
Acenaphthene-Based N-Heterocyclic Carbene Metal Complexes: Synthesis and Application in Catalysis


Abstract
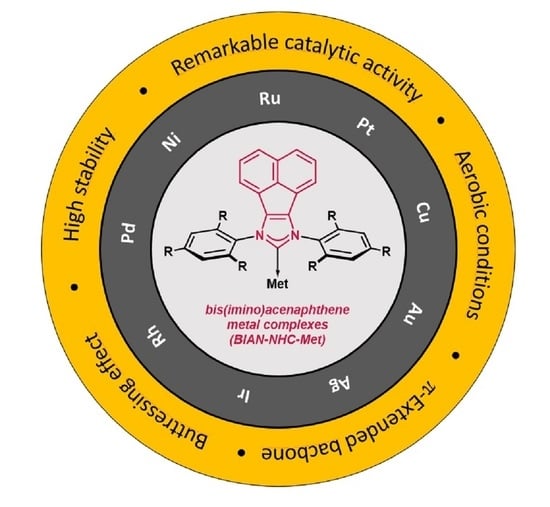
:
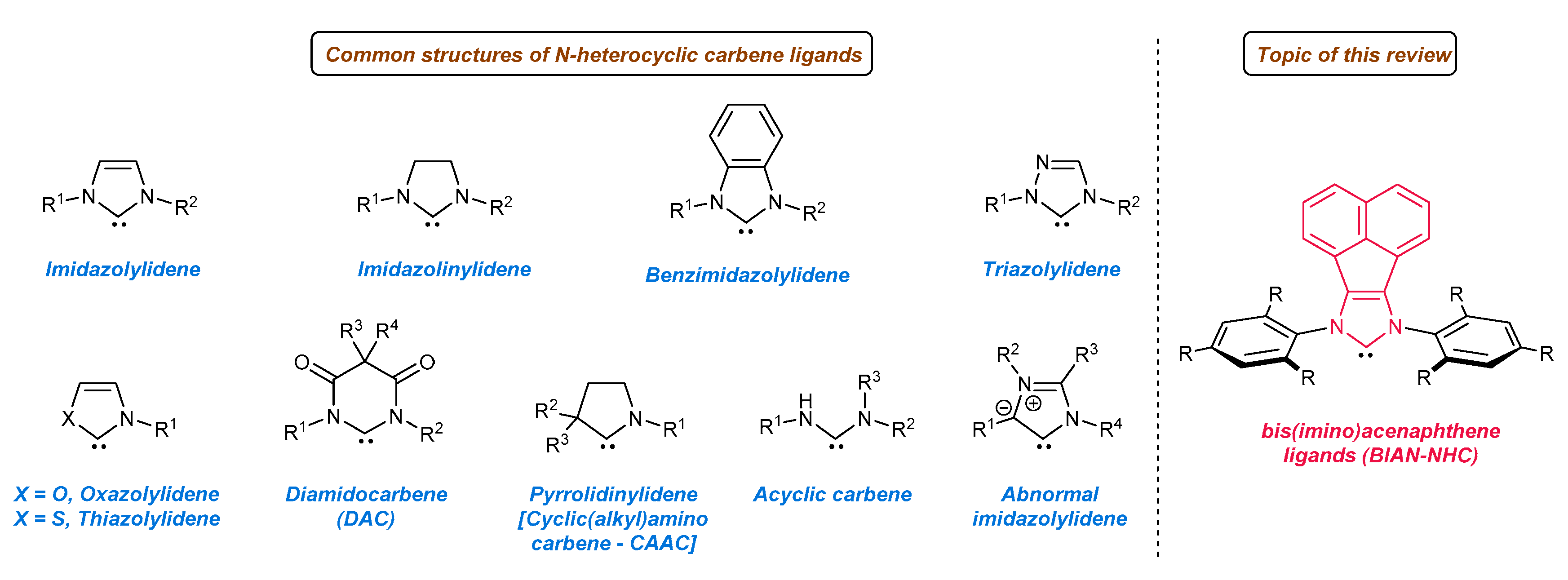
1. Introduction
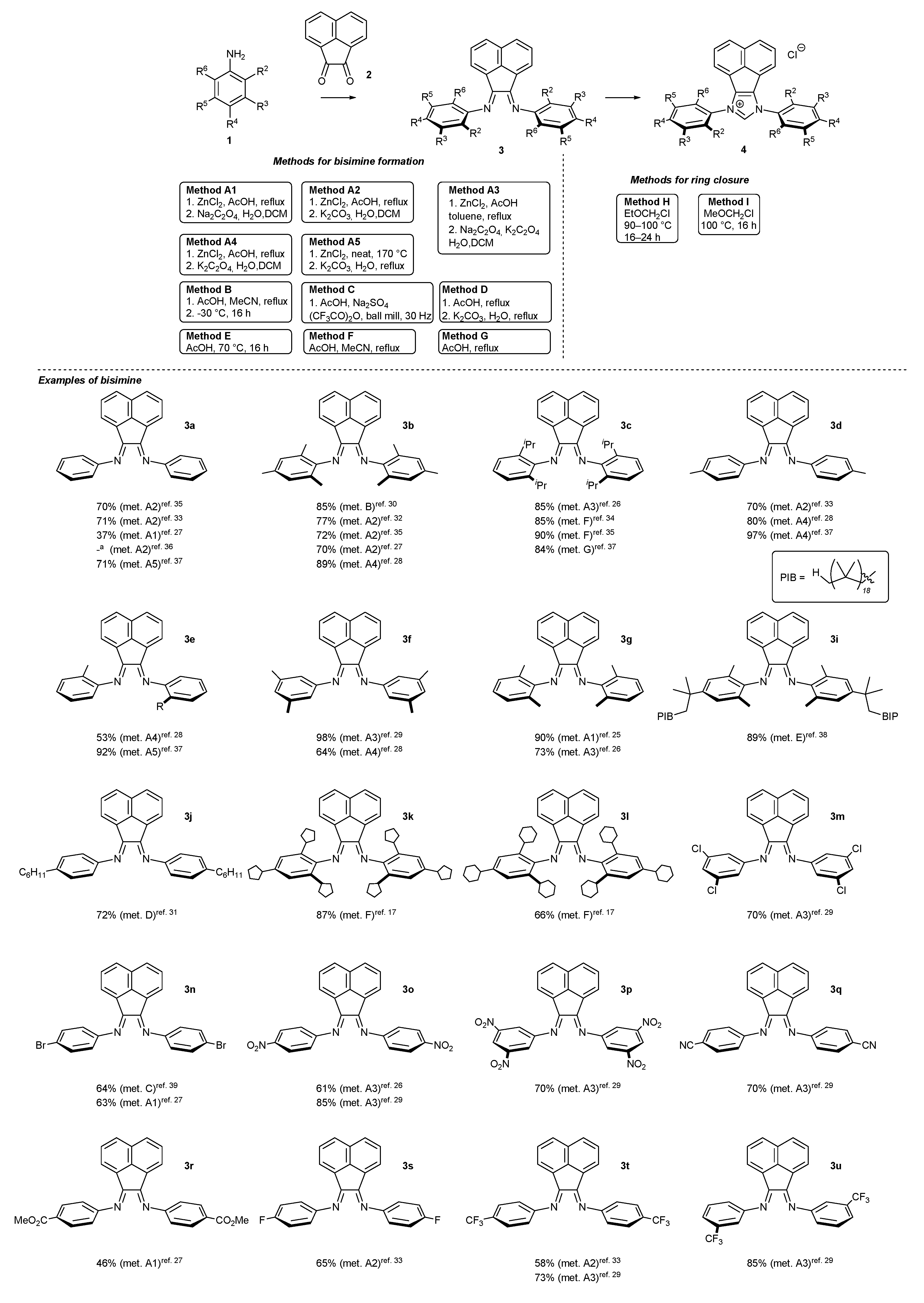
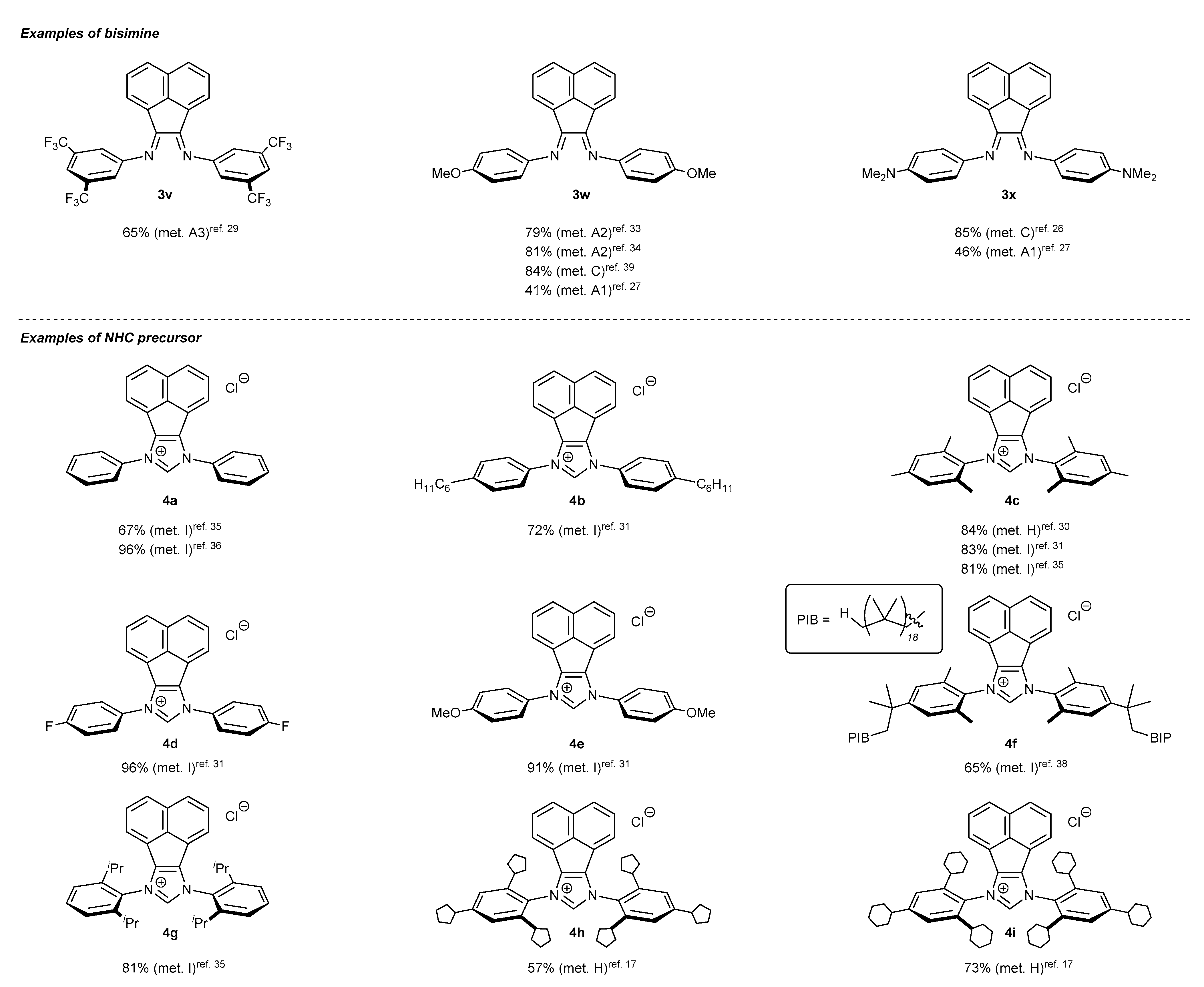
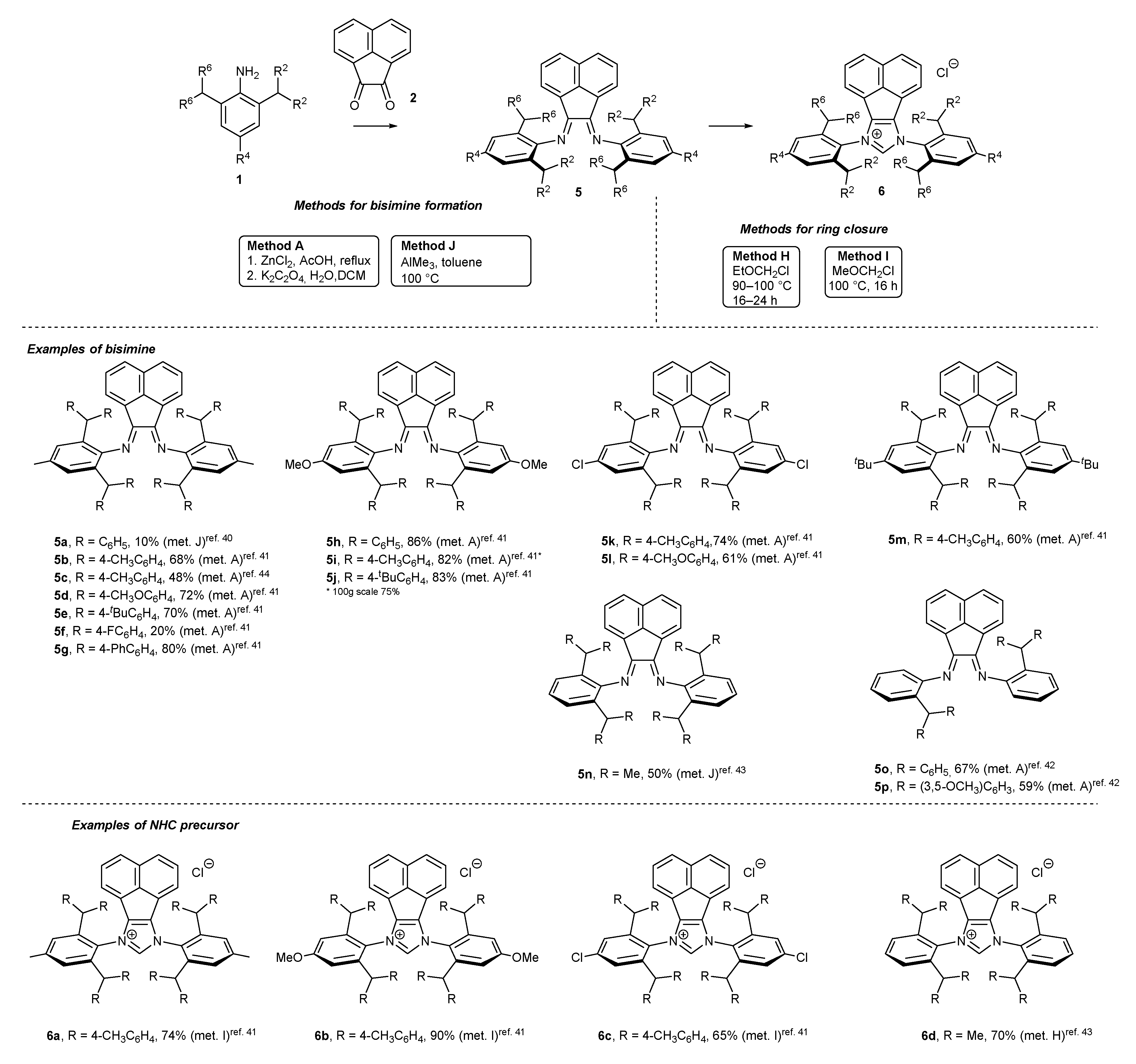
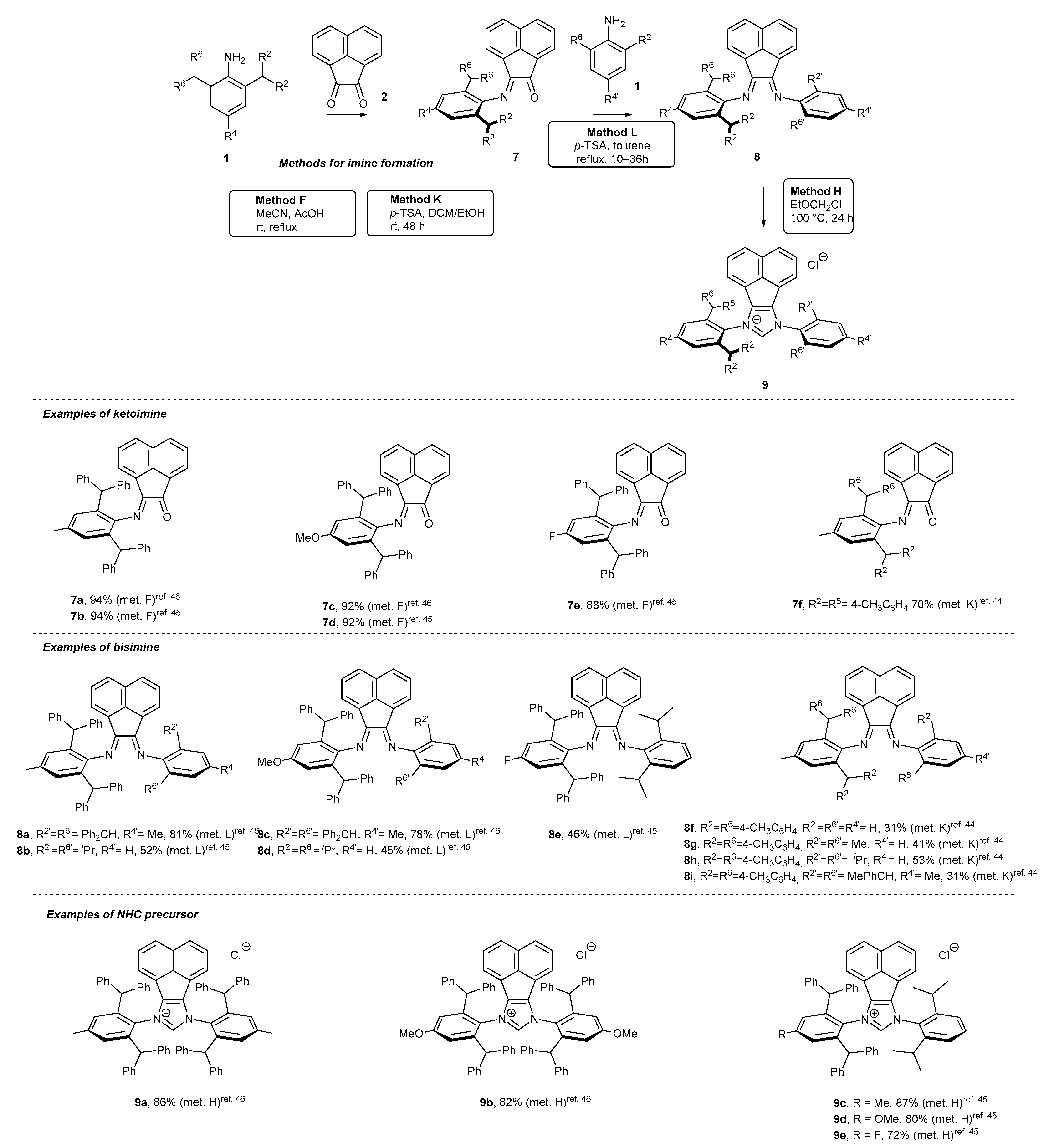
2. Synthetic Routes to BIAN-Type NHC Precursors
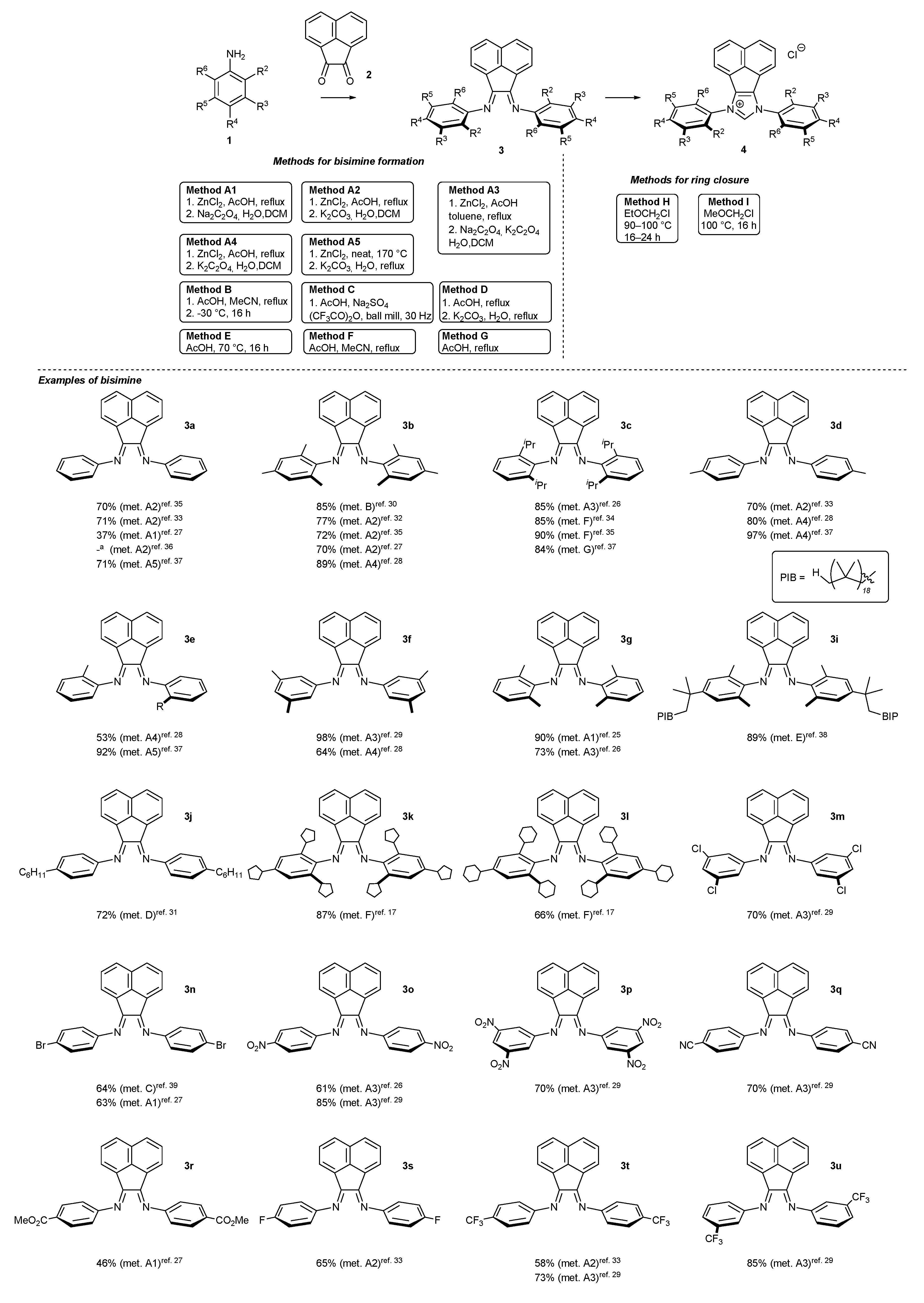
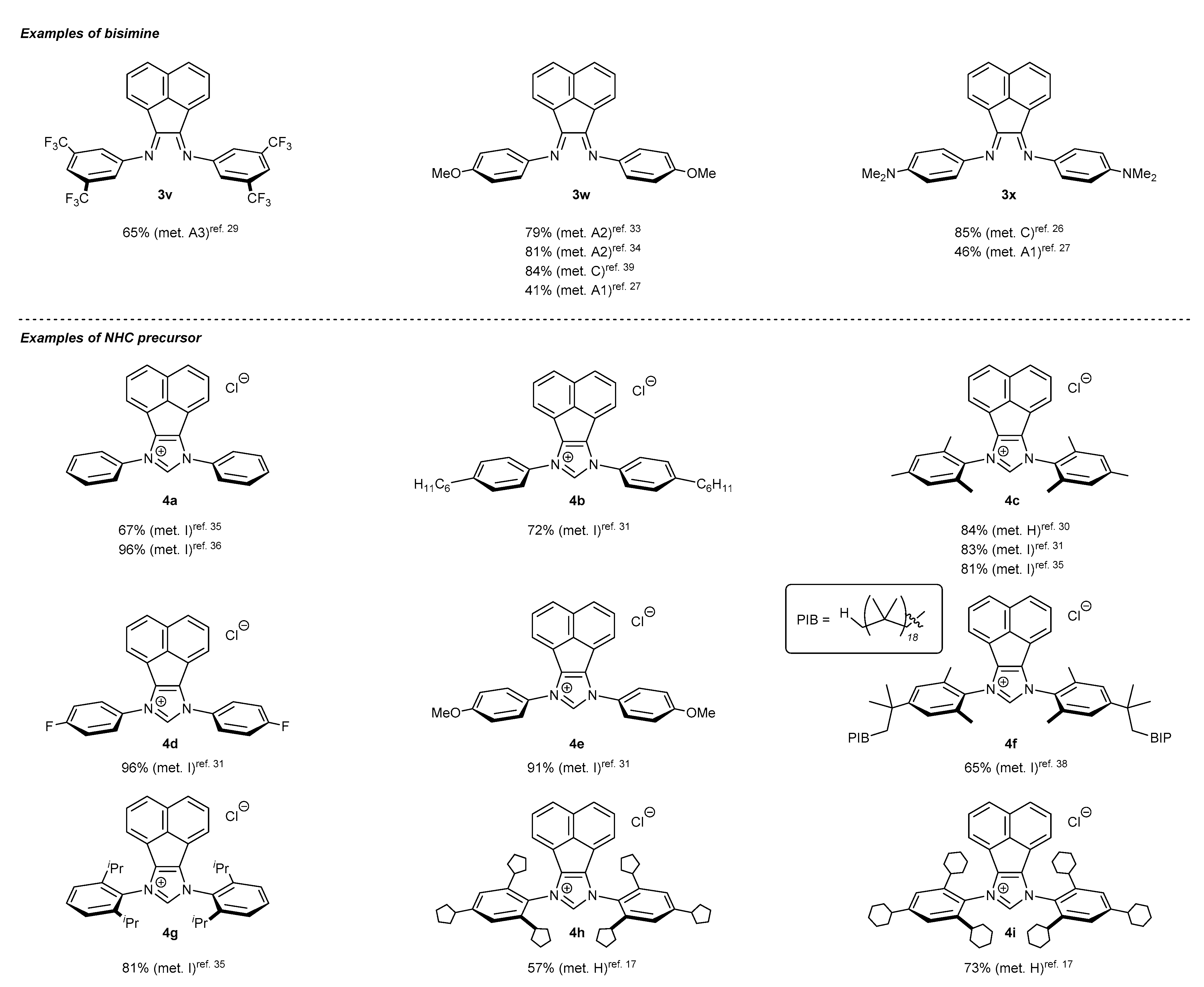
2.1. Synthesis of Monodentate Carbene Precursors
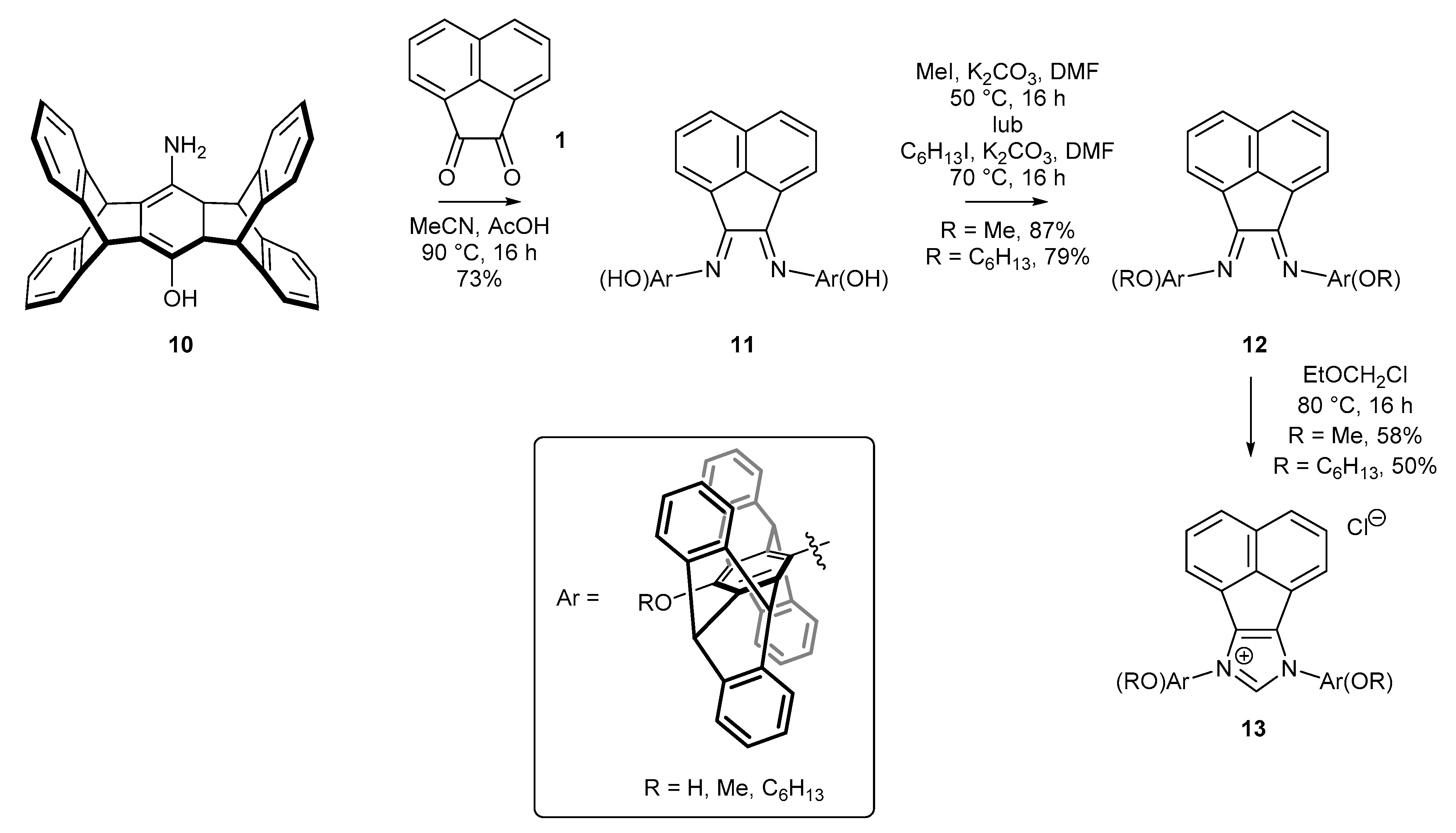
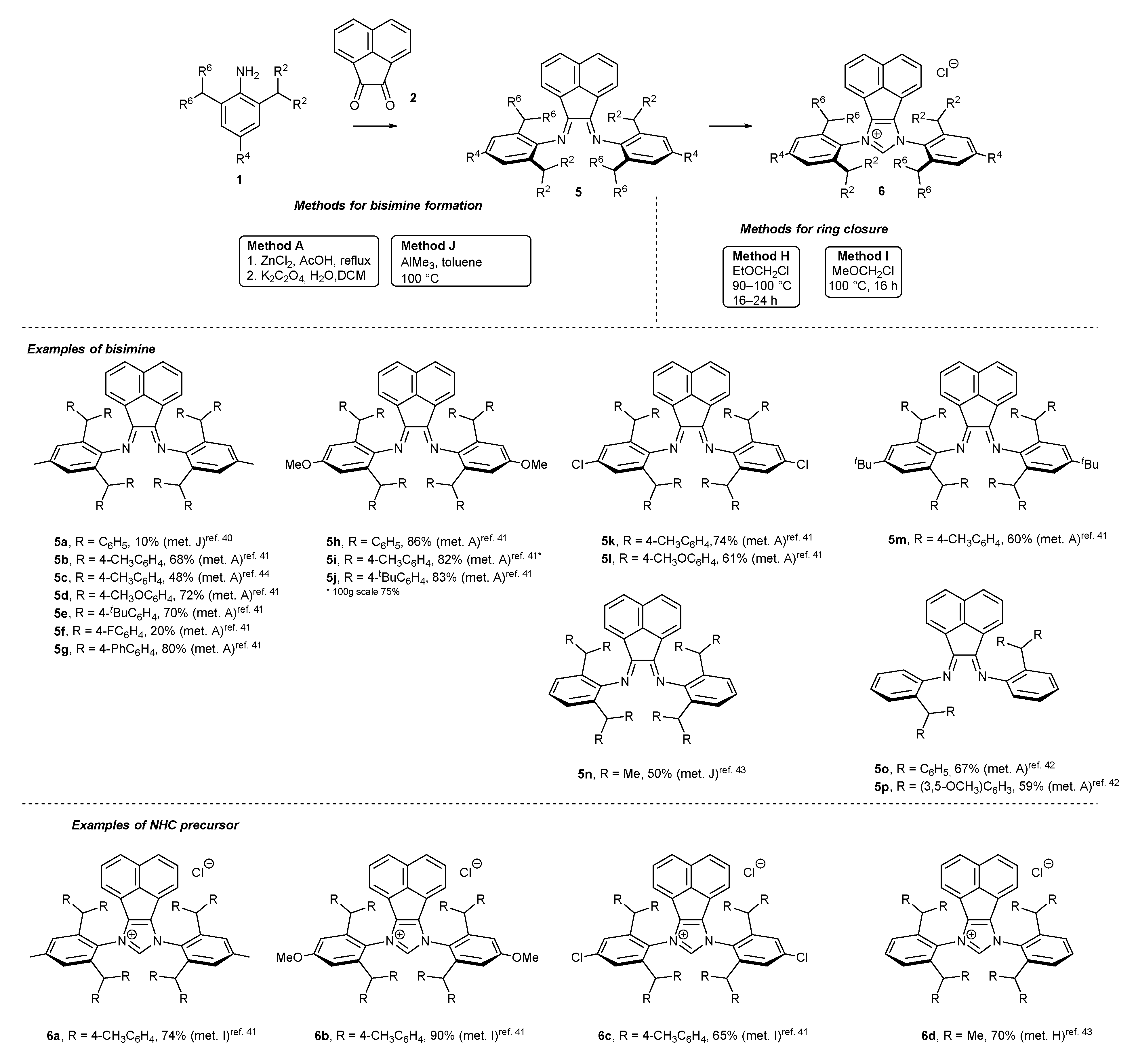
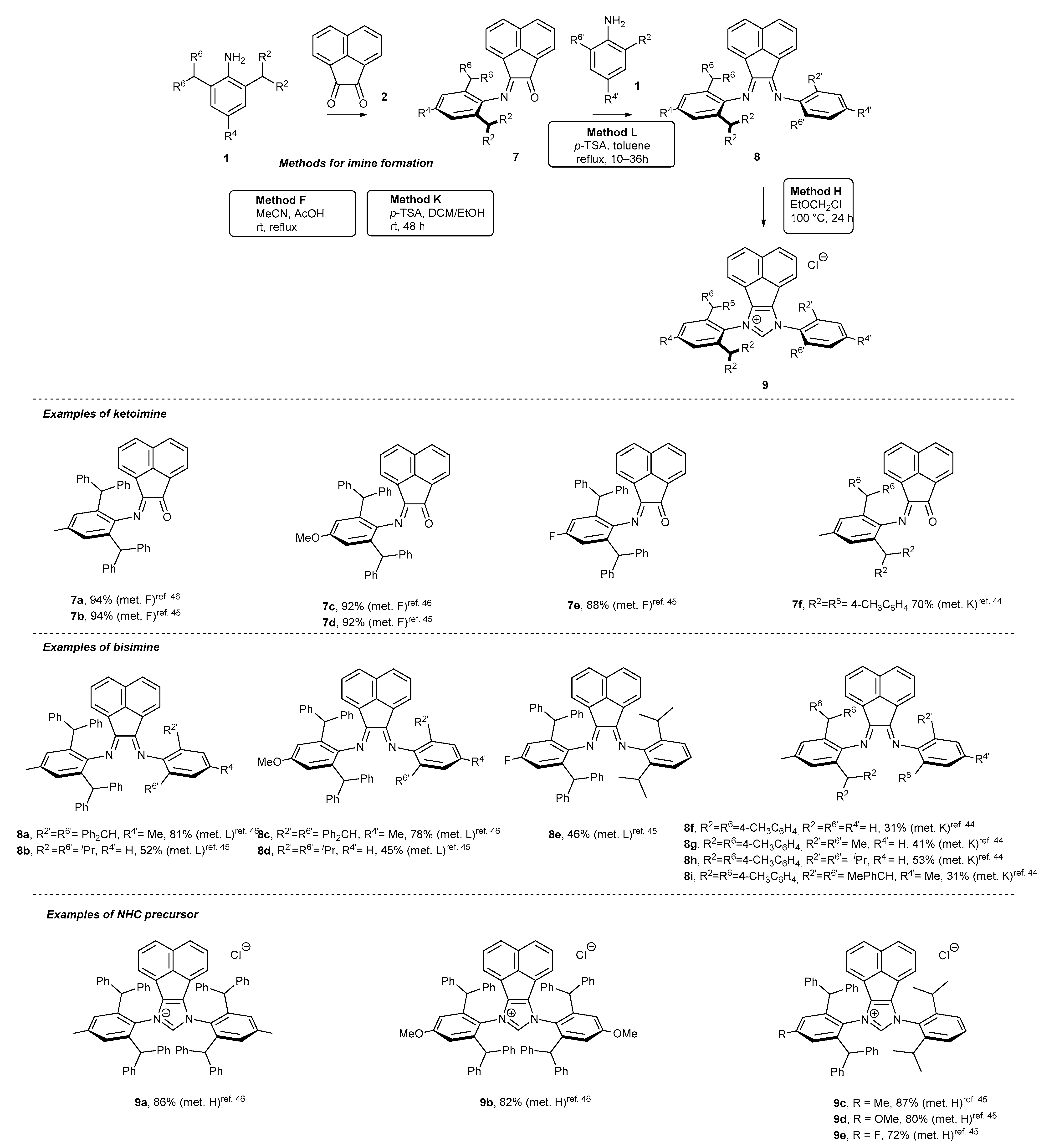
2.2. Synthesis of Polidentate Carbene Precursors
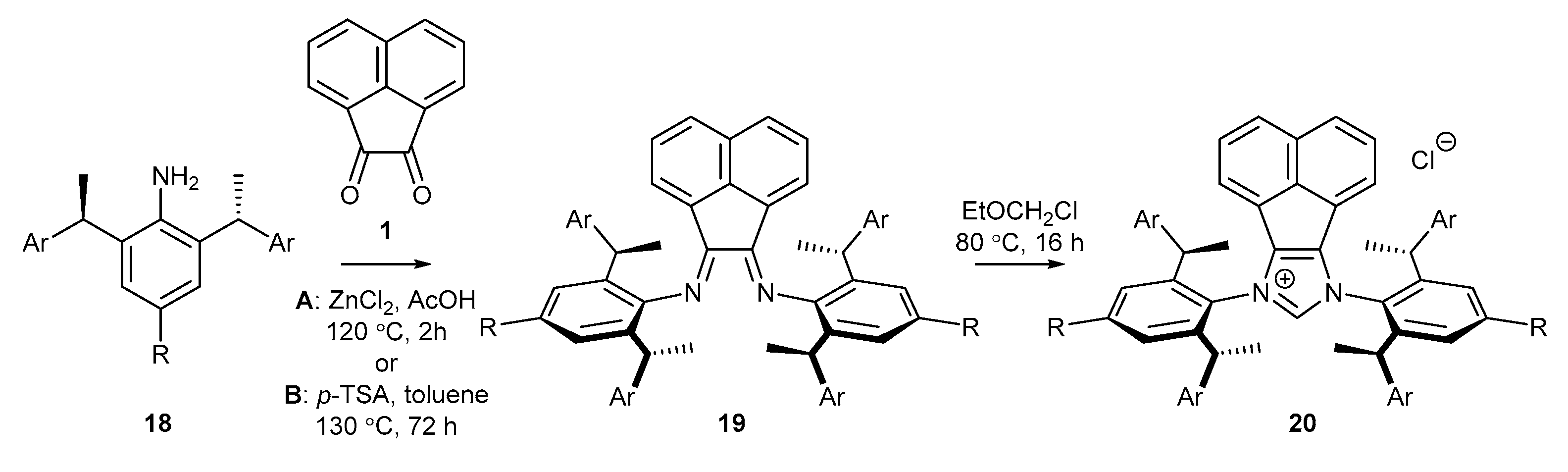
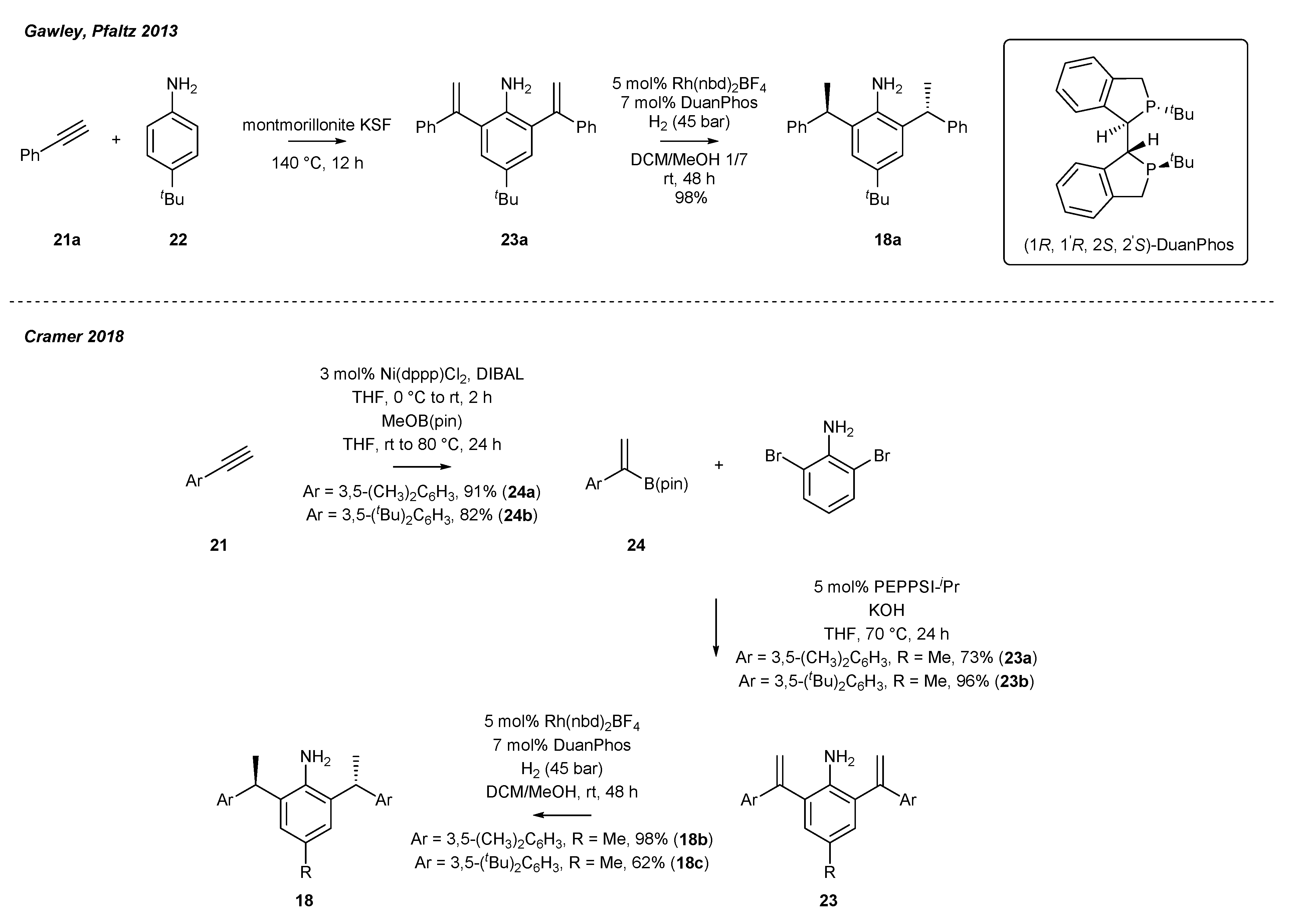
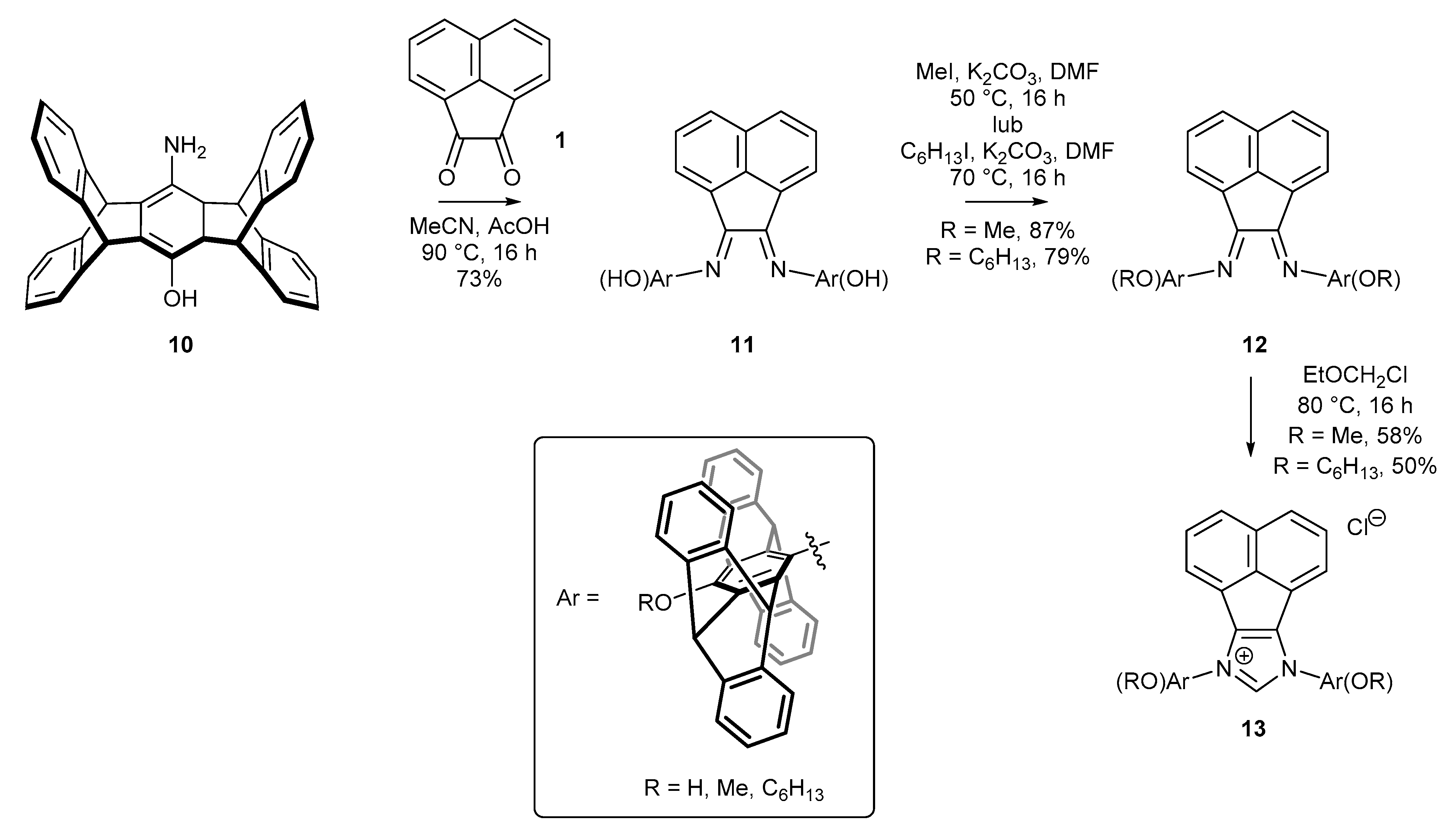
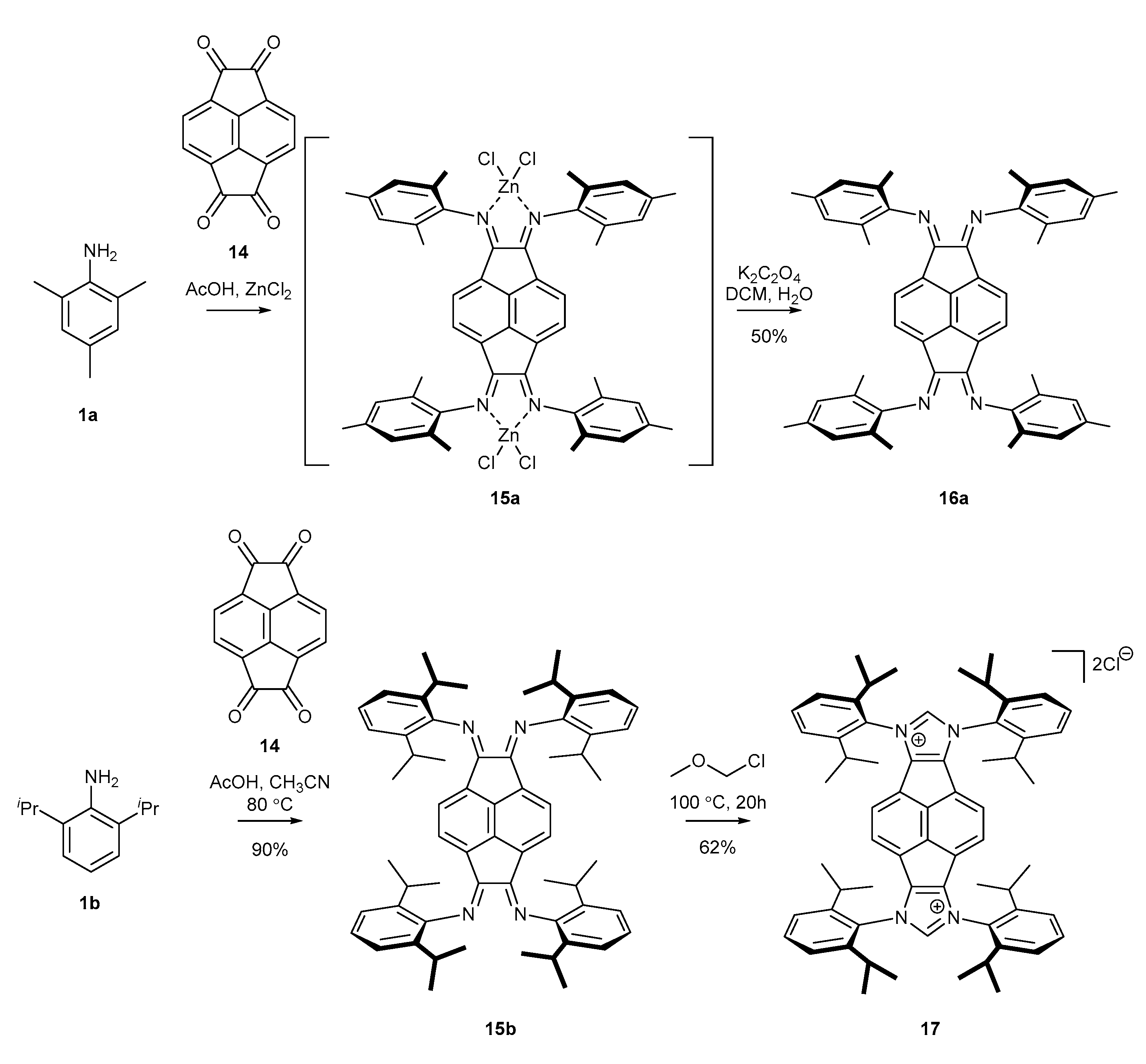
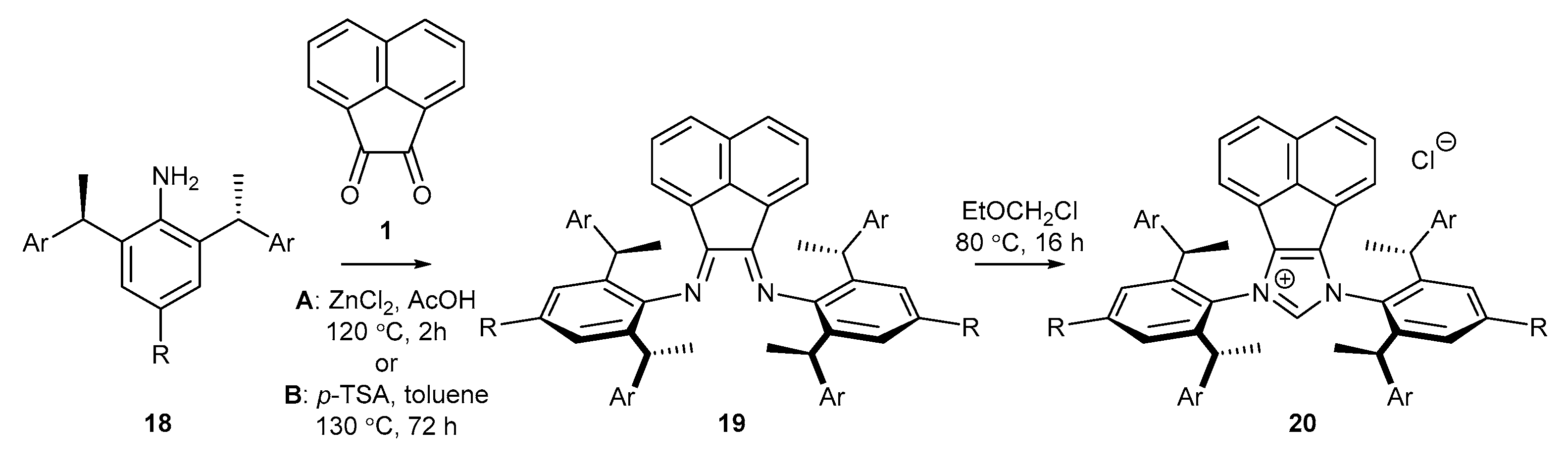
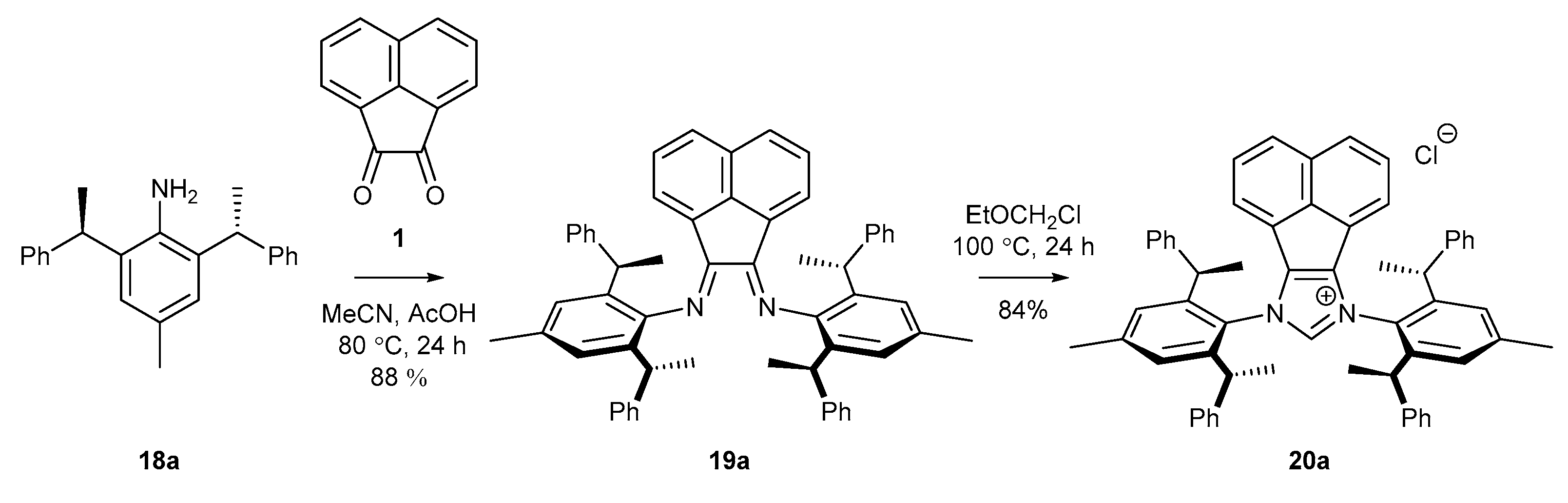
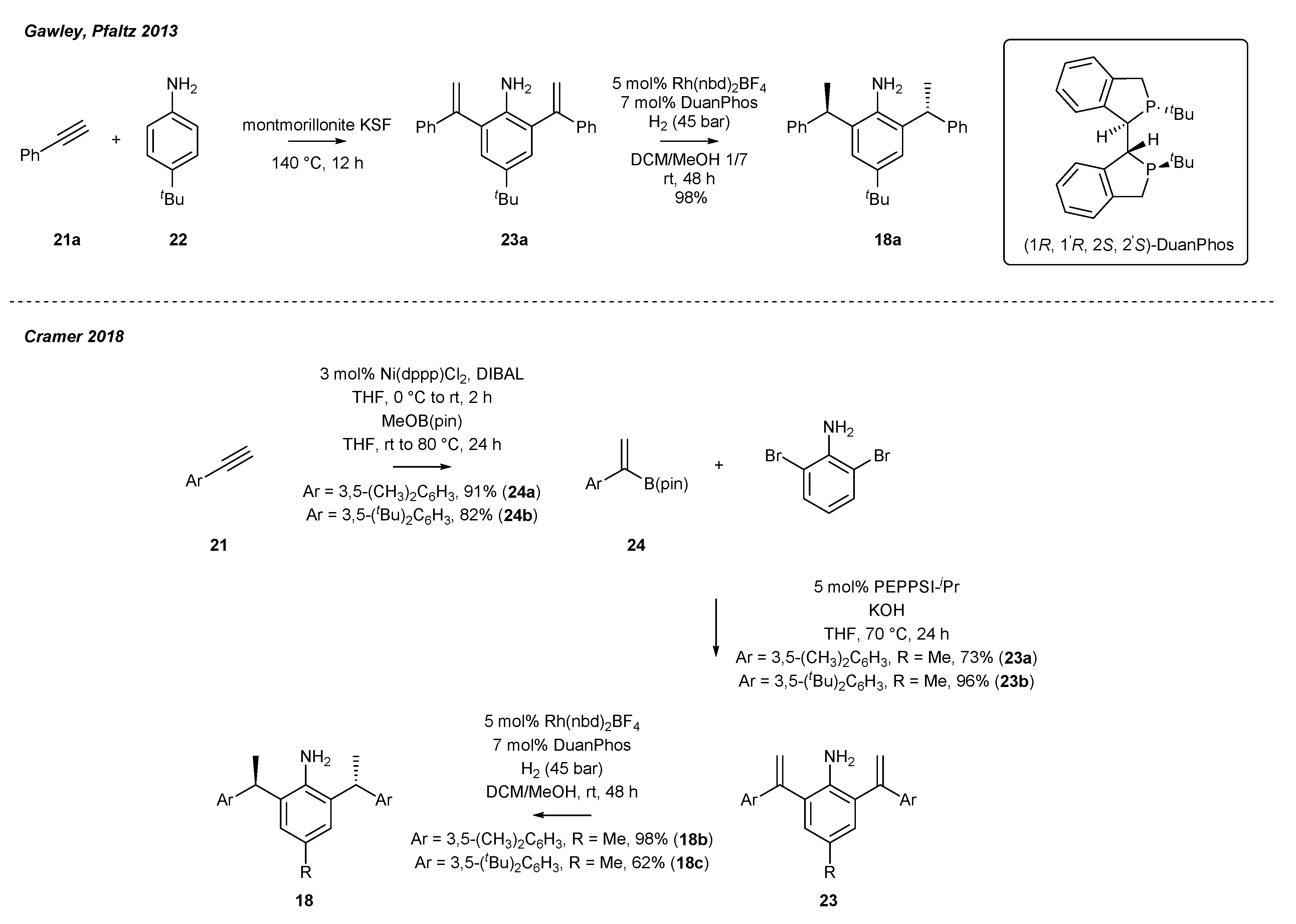
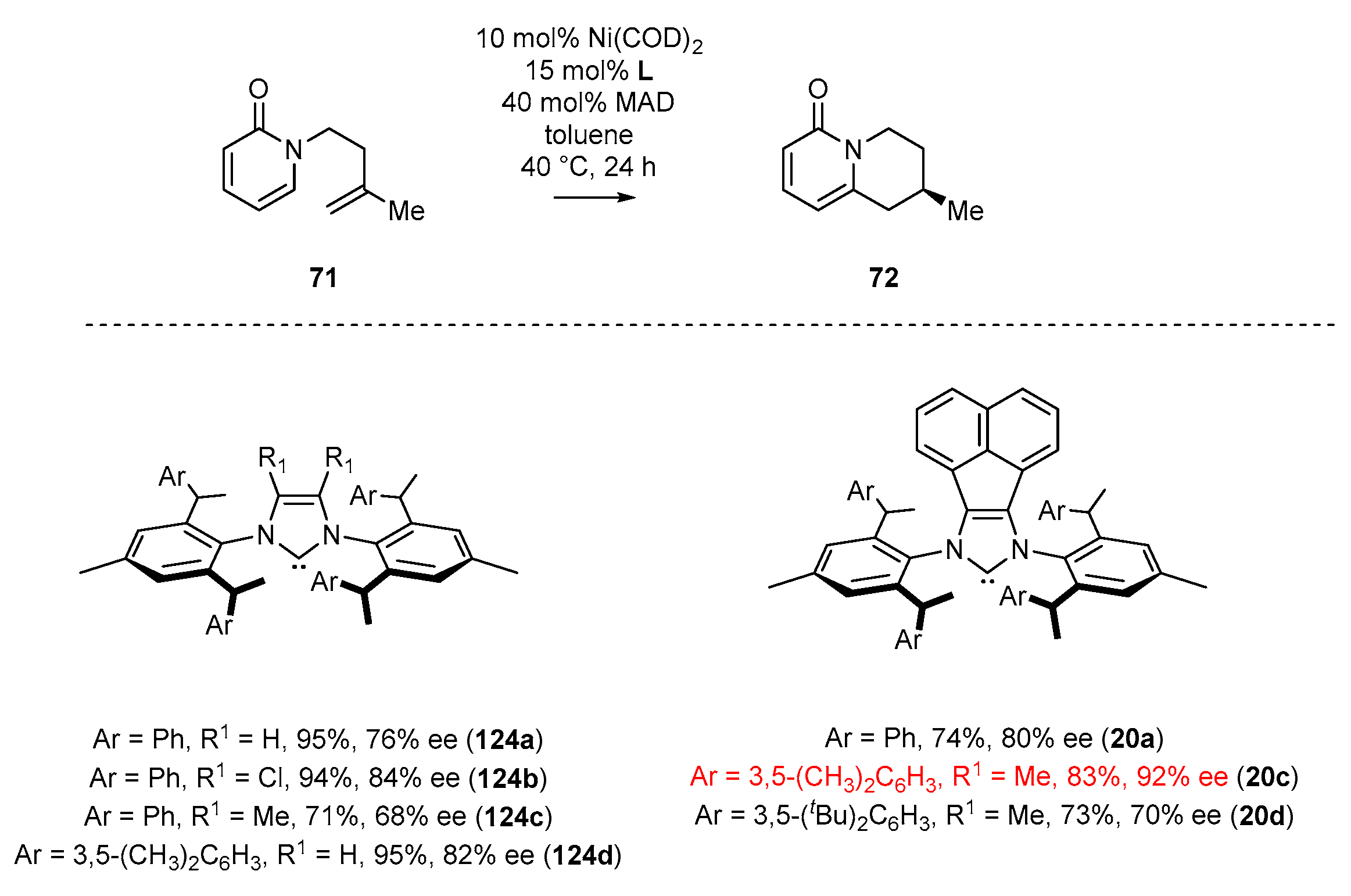
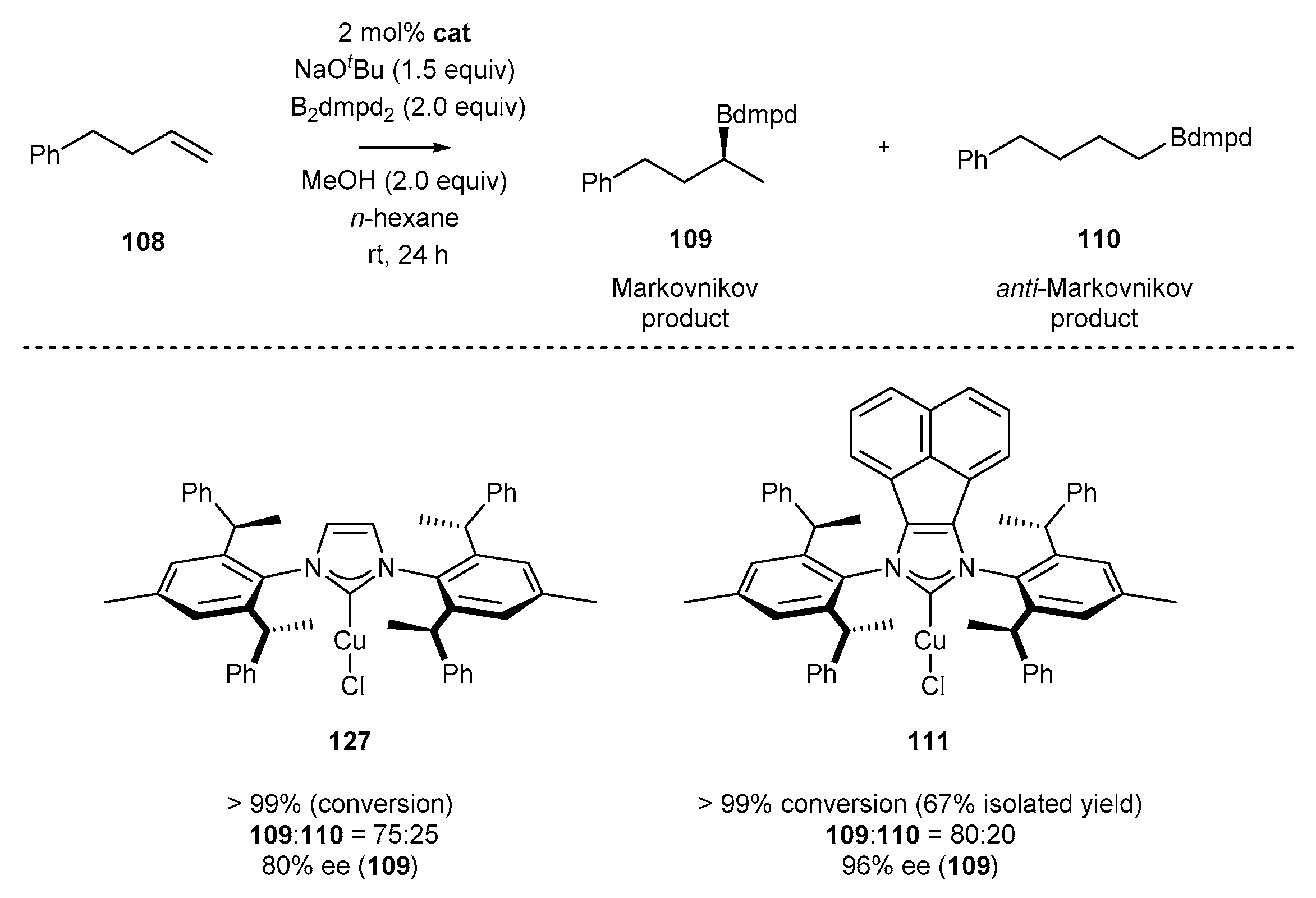
2.3. Synthesis of Chiral Carbene Precursors
3. Preparation of Metal Complexes and Their Application in Catalysis
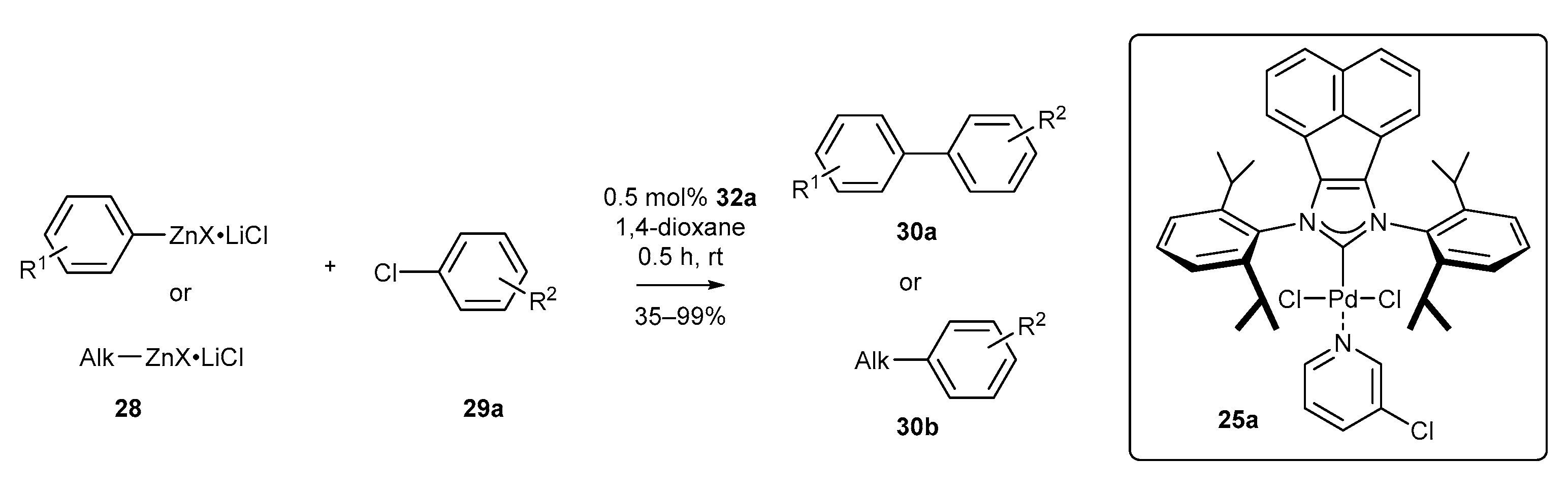
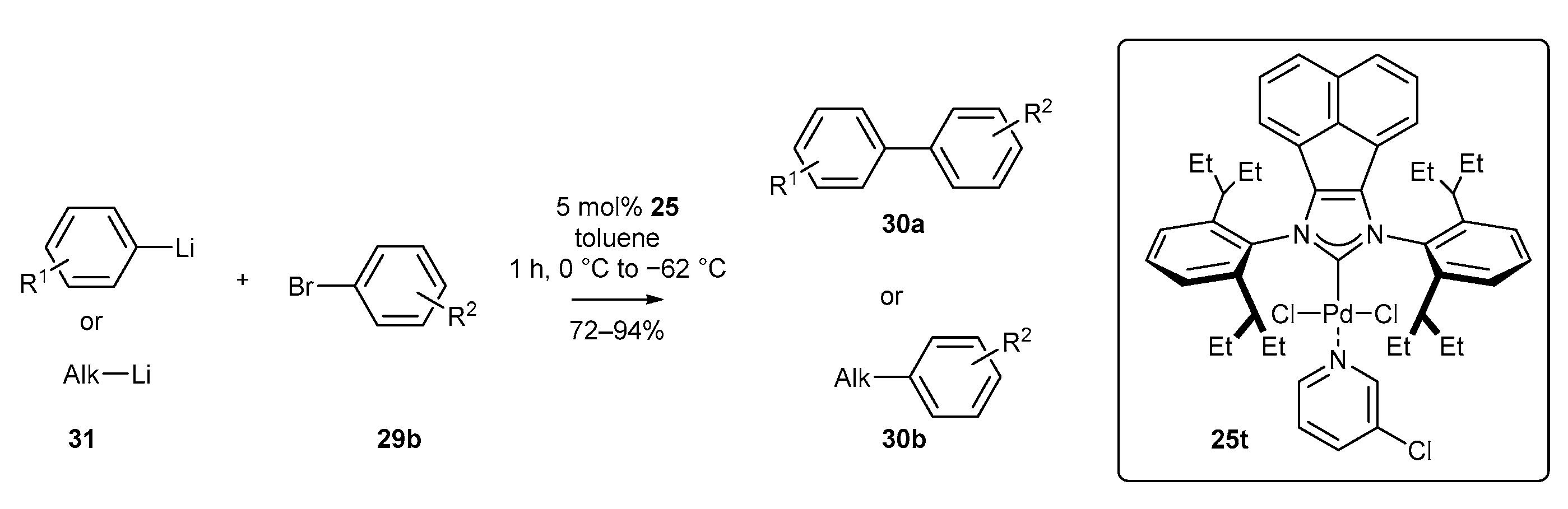
3.1. Synthesis of Palladium Complexes and Their Applications
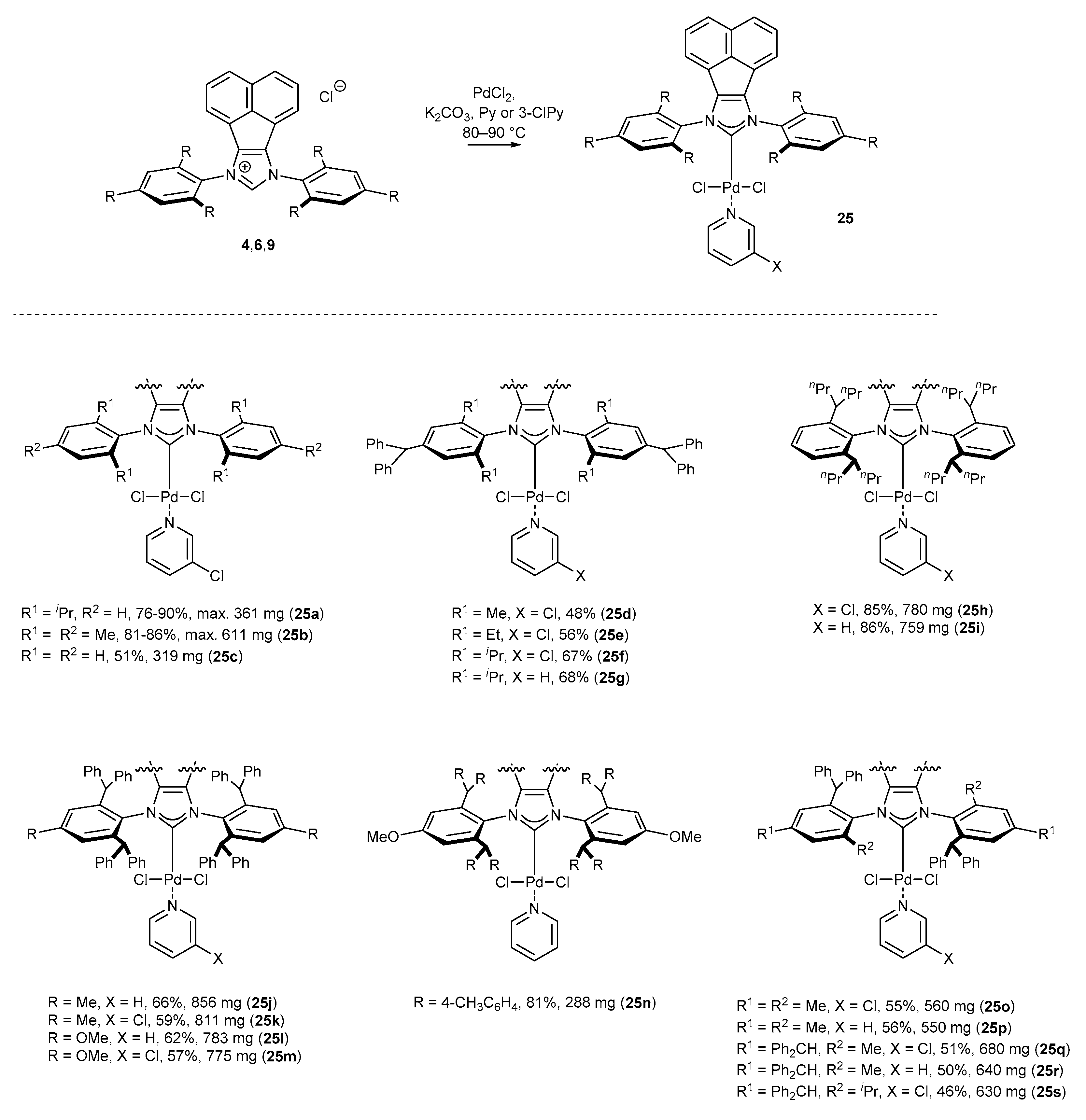
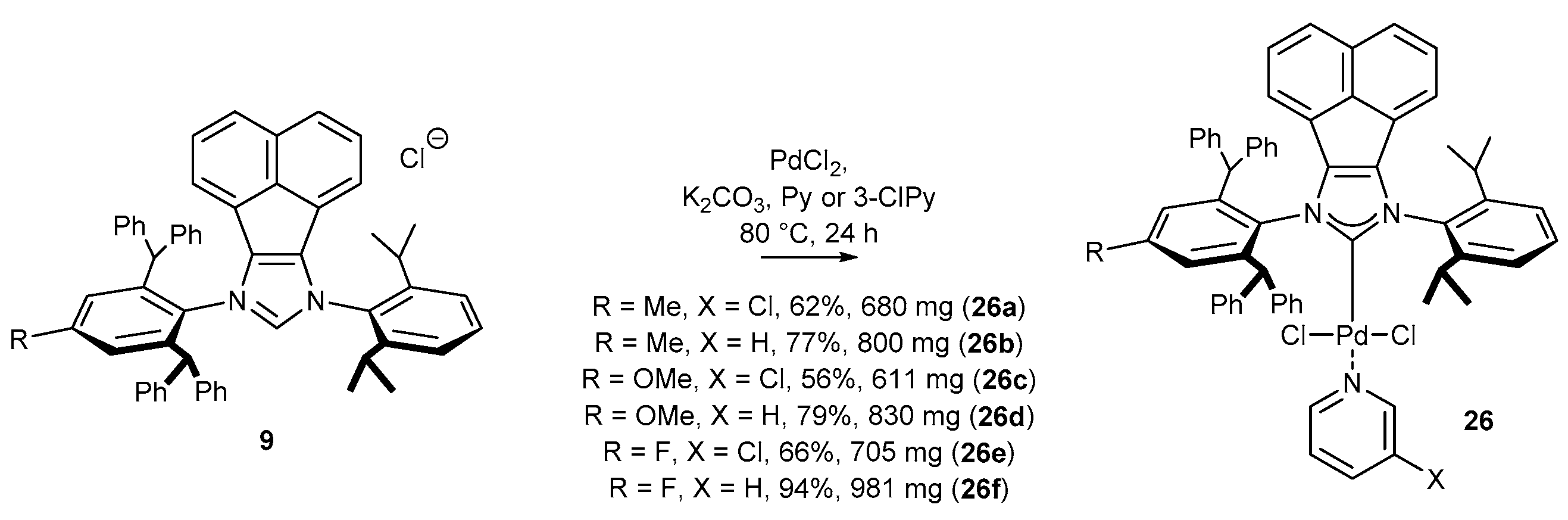
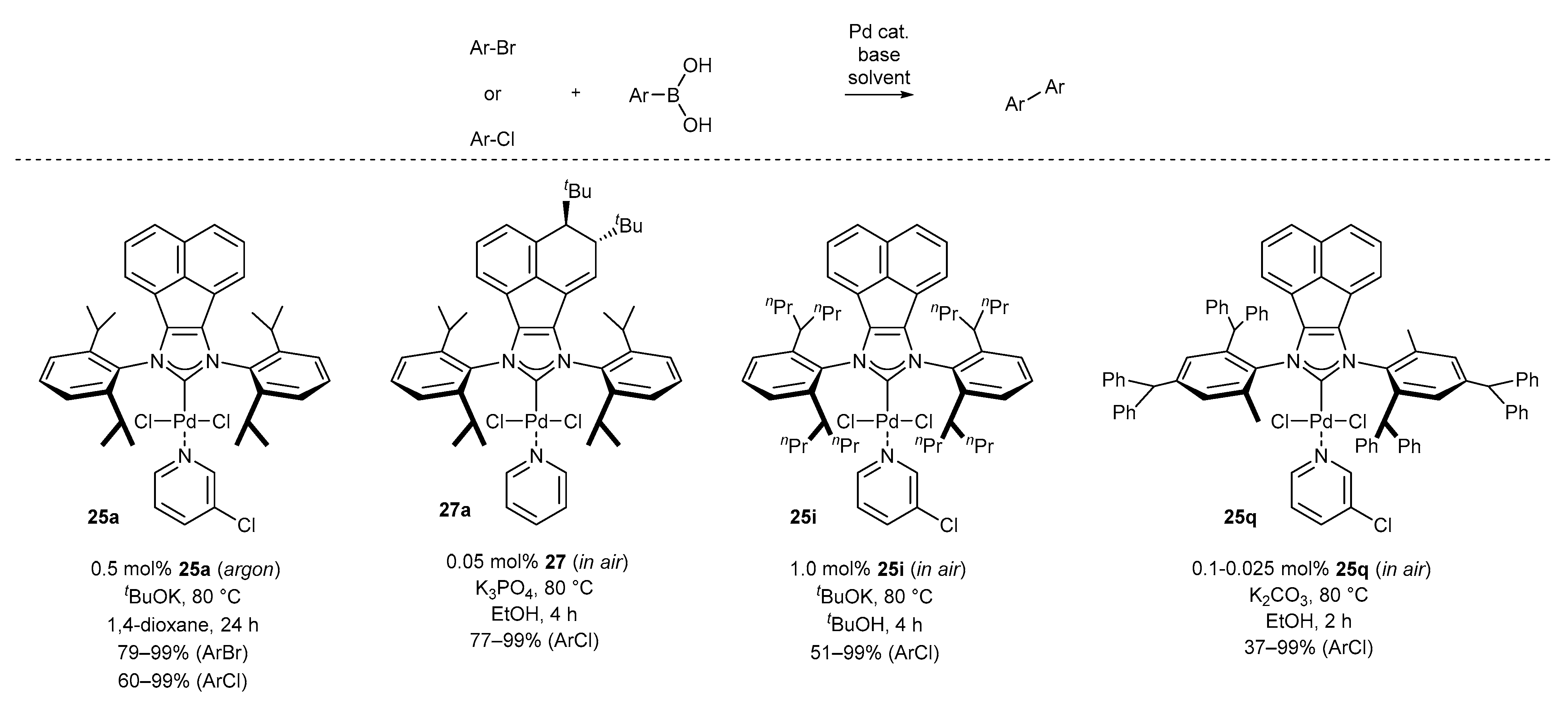
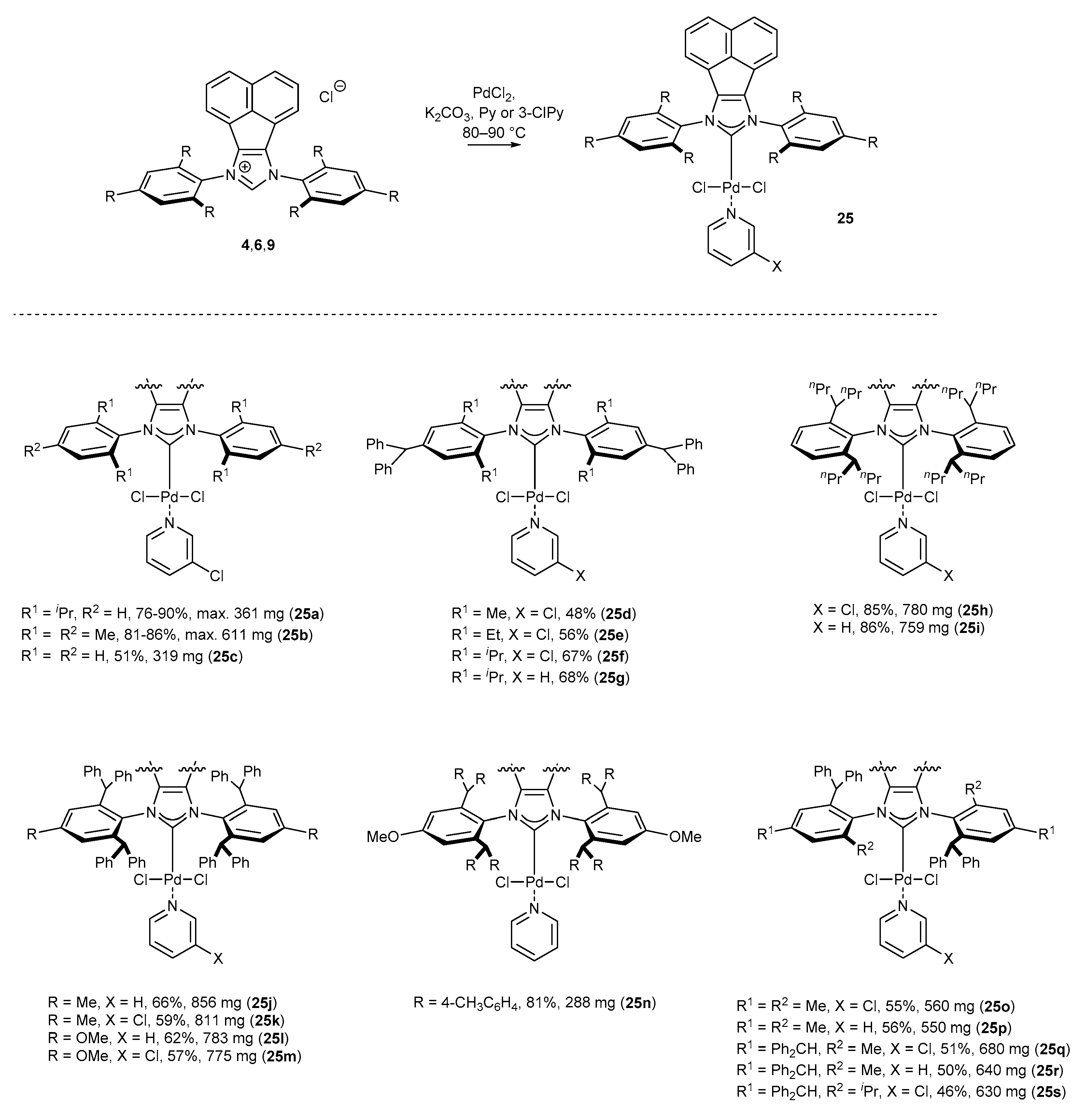
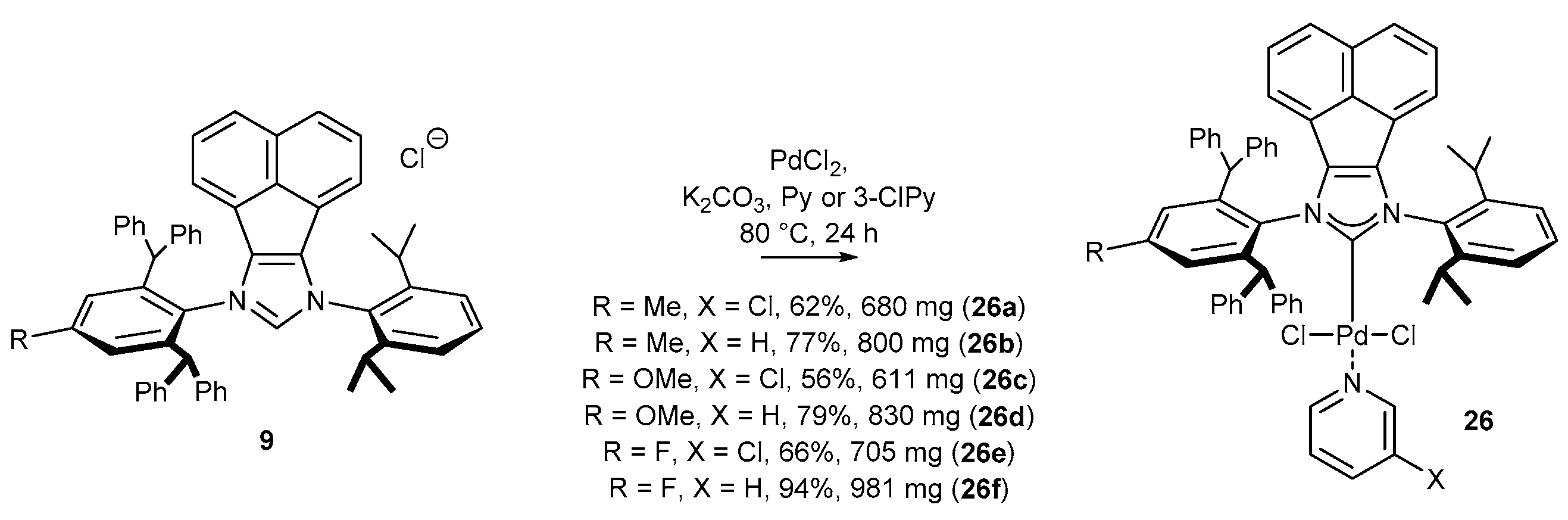
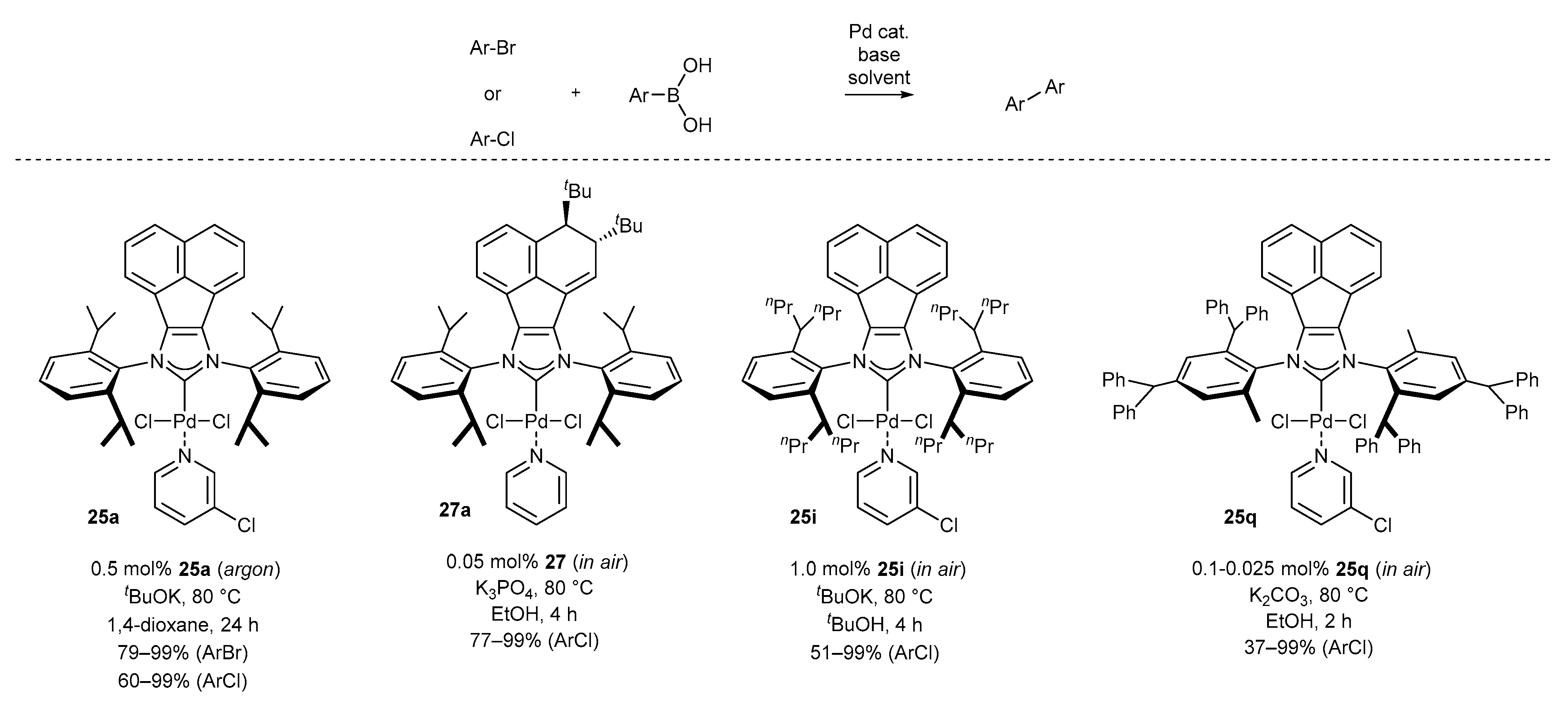
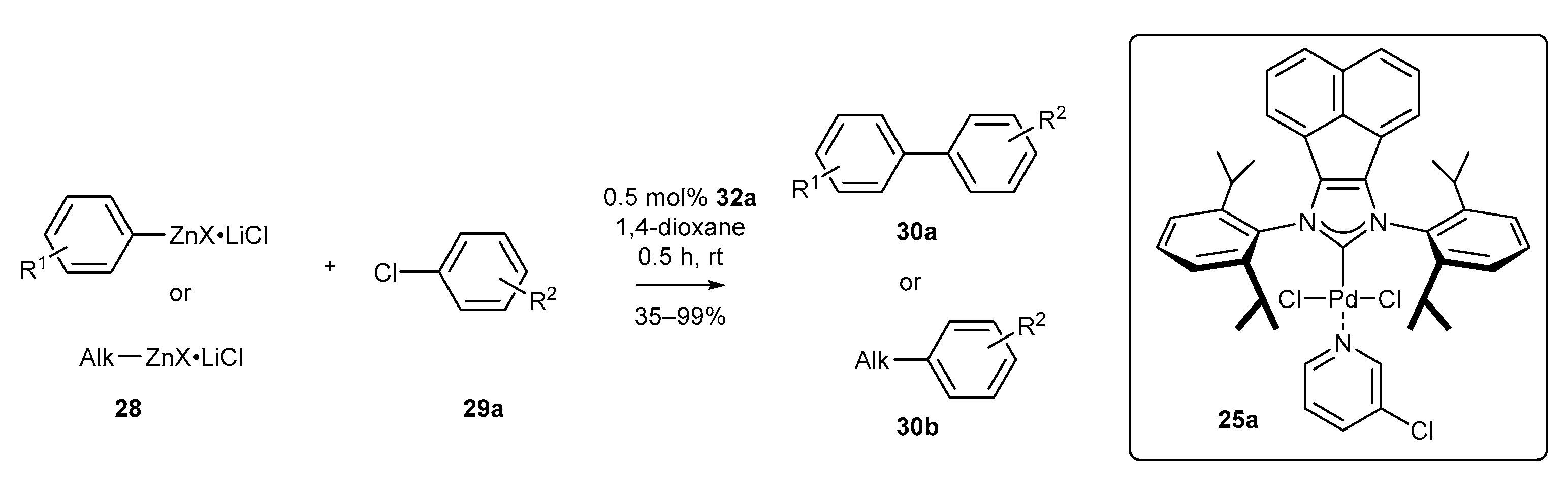
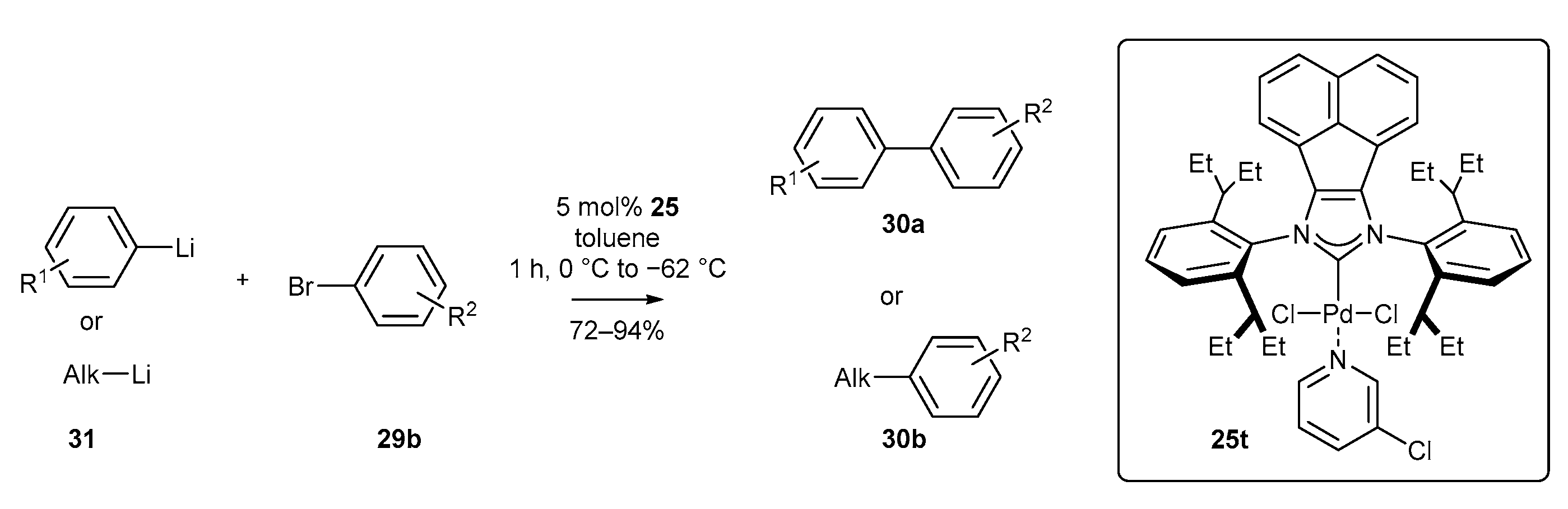
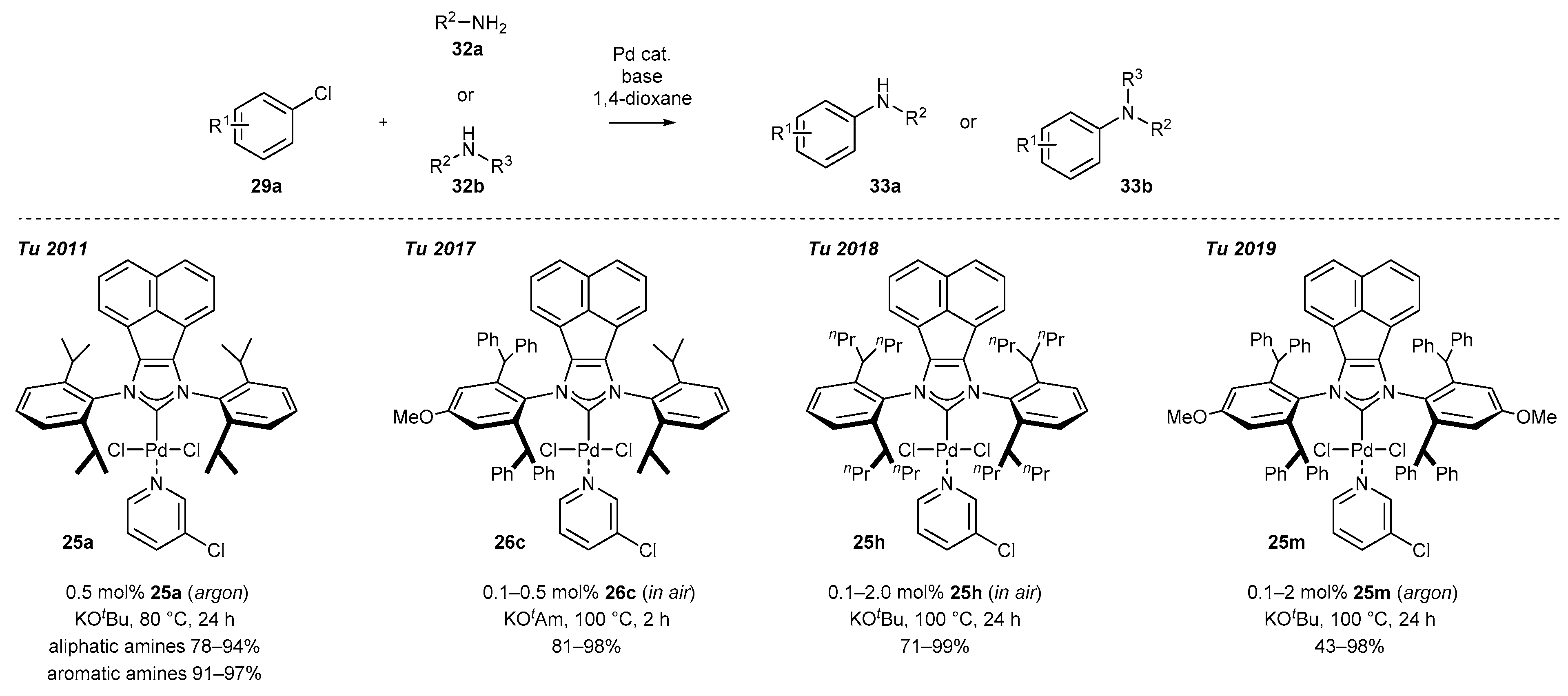
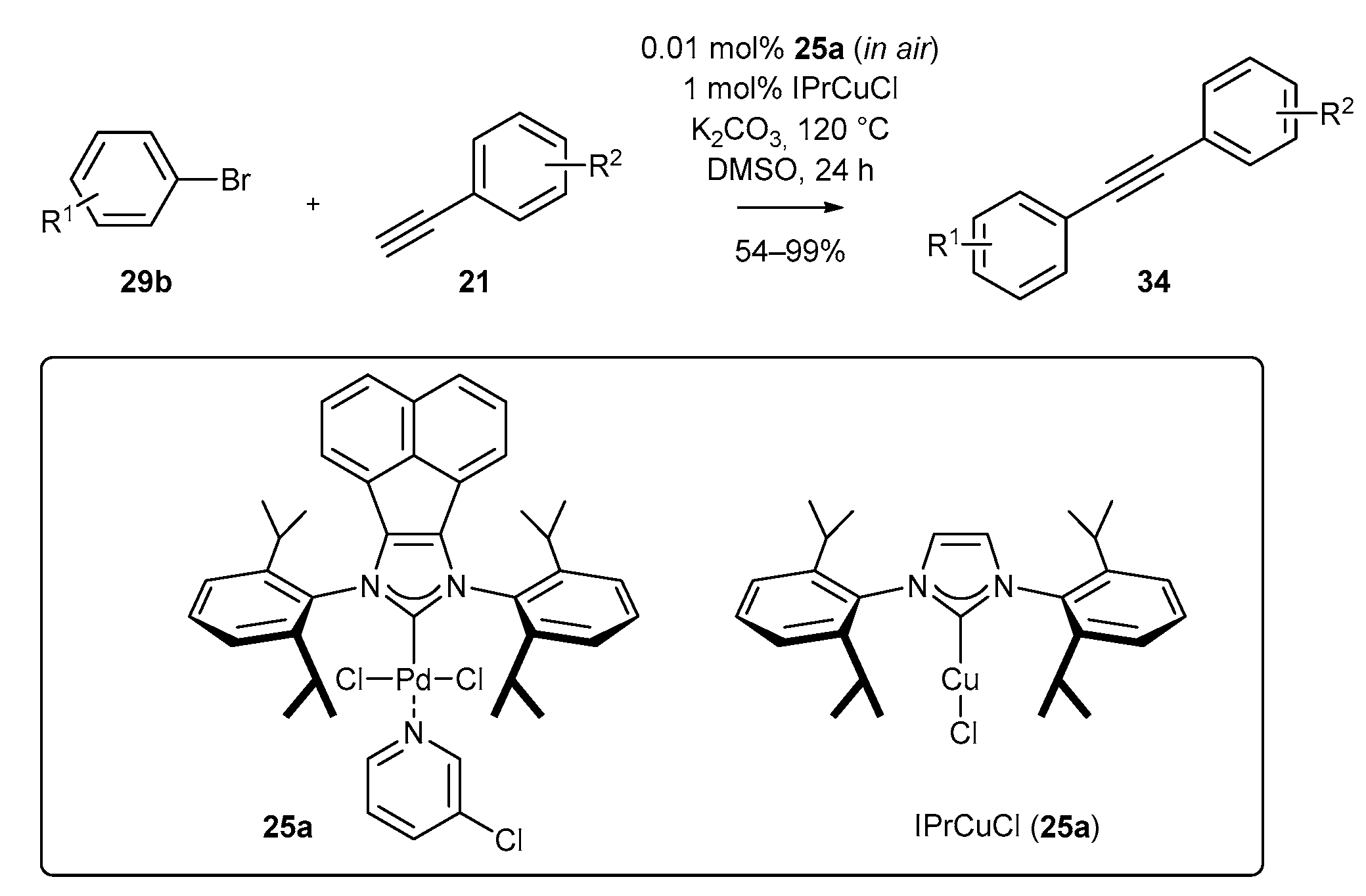
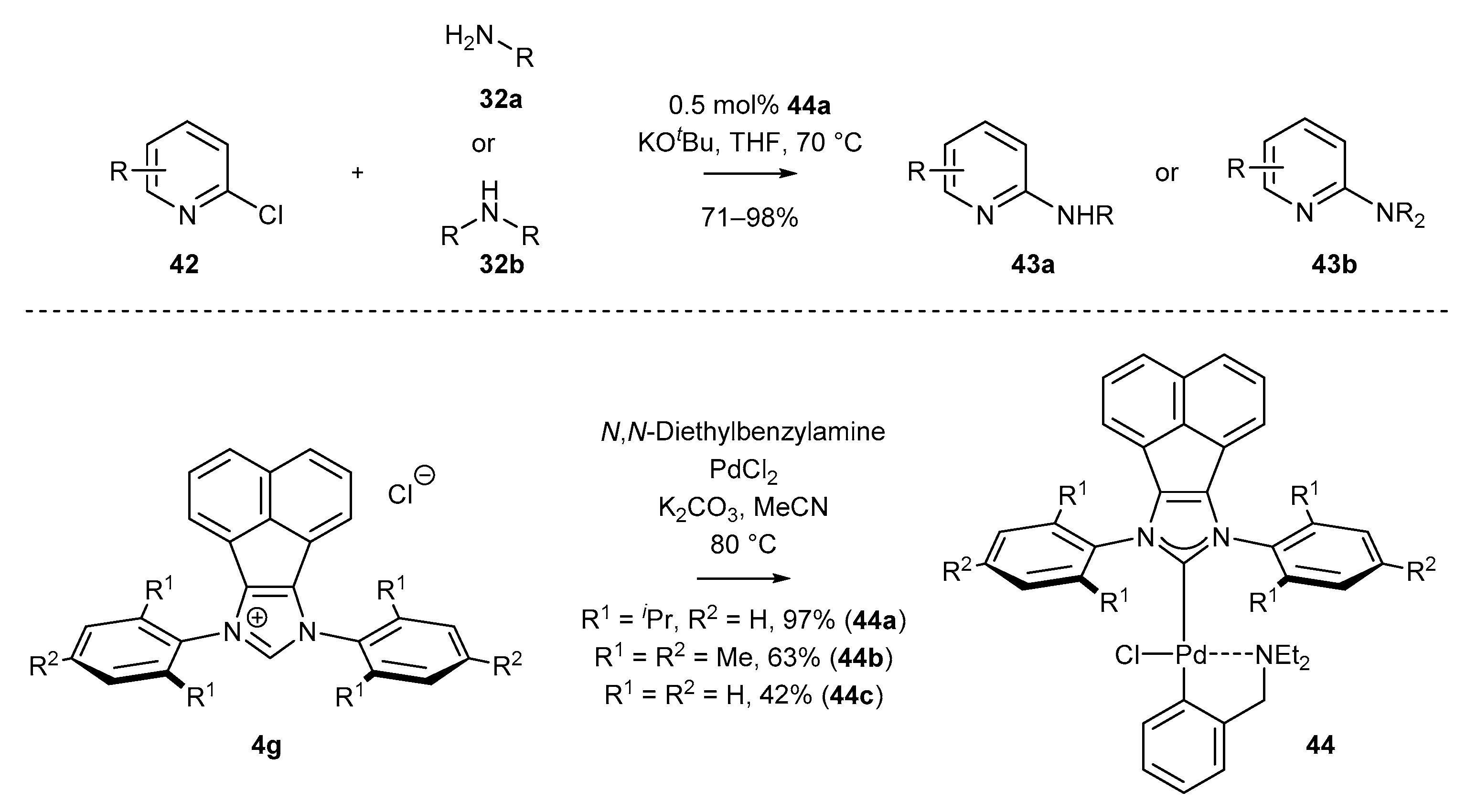
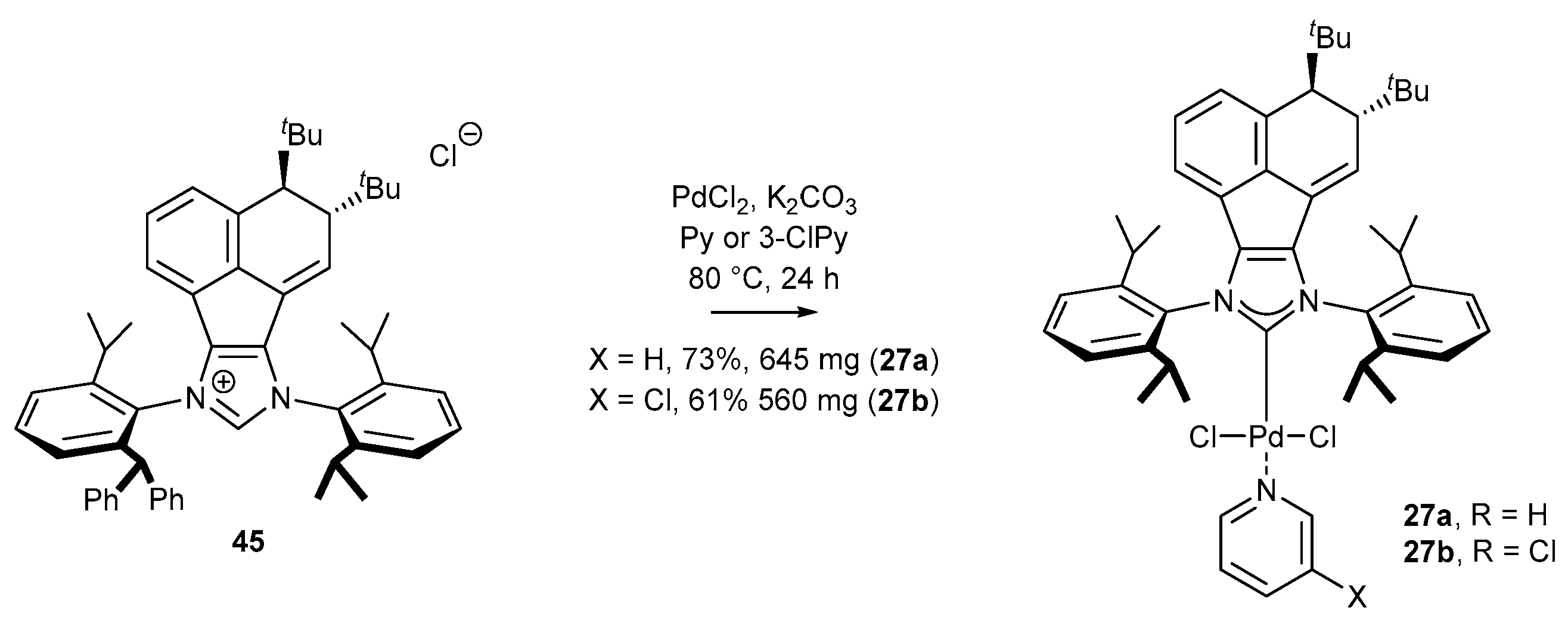
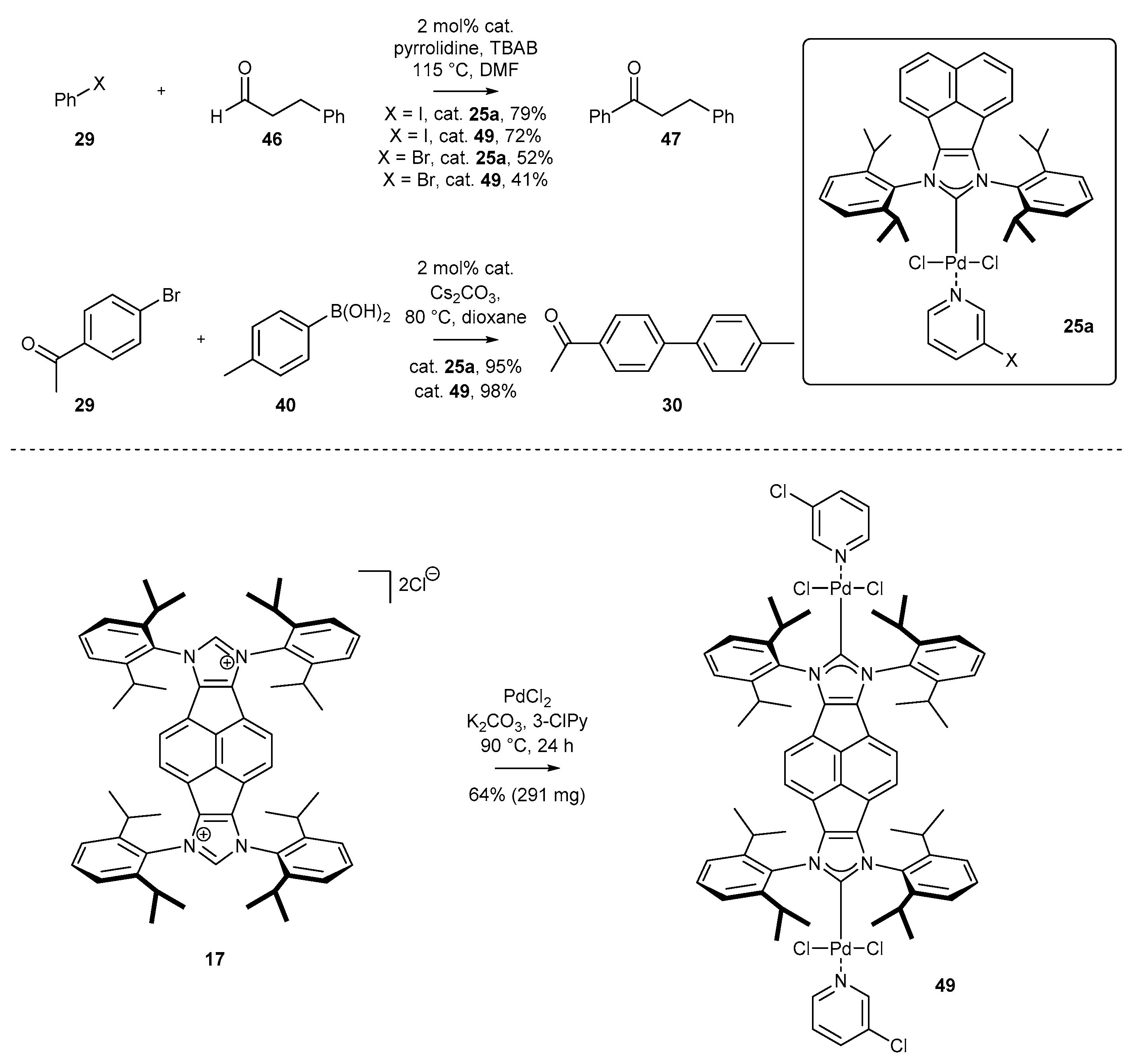
3.1.1. NHC-BIAN-PEPPSI-Type Complexes
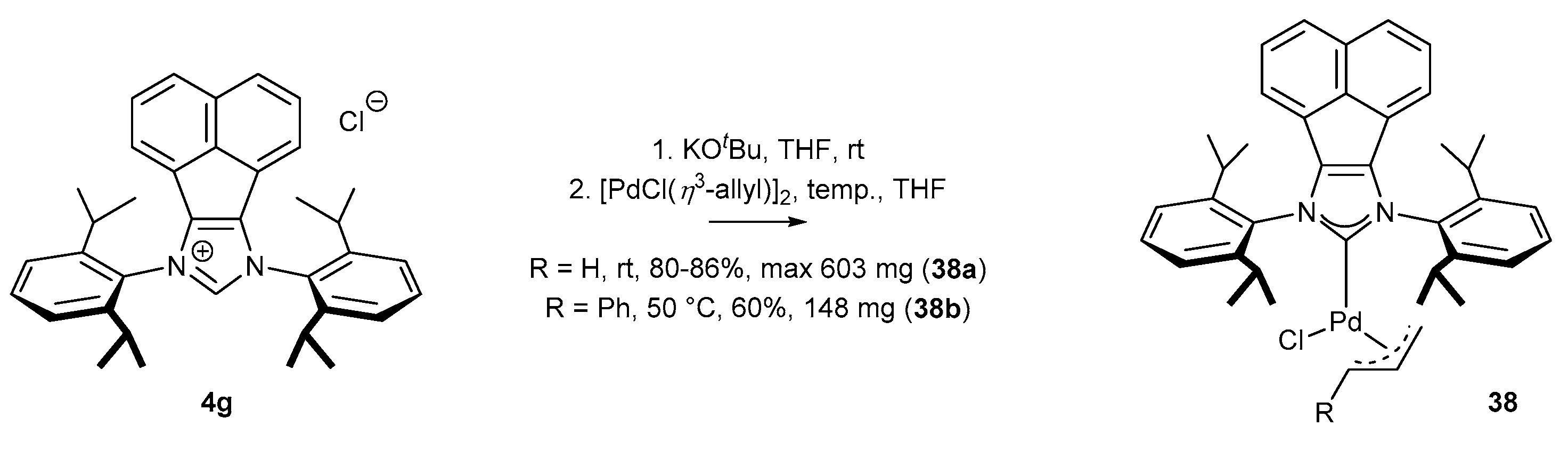
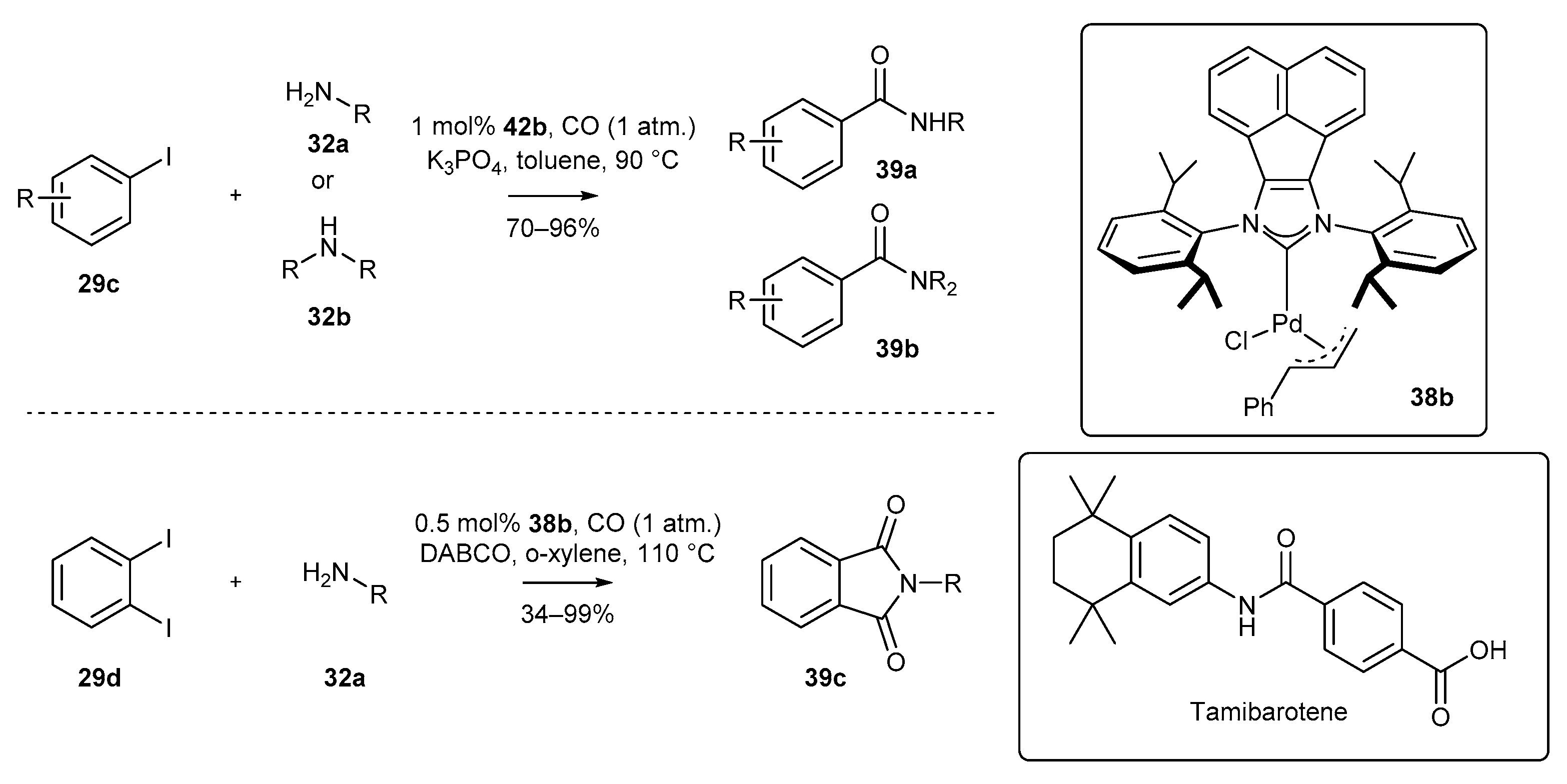
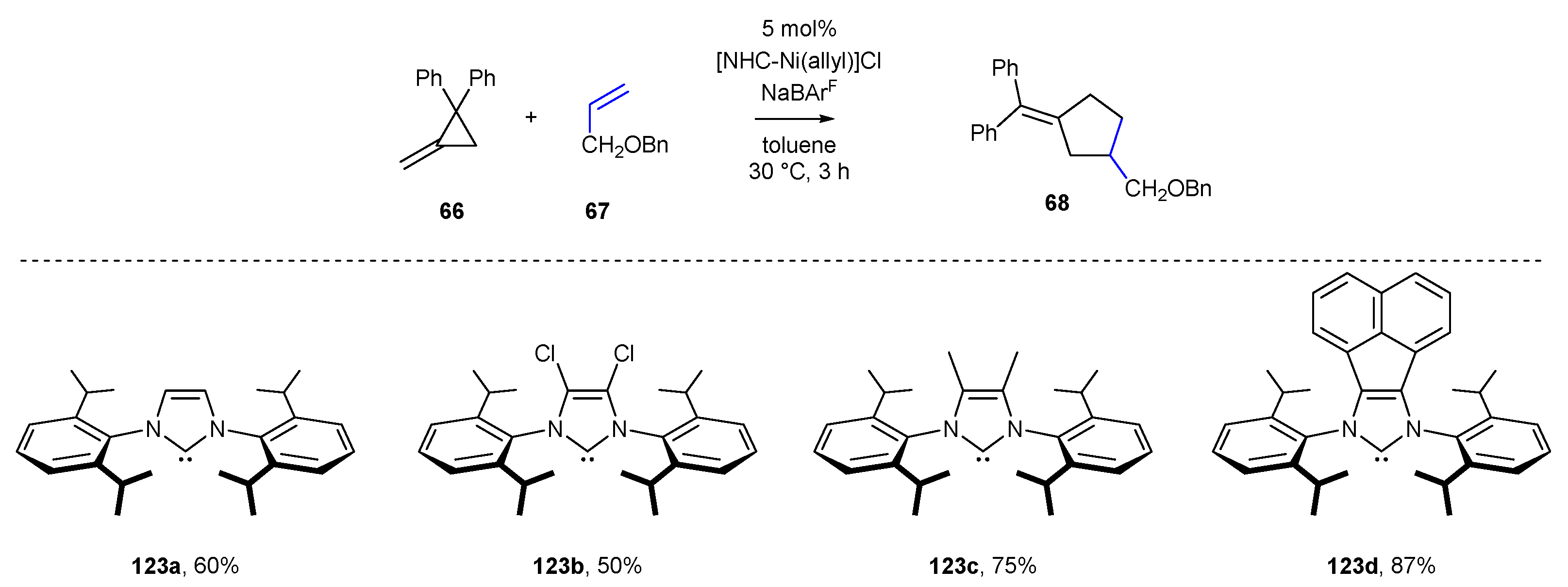
3.1.2. π-Allyl NHC-BIAN-Pd Complexes
3.1.3. Chelate Complexes
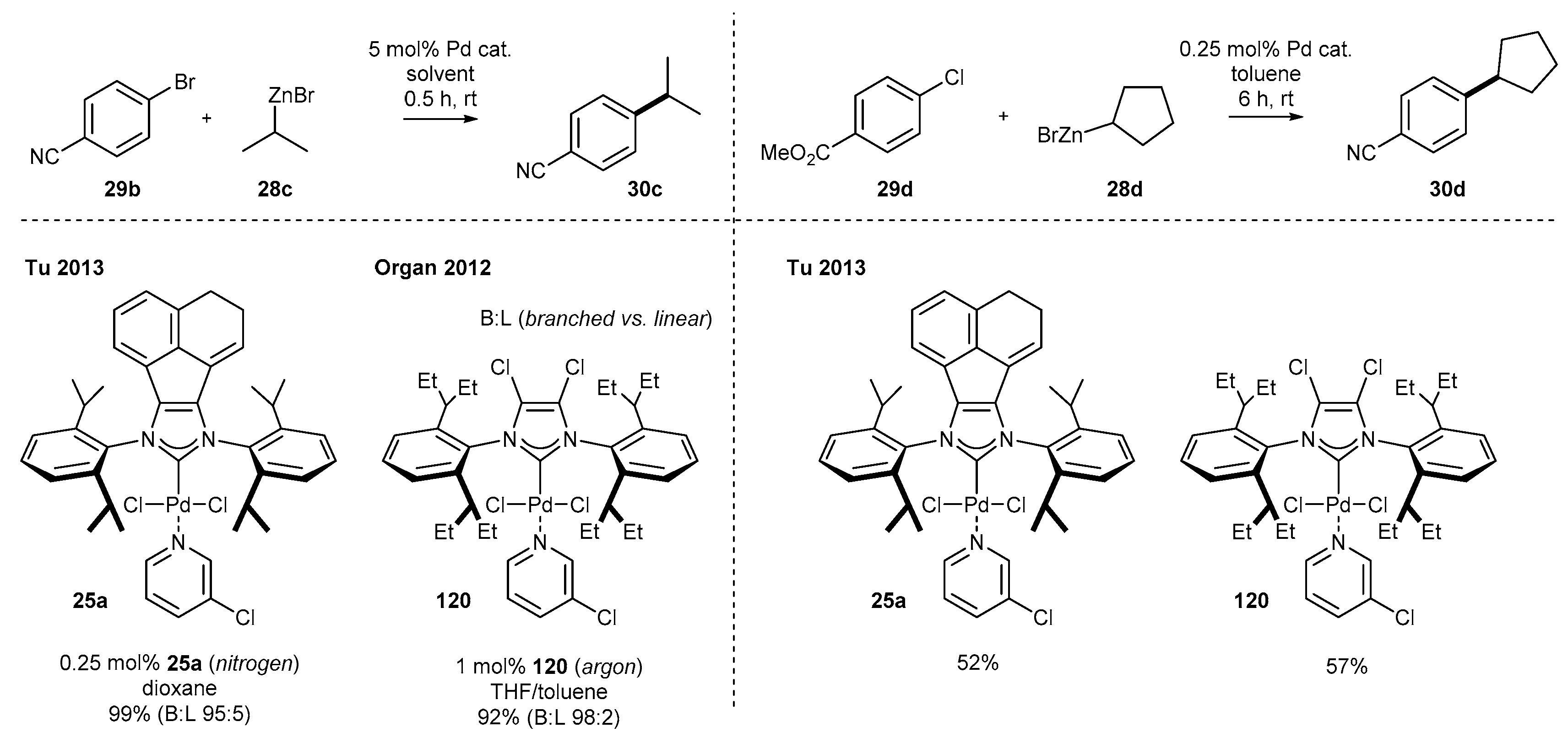
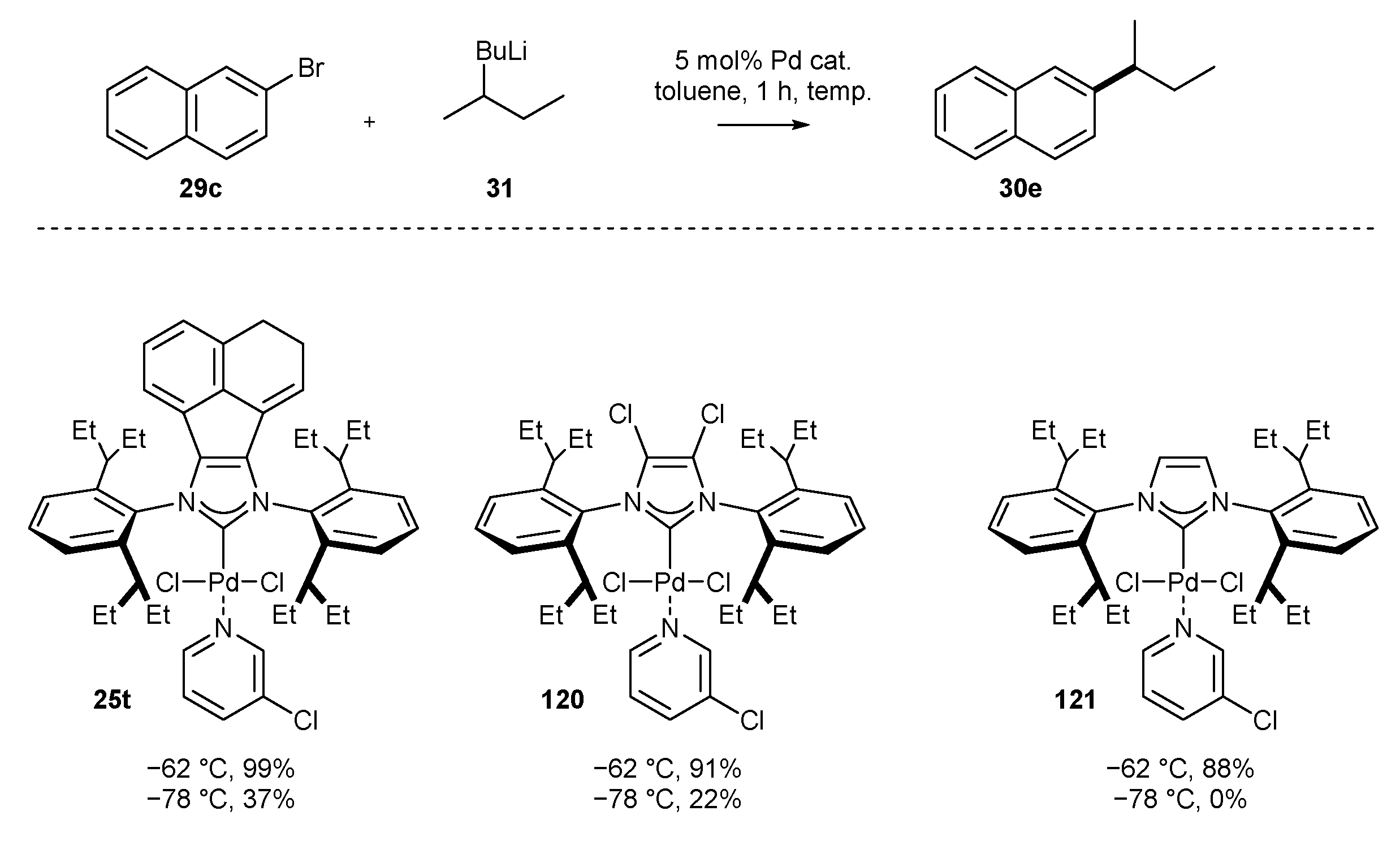
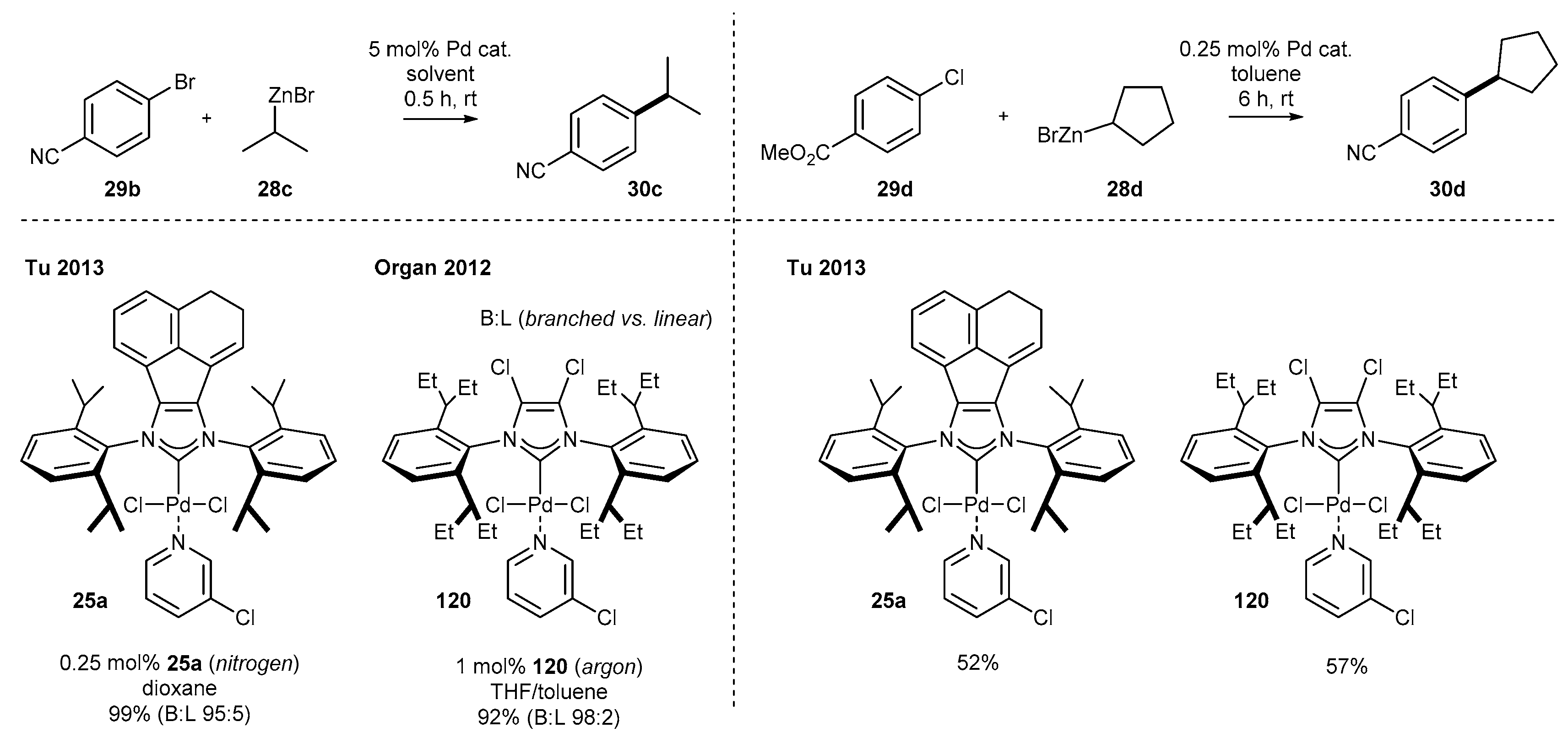
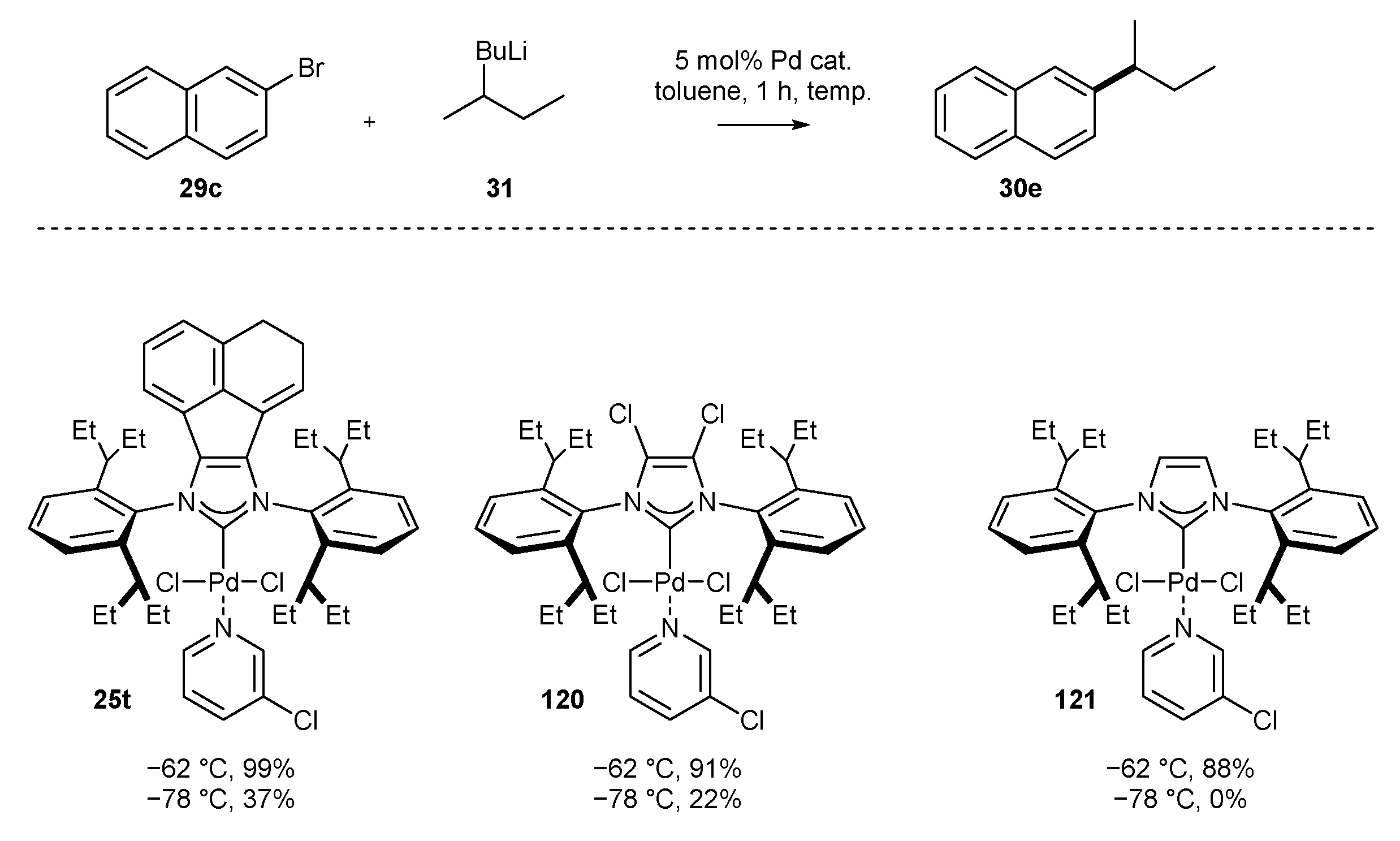
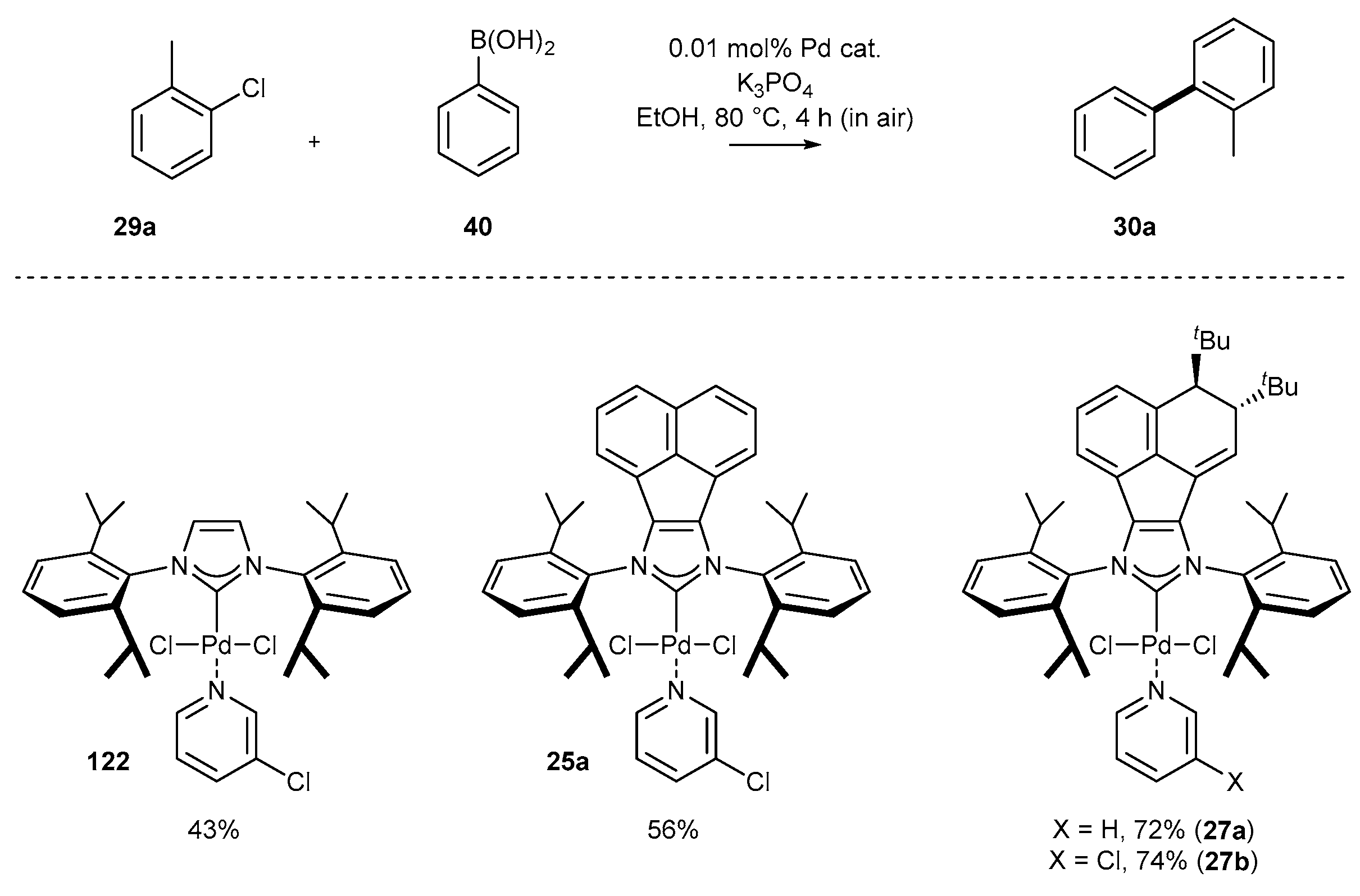
3.1.4. Skeleton Modifications of NHC-BIAN-Pd Complexes and Their Catalytic Activity
3.1.5. Polyfunctional NHC-BIAN-Pd Complexes
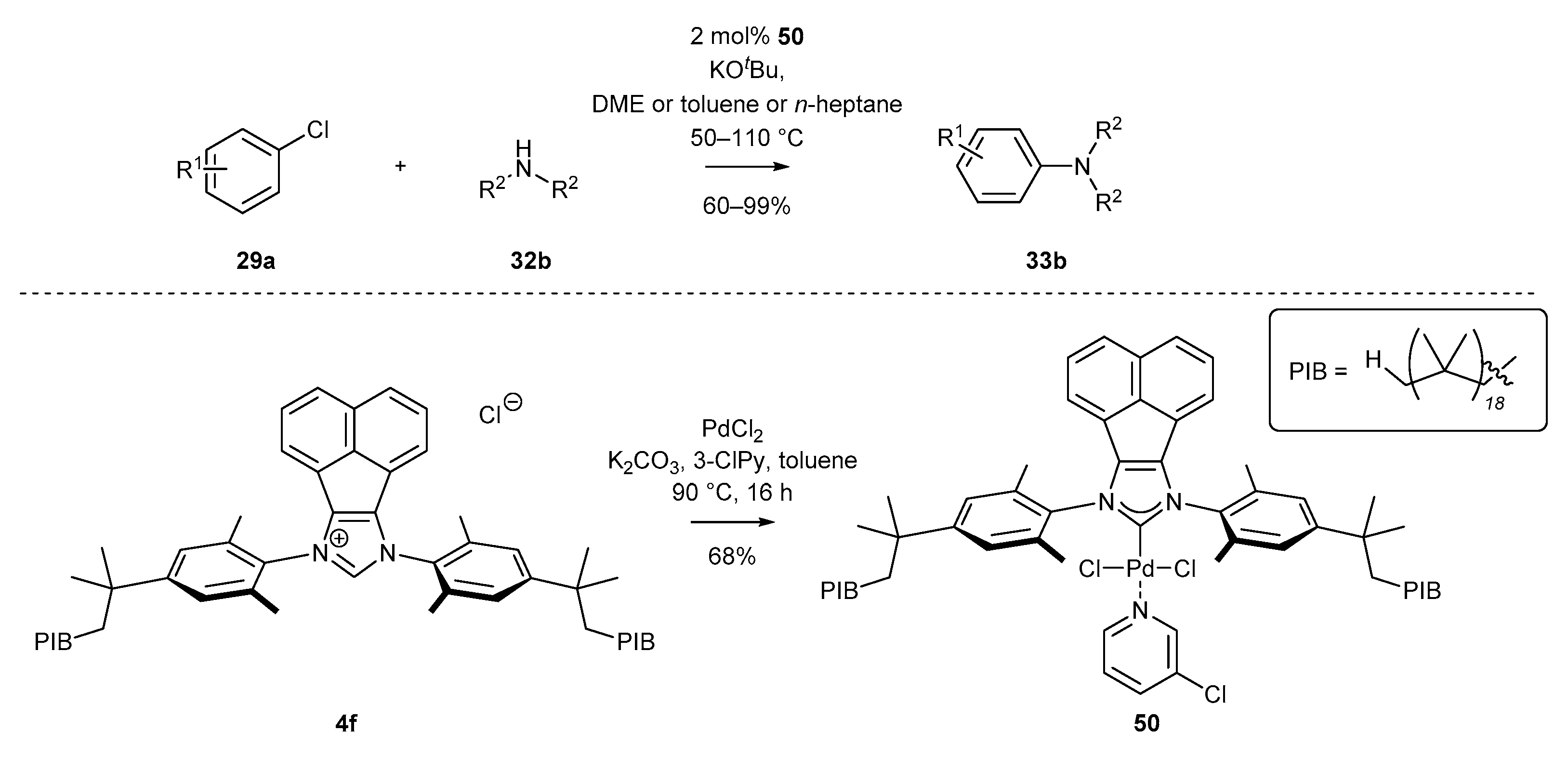
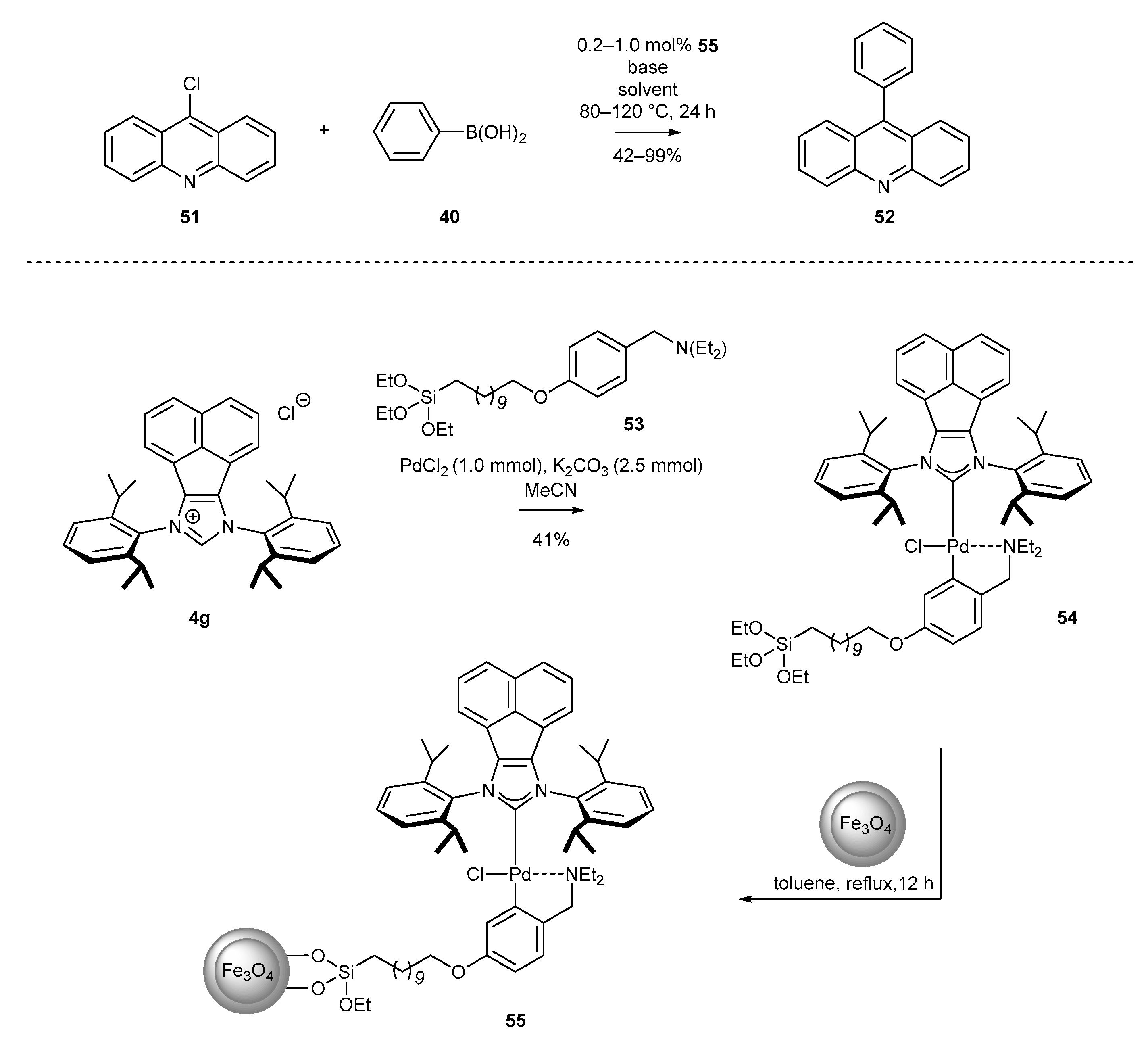
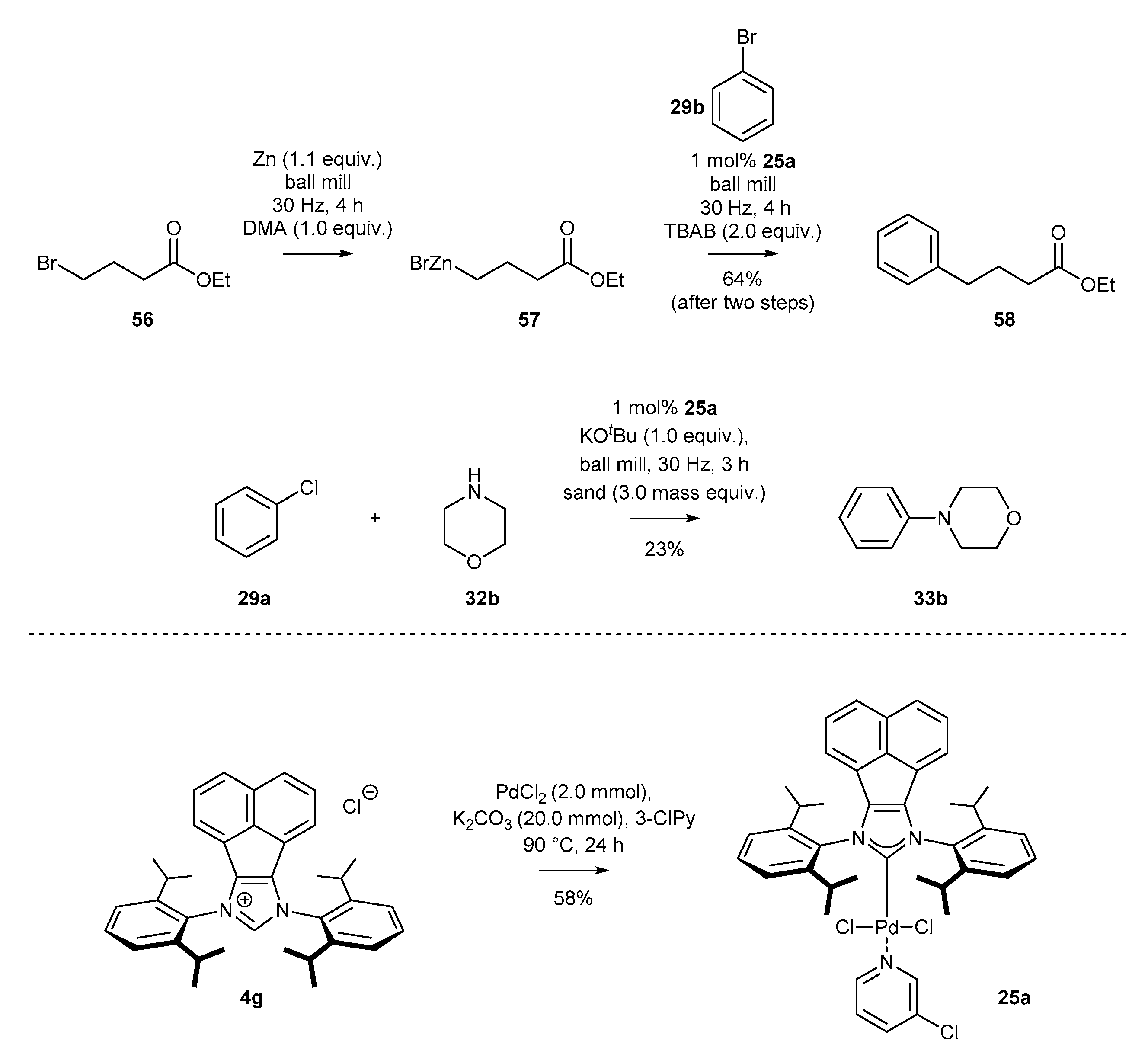
3.1.6. Heterogenization of NHC-BIAN-Pd Complexes
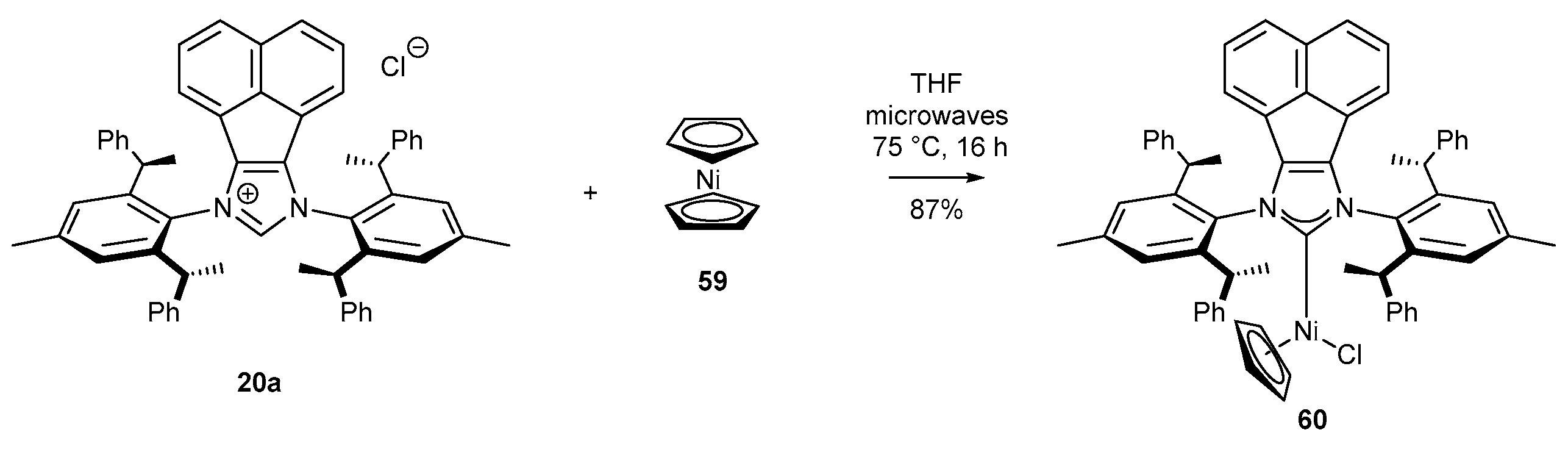
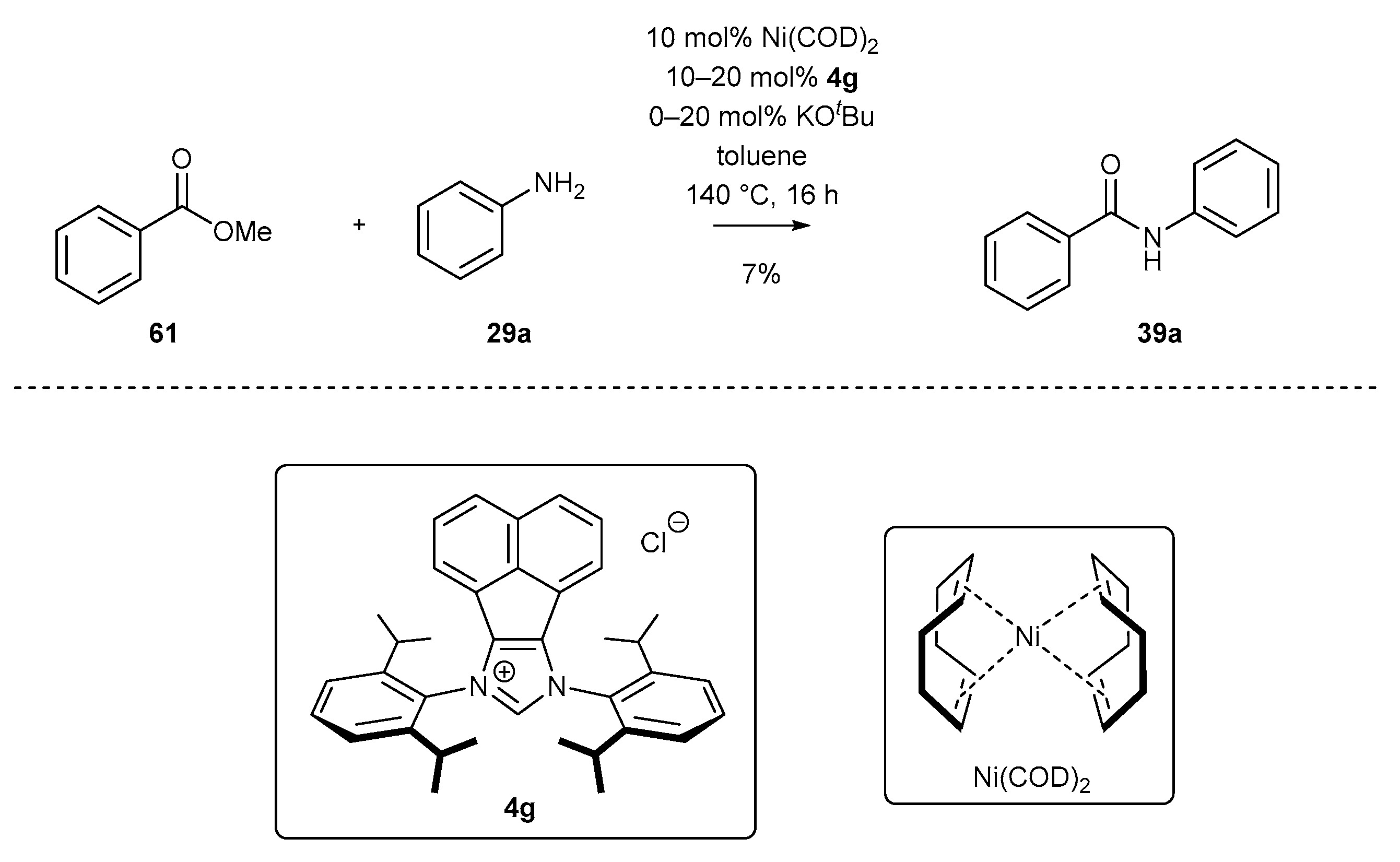
