
Hydrodesulfurization of 4,6-Dimethyldibenzothiophene and the Diesel Oil Fraction on NiMo Catalysts Supported over Proton-Exchanged AlMCM-41 and TiMCM-41 Extrudates

 , , ,
, , ,
Abstract
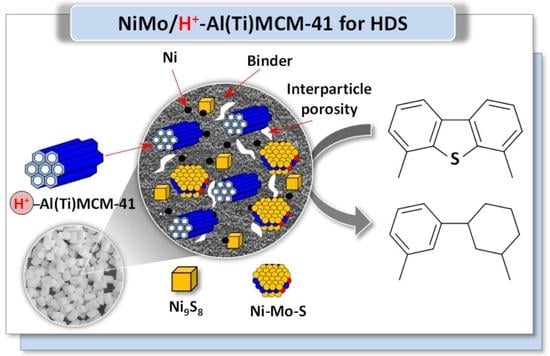
:
1. Introduction
2. Results and Discussion
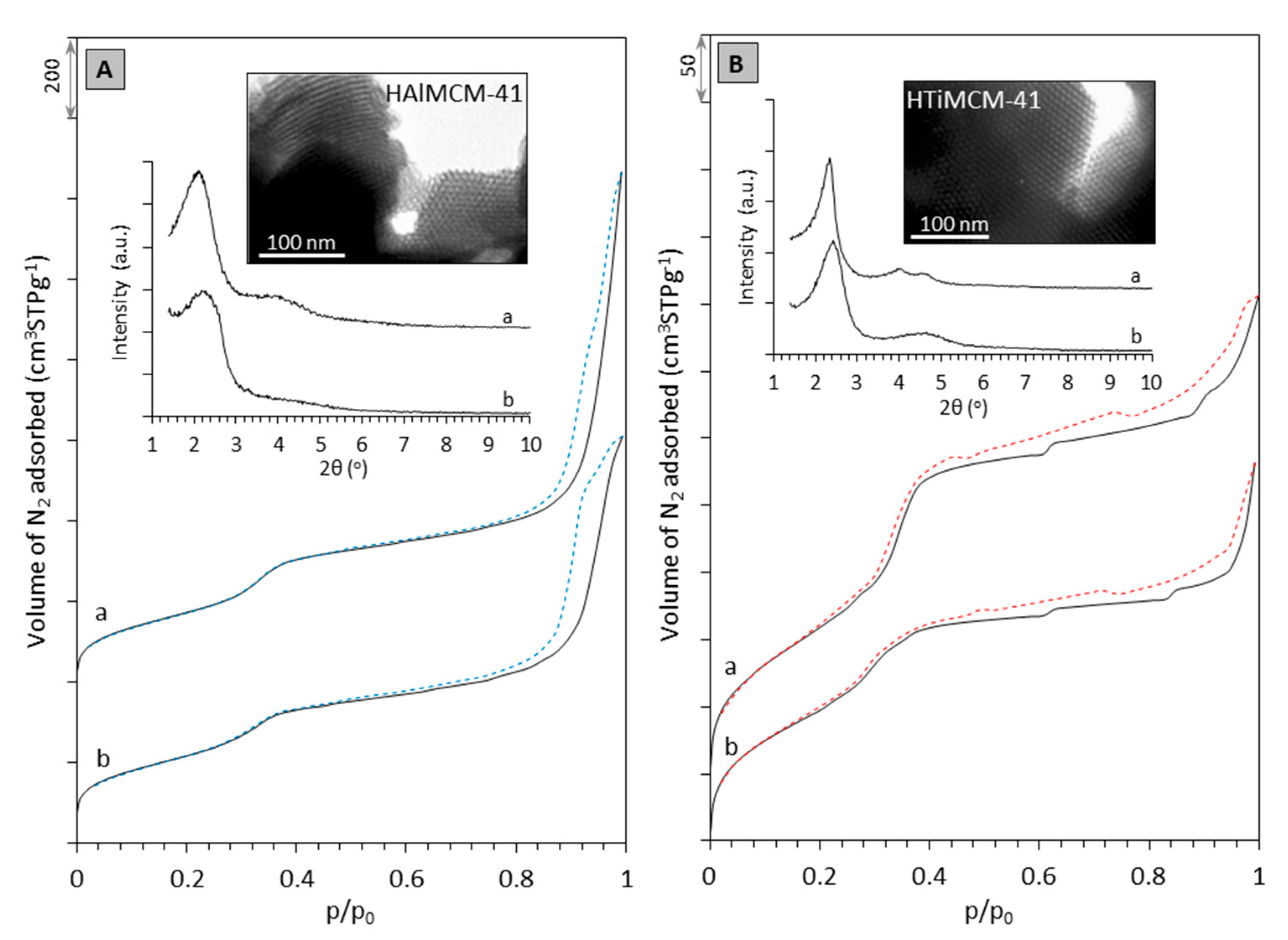
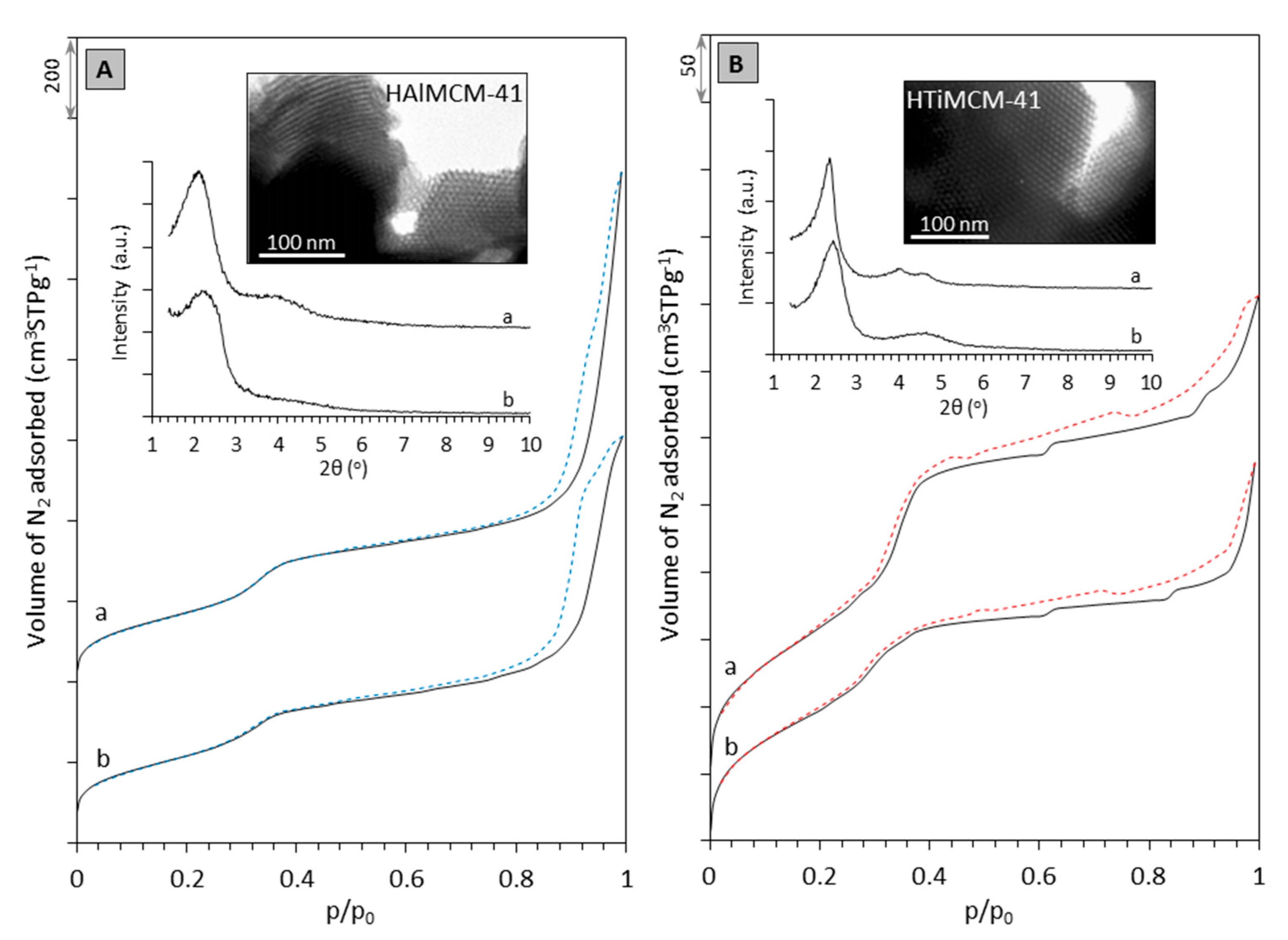
2.1. Characterization of Supports and Catalysts
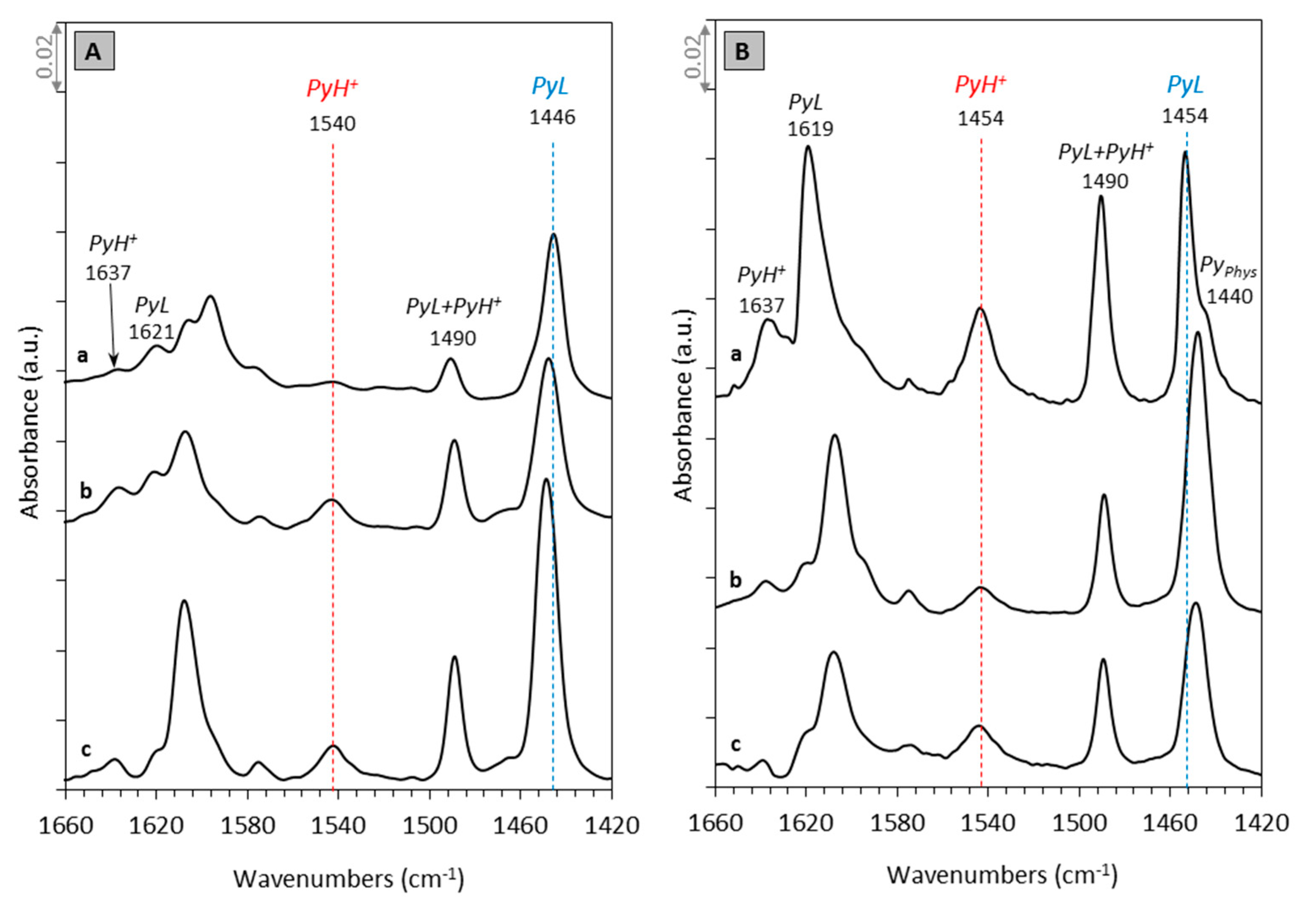
2.2. Acidity by Py–IR
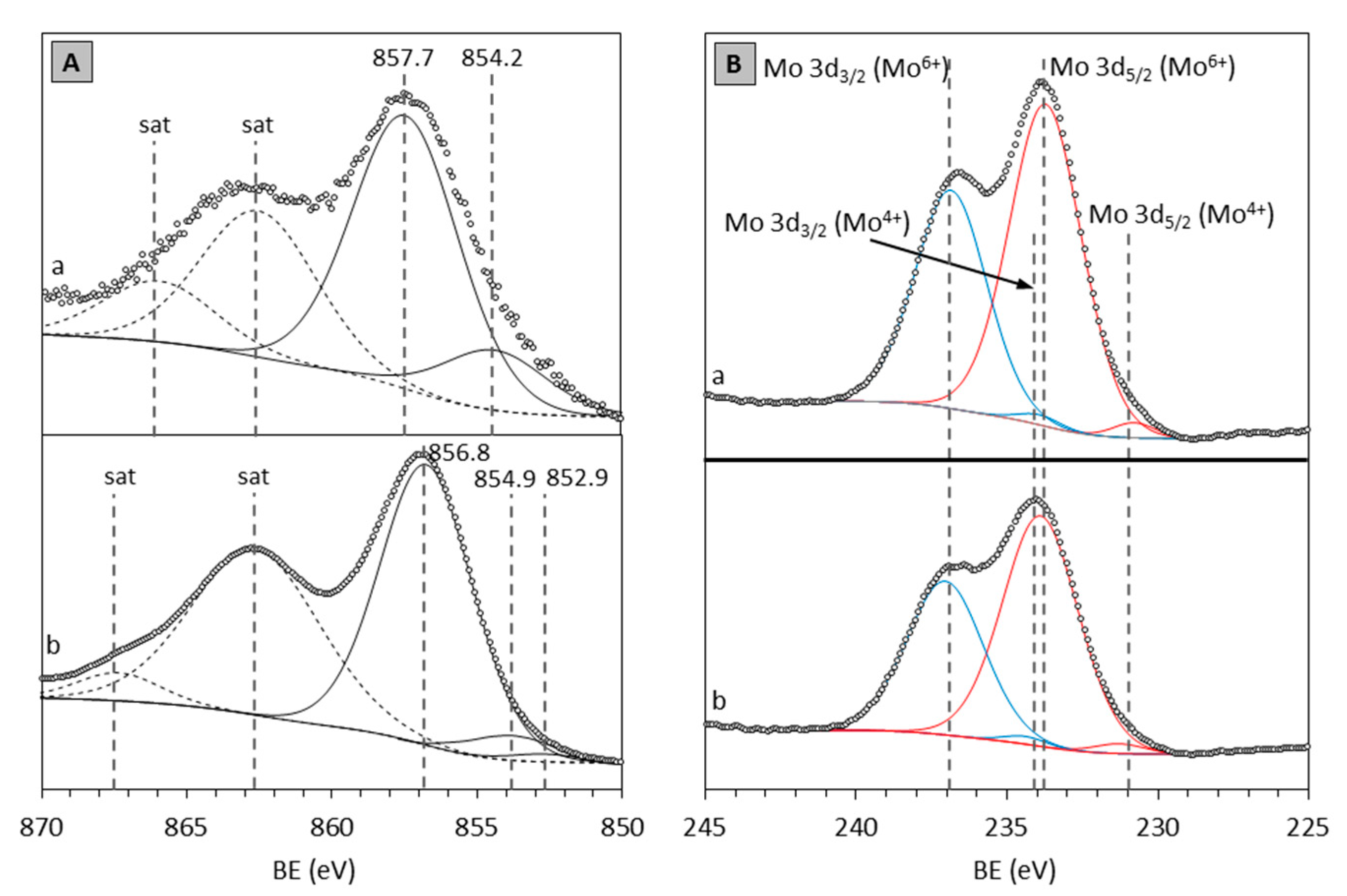
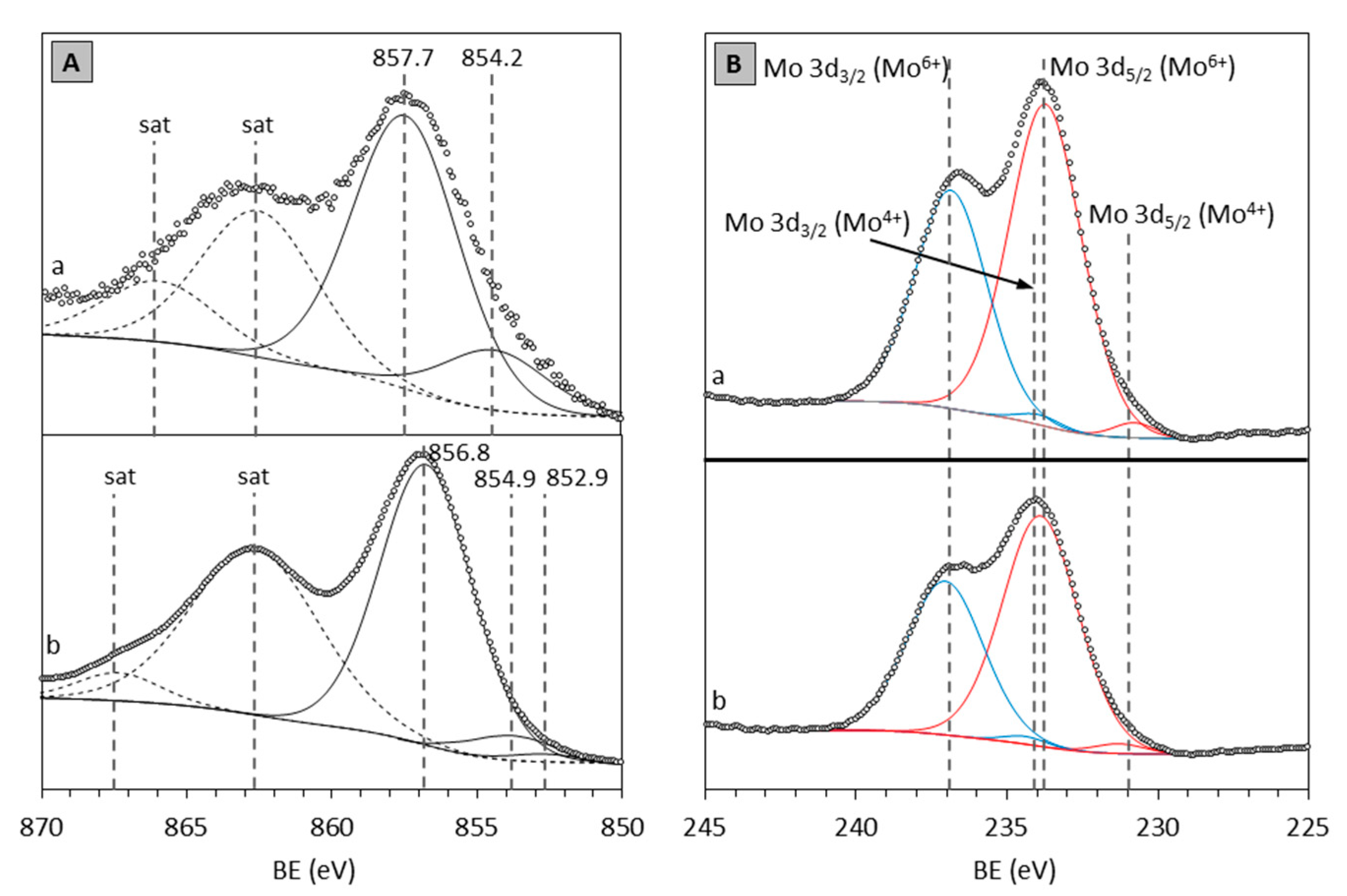
2.3. XPS
2.4. SEM
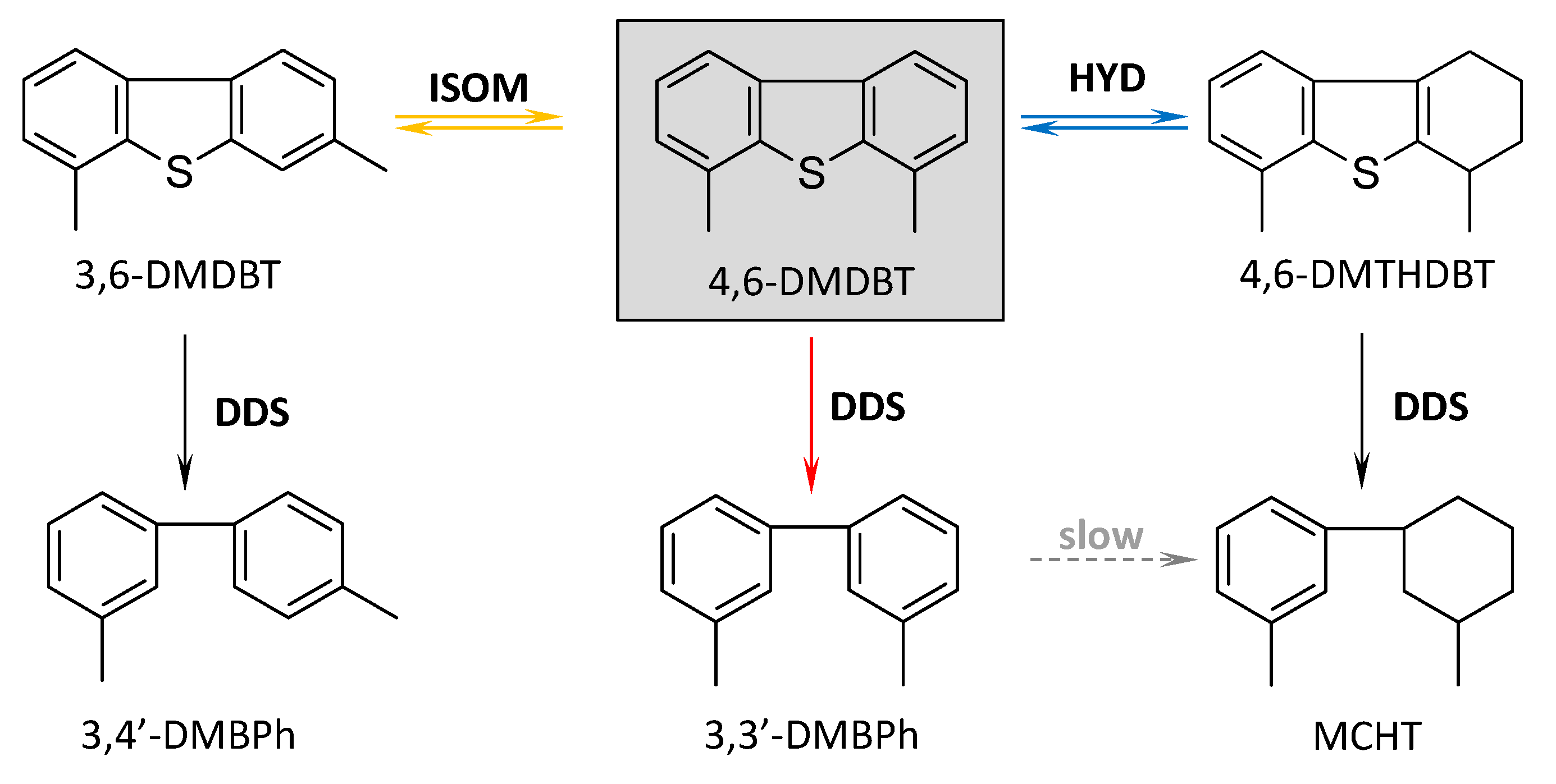
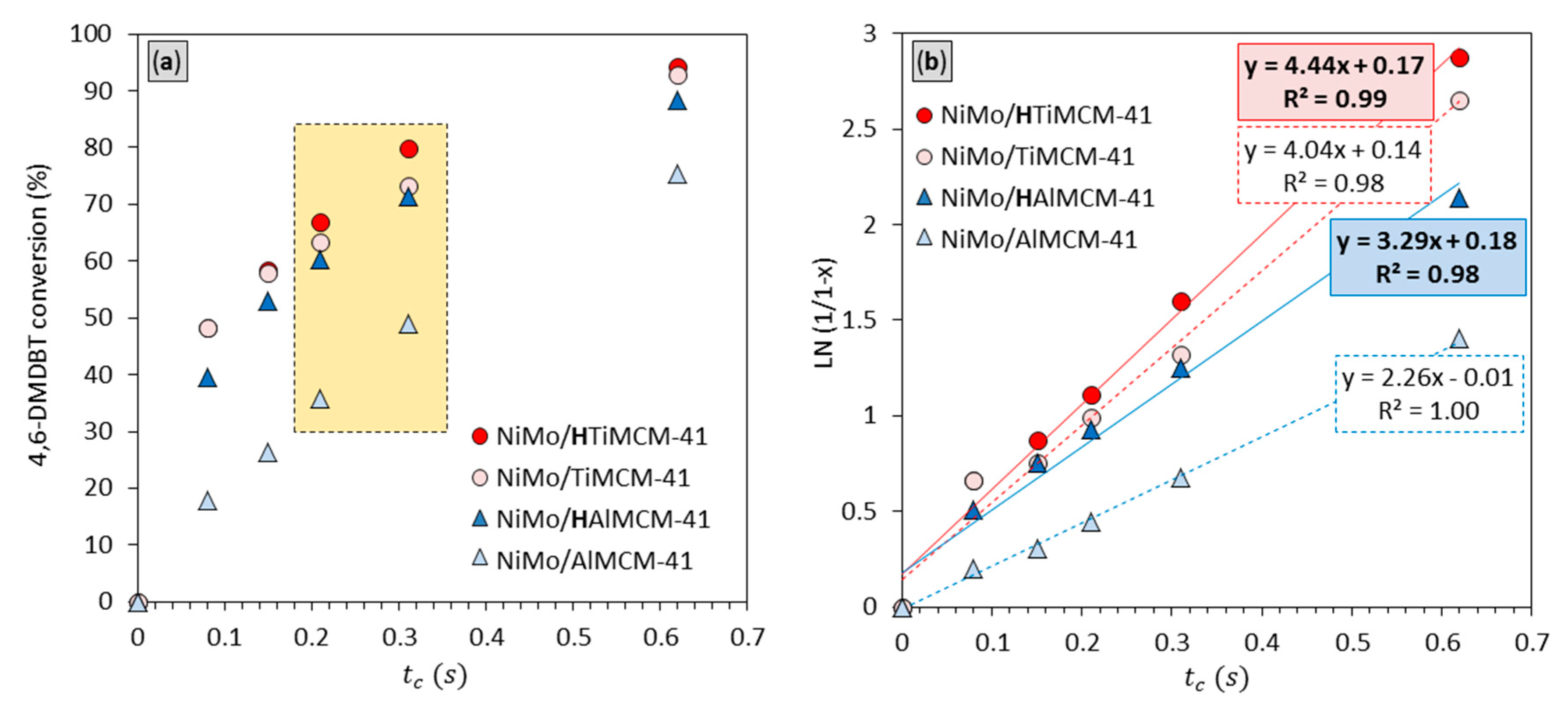
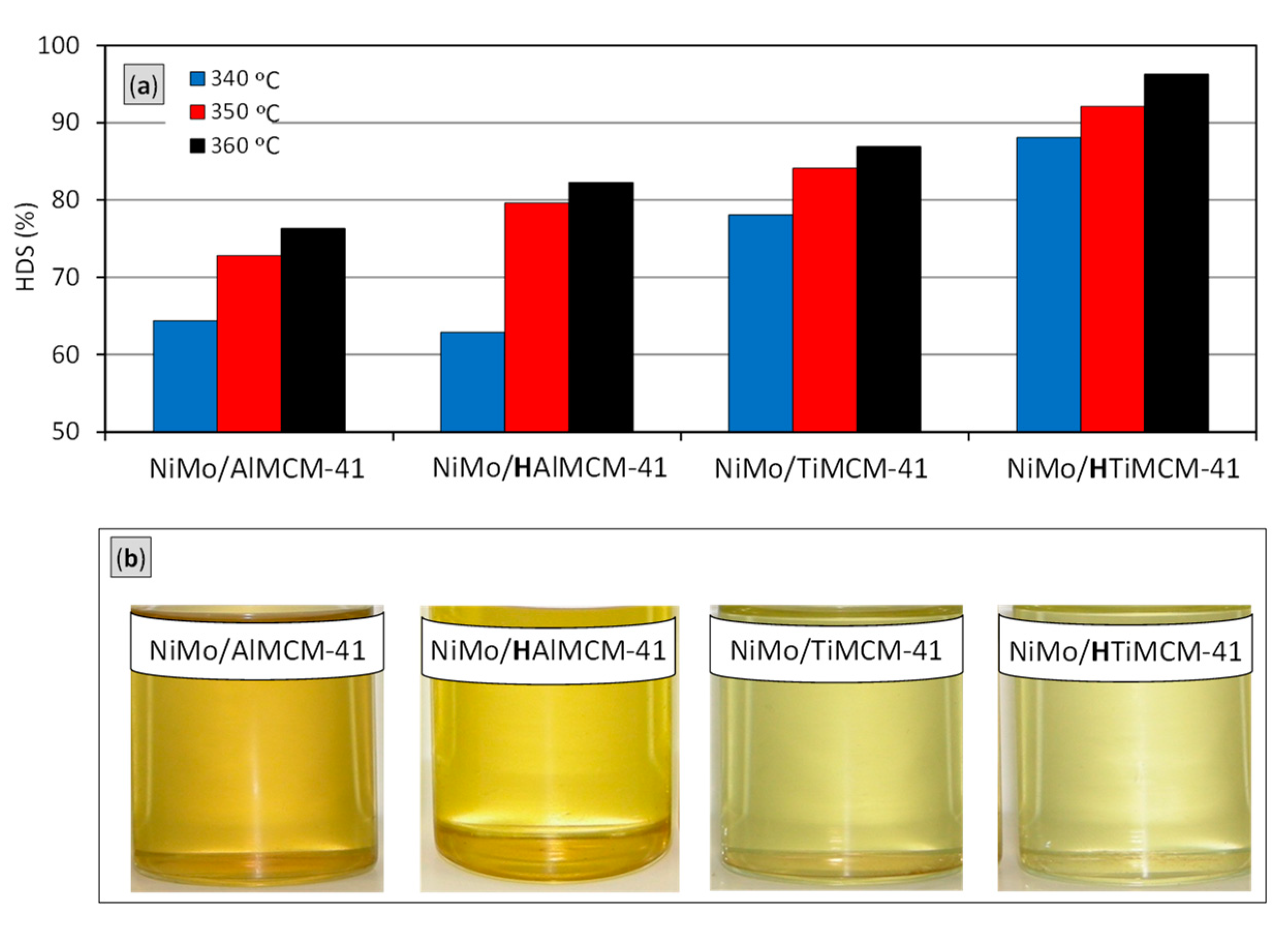
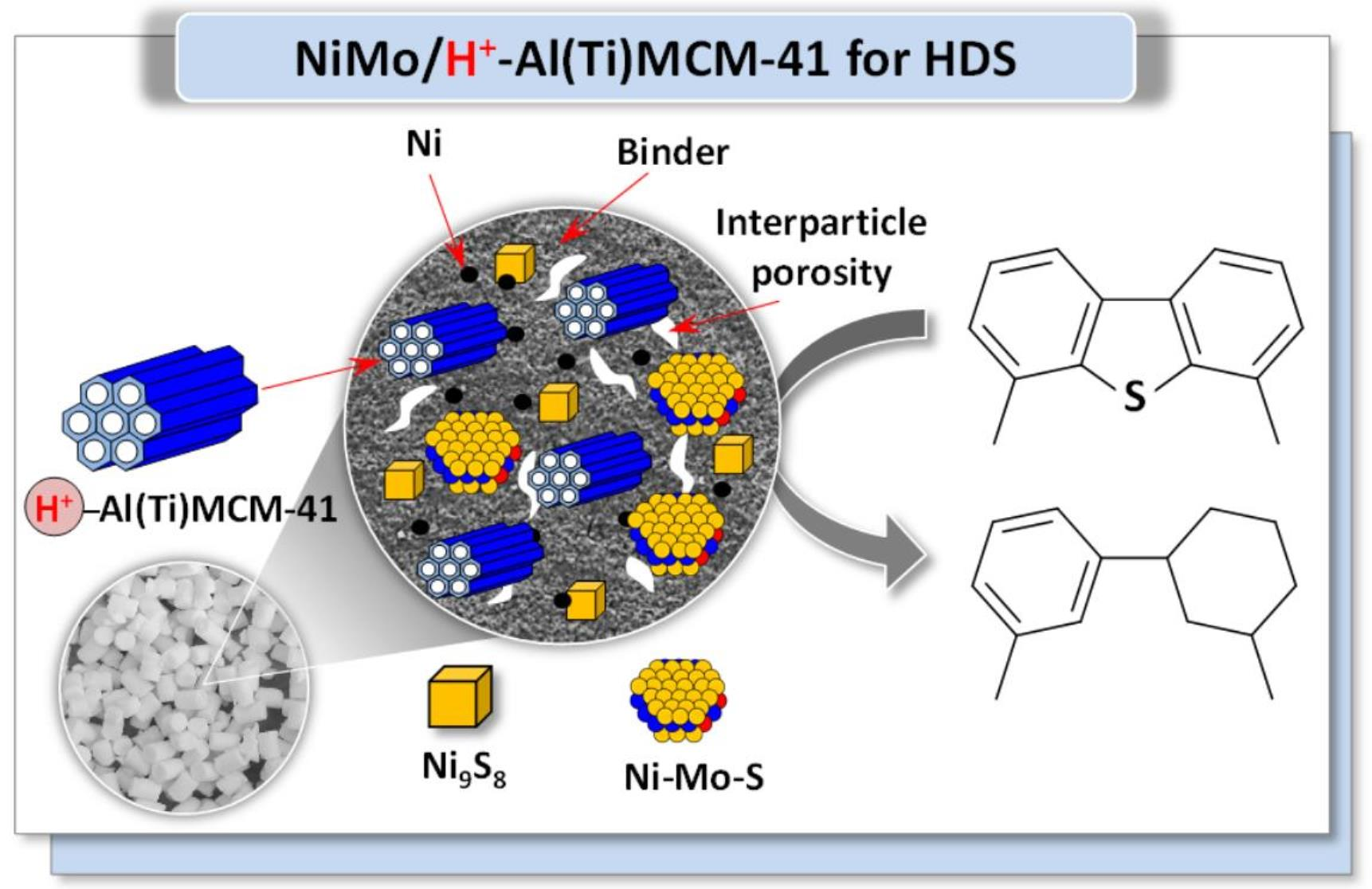
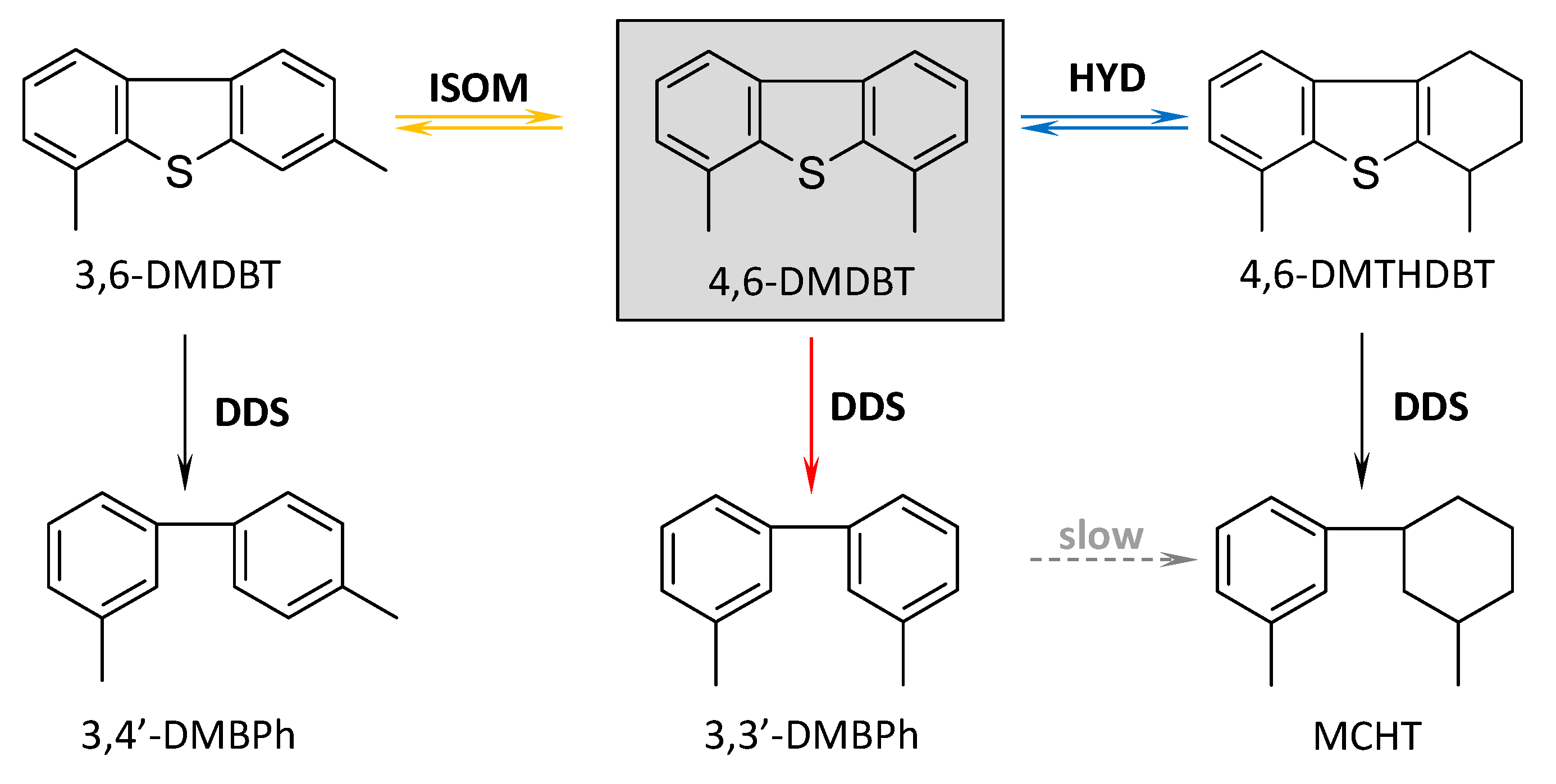
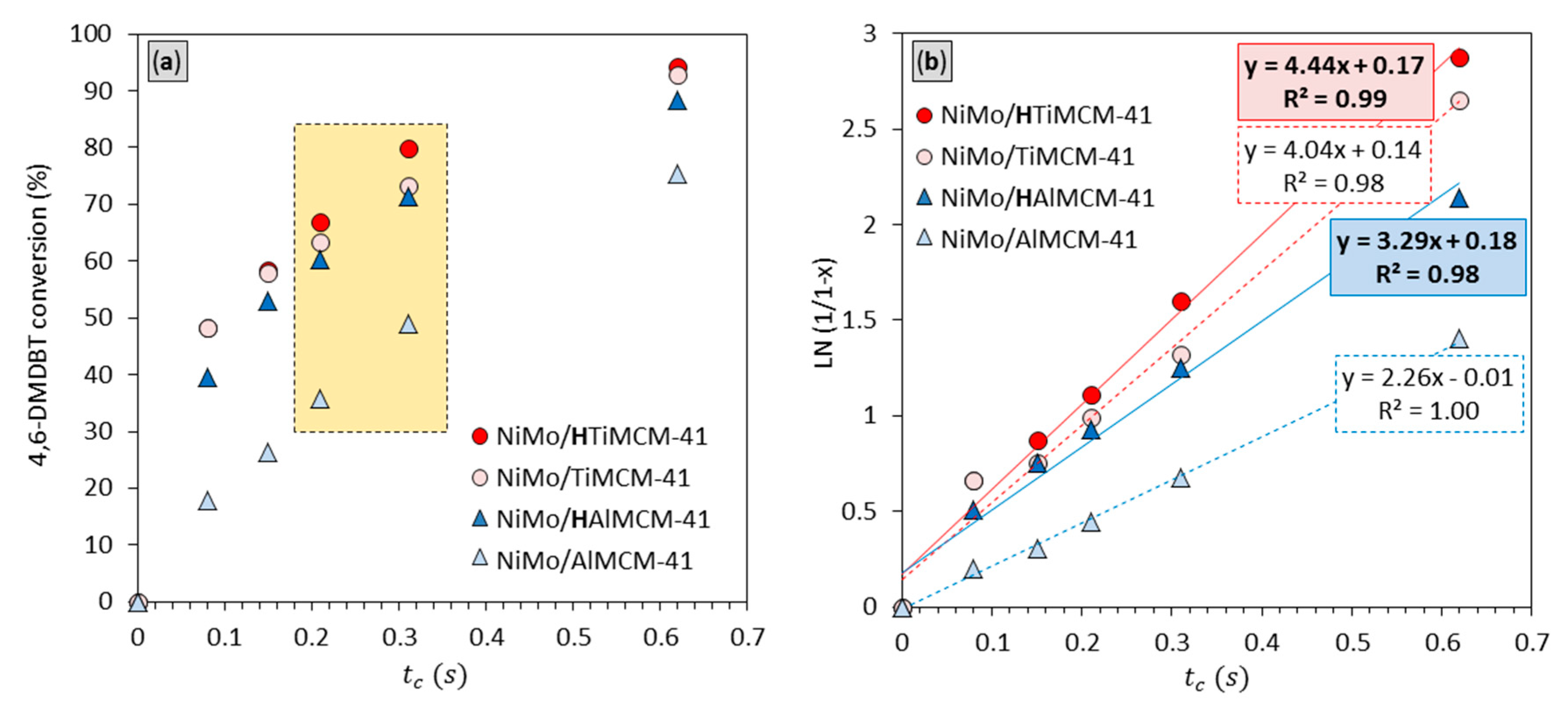
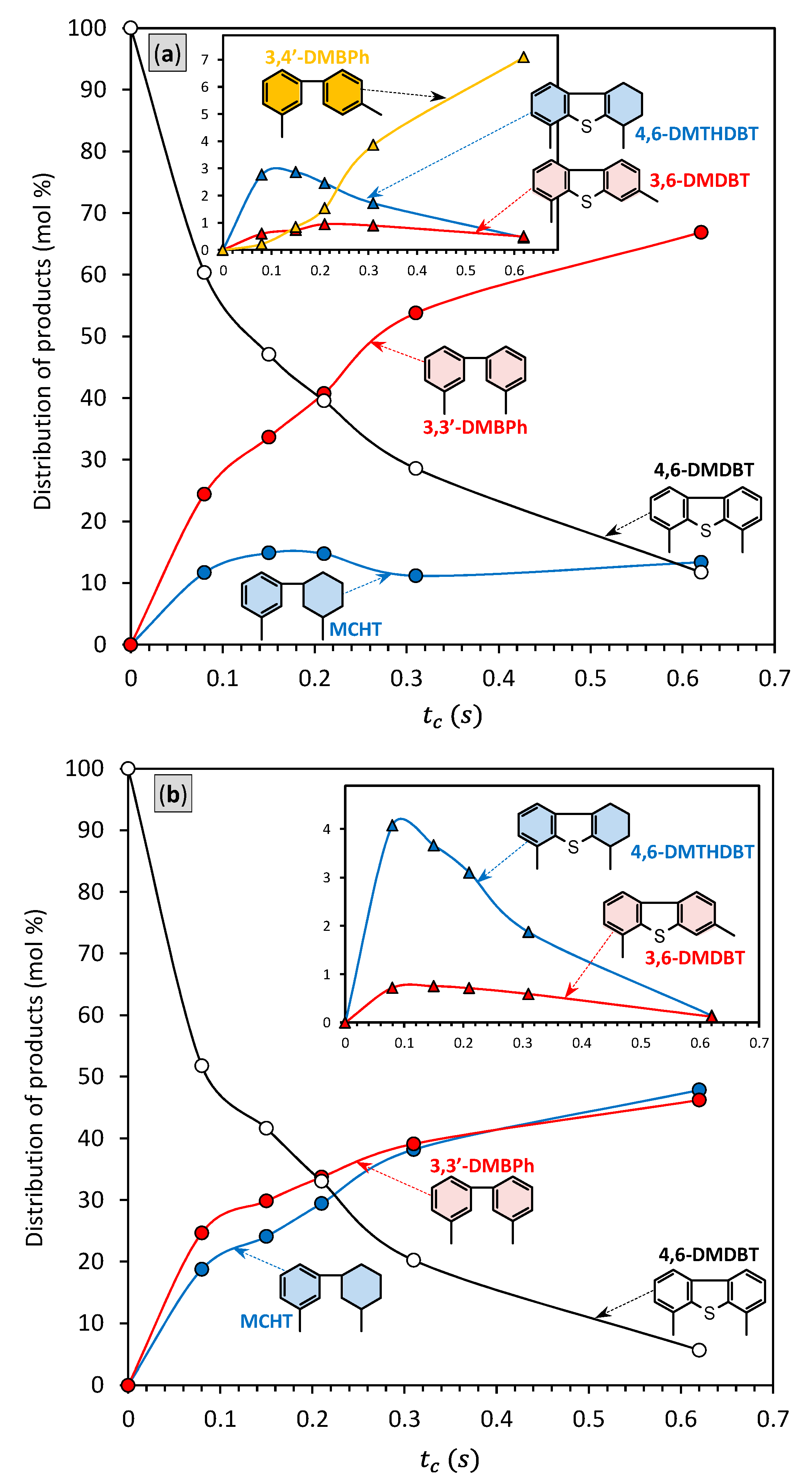
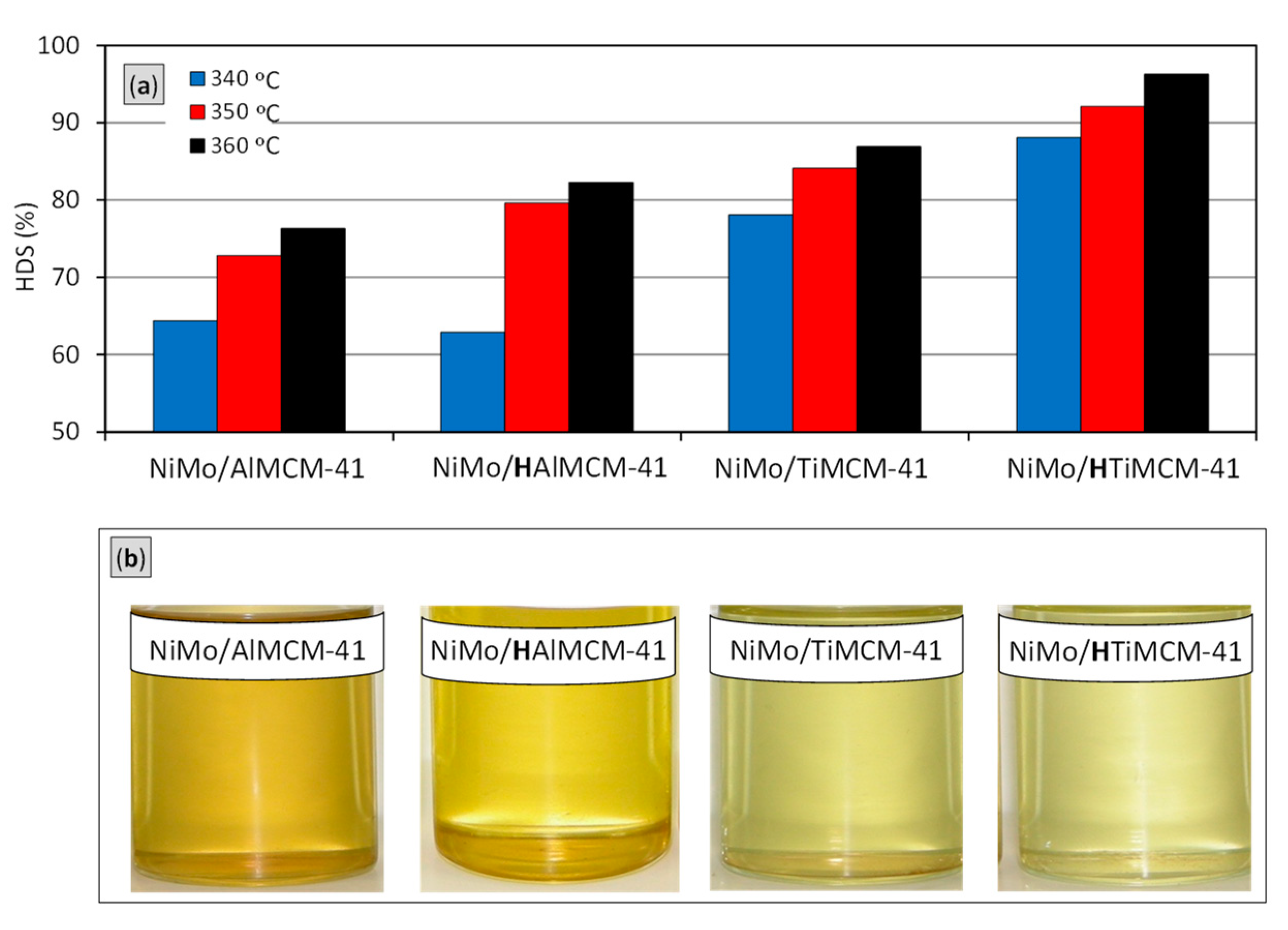
2.5. Catalytic Performance
3. Materials and Methods
3.1. Materials
3.2. Support and Catalyst Preparation
3.3. Characterization of Supports and Catalysts
3.4. Catalytic Activity Assessment
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alotaibi, F.M.; González-Cortés, S.; Alotibi, M.F.; Xiao, T.; Al-Megren, H.; Yang, G.; Edwards, P.P. Enhancing the production of light olefins from heavy crude oils: Turning challenges into opportunities. Catal. Today 2018, 317, 86–98. [Google Scholar] [CrossRef]
- Trongkaew, P.; Utistham, T.; Reubroycharoen, P.; Hinchiranan, N. Photocatalytic desulfurization of waste tire pyrolysis oil. Energies 2011, 4, 1880–1896. [Google Scholar] [CrossRef]
- Pieterse, J.A.Z.; Van Eijk, S.; Van Dijk, H.A.J.; Van Den Brink, R.W. On the potential of absorption and reactive adsorption for desulfurization of ultra low-sulfur commercial diesel in the liquid phase in the presence of fuel additive and bio-diesel. Fuel Process. Technol. 2011, 92, 616–623. [Google Scholar] [CrossRef]
- Stanislaus, A.; Marafi, A.; Rana, M.S. Recent advances in the science and technology of ultra low sulfur diesel (ULSD) production. Catal. Today 2010, 153, 1–68. [Google Scholar] [CrossRef]
- Shamsaee, B.H.; Mehri, F.; Rowshanzamir, S.; Ghamati, M.; Behrouzifar, A. Desulfurization of benzothiophene from model diesel fuel using experimental (dynamic electroreduction) and theoretical (DFT) approaches. Sep. Purif. Technol. 2019, 212, 505–514. [Google Scholar] [CrossRef]
- Tanimu, A.; Alhooshani, K. Advanced Hydrodesulfurization Catalysts: A Review of Design and Synthesis. Energy Fuels 2019, 33, 2810–2838. [Google Scholar] [CrossRef]
- Bej, S.K.; Maity, S.K.; Turaga, U.T. Catalyst: A Review of Recent Studies. Energy Fuels 2005, 18, 1227. [Google Scholar] [CrossRef]
- Hossain, M.N.; Park, H.C.; Choi, H.S. A comprehensive review on catalytic oxidative desulfurization of liquid fuel oil. Catalysts 2019, 9, 229. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Zhang, X.; Guo, R.; Ding, S.; Zheng, P.; Fan, J.; Mei, J.; Xu, C.; Duan, A. Synergistic effect of acidity and active phases for NiMo catalysts on dibenzothiophene hydrodesulfurization performance. Chem. Eng. J. 2020, 400, 125886. [Google Scholar] [CrossRef]
