natural products. Org. Chem. Front. 2020, 7, 3284–3311. [Google Scholar] [CrossRef]
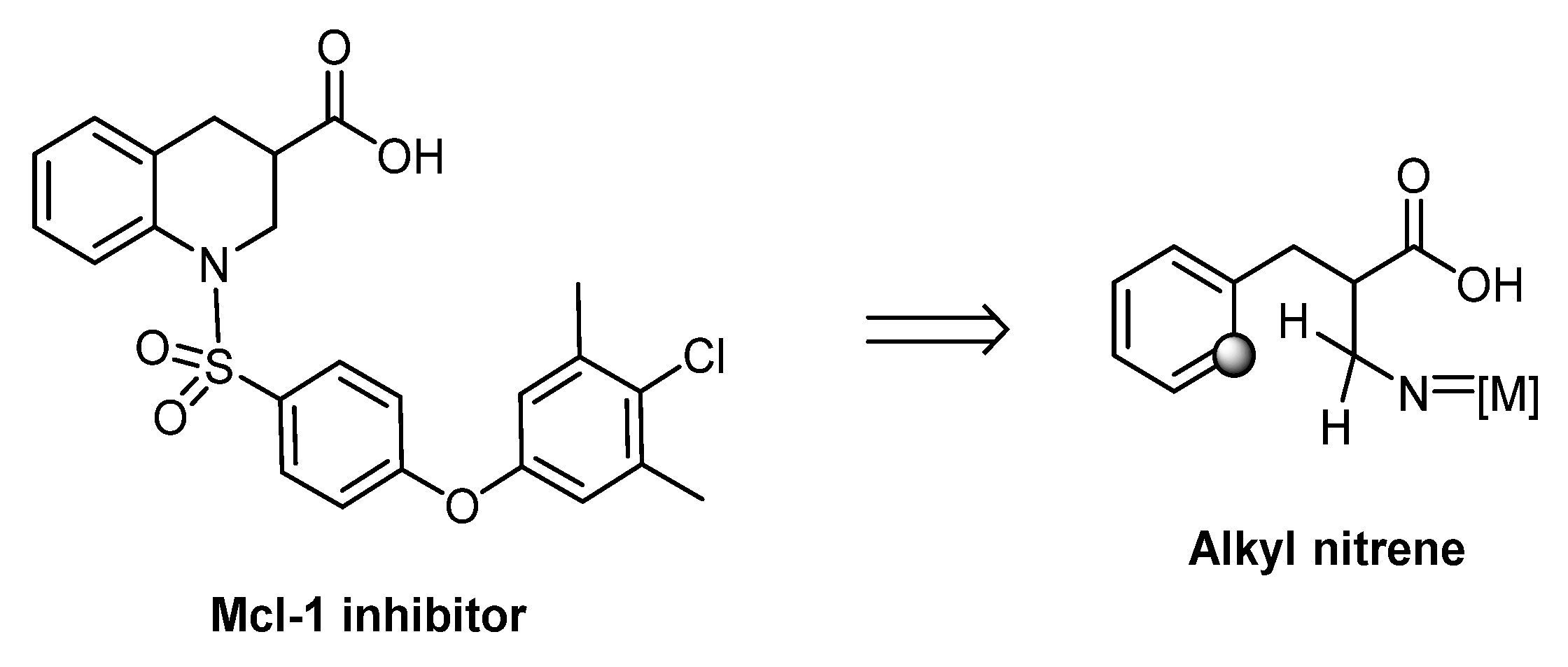
- Tak, R.K.; Amemiya, F.; Noda, H.; Shibasaki, M. Generation and application of Cu-bound alkyl nitrenes for the catalyst-controlled synthesis of cyclic β-amino acids. Chem. Sci. 2021, 12, 7809–7817. [Google Scholar] [CrossRef]
- Mittal, P.; Singh, S.; Sinha, R.; Shrivastava, A.; Singh, A.; Singh, I.K. A review on myeloid cell Leukemia 1 (MCL-1): Structural characteristics and application in cancer therapy. Int. J. Biol. Macromol. 2021, 187, 999–1018. [Google Scholar] [CrossRef] [PubMed]
- Pires, M.; Poeira, D.; Purificação, S.; Marques, M.M.B. Synthesis of substituted 4-, 5-, 6-, and 7-azaindoles from aminopyridines via. a cascade C–N cross-coupling/heck reaction. Org. Lett. 2016, 18, 3250–3253. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.S.; Seo, J.Y.; Yum, E.K. N-arylation of azaindoles in LiCl-mediated catalytic CuI reactions. Tetrahedron Lett. 2007, 48, 4831–4833. [Google Scholar] [CrossRef]
- Qian, G.; Hong, X.; Liu, B.; Mao, H.; Xu, B. Rhodium-catalyzed regioselective C–H chlorination of 7-azaindoles using 1,2-dichloroethane. Org. Lett. 2014, 16, 5294–5297. [Google Scholar] [CrossRef]
- Santos, A.S.; Mortinho, A.C.; Marques, M.M.B. Metal-catalyzed cross-coupling reactions on azaindole synthesis and functionalization. Molecules 2018, 23, 2673. [Google Scholar] [CrossRef]