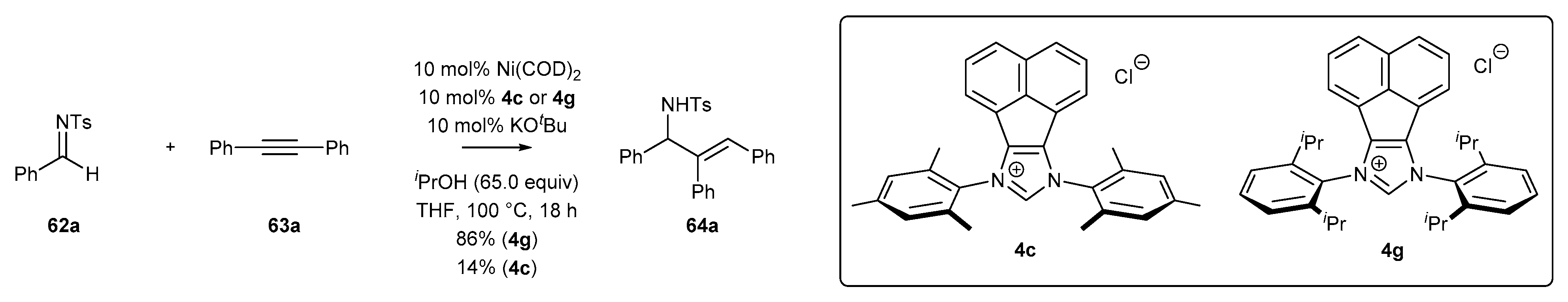
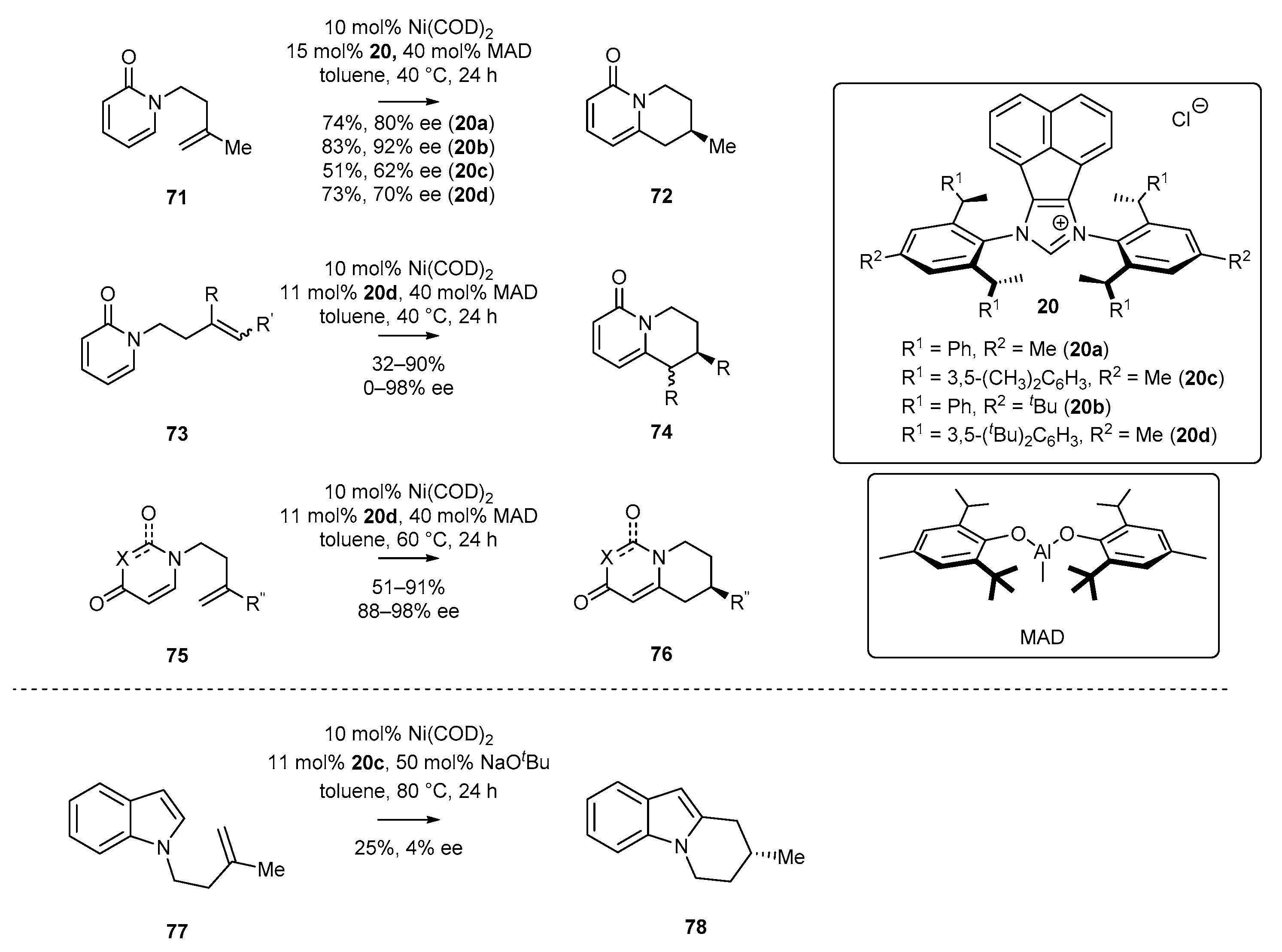
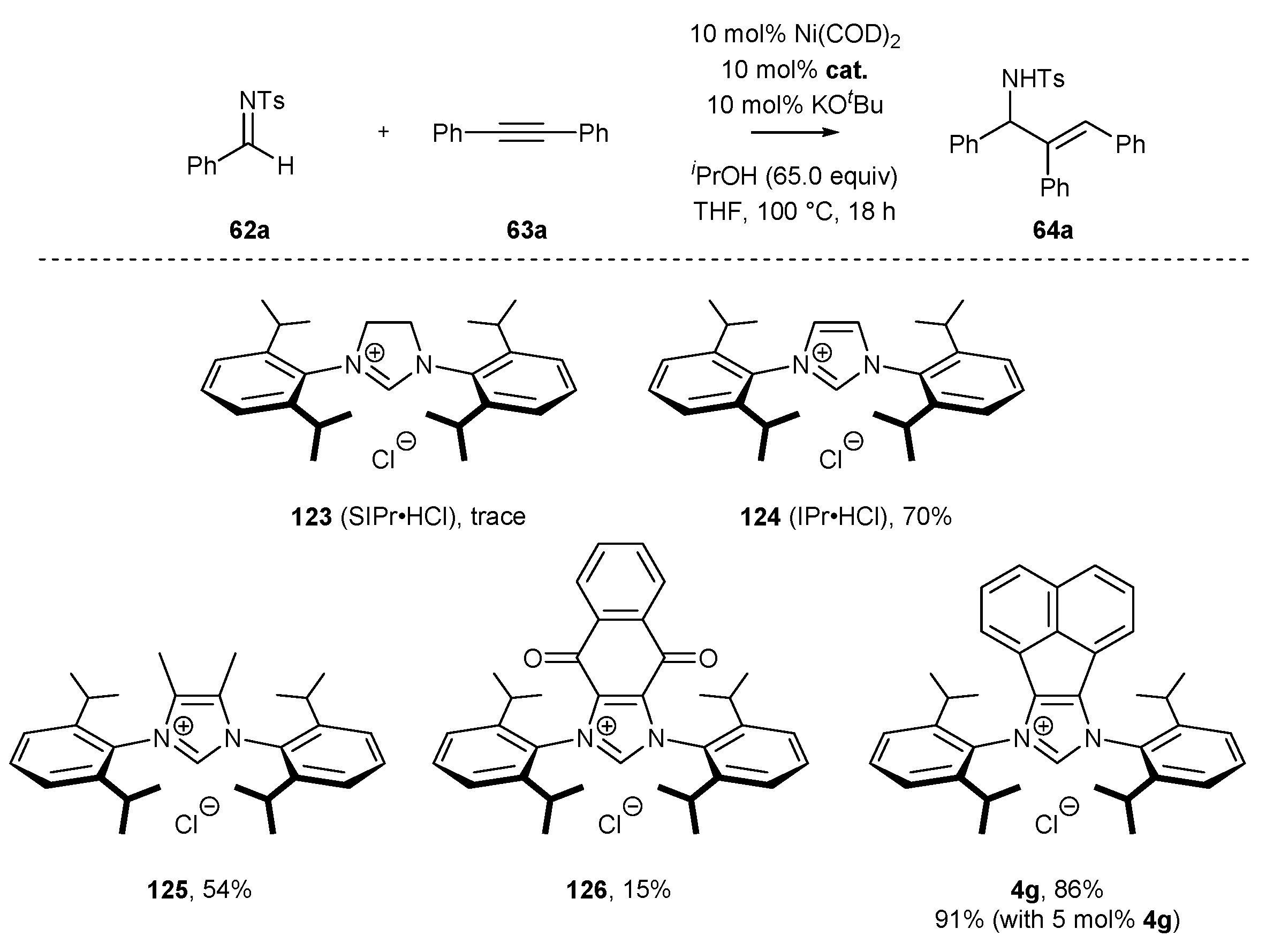
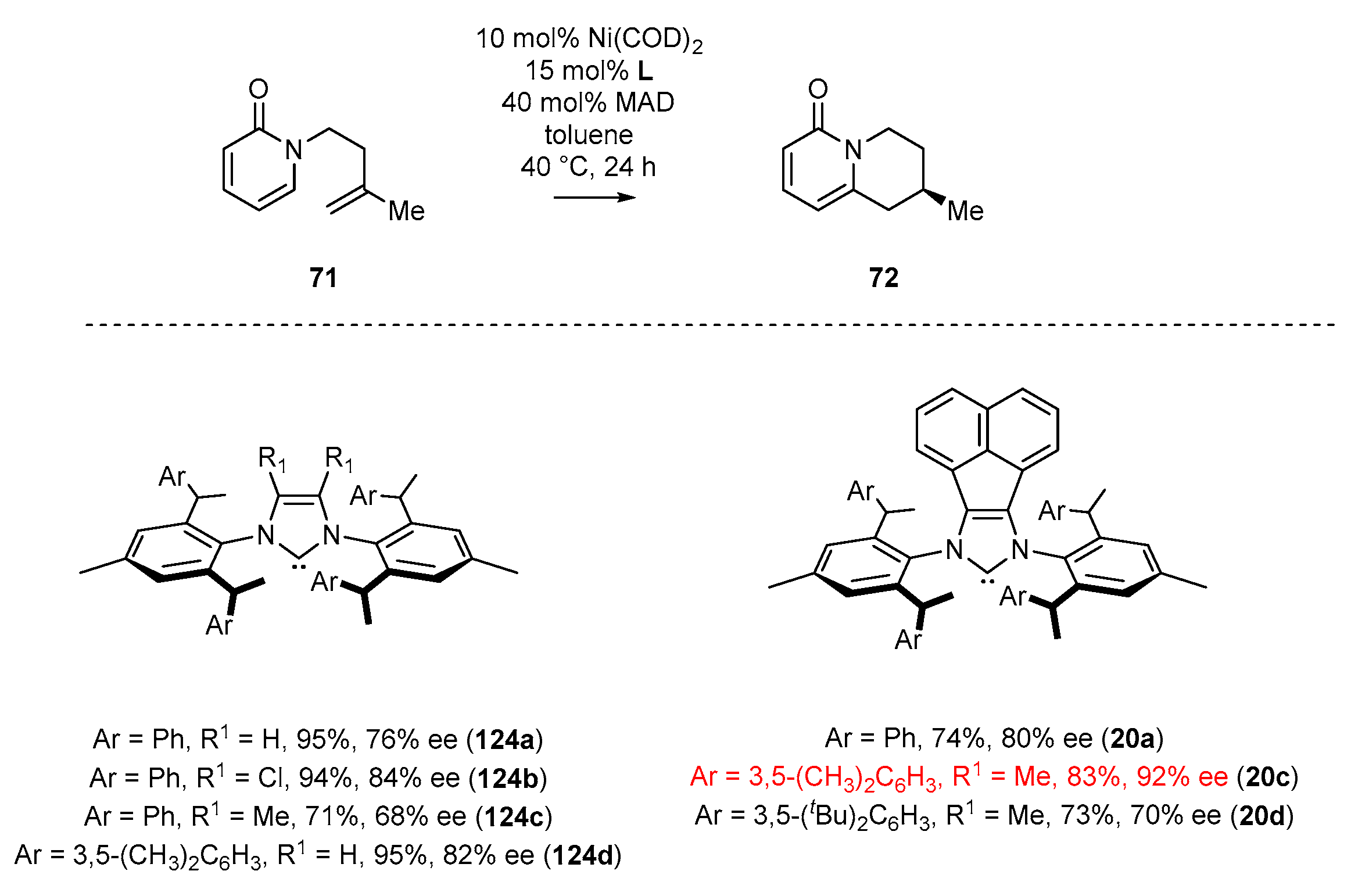
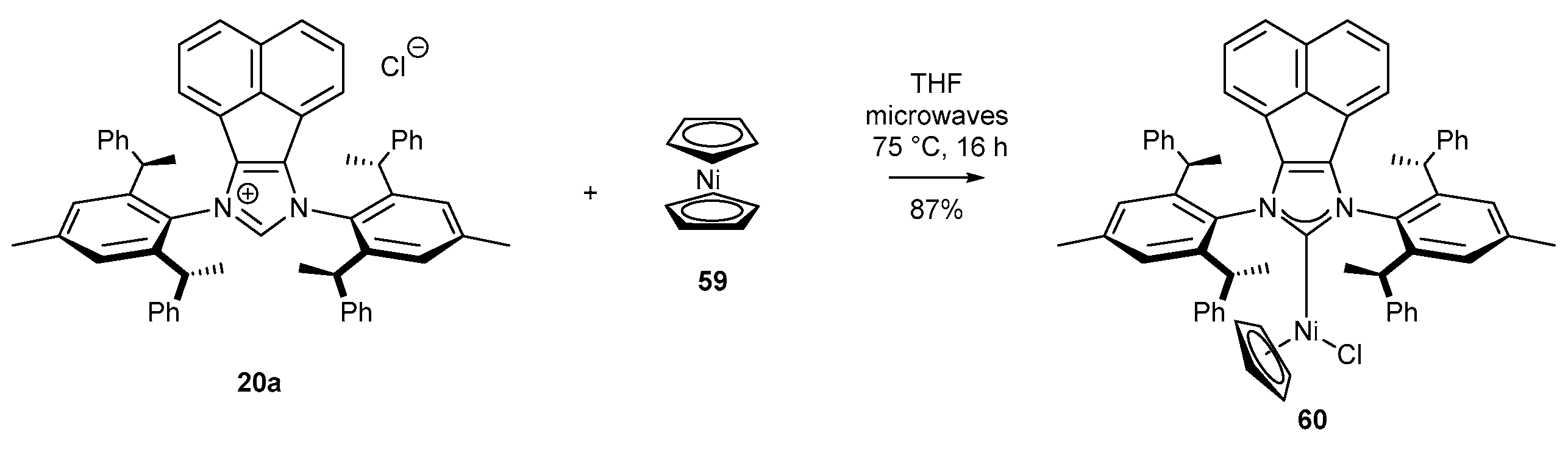
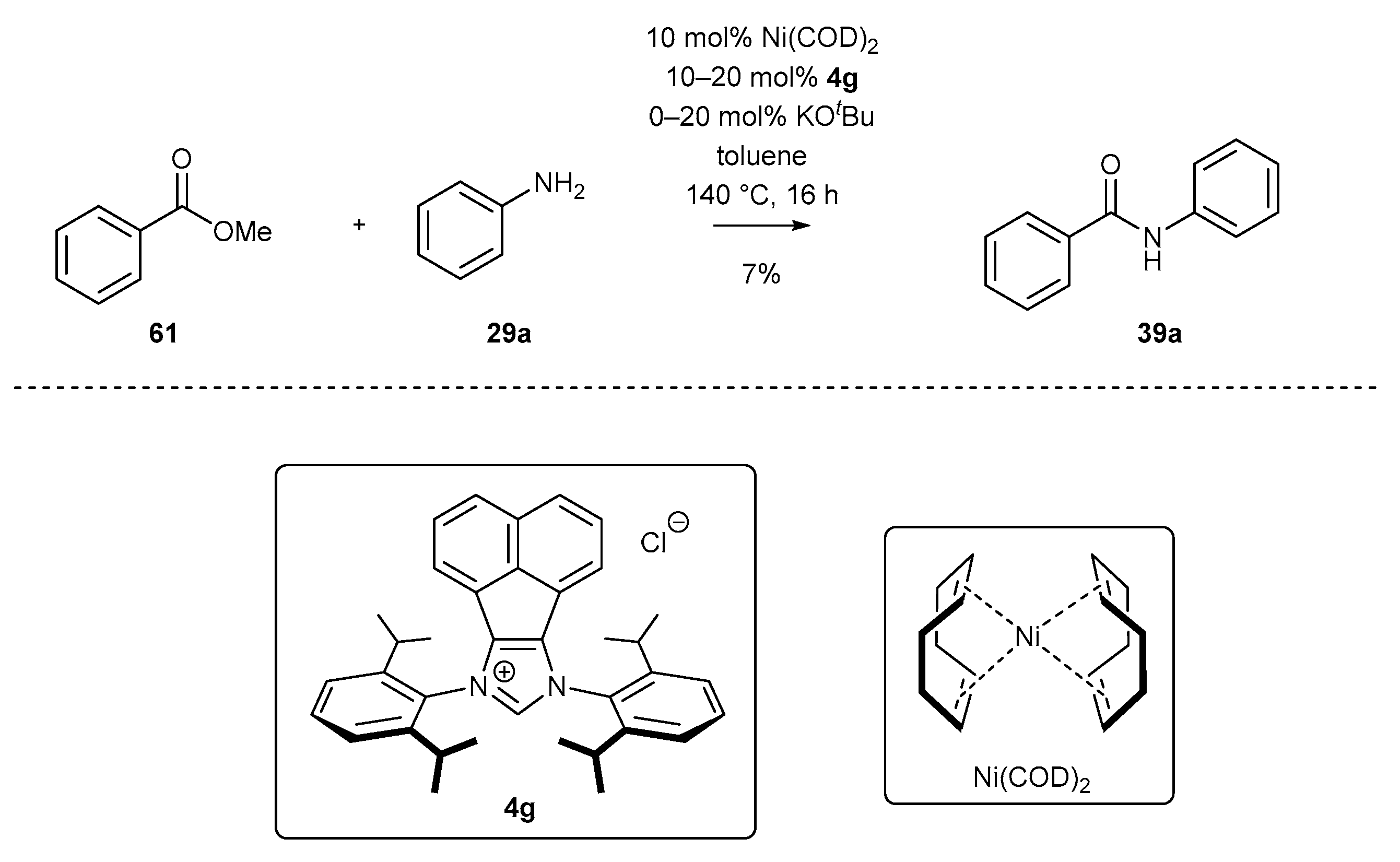
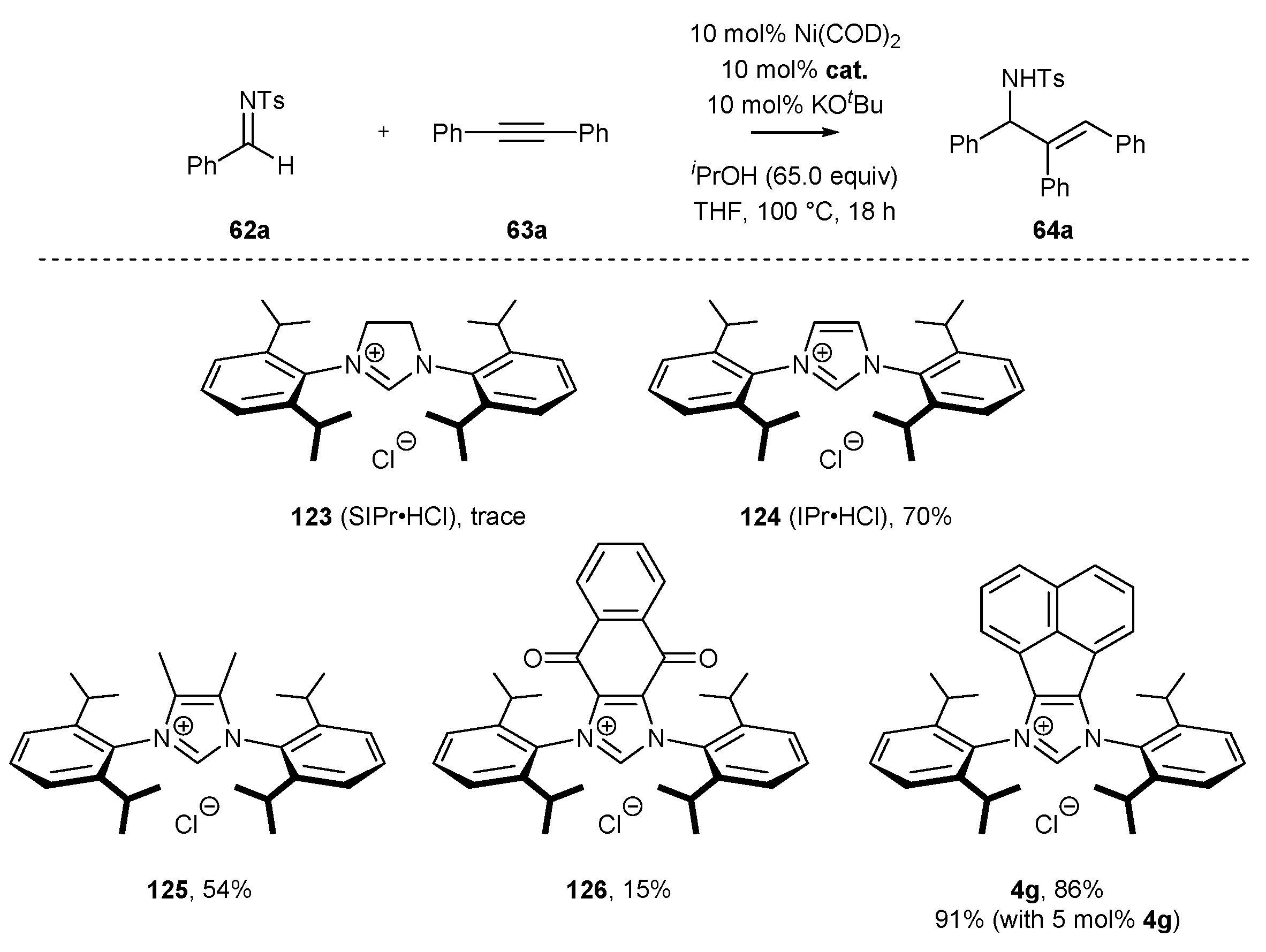
3.2. NHC-BIAN-Ni Complexes in Catalysis
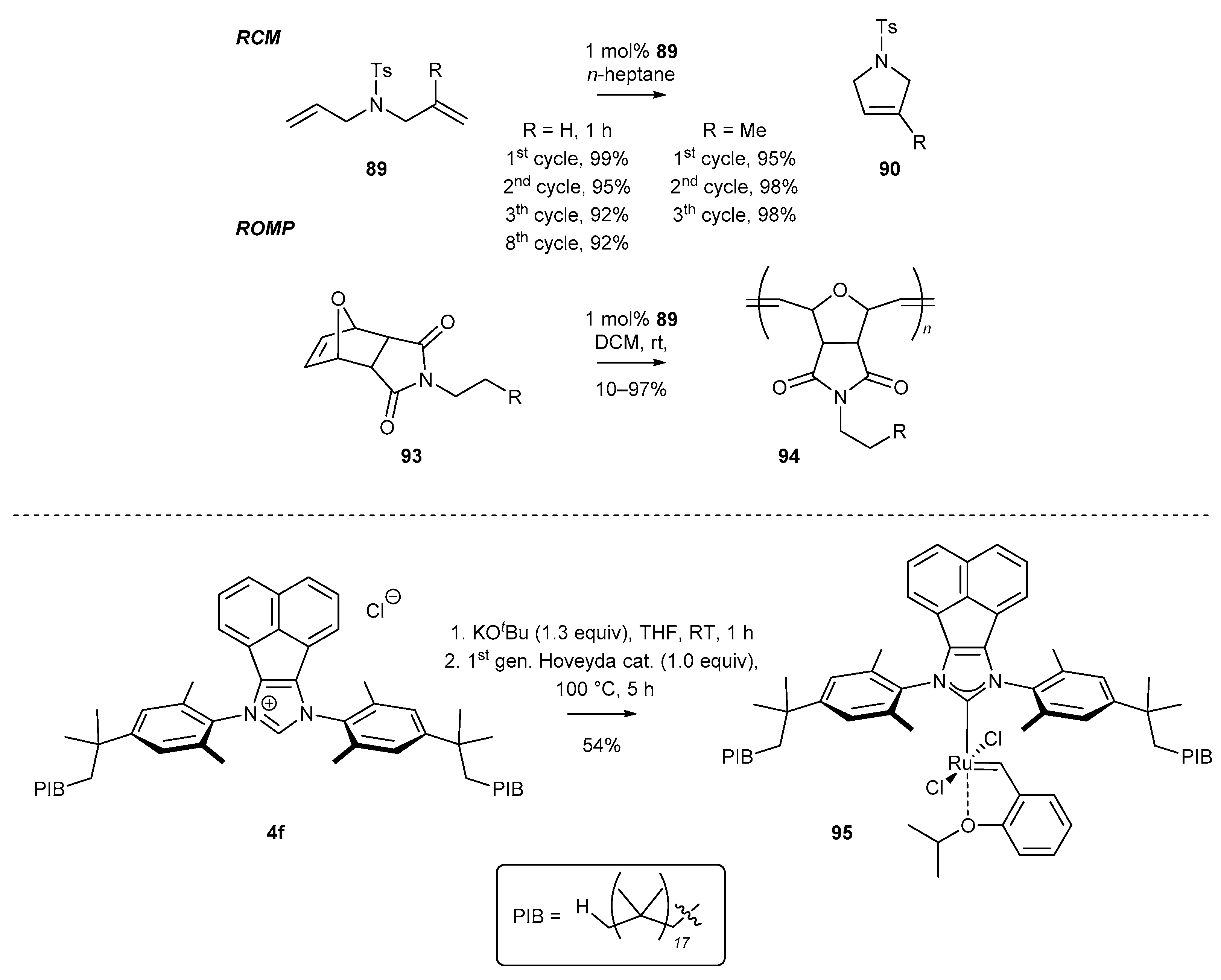
3.3. NHC-BIAN-Ru Complexes
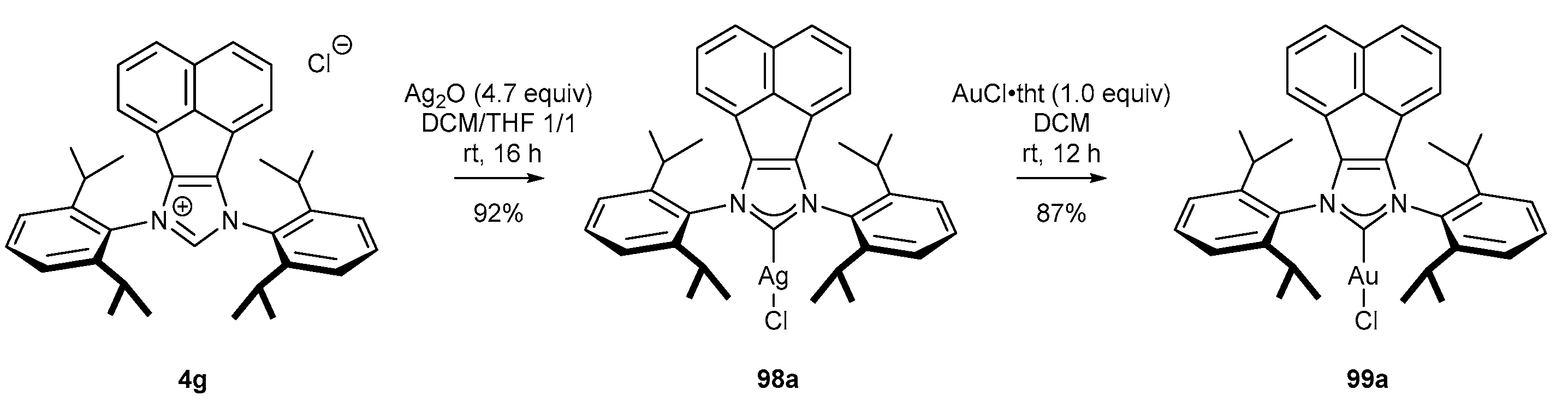
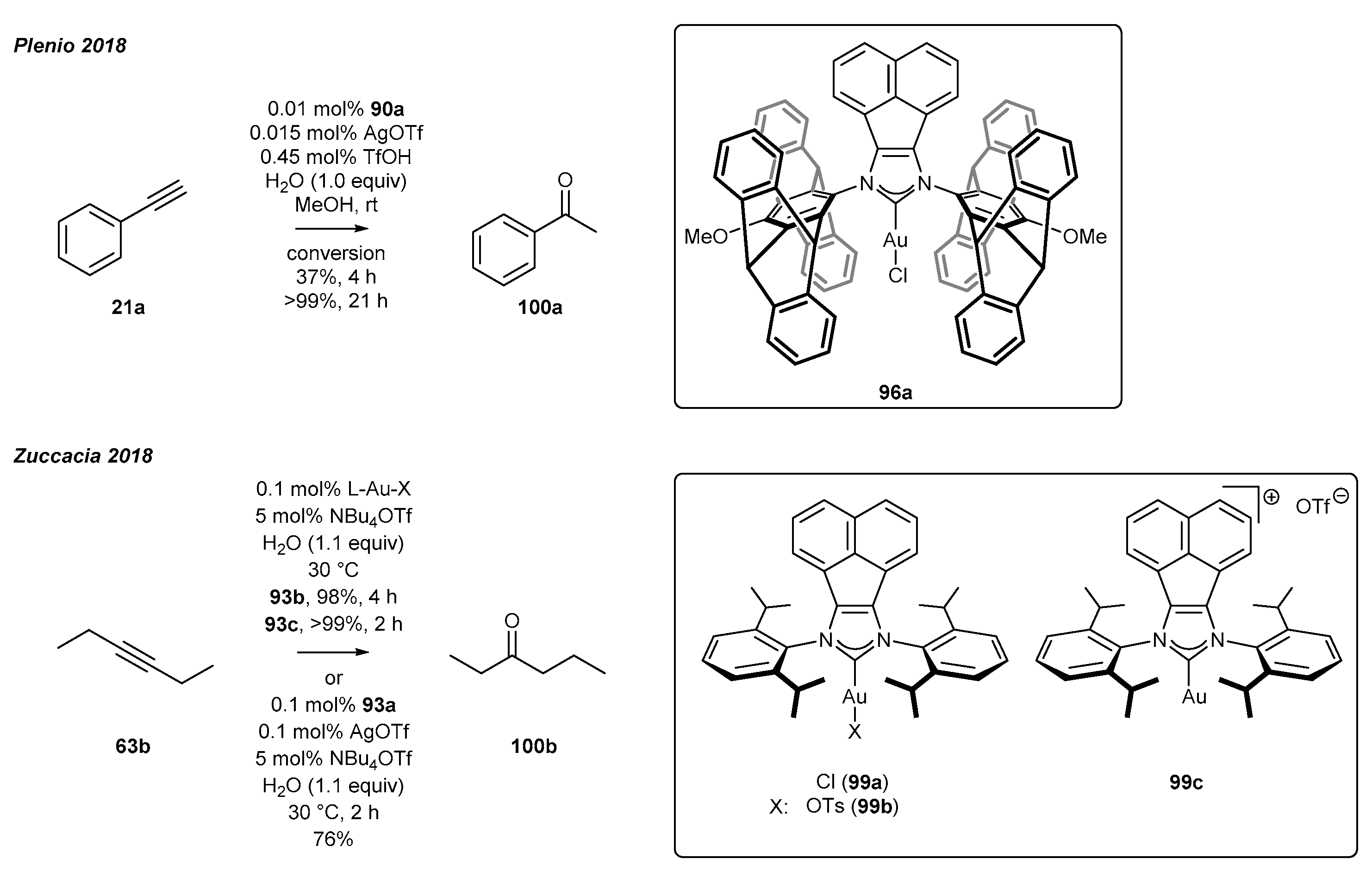
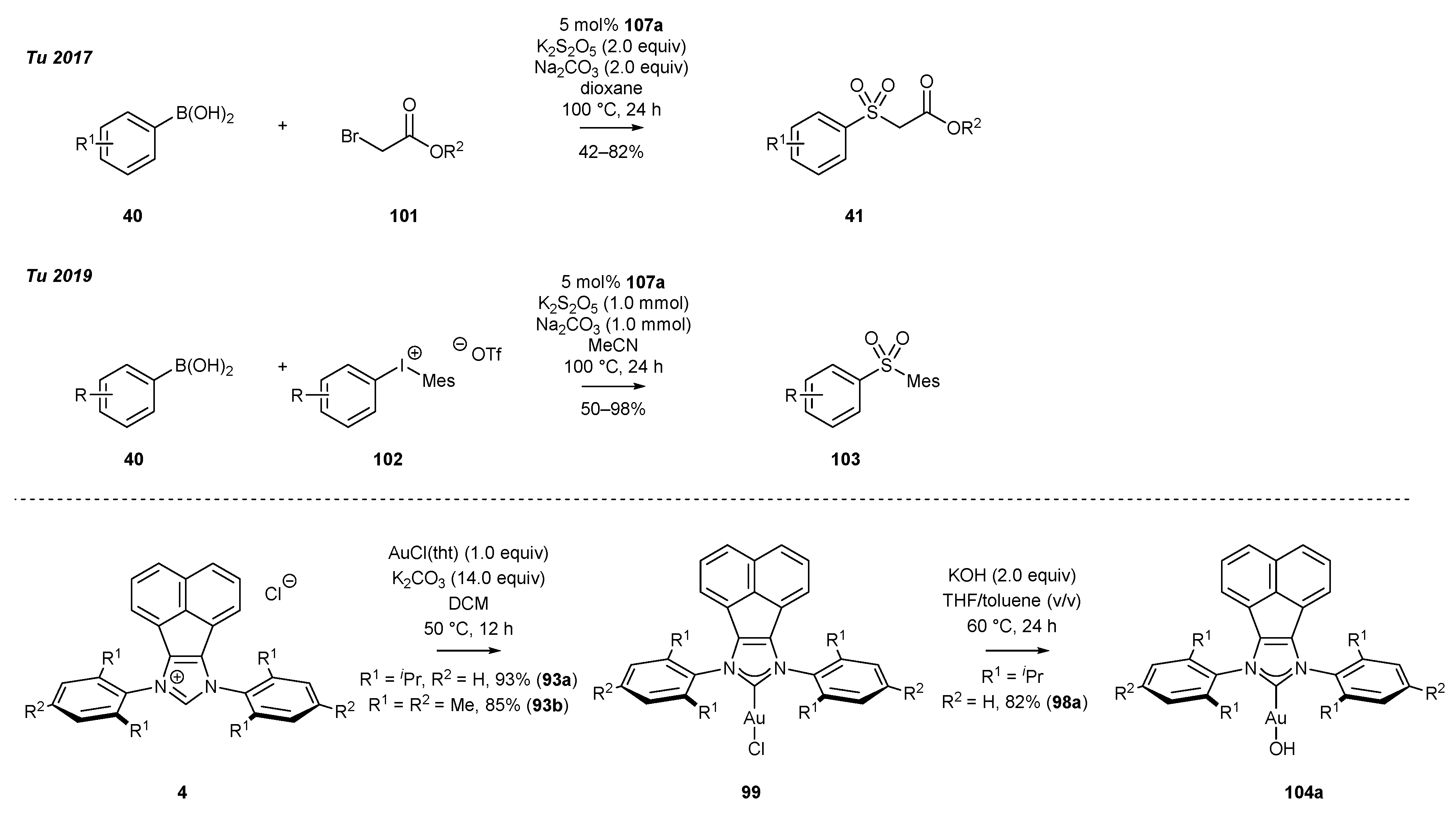
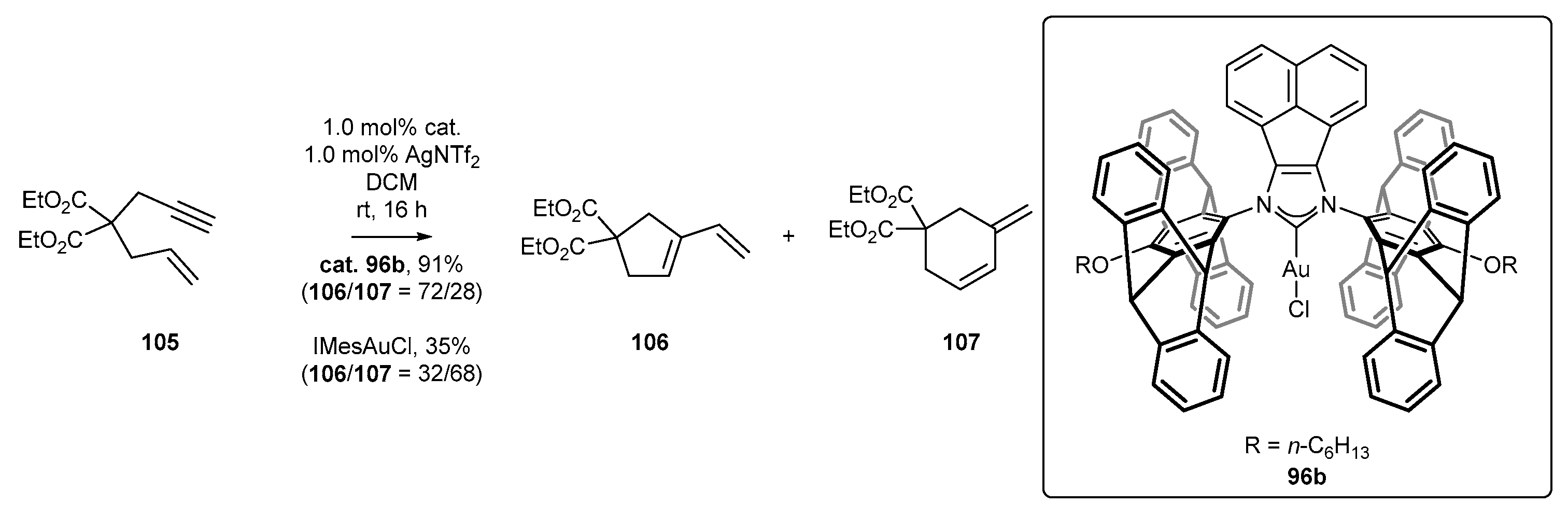
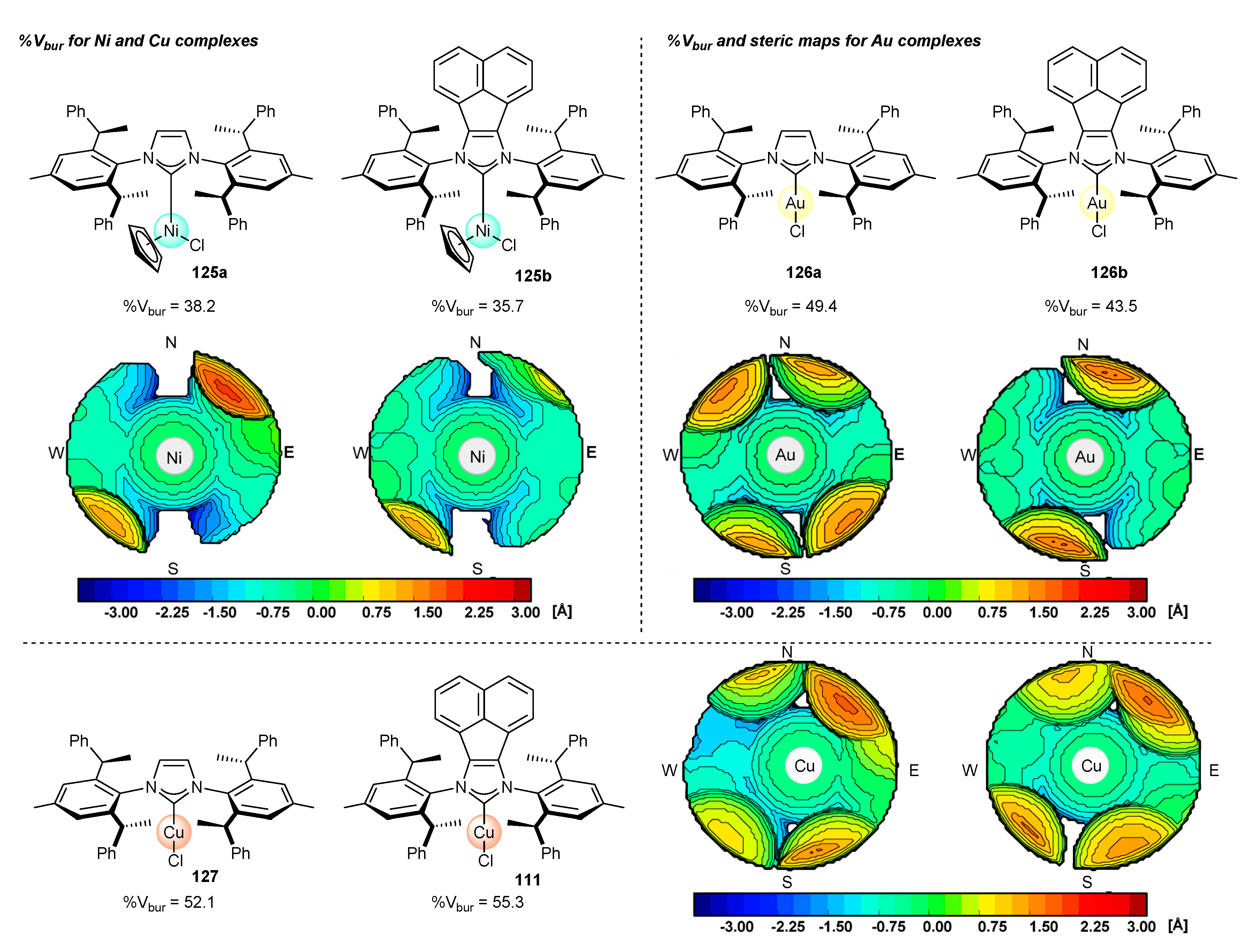
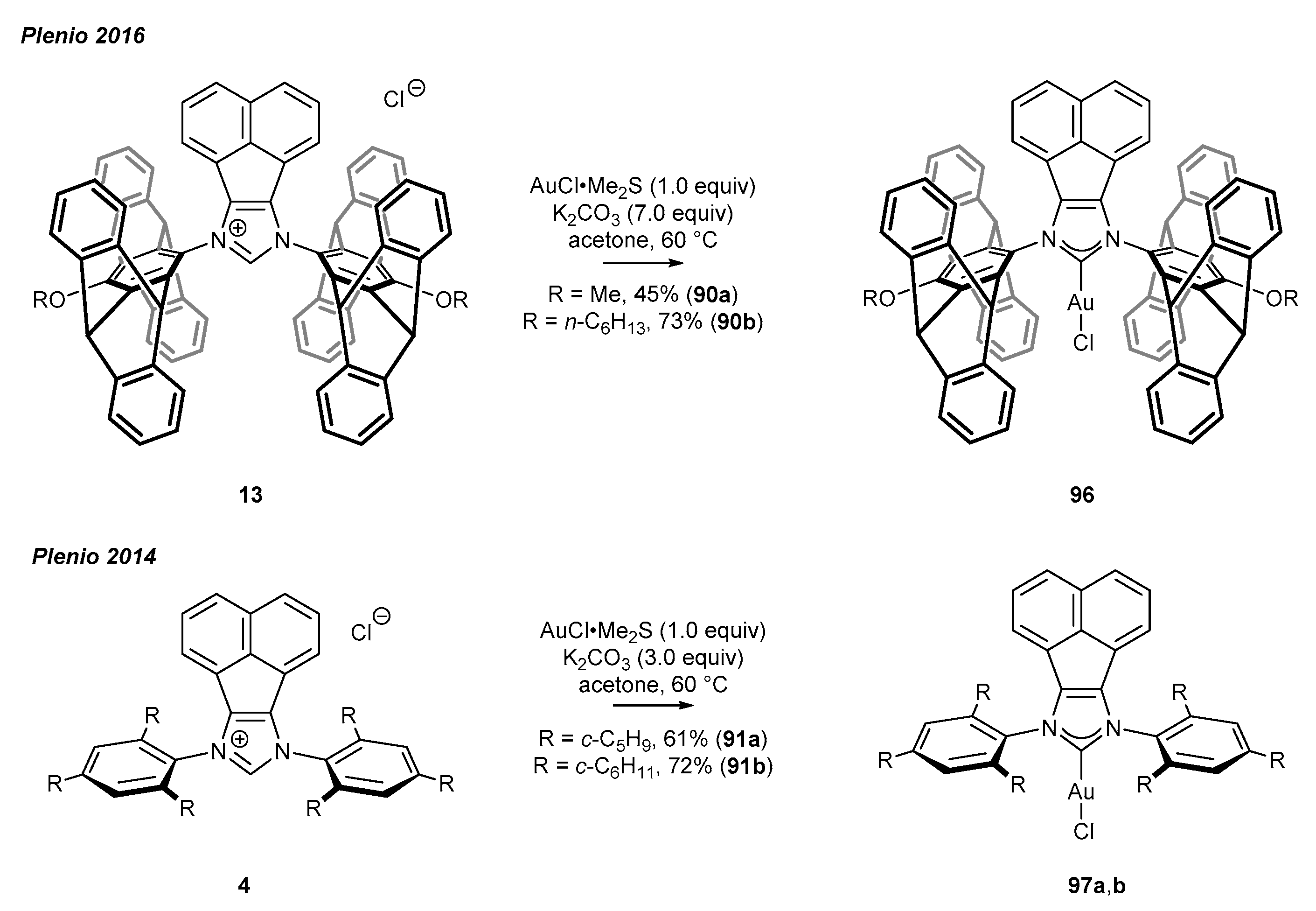
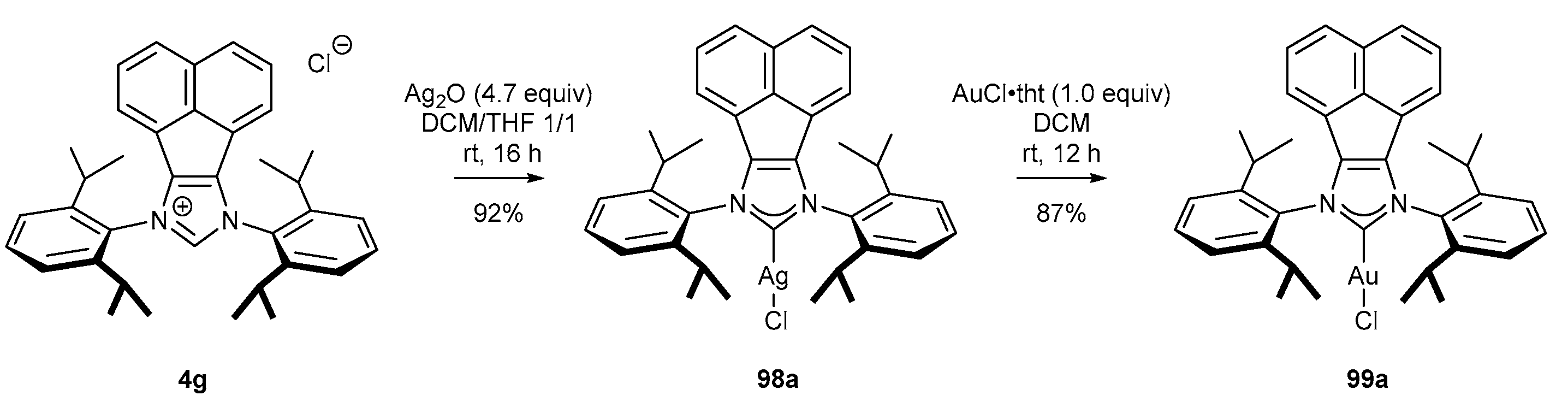
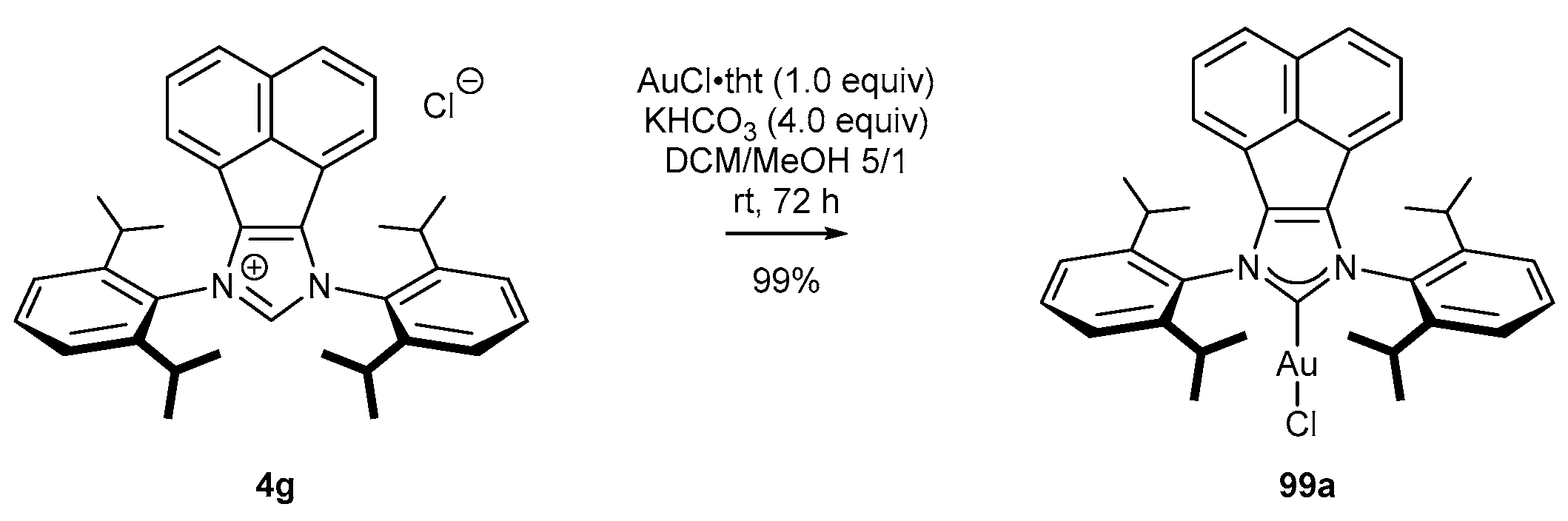
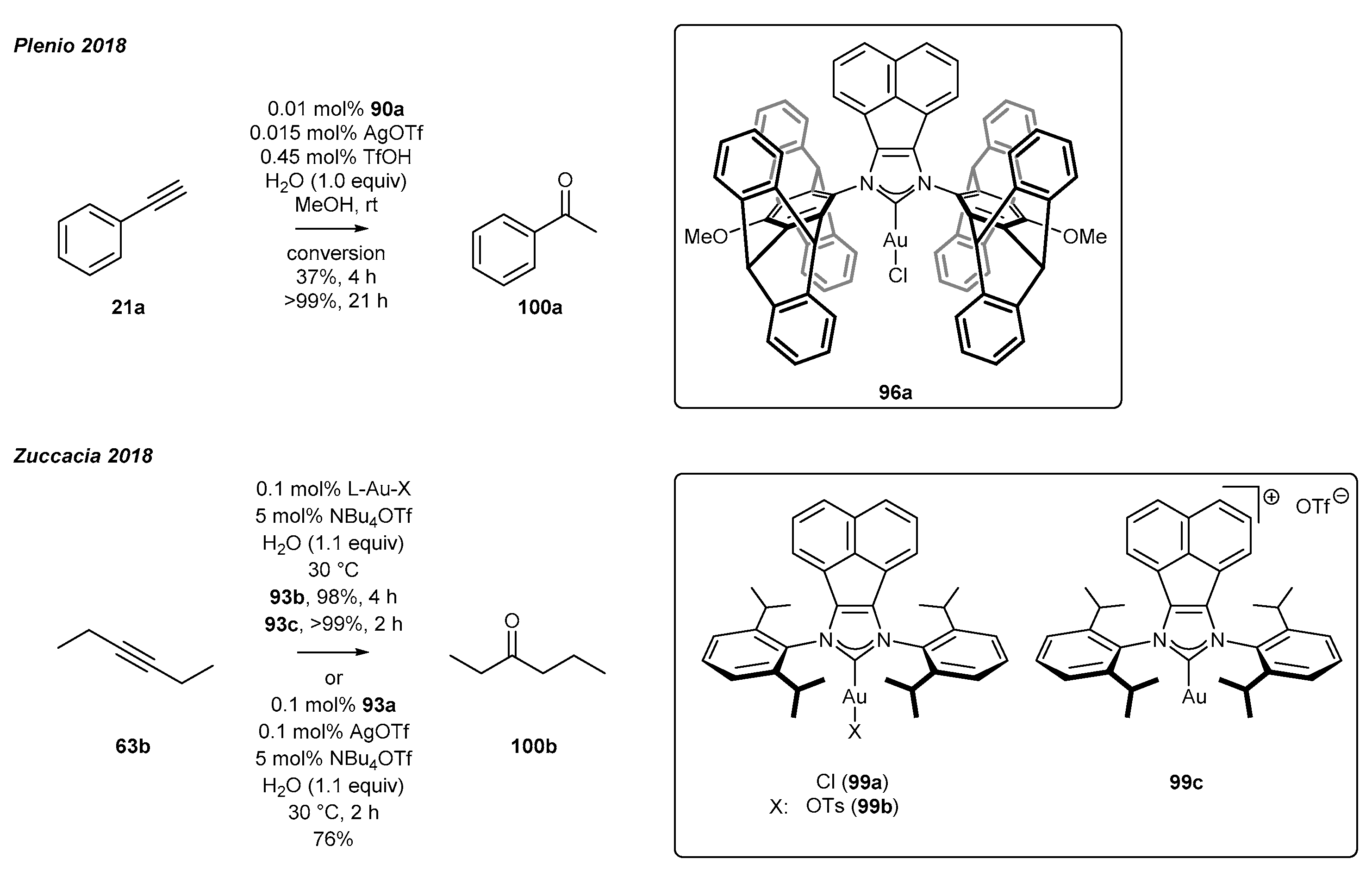
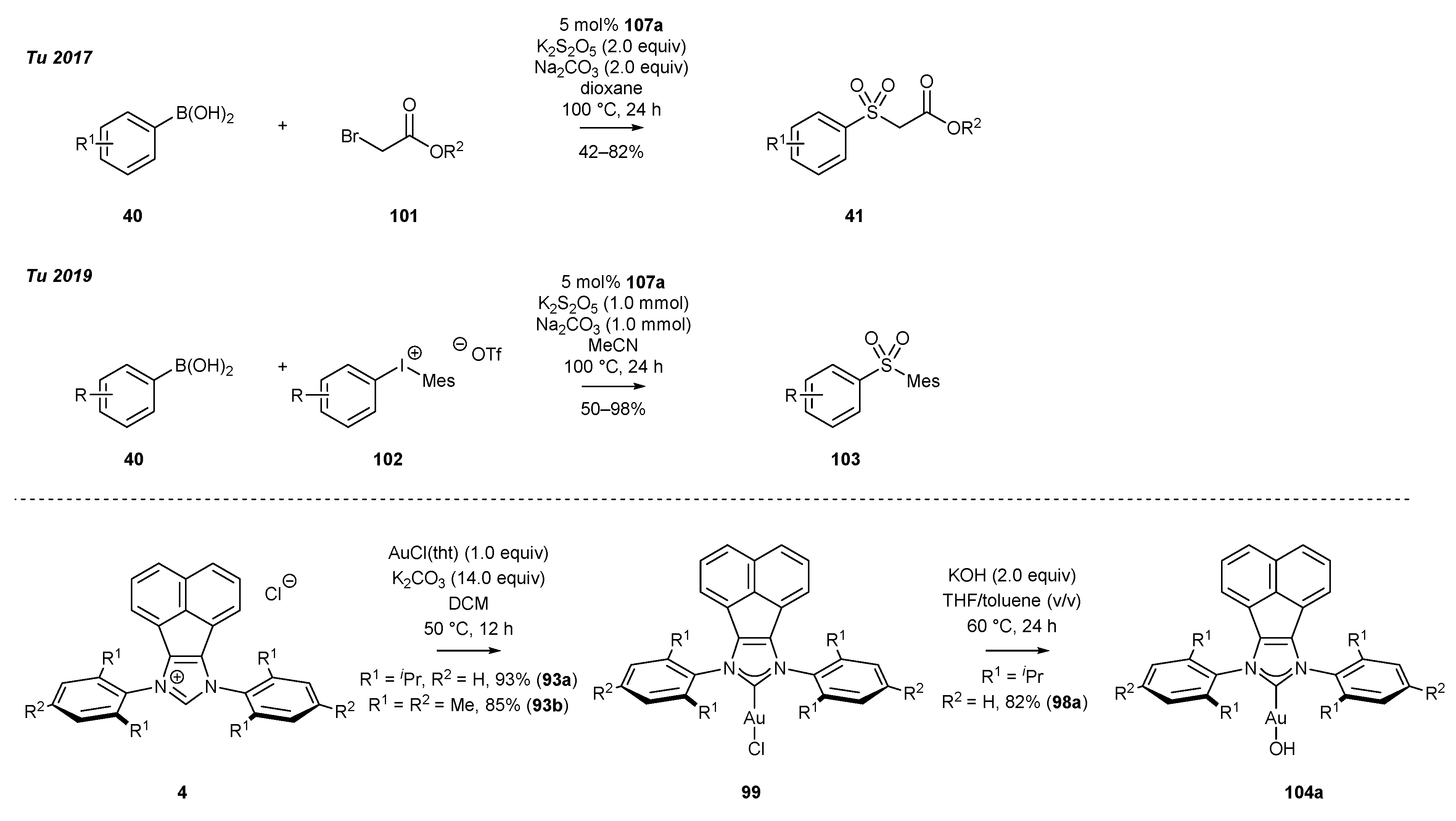
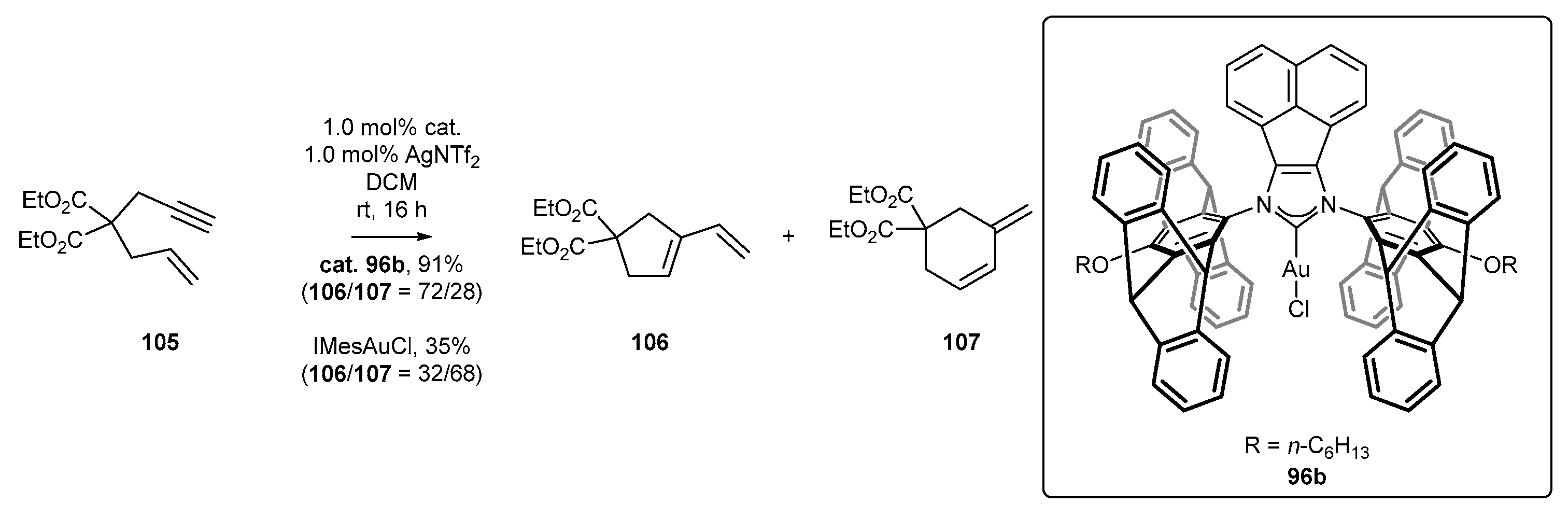
3.4. NHC-BIAN-Au Complexes
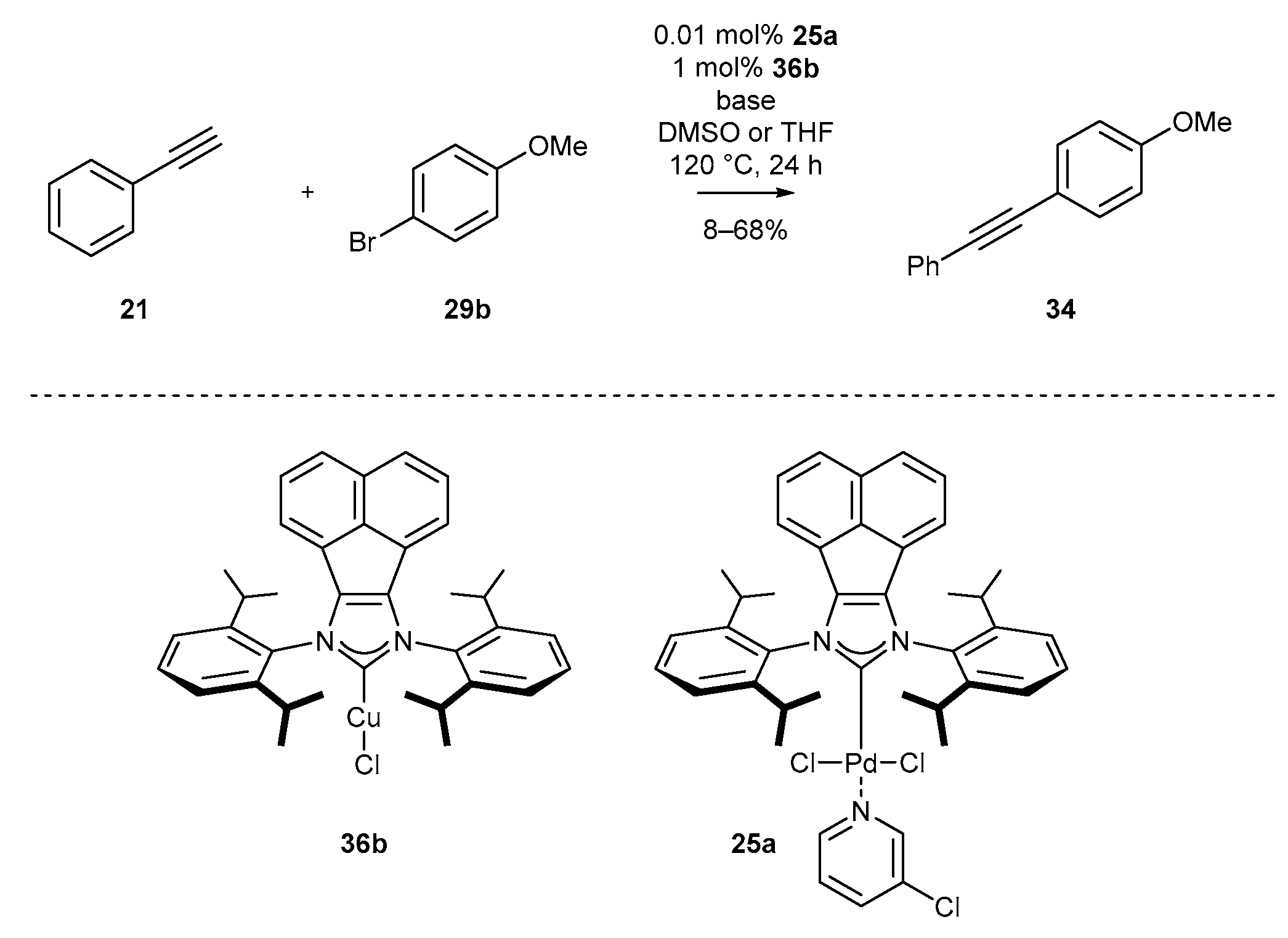
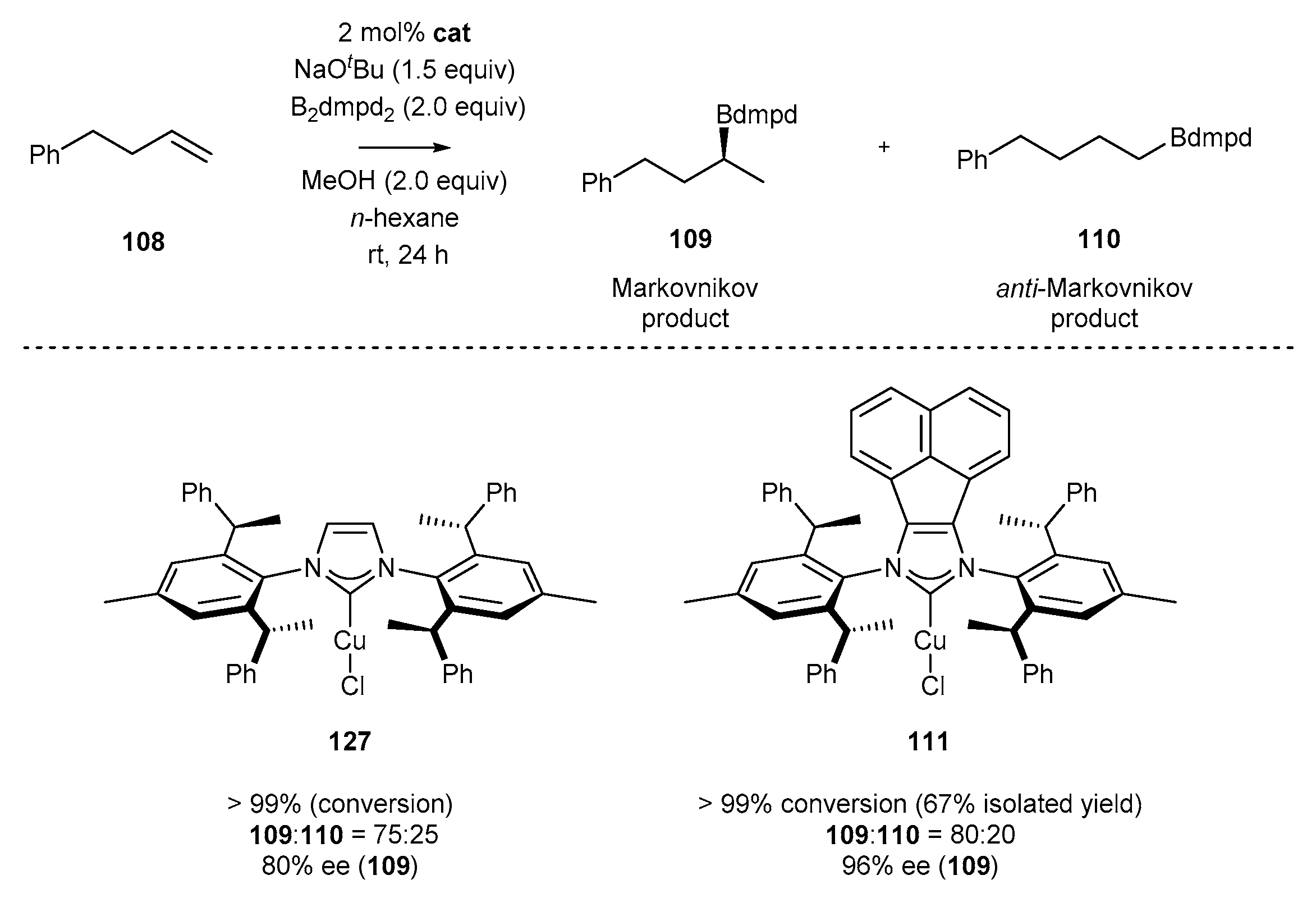
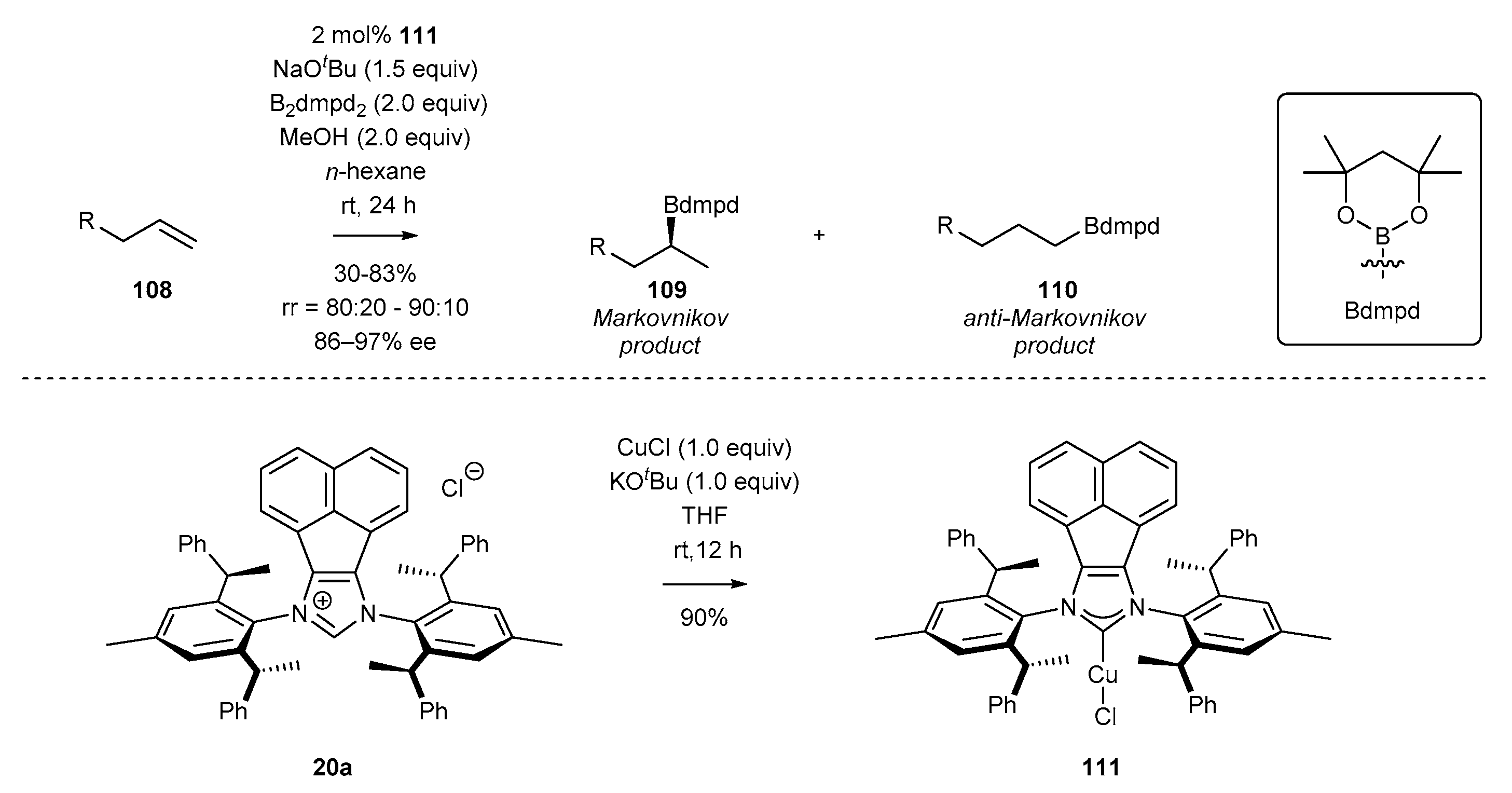
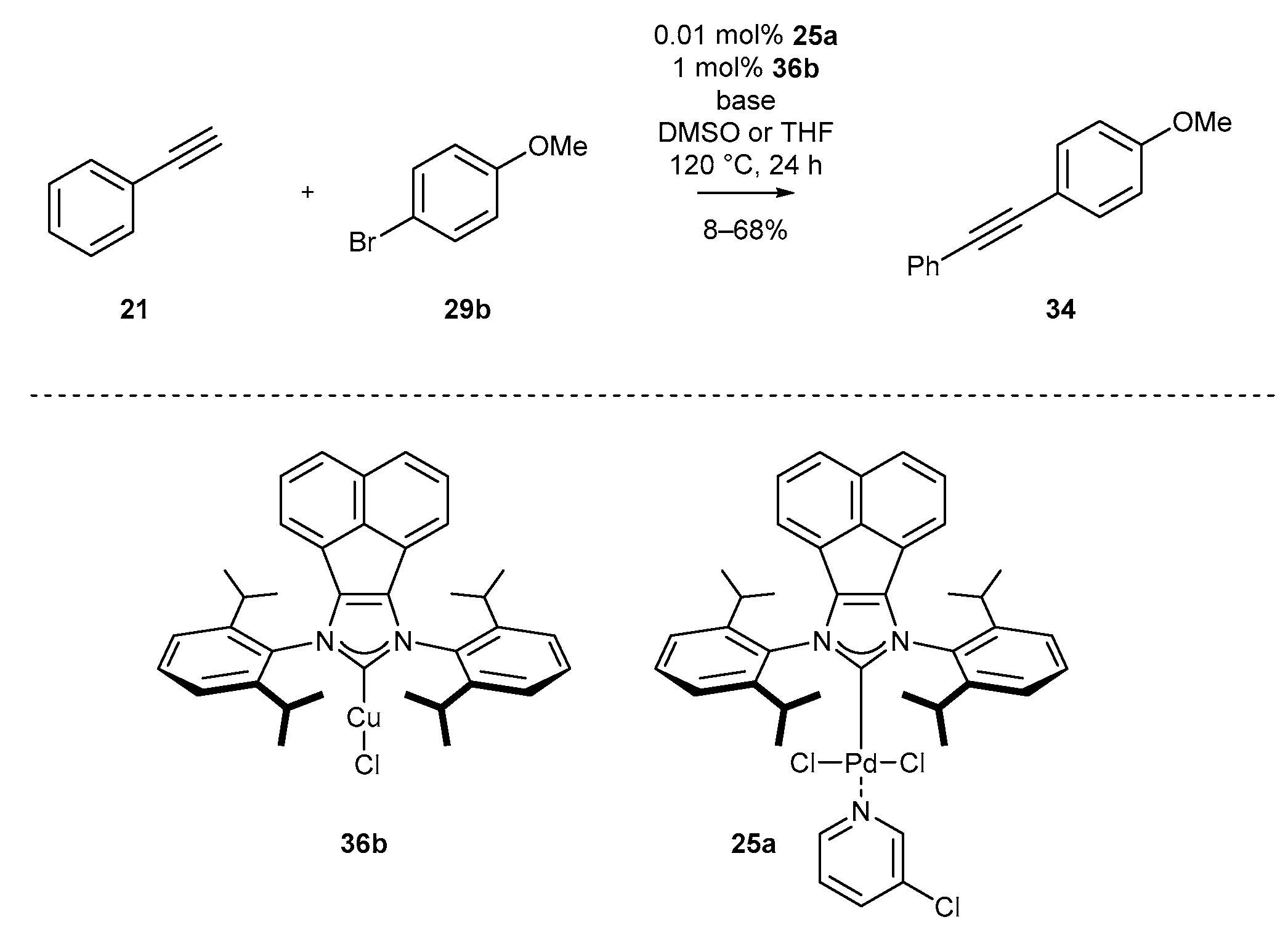
3.5. NHC-BIAN-Cu and NHC-BIAN-Pt
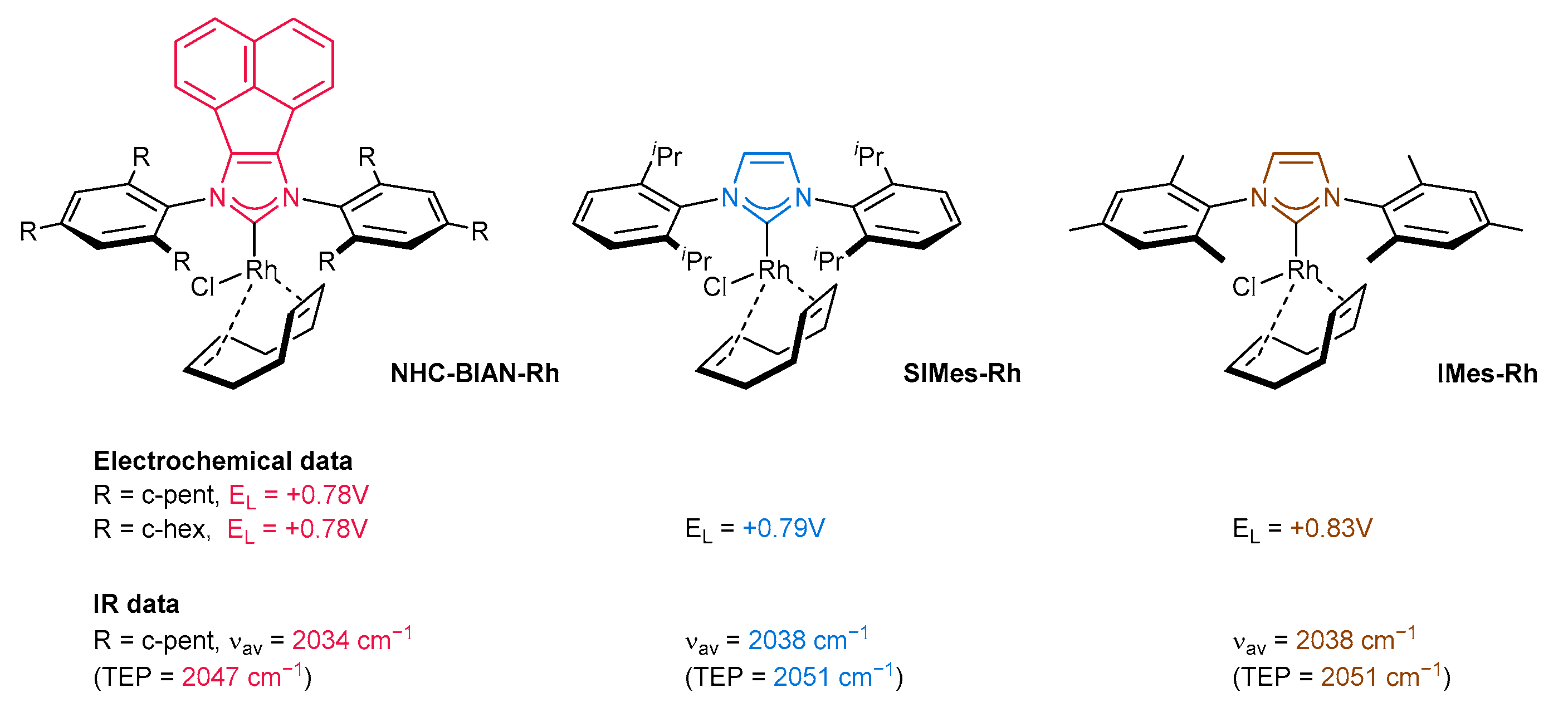
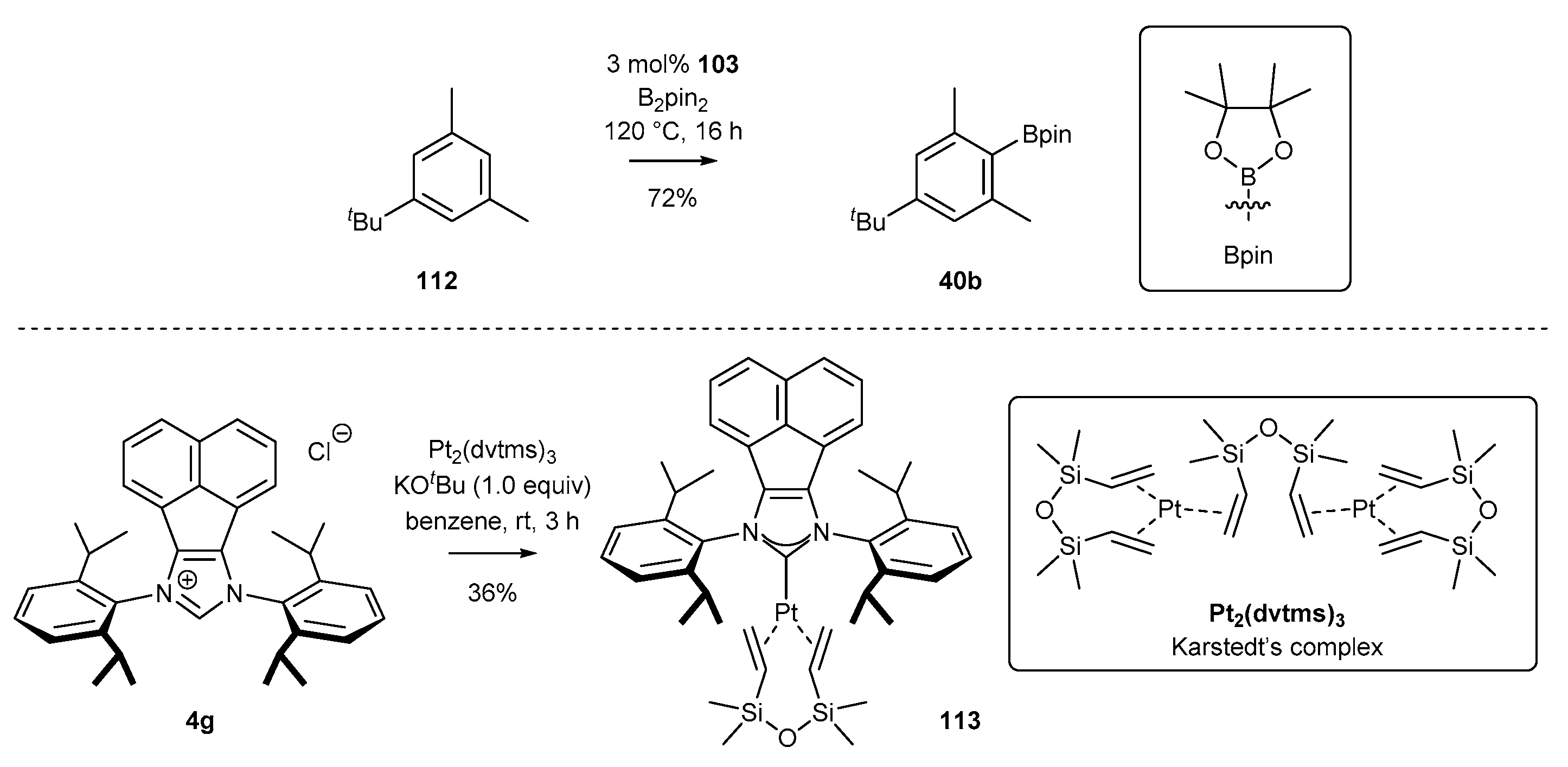
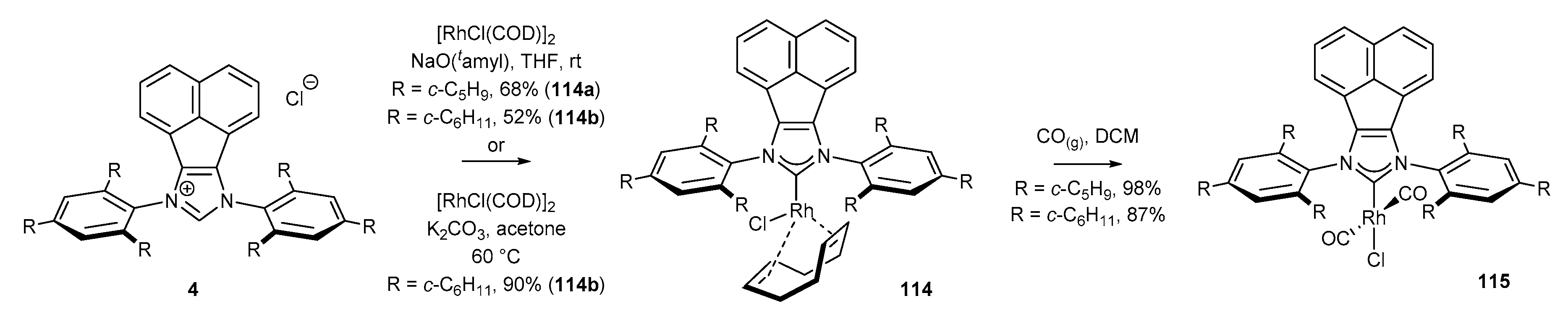
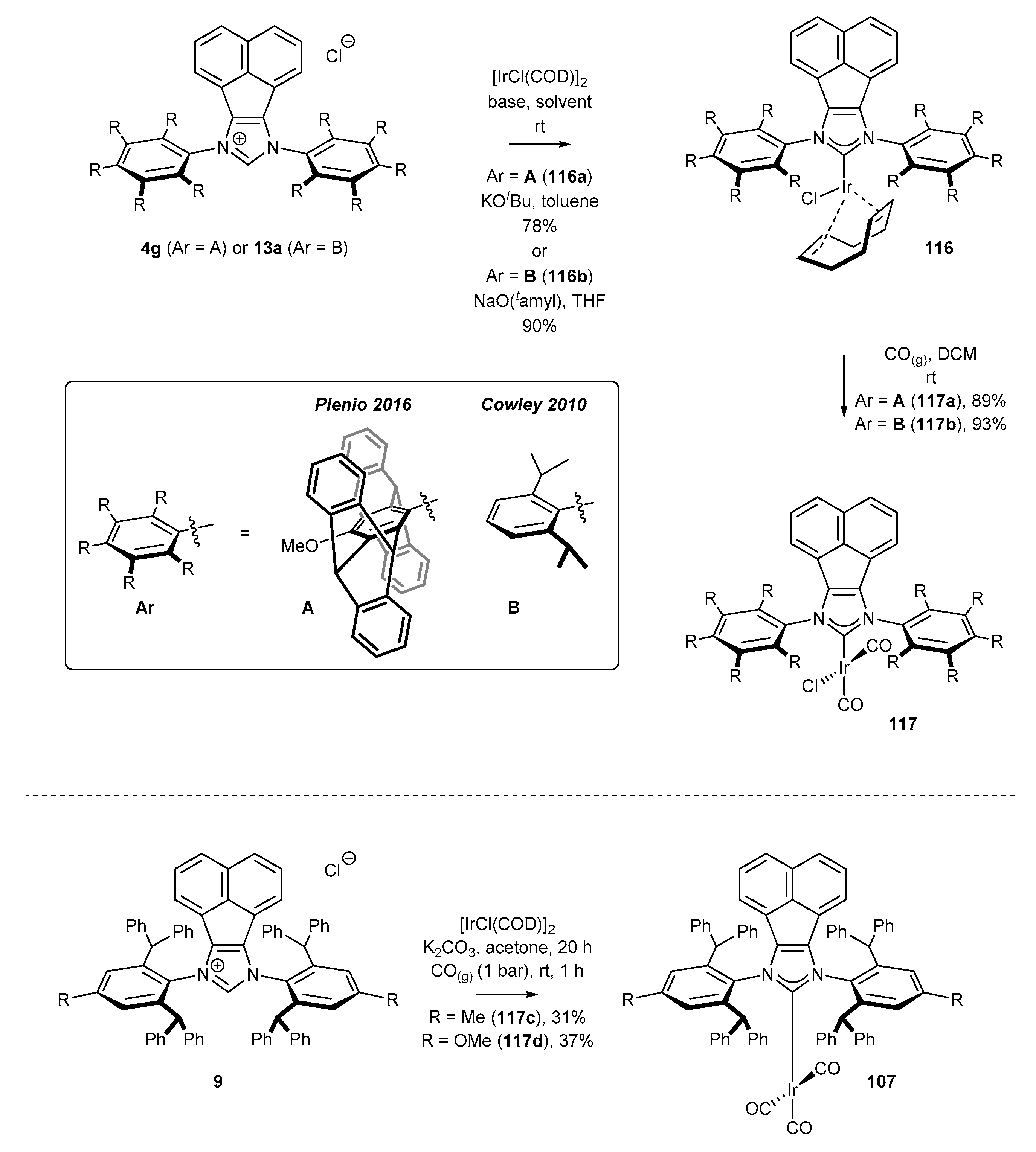
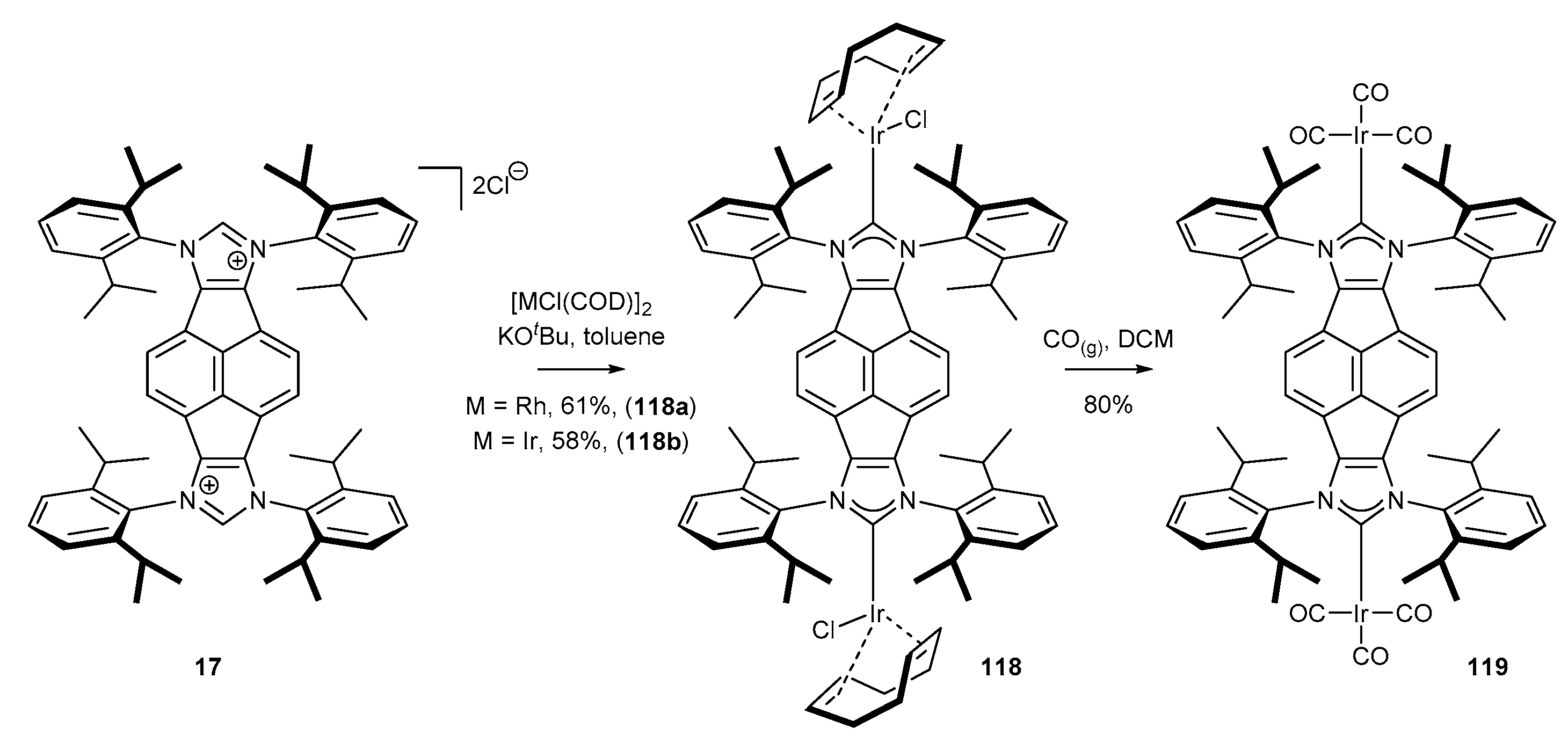
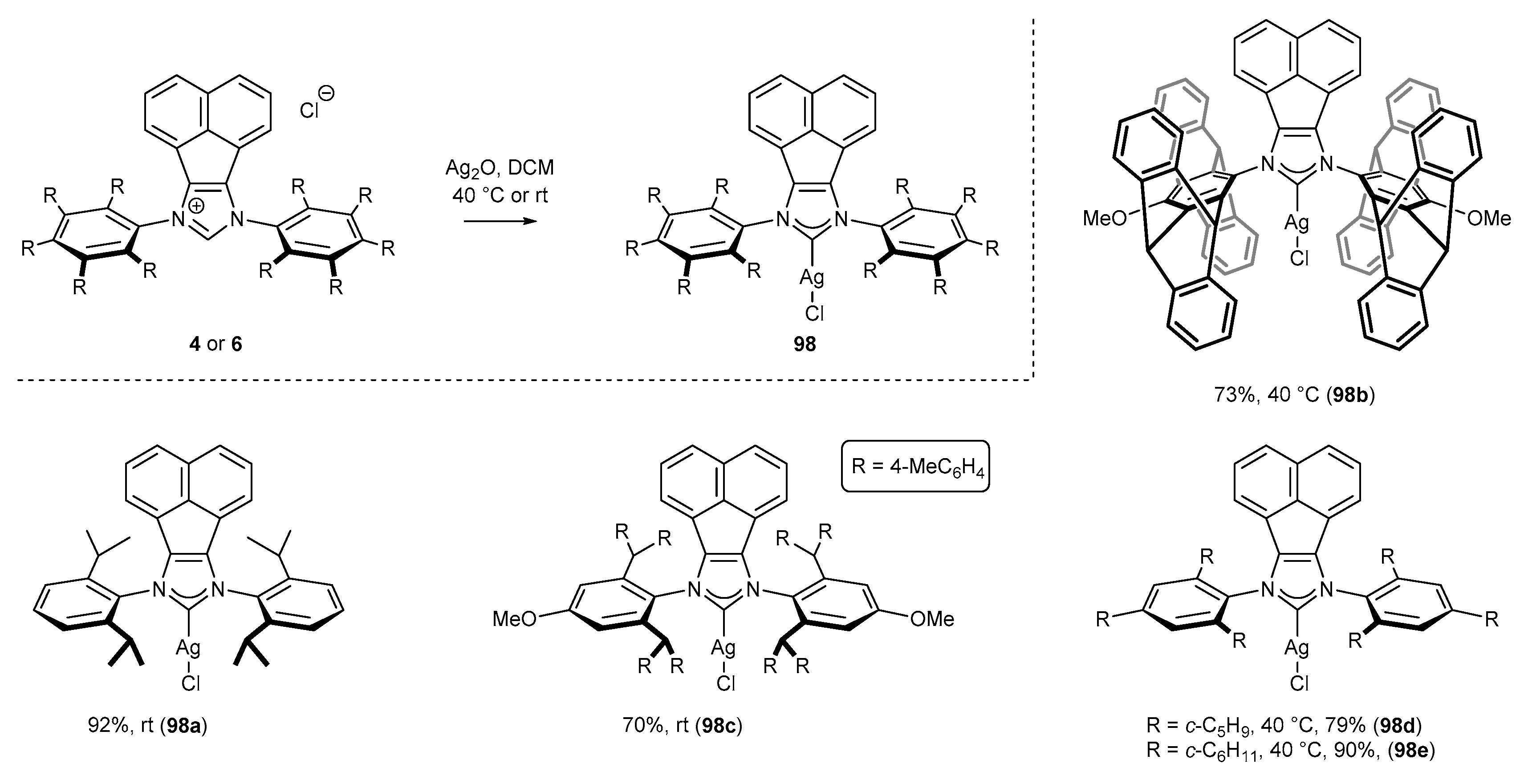
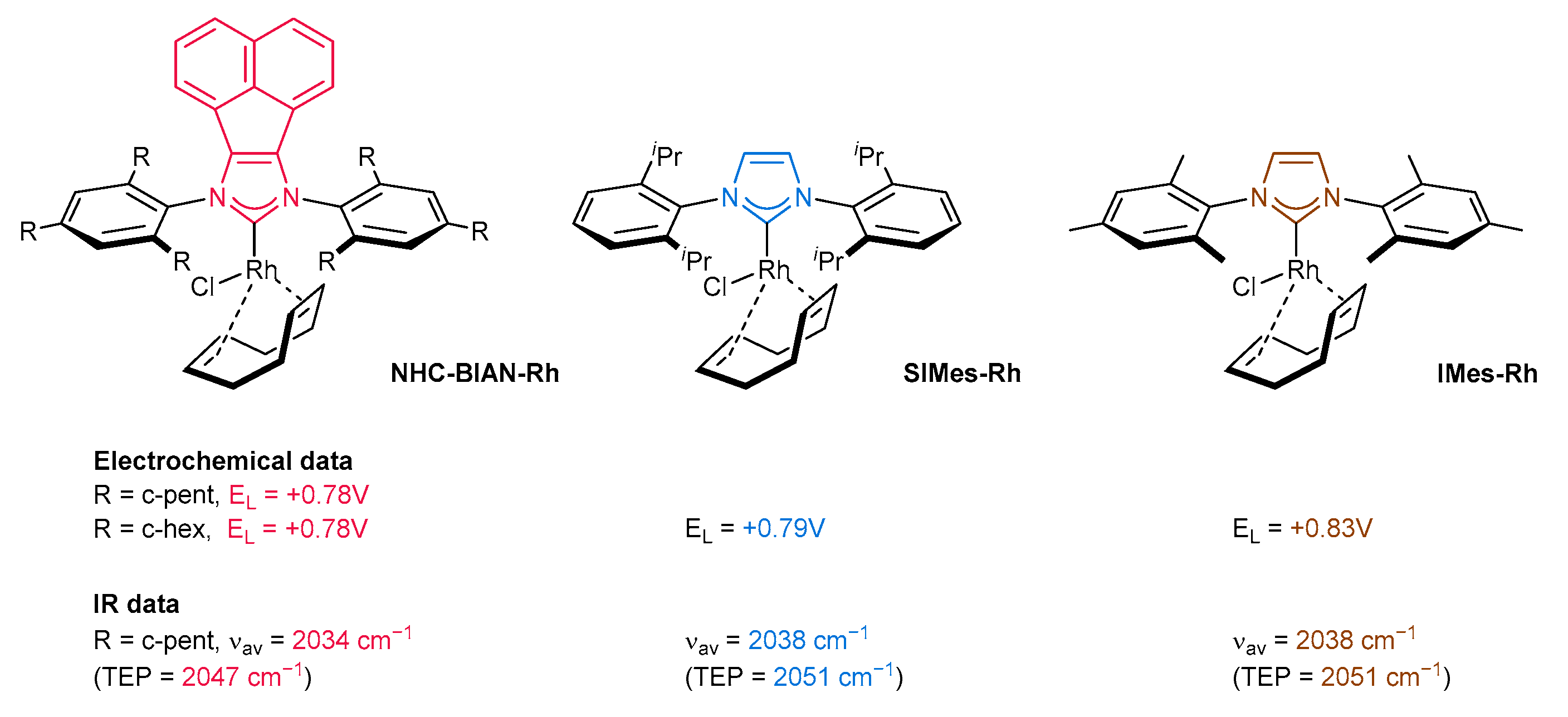
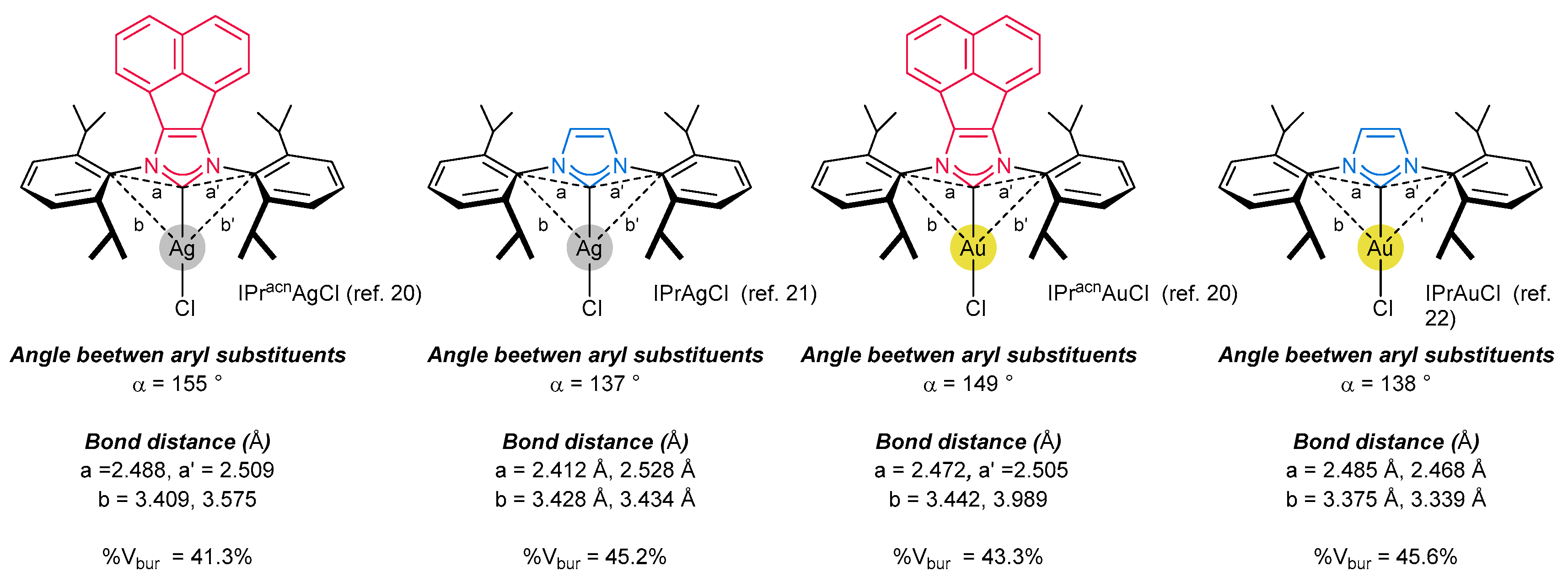
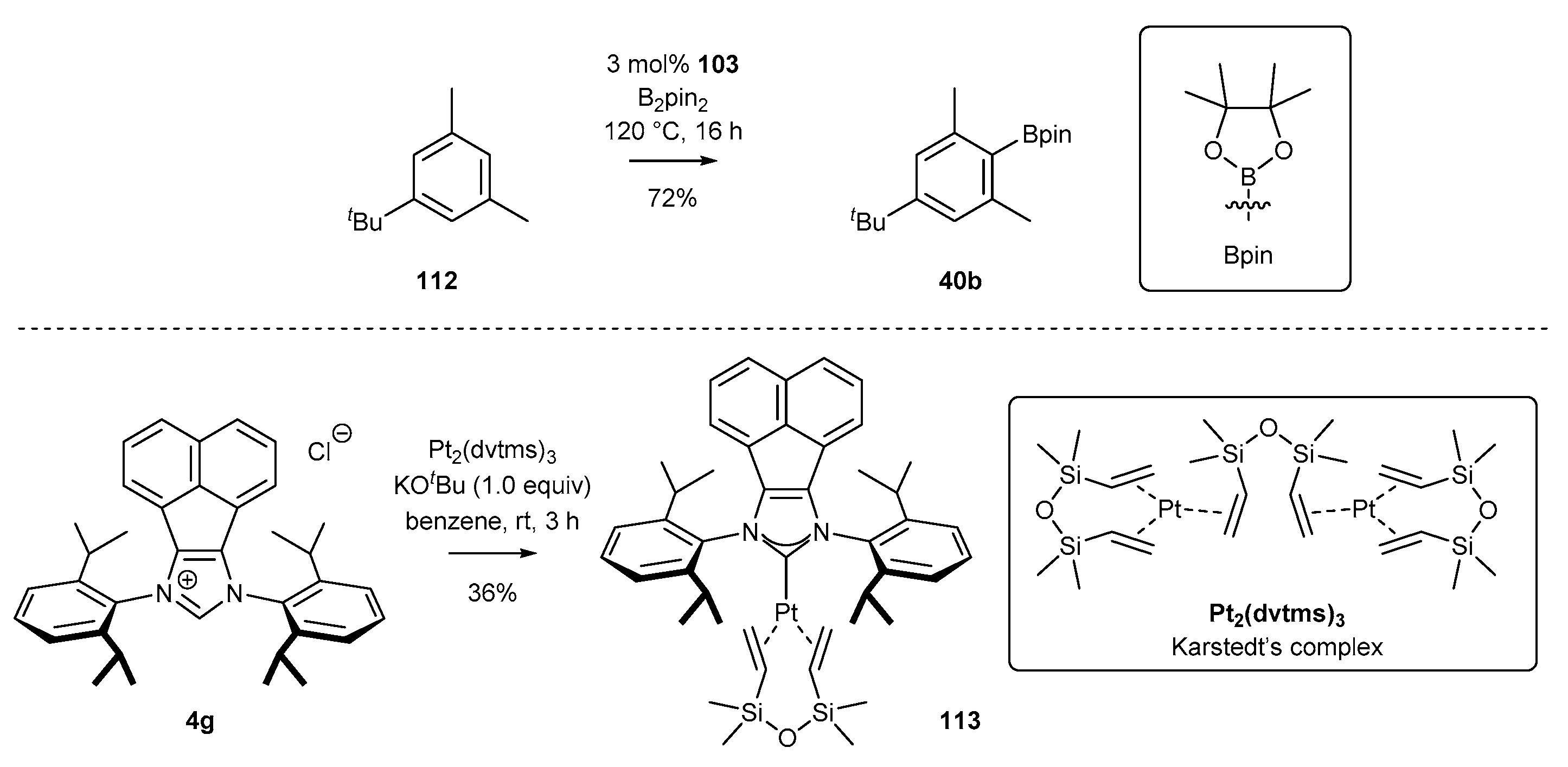
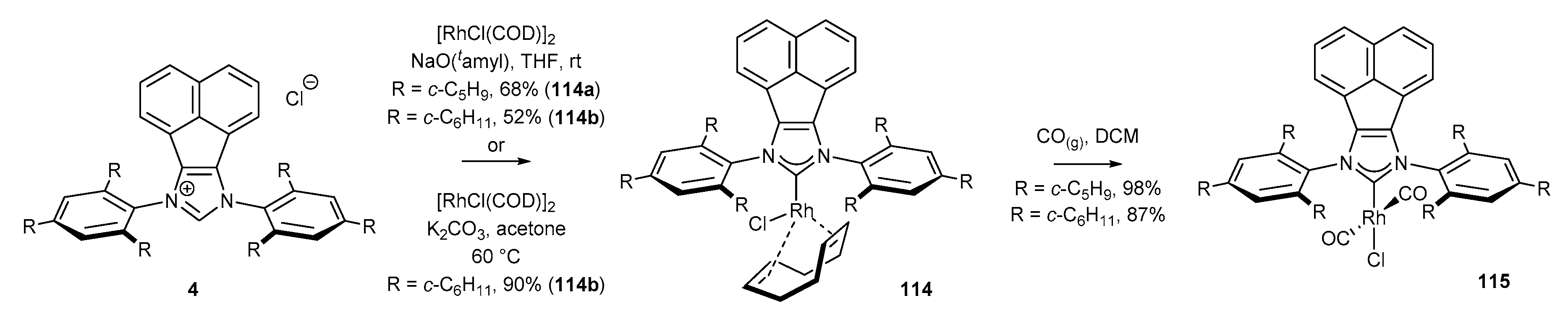
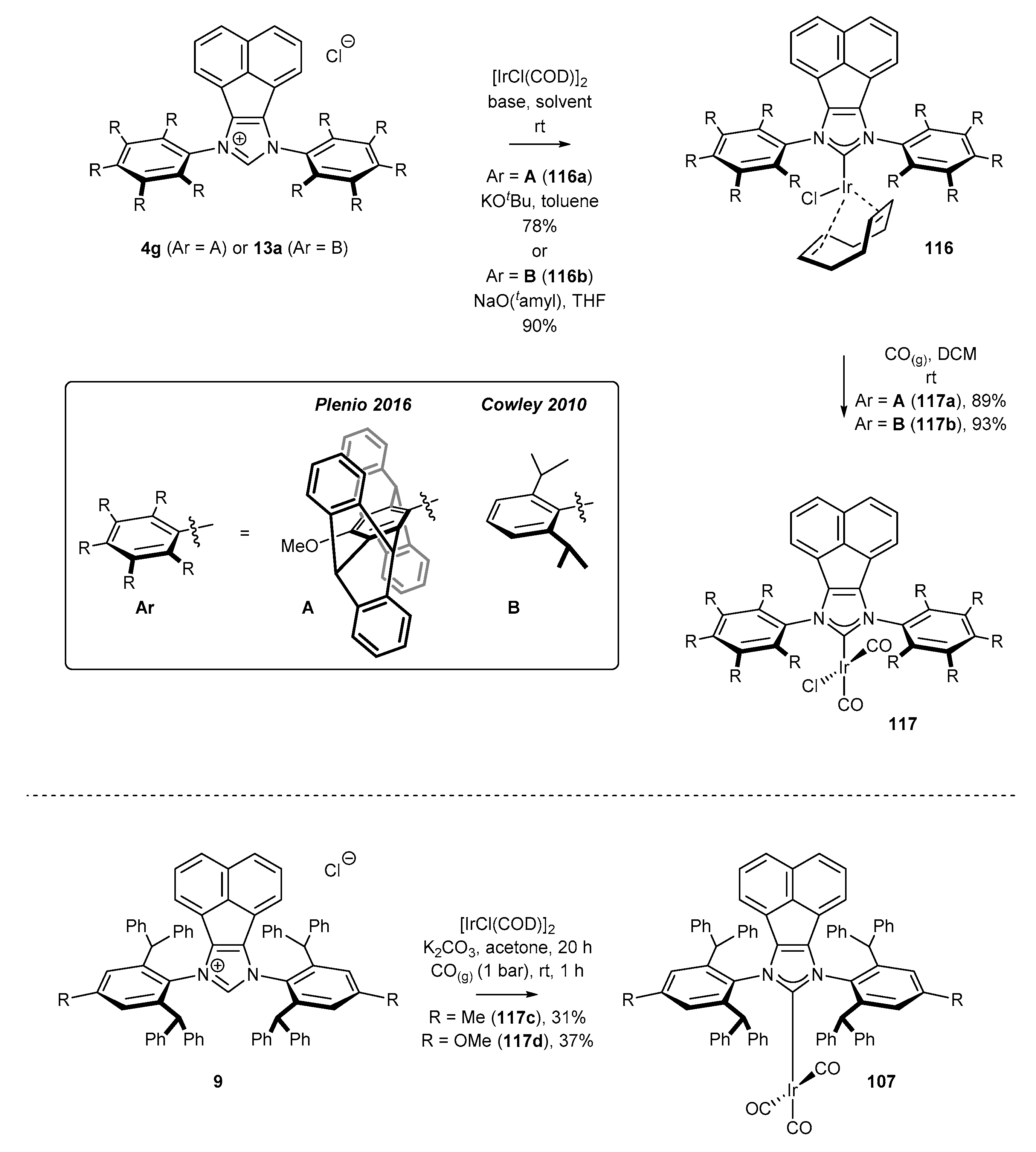
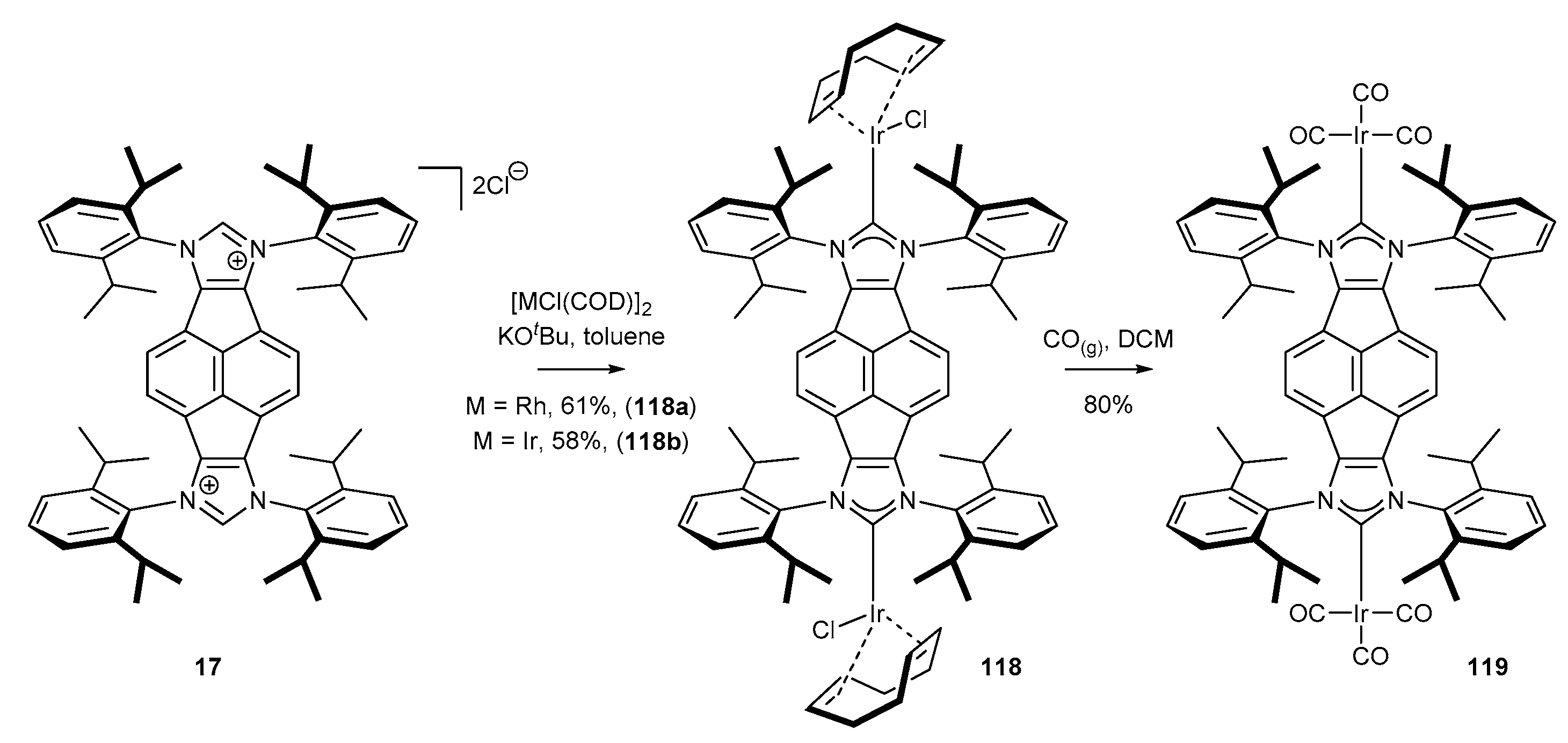
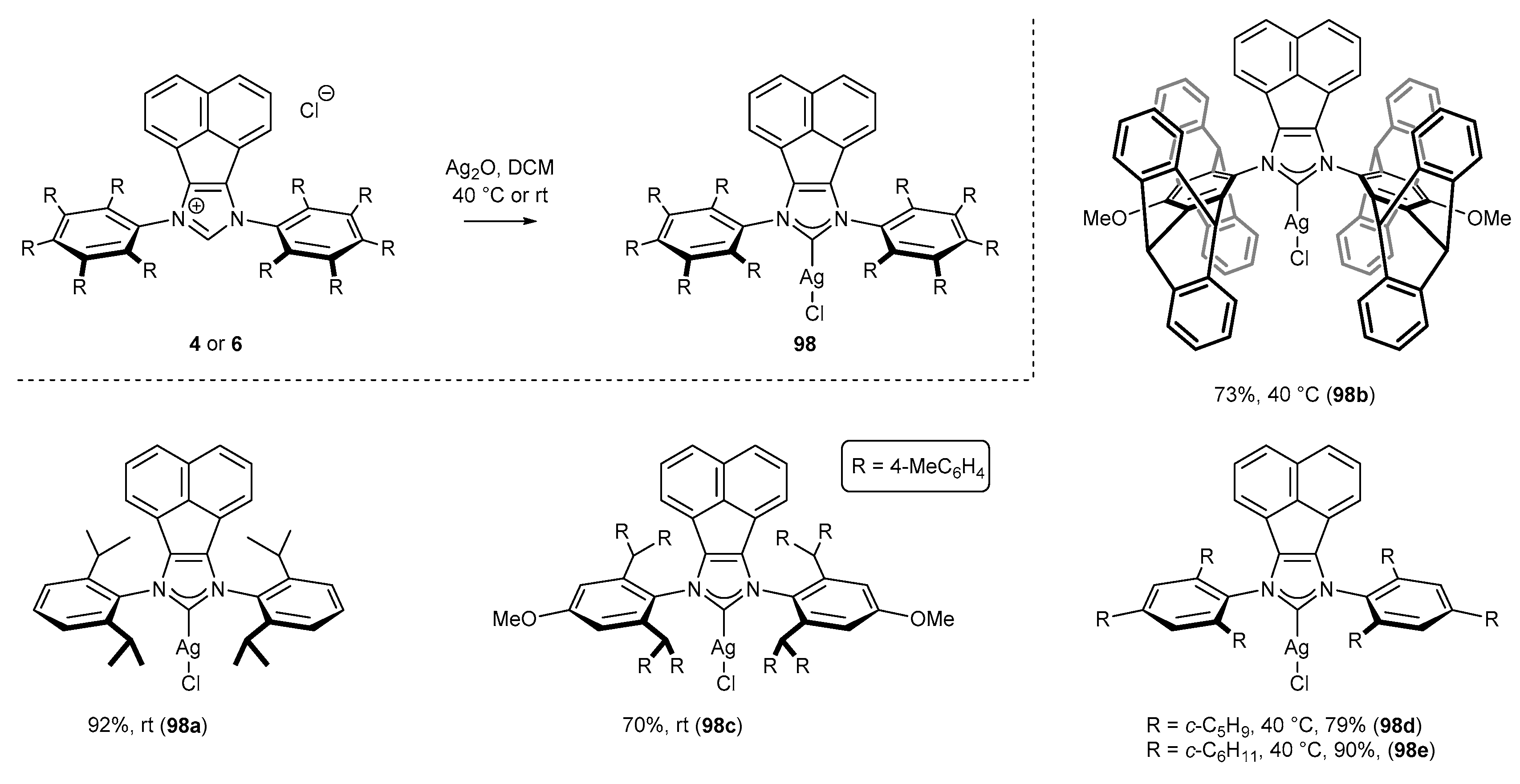
3.6. Synthesis of Other Metal Complexes (Rh, Ir, Ag)
3.7. Influence of Backbone Modification on Catalytic Activity
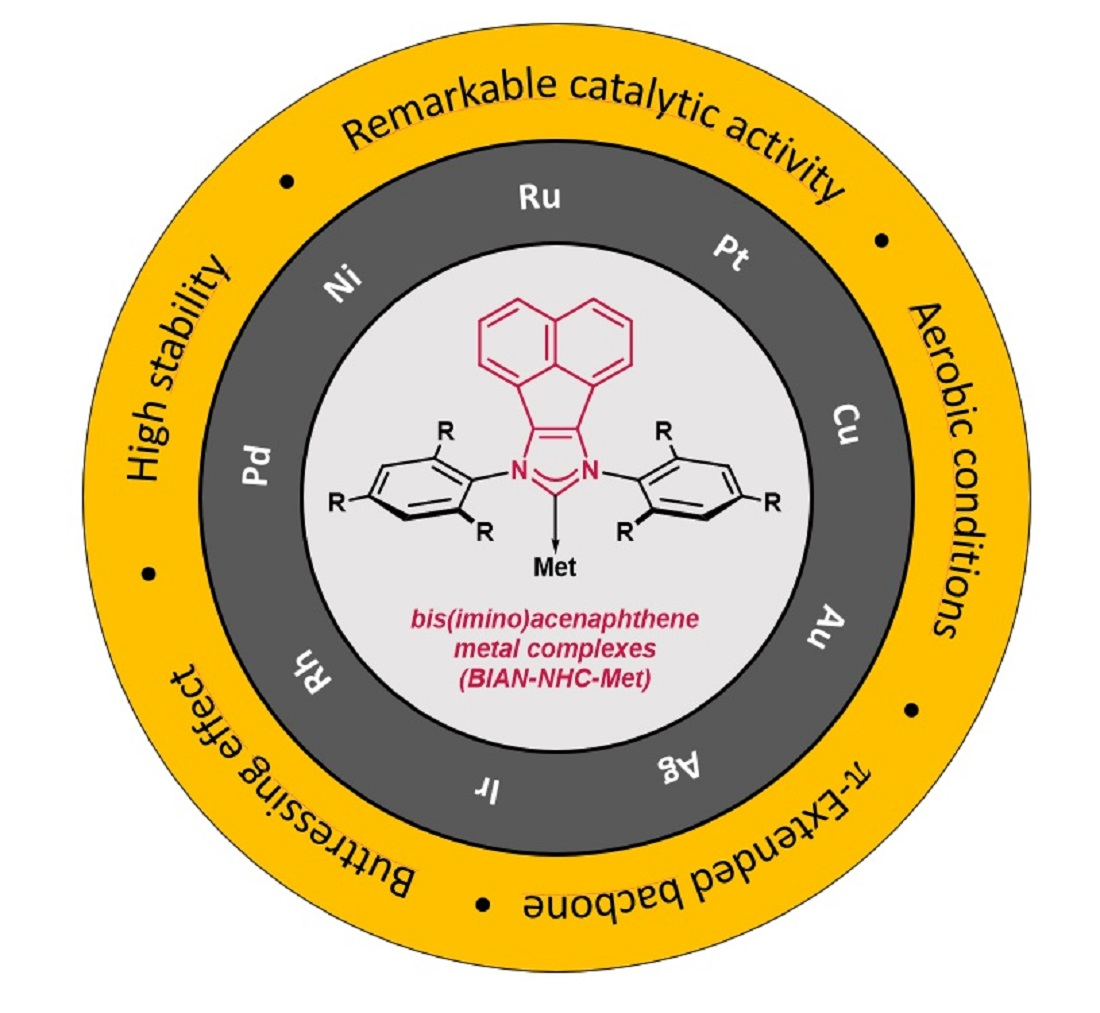
4. Conclusions and Outlook
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Zhao, Q.; Meng, G.; Nolan, S.P.; Szostak, M. N-Heterocyclic Carbene Complexes in C–H Activation Reactions. Chem. Rev. 2020, 120, 1981–2048. [Google Scholar] [CrossRef] [PubMed]
- Hudnall, T.W.; Ugarte, R.A.; Perera, T.A. N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools (2); The Royal Society of Chemistry: Cambridge, UK, 2017; pp. 178–237. [Google Scholar]
- Doddi, A.; Peters, M.; Tamm, M. N-Heterocyclic Carbene Adducts of Main Group Elements and Their Use as Ligands in Transition Metal Chemistry. Chem. Rev. 2019, 119, 6994–7112. [Google Scholar] [CrossRef]
- Michalak, M.; Kośnik, W. Chiral N-heterocyclic Carbene Gold Complexes: Synthesis and Applications in Catalysis. Catalysts 2019, 9, 890. [Google Scholar] [CrossRef] [Green Version]