- Esquivel, G.M.; Ramírez, J.; Gutiérrez-Alejandre, A. HDS of 4,6-DMDBT over NiW/Al-SBA15 catalysts. Catal. Today 2010, 148, 36–41. [Google Scholar] [CrossRef]
- Alonso-Pérez, M.O.; Pawelec, B.; Zepeda, T.A.; Alonso-Núñez, G.; Nava, R.; Navarro, R.M.; Huirache-Acuña, R. Effect of the titanium incorporation method on the morphology and HDS activity of supported ternary Ni–Mo–W/SBA-16 catalysts. Microporous Mesoporous Mater. 2021, 312, 110779. [Google Scholar] [CrossRef]
- Luo, Q.; Zhou, Q.; Lin, Y.; Wu, S.; Liu, H.; Du, C.; Zhong, Y.; Yang, C. Fast and deep oxidative desulfurization of dibenzothiophene with catalysts of MoO3-TiO2@MCM-22 featuring adjustable Lewis and Brønsted acid sites. Catal. Sci. Technol. 2019, 9, 6166–6179. [Google Scholar] [CrossRef]
- Han, W.; Nie, H.; Long, X.; Li, M.; Yang, Q.; Li, D. Effects of the support BrØnsted acidity on the hydrodesulfurization and hydrodenitrogention activity of sulfided NiMo/Al2O3 catalysts. Catal. Today 2017, 292, 58–66. [Google Scholar] [CrossRef]
- Brunet, S.; Lebeau, B.; Naboulsi, I.; Michelin, L.; Comparot, J.D.; Marichal, C.; Rigolet, S.; Bonne, M.; Blin, J. Effect of Mesostructured Zirconia Support on the Activity and Selectivity of 4,6-Dimethydibenzothiophene Hydrodesulfurization. Catalysts 2020, 10, 1162. [Google Scholar] [CrossRef]
- Liu, H.; Li, Y.; Yin, C.; Wu, Y.; Chai, Y.; Dong, D.; Li, X.; Liu, C. One-pot synthesis of ordered mesoporous NiMo-Al2O3 catalysts for dibenzothiophene hydrodesulfurization. Appl. Catal. B Environ. 2016, 198, 493–507. [Google Scholar] [CrossRef]
- Huirache-Acuña, R.; Pérez-Ayala, E.; Cervantes-Gaxiola, M.E.; Alonso-Nuñez, G.; Zepeda, T.A.; Rivera-Muñoz, E.M.; Pawelec, B. Dibenzothiophene hydrodesulfurization over ternary metallic NiMoW/Ti-HMS mesoporous catalysts. Catal. Commun. 2021, 148, 106162. [Google Scholar] [CrossRef]
- Méndez, F.J.; Franco-López, O.E.; Bokhimi, X.; Solís-Casados, D.A.; Escobar-Alarcón, L.; Klimova, T.E. Dibenzothiophene hydrodesulfurization with NiMo and CoMo catalysts supported on niobium-modified MCM-41. Appl. Catal. B Environ. 2017, 219, 479–491. [Google Scholar] [CrossRef]
- Méndez, F.J.; Franco-López, O.E.; Díaz, G.; Gómez-Cortés, A.; Bokhimi, X.; Klimova, T.E. On the role of niobium in nanostructured Mo/Nb-MCM-41 and NiMo/Nb-MCM-41 catalysts for hydrodesulfurization of dibenzothiophene. Fuel 2020, 280, 118550. [Google Scholar] [CrossRef]
- Rauscher, T.; Müller, C.I.; Schmidt, A.; Kieback, B.; Röntzsch, L. Ni-Mo-B alloys as cathode material for alkaline water electrolysis. Int. J. Hydrogen Energy 2016, 41, 2165–2176. [Google Scholar] [CrossRef]
- Jaroszewska, K.; Lewandowski, M.; Grzechowiak, J.R.; Szyja, B. Author’s personal copy Hydrodesulphurisation of 4, 6-dimethyldibenzothiophene over NiMo catalysts supported on Ti (Al) modified MCM-41. Catal. Today 2011, 176, 202–207. [Google Scholar] [CrossRef]