- Purificação, S.I.; Pires, M.; Rippel, R.; Santos, A.S.; Marques, M.M.B. One-pot synthesis of 1,2-disubstituted 4-, 5-, 6-, and 7-azaindoles from amino-o-halopyridines via. N-arylation/sonogashira/cyclization reaction. Org. Lett. 2017, 19, 5118–5121. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.S.; Martins, M.M.; Mortinho, A.C.; Silva, A.M.; Marques, M.M.B. Exploring the reactivity of halogen-free aminopyridines in one-pot palladium-catalyzed C–N cross-coupling/C–H functionalization. Tetrahedron Lett. 2020, 61, 152303. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, B.; Young, D.J.; Li, H.-X. Acceptorless dehydrogenative coupling with Ru-based catalysts for the synthesis of N-heteroaromatic compounds. Dalton Trans. 2020, 49, 15527–15547. [Google Scholar] [CrossRef]
- Mérour, J.-Y.; Buron, F.; Plé, K.; Bonnet, P.; Routier, S. The azaindole framework in the design of kinase inhibitors. Molecules 2014, 19, 19935–19979. [Google Scholar] [CrossRef]
- Rodriguez-Oliva, I.; Losada-Garcia, N.; Santos, A.S.; Marques, M.M.B.; Palomo, J.M. Palladium nanocatalysts for cascade C−N cross-coupling/heck reaction. Asian J. Org. Chem. 2021, 10, 872–878. [Google Scholar] [CrossRef]
- Martín, N.; Cirujano, F.G. Organic synthesis of high added value molecules with MOF catalysts. Org. Biomol. Chem. 2020, 18, 8058–8073. [Google Scholar] [CrossRef]
- Corma, A. Heterogeneous catalysis: Understanding for designing, and designing for applications. Angew. Chem. Int. Ed. 2016, 55, 6112–6113. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, A.I.; Llanes, P.; Pericàs, M.A. A versatile, immobilized gold catalyst for the reductive amination of aldehydes in batch and flow. React. Chem. Eng. 2018, 3, 714–721. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, Z.; Meanwell, N.A.; Kadow, J.F.; Wang, T. A general method for the preparation of 4- and 6-azaindoles. J. Org. Chem. 2002, 67, 2345–2347. [Google Scholar] [CrossRef] [PubMed]
- Popowycz, F.; Mérour, J.-Y.; Joseph, B. Synthesis and reactivity of 4-, 5- and 6-azaindoles. Tetrahedron 2007, 63, 8689–8707. [Google Scholar] [CrossRef]
- Guandalini, L.; Martini, E.; Gualtieri, F.; Romanelli, M.N.; Varani, K. Design, synthesis and preliminary pharmacological evaluation of rigid analogues of the nicotinic agonist 1,1-dimethyl-4-phenylpiperazinium iodide (DMPP). Arkivoc 2004, 5, 286–300. [Google Scholar] [CrossRef]
- Pires, M.; Purificação, S.; Santos, A.S.; Marques, M.M.B. The role of PEG on Pd- and Cu-catalyzed cross-coupling reactions. Synthesis 2017, 49, 2337–2350. [Google Scholar] [CrossRef]
- Dias Pires, M.J.; Poeira, D.L.; Marques, M.M.B. Metal-catalyzed cross-coupling reactions of aminopyridines. Eur. J. Org. Chem. 2015, 7197–7234. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
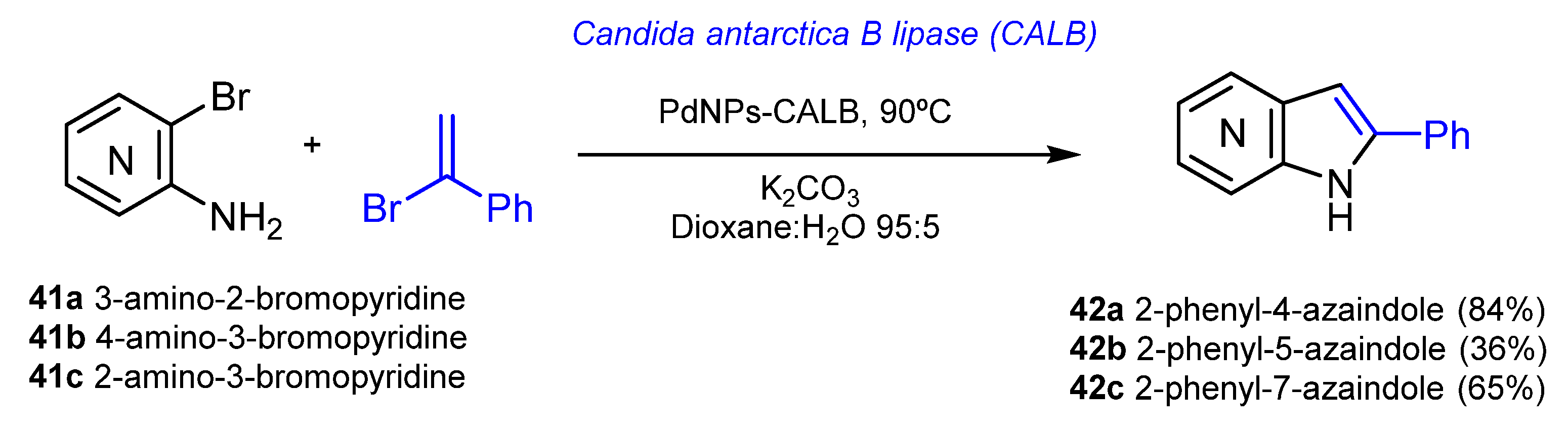{kind=link}
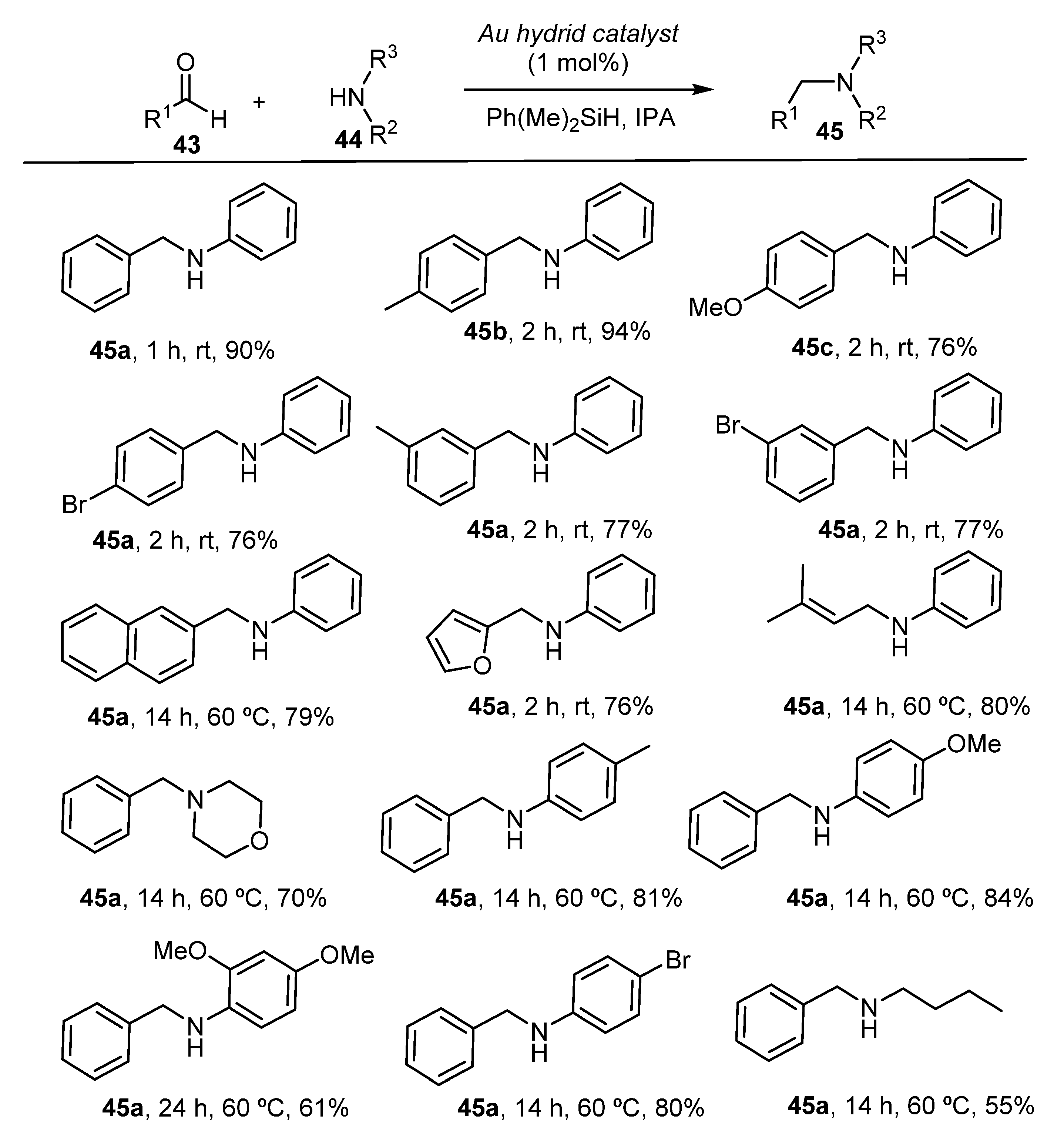{kind=link}
| Azaindole Synthesis | ||
|---|---|---|
| Classical Methods [57,58,59] | Metal-Catalyzed Methods [48,49,50,53] | |
| Advantages | Effective in attaining the product Moderated yields | One-pot reactions Less waste Compatible with water Mild conditions Atom economy |