- Bellemin-Laponnaz, S.; Dagorne, S. Group 1 and 2 and Early Transition Metal Complexes Bearing N-Heterocyclic Carbene Ligands: Coordination Chemistry, Reactivity, and Applications. Chem. Rev. 2014, 114, 8747–8774. [Google Scholar] [CrossRef] [PubMed]
- Danopoulos, A.A.; Simler, T.; Braunstein, P. N-Heterocyclic Carbene Complexes of Copper, Nickel, and Cobalt. Chem. Rev. 2019, 119, 3730–3961. [Google Scholar] [CrossRef] [PubMed]
- Peris, E. Smart N-Heterocyclic Carbene Ligands in Catalysis. Chem. Rev. 2018, 118, 9988–10031. [Google Scholar] [CrossRef]
- Janssen-Muller, D.; Schlepphorst, C.; Glorius, F. Privileged chiral N-heterocyclic carbene ligands for asymmetric transition-metal catalysis. Chem. Soc. Rev. 2017, 46, 4845–4854. [Google Scholar] [CrossRef]
- Enders, D.; Niemeier, O.; Henseler, A. Organocatalysis by N-Heterocyclic Carbenes. Chem. Rev. 2007, 107, 5606–5655. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Jin, Z.; Chi, Y.R. NHC-catalyzed covalent activation of heteroatoms for enantioselective reactions. Chem. Sci. 2021, 12, 5037–5043. [Google Scholar] [CrossRef]
- Das, T.K.; Biju, A.T. Progress in Heterocyclic Chemistry; Gribble, G.W., Joule, J.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 31, pp. 1–82. [Google Scholar]
- Chen, X.-Y.; Gao, Z.-H.; Ye, S. Bifunctional N-Heterocyclic Carbenes Derived from l-Pyroglutamic Acid and Their Applications in Enantioselective Organocatalysis. Acc. Chem. Res. 2020, 53, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Sau, S.C.; Hota, P.K.; Mandal, S.K.; Soleilhavoup, M.; Bertrand, G. Stable abnormal N-heterocyclic carbenes and their applications. Chem. Soc. Rev. 2020, 49, 1233–1252. [Google Scholar] [CrossRef] [PubMed]
- Valente, C.; Çalimsiz, S.; Hoi, K.H.; Mallik, D.; Sayah, M.; Organ, M.G. The Development of Bulky Palladium NHC Complexes for the Most-Challenging Cross-Coupling Reactions. Angew. Chem. Int. Ed. 2012, 51, 3314–3332. [Google Scholar] [CrossRef]
- Froese, R.D.J.; Lombardi, C.; Pompeo, M.; Rucker, R.P.; Organ, M.G. Designing Pd–N-Heterocyclic Carbene Complexes for High Reactivity and Selectivity for Cross-Coupling Applications. Acc. Chem. Res. 2017, 50, 2244–2253. [Google Scholar] [CrossRef]
- Savka, R.; Plenio, H. Metal Complexes of Very Bulky N,N′-Diarylimidazolylidene N-Heterocyclic Carbene (NHC) Ligands with 2,4,6-Cycloalkyl Substituents. Eur. J. Inorg. Chem. 2014, 2014, 6246–6253. [Google Scholar] [CrossRef]