- Nava, R.; Ortega, R.A.; Alonso, G.; Ornelas, C.; Pawelec, B.; Fierro, J.L.G. CoMo/Ti-SBA-15 catalysts for dibenzothiophene desulfurization. Catal. Today 2007, 127, 70–84. [Google Scholar] [CrossRef]
- Wang, A.; Wanga, Y.; Chen, Y.; Li, X.; Yao, P.; Kabe, T. Hydrodesulfurization of dibenzothiophene over Mo-based catalysts supported by siliceous MCM-41. Stud. Surf. Sci. Catal. 2002, 142, 795–798. [Google Scholar] [CrossRef]
- Wang, A.; Li, X.; Wang, Y.; Sun, Z.; Li, C.; Hu, Y. Hydrodesulfurizatton of dibenzothiophene over proton-exchanged siliceous MCM-41 supported bimetallic sulfides. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 2004; Volume 154, pp. 2930–2935. [Google Scholar]
- Trejda, M.; Ziolek, M. New iron containing mesoporous catalysts. Catal. Today 2005, 101, 109–116. [Google Scholar] [CrossRef]
- Sluban, M.; Cojocaru, B.; Parvulescu, V.I.; Iskra, J.; Cerc Korošec, R.; Umek, P. Protonated titanate nanotubes as solid acid catalyst for aldol condensation. J. Catal. 2017, 346, 161–169. [Google Scholar] [CrossRef]
- Camposeco, R.; Castillo, S.; Mejia-Centeno, I.; Navarrete, J.; Rodriguez-Gonzalez, V. Behavior of Lewis and Brönsted surface acidity featured by Ag, Au, Ce, La, Fe, Mn, Pd, Pt, V and W decorated on protonated titanate nanotubes. Microporous Mesoporous Mater. 2016, 236, 235–243. [Google Scholar] [CrossRef]
- Vutolkina, A.V.; Glotov, A.P.; Zanina, A.V.; Makhmutov, D.F.; Maximov, A.L.; Egazar’yants, S.V.; Karakhanov, E.A. Mesoporous Al-HMS and Al-MCM-41 supported Ni-Mo sulfide catalysts for HYD and HDS via in situ hydrogen generation through a WGSR. Catal. Today 2019, 329, 156–166. [Google Scholar] [CrossRef]
- Cánepa, A.L.; Vaschetti, V.M.; Pájaro, K.C.; Eimer, G.A.; Casuscelli, S.G.; Cortés Corberán, V. Selective oxidation of ethanol on V-MCM-41 catalysts. Catal. Today 2020, 356, 464–470. [Google Scholar] [CrossRef]
- Kwak, K.Y.; Kim, M.S.; Lee, D.W.; Cho, Y.H.; Han, J.; Kwon, T.S.; Lee, K.Y. Synthesis of cyclopentadiene trimer (tricyclopentadiene) over zeolites and Al-MCM-41: The effects of pore size and acidity. Fuel 2014, 137, 230–236. [Google Scholar] [CrossRef]
- Shestakova, P.; Popova, M.; Szegedi, Á.; Lazarova, H.; Nga Luong, T.K.; Trendafilova, I.; Mihály, J.; Parac-Vogt, T.N. Hybrid catalyst with combined Lewis and Brønsted acidity based on ZrIV substituted polyoxometalate grafted on mesoporous MCM-41 silica for esterification of renewable levulinic acid. Microporous Mesoporous Mater. 2021, 323, 111203. [Google Scholar] [CrossRef]
- Ziaei-Azad, H.; Kolle, J.M.; Al-Yasser, N.; Sayari, A. One-pot synthesis of large-pore AlMCM-41 aluminosilicates with high stability and adjustable acidity. Microporous Mesoporous Mater. 2018, 262, 166–174. [Google Scholar] [CrossRef]
- Araújo, R.S.; Maia, D.A.S.; Azevedo, D.C.S.; Cavalcante, C.L.; Rodríguez-Castellón, E.; Jimenez-Lopez, A. Assessment of surface acidity in mesoporous materials containing aluminum and titanium. Appl. Surf. Sci. 2009, 255, 6205–6209. [Google Scholar] [CrossRef]