| Disadvantages | Limited scope Stoichiometric reagents Strong acids High temperature Poor yields of some isomers | Catalysts may be expensive Moderate to high yields |
| Scope | Limited to few regioisomers | Access to all regioisomers |
| Energy efficiency | High energy consumption from traditional heating methods | Compatible with MW heating |
| Solvent | Hazardous and chlorinated | Compatible with water |
| N° of steps | At least 2 steps | Maximum 2 steps |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, A.S.; Raydan, D.; Cunha, J.C.; Viduedo, N.; Silva, A.M.S.; Marques, M.M.B. Advances in Green Catalysis for the Synthesis of Medicinally Relevant N-Heterocycles. Catalysts 2021, 11, 1108. https://doi.org/10.3390/catal11091108
Santos AS, Raydan D, Cunha JC, Viduedo N, Silva AMS, Marques MMB. Advances in Green Catalysis for the Synthesis of Medicinally Relevant N-Heterocycles. Catalysts. 2021; 11(9):1108. https://doi.org/10.3390/catal11091108
Chicago/Turabian StyleSantos, A. Sofia, Daniel Raydan, José C. Cunha, Nuno Viduedo, Artur M. S. Silva, and M. Manuel B. Marques. 2021. "Advances in Green Catalysis for the Synthesis of Medicinally Relevant N-Heterocycles" Catalysts 11, no. 9: 1108. https://doi.org/10.3390/catal11091108
APA StyleSantos, A. S., Raydan, D., Cunha, J. C., Viduedo, N., Silva, A. M. S., & Marques, M. M. B. (2021). Advances in Green Catalysis for the Synthesis of Medicinally Relevant N-Heterocycles. Catalysts, 11(9), 1108. https://doi.org/10.3390/catal11091108