- Huynh, H.V. Electronic Properties of N-Heterocyclic Carbenes and Their Experimental Determination. Chem. Rev. 2018, 118, 9457–9492. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, F.-S.; Szostak, M. BIAN-NHC Ligands in Transition-Metal-Catalysis: A Perfect Union of Sterically Encumbered, Electronically Tunable N-Heterocyclic Carbenes? Chem. Eur. J. 2021, 27, 4478–4499. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, K.V.; Butorac, R.R.; Abernethy, C.D.; Cowley, A.H. Synthesis and coordination compounds of a bis(imino)acenaphthene (BIAN)-supported N-heterocyclic carbene. Dalton Trans. 2010, 39, 7401–7408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Frémont, P.; Scott, N.M.; Stevens, E.D.; Ramnial, T.; Lightbody, O.C.; Macdonald, C.L.B.; Clyburne, J.A.C.; Abernethy, C.D.; Nolan, S.P. Synthesis of Well-Defined N-Heterocyclic Carbene Silver(I) Complexes. Organometallics 2005, 24, 6301–6309. [Google Scholar] [CrossRef]
- Fructos, M.R.; Belderrain, T.R.; de Frémont, P.; Scott, N.M.; Nolan, S.P.; Díaz-Requejo, M.M.; Pérez, P.J. A Gold Catalyst for Carbene-Transfer Reactions from Ethyl Diazoacetate. Angew. Chem. Int. Ed. 2005, 44, 5284–5288. [Google Scholar] [CrossRef]
- Falivene, L.; Credendino, R.; Poater, A.; Petta, A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps. Organometallics 2016, 35, 2286–2293. [Google Scholar] [CrossRef] [Green Version]
- Poater, A.; Cosenza, B.; Correa, A.; Giudice, S.; Ragone, F.; Scarano, V.; Cavallo, L. SambVca: A Web Application for the Calculation of the Buried Volume of N-Heterocyclic Carbene Ligands. Eur. J. Inorg. Chem. 2009, 2009, 1759–1766. [Google Scholar] [CrossRef]
- Gottumukkala, A.L.; Teichert, J.F.; Heijnen, D.; Eisink, N.; van Dijk, S.; Ferrer, C.; van den Hoogenband, A.; Minnaard, A.J. Pd-diimine: A highly selective catalyst system for the base-free oxidative Heck reaction. J. Org. Chem. 2011, 76, 3498–3501. [Google Scholar] [CrossRef]
- Wang, J.; Ganguly, R.; Yongxin, L.; Díaz, J.; Soo, H.S.; García, F. Synthesis and the Optical and Electrochemical Properties of Indium(III) Bis(arylimino)acenaphthene Complexes. Inorg. Chem. 2017, 56, 7811–7820. [Google Scholar] [CrossRef] [PubMed]
- Hasan, K.; Zysman-Colman, E. Synthesis, UV–Vis and CV properties of a structurally related series of bis(Arylimino)acenaphthenes (Ar-BIANs). J. Phys. Org. Chem. 2013, 26, 274–279. [Google Scholar] [CrossRef]
- Evans, D.A.; Lee, L.M.; Vargas-Baca, I.; Cowley, A.H. Aggregation-Induced Emission of Bis(imino)acenaphthene Zinc Complexes: Photophysical Tuning via Methylation of the Flanking Aryl Substituents. Organometallics 2015, 34, 2422–2428. [Google Scholar] [CrossRef]
- Gasperini, M.; Ragaini, F.; Cenini, S. Synthesis of Ar-BIAN Ligands (Ar-BIAN = Bis(aryl)acenaphthenequinonediimine) Having Strong Electron-Withdrawing Substituents on the Aryl Rings and Their Relative Coordination Strength toward Palladium(0) and -(II) Complexes. Organometallics 2002, 21, 2950–2957. [Google Scholar] [CrossRef]