- Ng, E.P.; Nur, H.; Wong, K.L.; Muhid, M.N.M.; Hamdan, H. Generation of Brönsted acidity in AlMCM-41 by sulphation for enhanced liquid phase tert-butylation of phenol. Appl. Catal. A Gen. 2007, 323, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Góra-Marek, K.; Datka, J. IR studies of OH groups in mesoporous aluminosilicates. Appl. Catal. A Gen. 2006, 302, 104–109. [Google Scholar] [CrossRef]
- Tekla, J.; Tarach, K.A.; Olejniczak, Z.; Girman, V.; Góra-Marek, K. Effective hierarchization of TS-1 and its catalytic performance in cyclohexene epoxidation. Microporous Mesoporous Mater. 2016, 233, 16–25. [Google Scholar] [CrossRef]
- Wada, E.; Kitano, M.; Nakajima, K.; Hara, M. Effect of preparation conditions on the structural and acid catalytic properties of protonated titanate nanotubes. J. Mater. Chem. A 2013, 1, 12768–12774. [Google Scholar] [CrossRef]
- Li, S.; Li, N.; Li, G.; Li, L.; Wang, A.; Cong, Y.; Wang, X.; Xu, G.; Zhang, T. Protonated titanate nanotubes as a highly active catalyst for the synthesis of renewable diesel and jet fuel range alkanes. Appl. Catal. B Environ. 2015, 170–171, 124–134. [Google Scholar] [CrossRef]
- Kitano, M.; Nakajima, K.; Kondo, J.N.; Hayashi, S.; Hara, M. Protonated Titanate Nanotubes as Solid Acid Catalyst. J. Am. Chem. Soc. 2010, 132, 6622–6623. [Google Scholar] [CrossRef]
- Camposeco, R.; Castillo, S.; Mejía-Centeno, I.; Navarrete, J.; Nava, N.; Rodríguez-González, V. Synthesis of protonated titanate nanotubes tailored by the washing step: Effect upon acid properties and photocatalytic activity. J. Photochem. Photobiol. A Chem. 2017, 341, 87–96. [Google Scholar] [CrossRef]
- Prasad Reddy, B.R.; Govardhana Reddy, P.V.; Kumar, D.P.; Reddy, B.N.; Shankar, M.V. Rapid synthesis of alkylaminophenols via the Petasis borono-Mannich reaction using protonated trititanate nanotubes as robust solid-acid catalysts. RSC Adv. 2016, 6, 14682–14691. [Google Scholar] [CrossRef]
- Kitano, M.; Wada, E.; Nakajima, K.; Hayashi, S.; Miyazaki, S.; Kobayashi, H.; Hara, M. Protonated titanate nanotubes with Lewis and Brønsted acidity: Relationship between nanotube structure and catalytic activity. Chem. Mater. 2013, 25, 385–393. [Google Scholar] [CrossRef]
- Tarach, K.A.; Śrębowata, A.; Kowalewski, E.; Gołąbek, K.; Kostuch, A.; Kruczała, K.; Girman, V.; Góra-Marek, K. Nickel loaded zeolites FAU and MFI: Characterization and activity in water-phase hydrodehalogenation of TCE. Appl. Catal. A Gen. 2018, 568, 64–75. [Google Scholar] [CrossRef]
- Li, M.; Fu, J.; Xing, S.; Yang, L.; Zhang, X.; Lv, P.; Wang, Z.; Yuan, Z. A novel catalyst with variable active sites for the direct hydrogenation of waste oils into jet fuel. Appl. Catal. B Environ. 2020, 260, 118114. [Google Scholar] [CrossRef]
- Navajas, A.; Jim, E.; Romero-sarria, F. Molybdenum Oxide for the Production of Biodiesel from Oil with High Free Fatty Acids Content. 1–14. Catalysts 2020, 10, 158. [Google Scholar] [CrossRef] [Green Version]