- Merino, E.; Poli, E.; Díaz, U.; Brunel, D. Synthesis and characterization of new ruthenium N-heterocyclic carbene Hoveyda II-type complexes. Study of reactivity in ring closing metathesis reactions. Dalton Trans. 2012, 41, 10913–10918. [Google Scholar] [CrossRef]
- Butorac, R.R.; Al-Deyab, S.S.; Cowley, A.H. Syntheses, structures and antimicrobial activities of bis(imino)acenaphthene (BIAN) imidazolium salts. Molecules 2011, 16, 3168–3178. [Google Scholar] [CrossRef]
- El-Ayaan, U.; Murata, F.; El-Derby, S.; Fukuda, Y. Synthesis, structural and solvent influence studies on solvatochromic mixed-ligand copper(II) complexes with the rigid nitrogen ligand: Bis[N-(2,4,6-trimethylphenyl)imino]acenaphthene. J. Mol. Struct. 2004, 692, 209–216. [Google Scholar] [CrossRef]
- Mak, C.S.K.; Wong, H.L.; Leung, Q.Y.; Tam, W.Y.; Chan, W.K.; Djurišić, A.B. The use of sublimable chlorotricarbonyl bis(phenylimino)acenaphthene rhenium(I) complexes as photosensitizers in bulk-heterojunction photovoltaic devices. J. Organomet. Chem. 2009, 694, 2770–2776. [Google Scholar] [CrossRef] [Green Version]
- Coventry, D.N.; Batsanov, A.S.; Goeta, A.E.; Howard, J.A.K.; Marder, T.B. Synthesis and molecular structures of α-diimines and their zinc and palladium dichloride complexes. Polyhedron 2004, 23, 2789–2795. [Google Scholar] [CrossRef]
- Tu, T.; Sun, Z.; Fang, W.; Xu, M.; Zhou, Y. Robust Acenaphthoimidazolylidene Palladium Complexes: Highly Efficient Catalysts for Suzuki–Miyaura Couplings with Sterically Hindered Substrates. Org. Lett. 2012, 14, 4250–4253. [Google Scholar] [CrossRef] [PubMed]
- Tronnier, A.; Pöthig, A.; Metz, S.; Wagenblast, G.; Münster, I.; Strassner, T. Enlarging the π System of Phosphorescent (C^C*) Cyclometalated Platinum(II) NHC Complexes. Inorg. Chem. 2014, 53, 6346–6356. [Google Scholar] [CrossRef] [PubMed]
- van Asselt, R.; Elsevier, C.J.; Smeets, W.J.J.; Spek, A.L.; Benedix, R. Synthesis and characterization of rigid bidentate nitrogen ligands and some examples of coordination to divalent palladium. X-ray crystal structures of bis (p-tolylimino) acenaphthene and methylchloro [bis(o,o′-diisopropylphenyl-imino) acenaphthene] palladium (II). Recl. Trav. Chim. Pays-Bas 1994, 113, 88–98. [Google Scholar] [CrossRef]
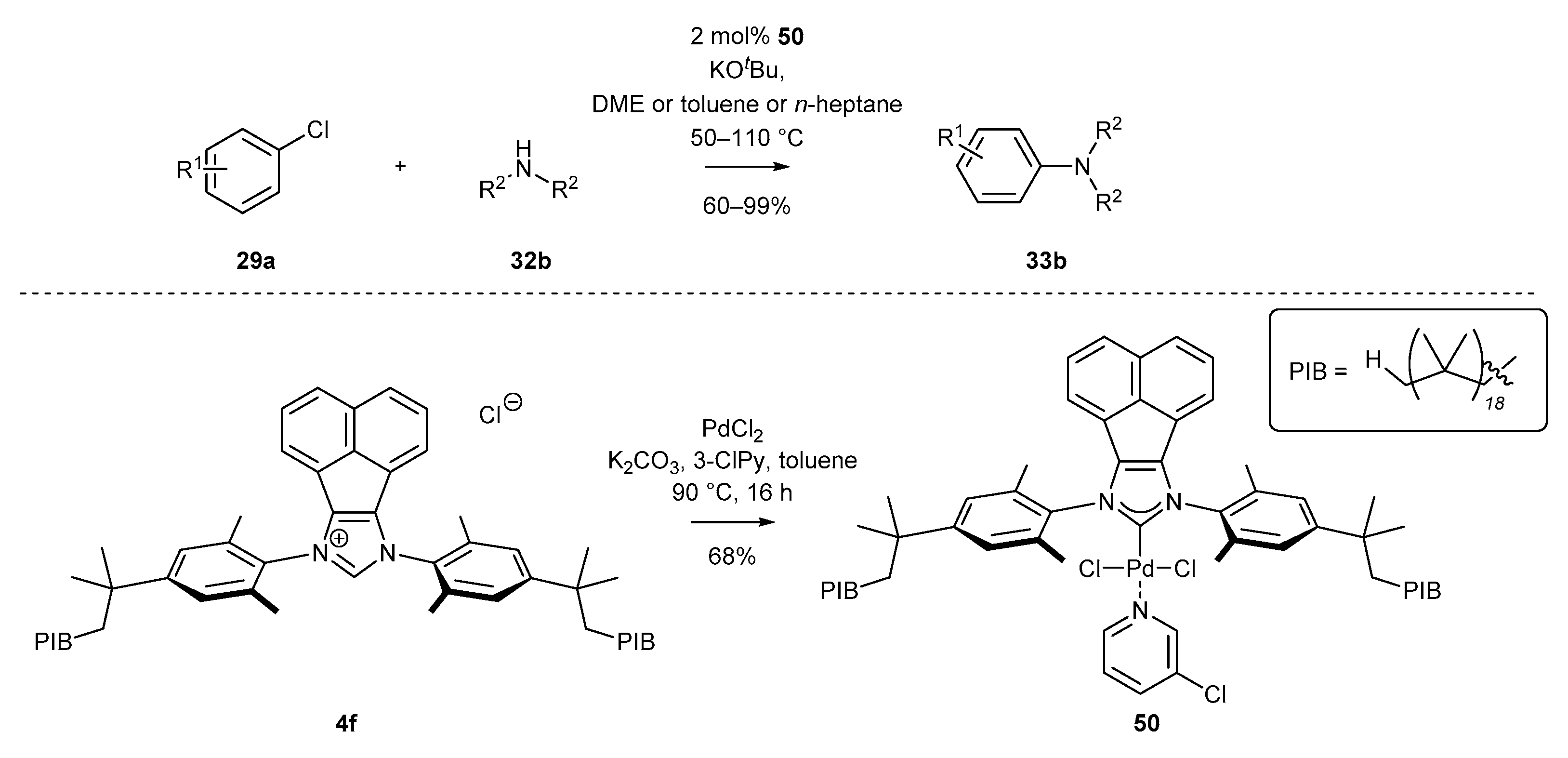
- Balogh, J.; Hlil, A.R.; El-Zoghbi, I.; Rafique, M.G.; Chouikhi, D.; Al-Hashimi, M.; Bazzi, H.S. Phase-Separable Polyisobutylene Palladium-PEPPSI Precatalysts: Synthesis and Application in Buchwald–Hartwig Amination. Macromol. Rapid. Comm. 2017, 38, 1700214. [Google Scholar] [CrossRef]
- Wang, J.; Ganguly, R.; Yongxin, L.; Díaz, J.; Soo, H.S.; García, F. A multi-step solvent-free mechanochemical route to indium(iii) complexes. Dalton Trans. 2016, 45, 7941–7946. [Google Scholar] [CrossRef] [Green Version]
- Rhinehart, J.L.; Mitchell, N.E.; Long, B.K. Enhancing α-Diimine Catalysts for High-Temperature Ethylene Polymerization. ACS Catal. 2014, 4, 2501–2504. [Google Scholar] [CrossRef]
- Guo, L.; Kong, W.; Xu, Y.; Yang, Y.; Ma, R.; Cong, L.; Dai, S.; Liu, Z. Large-scale synthesis of novel sterically hindered acenaphthene-based α-diimine ligands and their application in coordination chemistry. J. Organomet. Chem. 2018, 859, 58–67. [Google Scholar] [CrossRef]
- Ma, X.; Hu, X.; Zhang, Y.; Mu, H.; Cui, L.; Jian, Z. Preparation and in situ chain-end-functionalization of branched ethylene oligomers by monosubstituted α-diimine nickel catalysts. Polym. Chem. 2019, 10, 2596–2607. [Google Scholar] [CrossRef]
- Lu, D.-D.; He, X.-X.; Liu, F.-S. Bulky Yet Flexible Pd-PEPPSI-IPentAn for the Synthesis of Sterically Hindered Biaryls in Air. J. Org. Chem. 2017, 82, 10898–10911. [Google Scholar] [CrossRef]
- Gong, Y.; Li, S.; Gong, Q.; Zhang, S.; Liu, B.; Dai, S. Systematic Investigations of Ligand Steric Effects on α-Diimine Nickel Catalyzed Olefin Polymerization and Copolymerization. Organometallics 2019, 38, 2919–2926. [Google Scholar] [CrossRef]
- Lan, X.-B.; Li, Y.; Li, Y.-F.; Shen, D.-S.; Ke, Z.; Liu, F.-S. Flexible Steric Bulky Bis(Imino)acenaphthene (BIAN)-Supported N-Heterocyclic Carbene Palladium Precatalysts: Catalytic Application in Buchwald–Hartwig Amination in Air. J. Org. Chem. 2017, 82, 2914–2925. [Google Scholar] [CrossRef]
- Zhang, F.-Y.; Lan, X.-B.; Xu, C.; Yao, H.-G.; Li, T.; Liu, F.-S. Rigid hindered N-heterocyclic carbene palladium precatalysts: Synthesis, characterization and catalytic amination. Org. Chem. Front. 2019, 6, 3292–3299. [Google Scholar] [CrossRef]
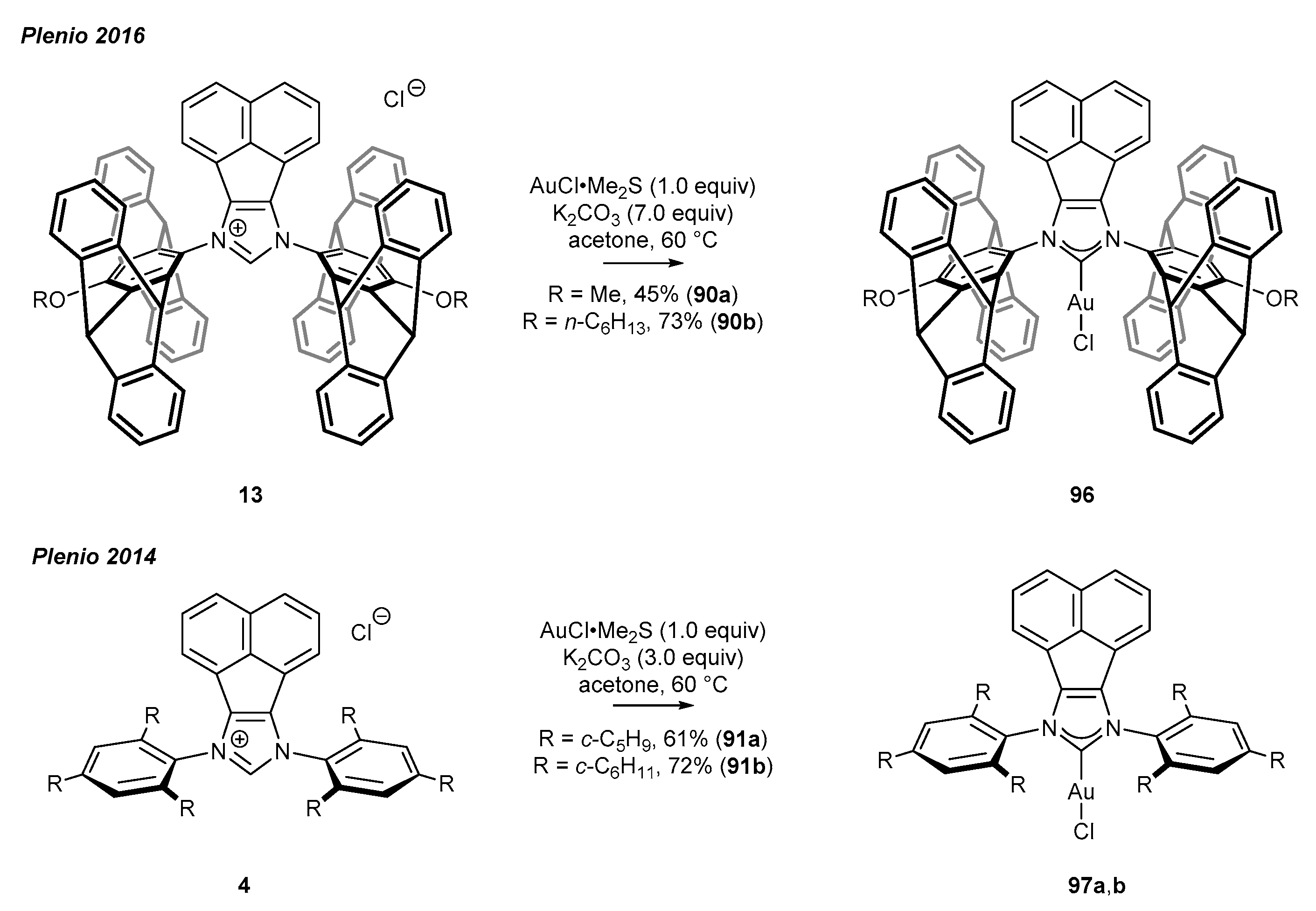
- Savka, R.; Foro, S.; Plenio, H. Pentiptycene-based concave NHC-metal complexes. Dalton Trans. 2016, 45, 11015–11024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyatos, M.; Mata, J.A.; Peris, E. Complexes with Poly(N-heterocyclic carbene) Ligands: Structural Features and Catalytic Applications. Chem. Rev. 2009, 109, 3677–3707. [Google Scholar] [CrossRef]
- Guisado-Barrios, G.; Hiller, J.; Peris, E. Pyracene-Linked Bis-Imidazolylidene Complexes of Palladium and Some Catalytic Benefits Produced by Bimetallic Catalysts. Chem. Eur. J. 2013, 19, 10405–10411. [Google Scholar] [CrossRef]
- Vasudevan, K.V.; Findlater, M.; Cowley, A.H. Synthesis and reactivity of tetrakis(imino)pyracene (TIP) ligands; bifunctional analogues of the BIAN ligand class. Chem. Commun. 2008, 1918–1919. [Google Scholar] [CrossRef] [PubMed]