- Lakiss, L.; Gilson, J.P.; Valtchev, V.; Mintova, S.; Vicente, A.; Vimont, A.; Bedard, R.; Abdo, S.; Bricker, J. Zeolites in a good shape: Catalyst forming by extrusion modifies their performances. Microporous Mesoporous Mater. 2020, 299. [Google Scholar] [CrossRef]
- Zaki, M.I.; Hasan, M.A.; Al-Sagheer, F.A.; Pasupulety, L. In situ FTIR spectra of pyridine adsorbed on SiO2-Al2O3, TiO2, ZrO2 and CeO2: General considerations for the identification of acid sites on surfaces of finely divided metal oxides. Colloids Surf. A Physicochem. Eng. Asp. 2001, 190, 261–274. [Google Scholar] [CrossRef]
- Martin, C.; Martin, I.; Delmoral, C.; Rives, V. FT-IR Assessment Through Pyridine Adsorption of the Surface Acidity of Alkali-Doped MoO3/TiO2. J. Catal. 1994, 146, 415–421. [Google Scholar] [CrossRef]
- Daoura, O.; Fornasieri, G.; Boutros, M.; El Hassan, N.; Beaunier, P.; Thomas, C.; Selmane, M.; Miche, A.; Sassoye, C.; Ersen, O.; et al. One-pot prepared mesoporous silica SBA-15-like monoliths with embedded Ni particles as selective and stable catalysts for methane dry reforming. Appl. Catal. B Environ. 2021, 280, 119417. [Google Scholar] [CrossRef]
- Shi, L.Y.; Li, Y.X.; Xue, D.M.; Tan, P.; Jiang, Y.; Liu, X.Q.; Sun, L.B. Fabrication of highly dispersed nickel in nanoconfined spaces of as-made SBA-15 for dry reforming of methane with carbon dioxide. Chem. Eng. J. 2020, 390, 124491. [Google Scholar] [CrossRef]
- Fang, K.; Ren, J.; Sun, Y. Effect of nickel precursors on the performance of Ni/AlMCM-41 catalysts for n-dodecane hydroconversion. J. Mol. Catal. A Chem. 2005, 229, 51–58. [Google Scholar] [CrossRef]
- Lim, Z.Y.; Ma, X.; Chen, B. Enhanced porosity of Ni@HSZ for dry reforming of methane. New J. Chem. 2020, 44, 1707–1710. [Google Scholar] [CrossRef]
- Wojcieszak, R.; Monteverdi, S.; Mercy, M.; Nowak, I.; Ziolek, M.; Bettahar, M.M. Nickel containing MCM-41 and AlMCM-41 mesoporous molecular sieves: Characteristics and activity in the hydrogenation of benzene. Appl. Catal. A Gen. 2004, 268, 241–253. [Google Scholar] [CrossRef]
- Afshar Taromi, A.; Kaliaguine, S. Hydrodeoxygenation of triglycerides over reduced mesostructured Ni/Γ-alumina catalysts prepared via one-pot sol-gel route for green diesel production. Appl. Catal. A Gen. 2018, 558, 140–149. [Google Scholar] [CrossRef]
- Patil, R.B.; House, S.D.; Mantri, A.; Yang, J.C.; McKone, J.R. Direct Observation of Ni-Mo Bimetallic Catalyst Formation via Thermal Reduction of Nickel Molybdate Nanorods. ACS Catal. 2020, 10, 10390–10398. [Google Scholar] [CrossRef]
- Wang, F.; Yu, F.; Wei, Y.; Li, A.; Xu, S.; Lu, X. Promoting hydrocarbon production from fatty acid pyrolysis using transition metal or phosphorus modified Al-MCM-41 catalyst. J. Anal. Appl. Pyrolysis 2021, 156, 105146. [Google Scholar] [CrossRef]