- Prades, A.; Peris, E.; Alcarazo, M. Pyracenebis(imidazolylidene): A New Janus-Type Biscarbene and Its Coordination to Rhodium and Iridium. Organometallics 2012, 31, 4623–4626. [Google Scholar] [CrossRef]
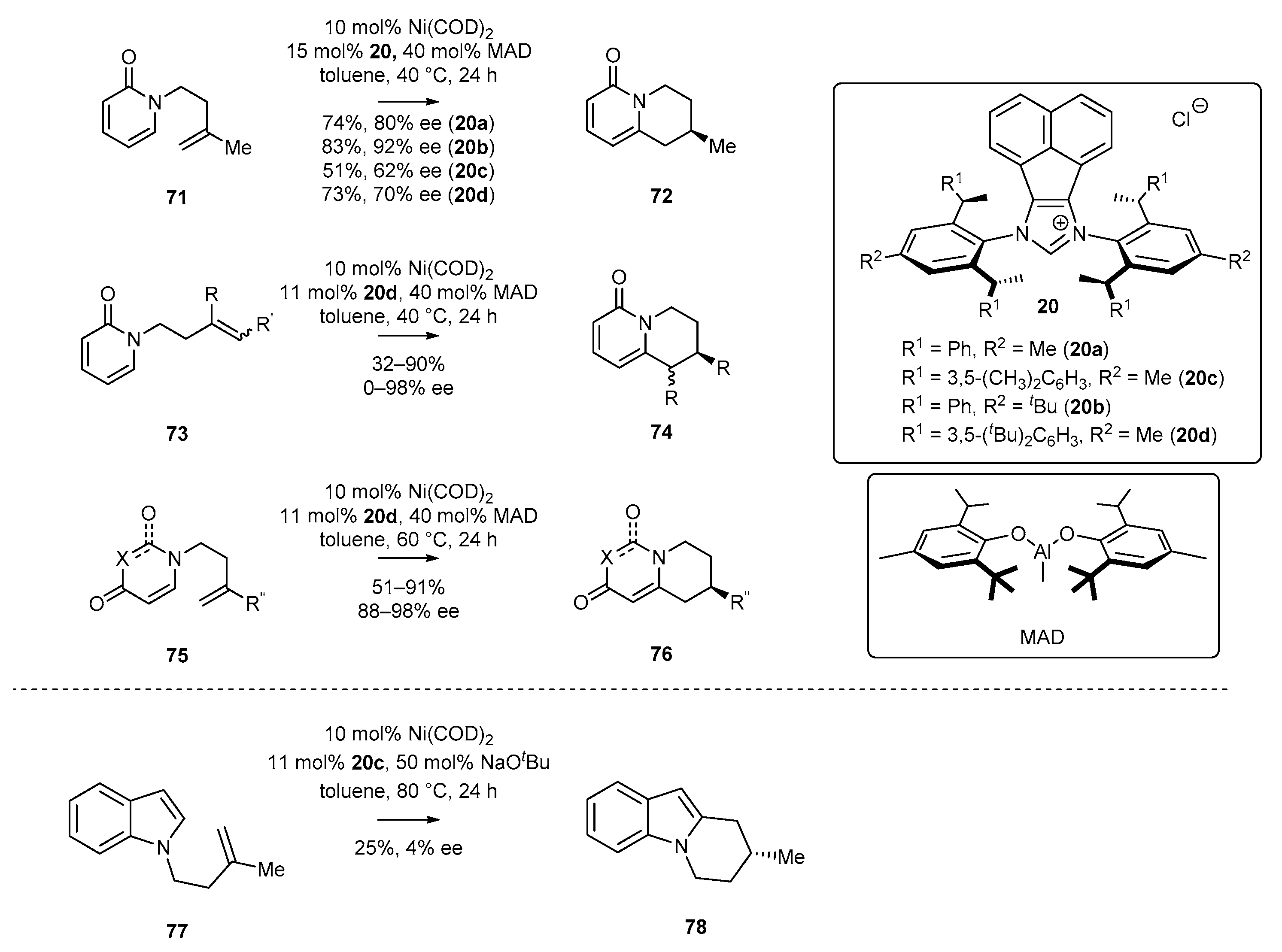
- Diesel, J.; Finogenova, A.M.; Cramer, N. Nickel-Catalyzed Enantioselective Pyridone C–H Functionalizations Enabled by a Bulky N-Heterocyclic Carbene Ligand. J. Am. Chem. Soc. 2018, 140, 4489–4493. [Google Scholar] [CrossRef]
- Cai, Y.; Yang, X.-T.; Zhang, S.-Q.; Li, F.; Li, Y.-Q.; Ruan, L.-X.; Hong, X.; Shi, S.-L. Copper-Catalyzed Enantioselective Markovnikov Protoboration of α-Olefins Enabled by a Buttressed N-Heterocyclic Carbene Ligand. Angew. Chem. Int. Ed. 2018, 57, 1376–1380. [Google Scholar] [CrossRef] [PubMed]
- Diesel, J.; Grosheva, D.; Kodama, S.; Cramer, N. A Bulky Chiral N-Heterocyclic Carbene Nickel Catalyst Enables Enantioselective C−H Functionalizations of Indoles and Pyrroles. Angew. Chem. Int. Ed. 2019, 58, 11044–11048. [Google Scholar] [CrossRef]
- Albright, A.; Eddings, D.; Black, R.; Welch, C.J.; Gerasimchuk, N.N.; Gawley, R.E. Design and Synthesis of C2-Symmetric N-Heterocyclic Carbene Precursors and Metal Carbenoids. J. Org. Chem. 2011, 76, 7341–7351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spahn, E.; Albright, A.; Shevlin, M.; Pauli, L.; Pfaltz, A.; Gawley, R.E. Double-Asymmetric Hydrogenation Strategy for the Reduction of 1,1-Diaryl Olefins Applied to an Improved Synthesis of CuIPhEt, a C2-Symmetric N-Heterocyclic Carbenoid. J. Org. Chem. 2013, 78, 2731–2735. [Google Scholar] [CrossRef] [Green Version]
- Organ, M.G.; Avola, S.; Dubovyk, I.; Hadei, N.; Kantchev, E.A.B.; O’Brien, C.J.; Valente, C. A User-Friendly, All-Purpose Pd–NHC (NHC=N-Heterocyclic Carbene) Precatalyst for the Negishi Reaction: A Step Towards a Universal Cross-Coupling Catalyst. Chem. Eur. J. 2006, 12, 4749–4755. [Google Scholar] [CrossRef]
- O’Brien, C.J.; Kantchev, E.A.B.; Valente, C.; Hadei, N.; Chass, G.A.; Lough, A.; Hopkinson, A.C.; Organ, M.G. Easily Prepared Air- and Moisture-Stable Pd–NHC (NHC=N-Heterocyclic Carbene) Complexes: A Reliable, User-Friendly, Highly Active Palladium Precatalyst for the Suzuki–Miyaura Reaction. Chem. Eur. J. 2006, 12, 4743–4748. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Fang, W.; Jiang, J. A highly efficient precatalyst for amination of aryl chlorides: Synthesis, structure and application of a robust acenaphthoimidazolylidene palladium complex. Chem. Commun. 2011, 47, 12358–12360. [Google Scholar] [CrossRef]
- Liu, Z.; Dong, N.; Xu, M.; Sun, Z.; Tu, T. Mild Negishi Cross-Coupling Reactions Catalyzed by Acenaphthoimidazolylidene Palladium Complexes at Low Catalyst Loadings. J. Org. Chem. 2013, 78, 7436–7444. [Google Scholar] [CrossRef]