- Afshar Taromi, A.; Kaliaguine, S. Green diesel production via continuous hydrotreatment of triglycerides over mesostructured Γ-alumina supported NiMo/CoMo catalysts. Fuel Process. Technol. 2018, 171, 20–30. [Google Scholar] [CrossRef]
- Cortés, J.C.; Rodríguez, C.; Molina, R.; Moreno, S. Hydrocracking of 1-methylnaphtalene (1MN) over modified clays-supported NiMoS and NiWS catalyst. Fuel 2021, 295, 120612. [Google Scholar] [CrossRef]
- Chong, C.C.; Bukhari, S.N.; Cheng, Y.W.; Setiabudi, H.D.; Jalil, A.A.; Phalakornkule, C. Robust Ni/Dendritic fibrous SBA-15 (Ni/DFSBA-15) for methane dry reforming: Effect of Ni loadings. Appl. Catal. A Gen. 2019, 584, 117174. [Google Scholar] [CrossRef]
- Gundeboina, R.; Gadasandula, S.; Velisoju, V.K.; Gutta, N.; Kotha, L.R.; Aytam, H.P. Ni-Al-Ti Hydrotalcite Based Catalyst for the Selective Hydrogenation of Biomass-Derived Levulinic Acid to γ-Valerolactone. ChemistrySelect 2019, 4, 202–210. [Google Scholar] [CrossRef]
- Ganiyu, S.A.; Ali, S.A.; Alhooshani, K. Synthesis of a Ti-SBA-15-nimo hydrodesulfurization catalyst: The effect of the hydrothermal synthesis temperature of nimo and molybdenum loading on the catalytic activity. Ind. Eng. Chem. Res. 2017, 56, 5201–5209. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, H.; Ding, R.; Wang, L.; Liu, Z.; Lv, B. Highly efficient oxidative desulfurization of dibenzothiophene using Ni modified MoO3 catalyst. Appl. Catal. A Gen. 2020, 589. [Google Scholar] [CrossRef]
- Puello-Polo, E.; Pájaro, Y.; Márquez, E. Effect of the Gallium and Vanadium on the Dibenzothiophene Hydrodesulfurization and Naphthalene Hydrogenation Activities Using Sulfided NiMo-V2O5/Al2O3-Ga2O3. Catalysts 2020, 10, 894. [Google Scholar] [CrossRef]
- Méndez, F.J.; Bravo-Ascención, G.; González-Mota, M.; Puente-Lee, I.; Bokhimi, X.; Klimova, T.E. NiMo catalysts supported on Al, Nb, Ti or Zr-containing MCM-41 for dibenzothiophene hydrodesulfurization. Catal. Today 2020, 349, 217–227. [Google Scholar] [CrossRef]
- Zepeda, T.A.; Pawelec, B.; Fierro, J.L.G.; Halachev, T. Effect of Ti on the catalytic properties of CoMo/Ti(x)-HMS catalysts in the reaction of hydrodesulfurization of 4-ethyl-6-methyl dibenzothiophene. J. Catal. 2006, 242, 254–269. [Google Scholar] [CrossRef]
- Klimova, T.; Reyes, J.; Gutiérrez, O.; Lizama, L. Novel bifunctional NiMo/Al-SBA-15 catalysts for deep hydrodesulfurization: Effect of support Si/Al ratio. Appl. Catal. A Gen. 2008, 335, 159–171. [Google Scholar] [CrossRef]
- Gao, Q.; Zhang, Y.; Zhou, K.; Wu, H.; Guo, J.; Zhang, L.; Duan, A.; Zhao, Z.; Zhang, F.; Zhou, Y. Synthesis of ZSM-5/KIT-6 with a tunable pore structure and its catalytic application in the hydrodesulfurization of dibenzothiophene and diesel oil. RSC Adv. 2018, 8, 28879–28890. [Google Scholar] [CrossRef] [Green Version]
- Tio, S.; Reddy, B.M.; Chowdhury, B.; Smirniotis, P.G. An XPS study of the dispersion of MoO3 on. Appl. Catal. 2001, 211, 19–30. [Google Scholar]
- Vozka, P.; Orazgaliyeva, D.; Šimáček, P.; Blažek, J.; Kilaz, G. Activity comparison of Ni-Mo/Al2O3 and Ni-Mo/TiO2 catalysts in hydroprocessing of middle petroleum distillates and their blend with rapeseed oil. Fuel Process. Technol. 2017, 167, 684–694. [Google Scholar] [CrossRef]
- Choi, J.G.; Thompson, L.T. XPS study of as-prepared and reduced molybdenum oxides. Appl. Surf. Sci. 1996, 93, 143–149. [Google Scholar] [CrossRef]
- Zhou, W.; Yang, L.; Liu, L.; Chen, Z.; Zhou, A.; Zhang, Y.; He, X.; Shi, F.; Zhao, Z. Synthesis of novel NiMo catalysts supported on highly ordered TiO2-Al2O3 composites and their superior catalytic performance for 4,6-dimethyldibenzothiophene hydrodesulfurization. Appl. Catal. B Environ. 2020, 268, 118428. [Google Scholar] [CrossRef]
- Ganiyu, S.A.; Ali, S.A.; Alhooshani, K. Simultaneous HDS of DBT and 4,6-DMDBT over single-pot Ti-SBA-15-NiMo catalysts: Influence of Si/Ti ratio on the structural properties, dispersion and catalytic activity. RSC Adv. 2017, 7, 21943–21952. [Google Scholar] [CrossRef] [Green Version]
- Morales-Ortuño, J.C.; Ortega-Domínguez, R.A.; Hernández-Hipólito, P.; Bokhimi, X.; Klimova, T.E. HDS performance of NiMo catalysts supported on nanostructured materials containing titania. Catal. Today 2016, 271, 127–139. [Google Scholar] [CrossRef]
- Castillo-Villalón, P.; Ramírez, J.; Cuevas, R.; Vázquez, P.; Castañeda, R. Influence of the support on the catalytic performance of Mo, CoMo, and NiMo catalysts supported on Al2O3 and TiO2 during the HDS of thiophene, dibenzothiophene, or 4,6-dimethyldibenzothiophene. Catal. Today 2016, 259, 140–149. [Google Scholar] [CrossRef]
- Hensen, E.J.M.; Kooyman, P.J.; Van der Meer, Y.; Van der Kraan, A.M.; De Beer, V.H.J.; Van Veen, J.A.R.; Van Santen, R.A.; Kooyman, P.J.; Van der Meer, Y.; Van der Kraan, A.M. The relation between morphology and hydrotreating activity for supported MoS2 particles. J. Catal. 2001, 199, 224–235. [Google Scholar] [CrossRef]
- Zhou, W.; Liu, M.; Zhou, Y.; Wei, Q.; Zhang, Q.; Ding, S.; Zhang, Y.; Yu, T.; You, Q. 4,6-Dimethyldibenzothiophene Hydrodesulfurization on Nickel-Modified USY-Supported NiMoS Catalysts: Effects of Modification Method. Energy Fuels 2017, 31, 7445–7455. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, G.; Mei, J.; Xiao, C.; Hu, D.; Wang, A.; Song, Y.; Ni, Y.; Jiang, G.; Duan, A. The Influence of Pore Structure and Acidity on the Hydrodesulfurization of Dibenzothiophene over NiMo-Supported Catalysts. ACS Omega 2020, 5, 15576–15585. [Google Scholar] [CrossRef] [PubMed]
- Lecrenay, E.; Sakanishi, K.; Mochida, I. Catalytic hydrodesulfurization of gas oil and model sulfur compounds over commercial and laboratory-made CoMo and NiMo catalysts: Activity and reaction scheme. Catal. Today 1997, 39, 13–20. [Google Scholar] [CrossRef]
- Pawelec, B.; Fierro, J.L.G.; Montesinos, A.; Zepeda, T.A. Influence of the acidity of nanostructured CoMo/P/Ti-HMS catalysts on the HDS of 4,6-DMDBT reaction pathways. Appl. Catal. B Environ. 2008, 80, 1–14. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, C.; Zhao, X.; Liu, C. Synthesis of bifunctional highly-loaded NiMoW catalysts and their catalytic performance of 4,6-DMDBT HDS. Catal. Commun. 2017, 88, 13–17. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | a | b | |
|---|---|---|---|
| H-AlMCM-41 | 534.3 | 0.36 | 2.8 |
| NiMo/HAlMCM-41 | 329.1 | 0.44 | 2.8 |
| H-TiMCM-41 | 745.9 | 0.48 | 2.8 |
| NiMo/HTiMCM-41 | 483.4 | 0.58 | 2.6 |
| Sample | a | b | c | ||
|---|---|---|---|---|---|
| A260/A0 d | A260/A0 d | ||||
| H-AlMCM-41 | 18 | 0.23 | 150 | 0.42 | 168 |
| NiMo/HAlMCM-41 | 41 | 0.19 | 250 | 0.42 | 291 |
| NiMo/AlMCM-41 | 37 | 0.47 | 140 | 0.74 | 177 |
| H-TiMCM-41 | 220 | 0.39 | 250 | 0.60 | 470 |
| NiMo/HTiMCM-41 | 100 | 0.40 | 200 | 0.48 | 300 |
| NiMo/TiMCM-41 | 32 | 0.50 | 245 | 0.54 | 277 |
| Catalyst | Mo 3d3/2 (eV) | Mo 3d5/2 (eV) | ||
|---|---|---|---|---|
| Mo4+ | Mo6+ | Mo4+ | Mo6+ | |
| NiMo/HAlMCM-41 | 232.9 | 235.5 | 229.8 | 232.6 |
| NiMo/HAlMCM-41 * | 233.7 | 237.1 | 230.8 | 234.1 |
| NiMo/HTiMCM-41 | 233.4 | 236.2 | 230.2 | 233.1 |
| NiMo/HTiMCM-41 * | 234.7 | 237.3 | 231.5 | 234.1 |
| Catalyst | a | Conversion (%) | Product Distribution (mol%) b | ||||
|---|---|---|---|---|---|---|---|
MCHT | 3,3′-DMBPh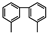 | 3,4′-DMBPh | 4,6-DMTHDBT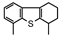 | 3,6-DMDBT | |||
| NiMo/HAlMCM-41 | 3.29 | 88.3 | 13.36 | 66.88 | 7.09 | 0.45 | 0.49 |
| NiMo/AlMCM-41 | 2.26 | 75.4 | 34.75 | 37.11 | n.d. | 2.60 | 0.94 |
| NiMo/HTiMCM-41 | 4.44 | 94.4 | 47.86 | 46.23 | n.d. | 0.14 | 0.12 |
| NiMo/TiMCM-41 | 4.04 | 93.0 | 65.11 | 27.17 | n.d. | 0.56 | 0.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaroszewska, K.; Lewandowski, M.; Góra-Marek, K.; Grzechowiak, J.; Djéga-Mariadassou, G. Hydrodesulfurization of 4,6-Dimethyldibenzothiophene and the Diesel Oil Fraction on NiMo Catalysts Supported over Proton-Exchanged AlMCM-41 and TiMCM-41 Extrudates. Catalysts 2021, 11, 1086. https://doi.org/10.3390/catal11091086
Jaroszewska K, Lewandowski M, Góra-Marek K, Grzechowiak J, Djéga-Mariadassou G. Hydrodesulfurization of 4,6-Dimethyldibenzothiophene and the Diesel Oil Fraction on NiMo Catalysts Supported over Proton-Exchanged AlMCM-41 and TiMCM-41 Extrudates. Catalysts. 2021; 11(9):1086. https://doi.org/10.3390/catal11091086
Chicago/Turabian StyleJaroszewska, Karolina, Marek Lewandowski, Kinga Góra-Marek, Jolanta Grzechowiak, and Gérald Djéga-Mariadassou. 2021. "Hydrodesulfurization of 4,6-Dimethyldibenzothiophene and the Diesel Oil Fraction on NiMo Catalysts Supported over Proton-Exchanged AlMCM-41 and TiMCM-41 Extrudates" Catalysts 11, no. 9: 1086. https://doi.org/10.3390/catal11091086
APA StyleJaroszewska, K., Lewandowski, M., Góra-Marek, K., Grzechowiak, J., & Djéga-Mariadassou, G. (2021). Hydrodesulfurization of 4,6-Dimethyldibenzothiophene and the Diesel Oil Fraction on NiMo Catalysts Supported over Proton-Exchanged AlMCM-41 and TiMCM-41 Extrudates. Catalysts, 11(9), 1086. https://doi.org/10.3390/catal11091086