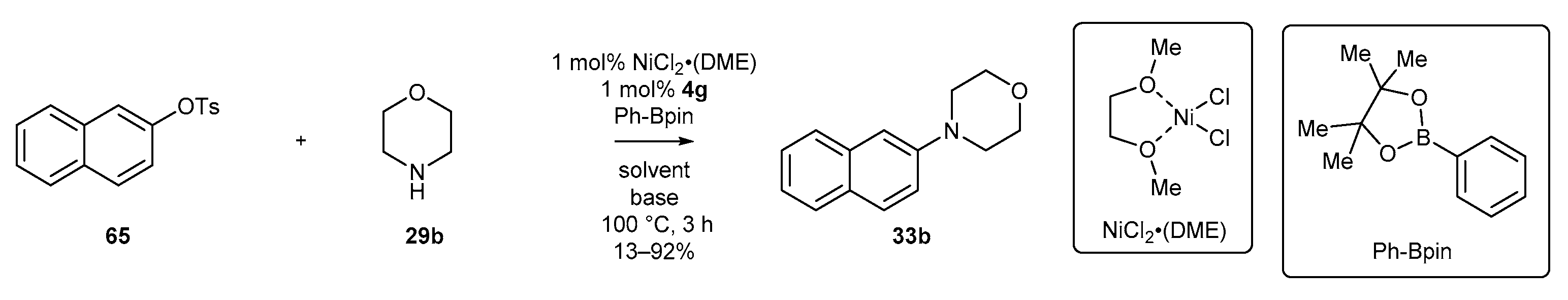
- Jiang, J.; Zhu, H.; Shen, Y.; Tu, T. Acenaphthoimidazolium chloride-enabled nickel-catalyzed amination of bulky aryl tosylates. Org. Chem. Front. 2014, 1, 1172–1175. [Google Scholar] [CrossRef]
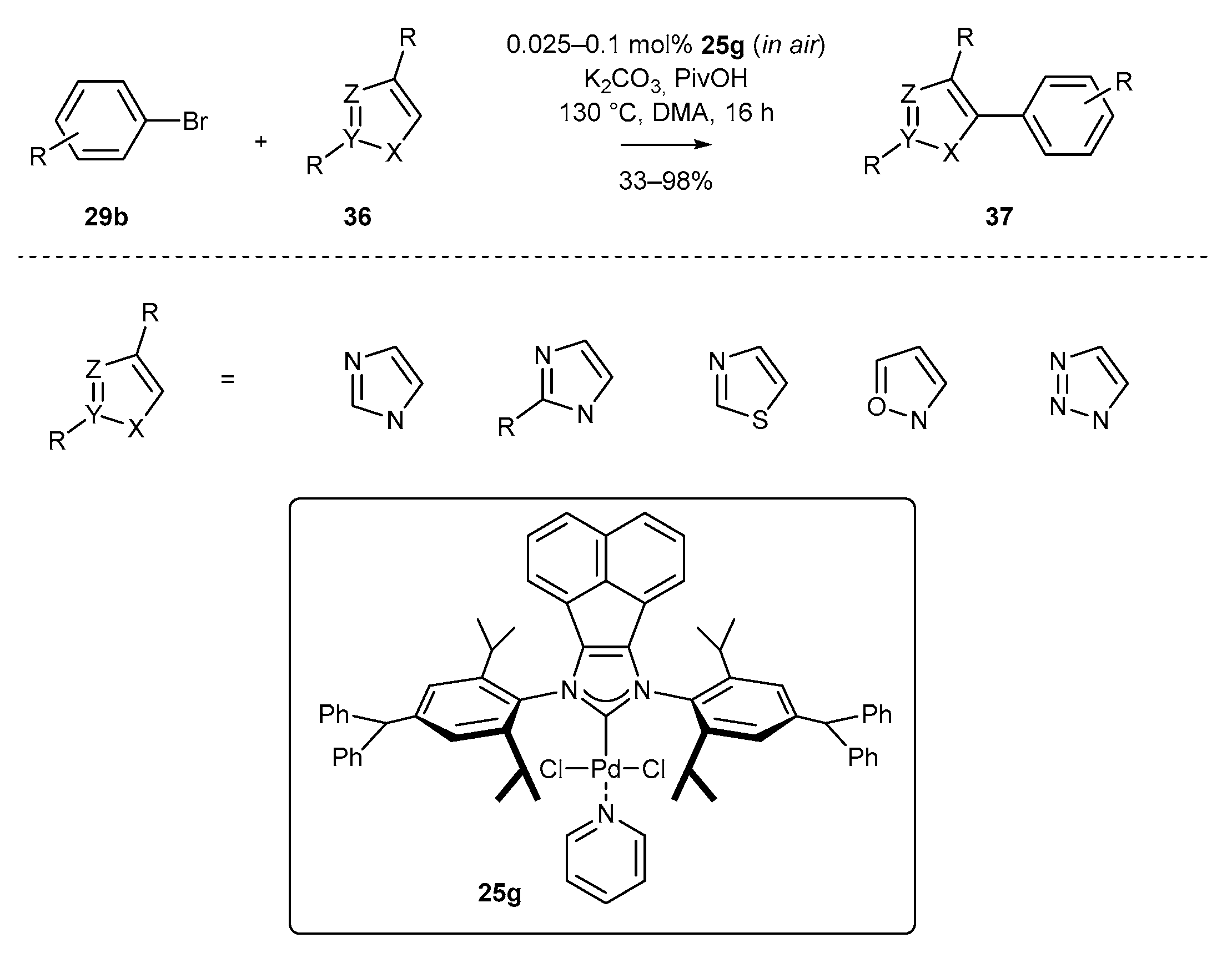
- Hu, L.-Q.; Deng, R.-L.; Li, Y.-F.; Zeng, C.-J.; Shen, D.-S.; Liu, F.-S. Developing Bis(imino)acenaphthene-Supported N-Heterocyclic Carbene Palladium Precatalysts for Direct Arylation of Azoles. Organometallics 2018, 37, 214–226. [Google Scholar] [CrossRef]
- Huang, F.-D.; Xu, C.; Lu, D.-D.; Shen, D.-S.; Li, T.; Liu, F.-S. Pd-PEPPSI-IPentAn Promoted Deactivated Amination of Aryl Chlorides with Amines under Aerobic Conditions. J. Org. Chem. 2018, 83, 9144–9155. [Google Scholar] [CrossRef]
- Ouyang, J.-S.; Li, Y.-F.; Huang, F.-D.; Lu, D.-D.; Liu, F.-S. The Highly Efficient Suzuki–Miyaura Cross-Coupling of (Hetero)aryl Chlorides and (Hetero)arylboronic Acids Catalyzed by “Bulky-yet-Flexible” Palladium–PEPPSI Complexes in Air. ChemCatChem 2018, 10, 371–375. [Google Scholar] [CrossRef]
- Leuthäußer, S.; Schmidts, V.; Thiele, C.M.; Plenio, H. π-Face Donor Properties of N-Heterocyclic Carbenes in Grubbs II Complexes. Chem. Eur. J. 2008, 14, 5465–5481. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.H.; Konstandaras, N.; Cole, M.L.; Harper, J.B. Targeted and Systematic Approach to the Study of pKa Values of Imidazolium Salts in Dimethyl Sulfoxide. J. Org. Chem. 2017, 82, 7324–7331. [Google Scholar] [CrossRef]
- Rayner, P.; Norcott, P.; Appleby, K.; Iali, W.; John, R.; Hart, S.; Whitwood, A.; Duckett, S. Fine-tuning the efficiency of para-hydrogen-induced hyperpolarization by rational N-heterocyclic carbene design. Nat. Commun. 2018, 9, 4251. [Google Scholar] [CrossRef] [PubMed]
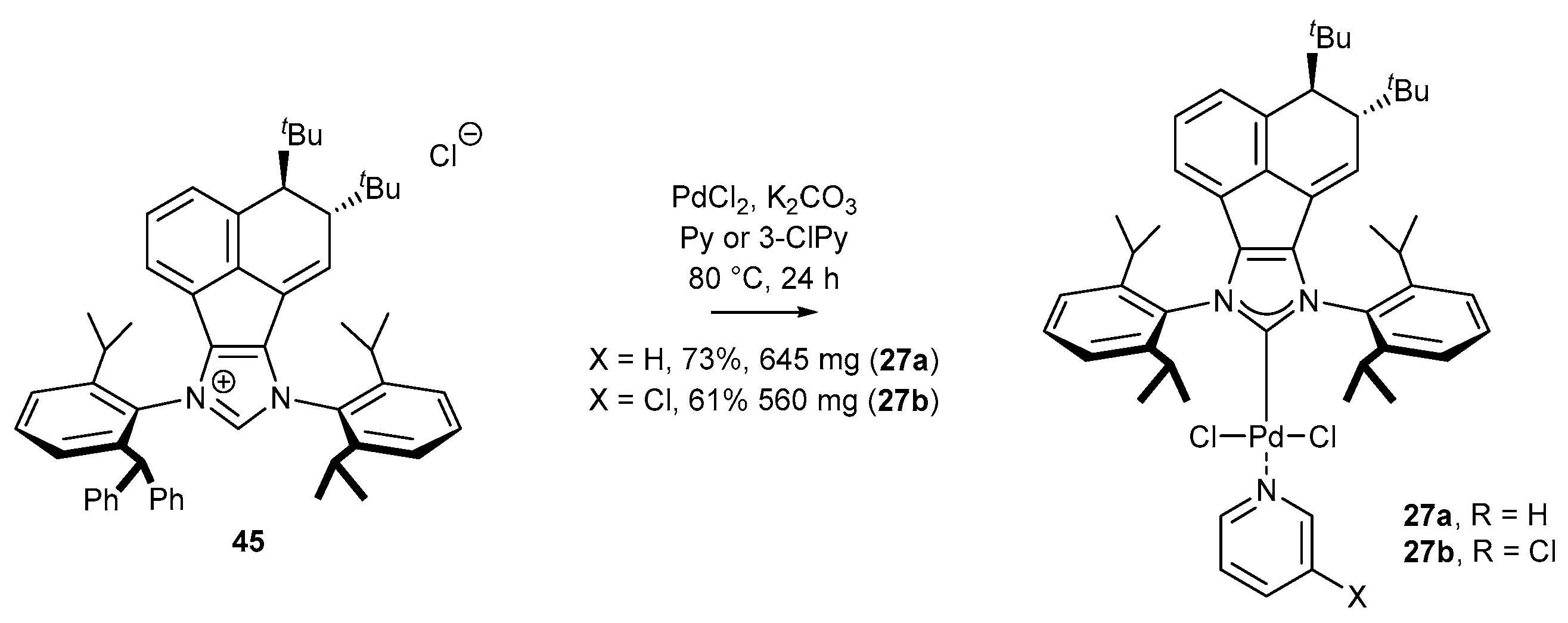
- Lan, X.-B.; Chen, F.-M.; Ma, B.-B.; Shen, D.-S.; Liu, F.-S. Pd-PEPPSI Complexes Bearing Bulky [(1,2-Di-(tert-butyl)acenaphthyl] (DtBu-An) on N-Heterocarbene Backbones: Highly Efficient for Suzuki–Miyaura Cross-Coupling under Aerobic Conditions. Organometallics 2016, 35, 3852–3860. [Google Scholar] [CrossRef]
- Sinha, N.; Heijnen, D.; Feringa, B.L.; Organ, M.G. Murahashi Cross-Coupling at −78 °C: A One-Pot Procedure for Sequential C−C/C−C, C−C/C−N, and C−C/C−S Cross-Coupling of Bromo-Chloro-Arenes. Chem. Eur. J. 2019, 25, 9180–9184. [Google Scholar] [CrossRef]
- Shen, A.; Wu, Z.; Fang, Y.; Yang, J.; Zhu, H.; Tu, T. A Concerted Catalytic System for Sonogashira Coupling Reactions: Combination of N-Heterocyclic Carbene Palladium and Copper Complexes. Asian J. Org. Chem. 2018, 7, 1113–1117. [Google Scholar] [CrossRef]
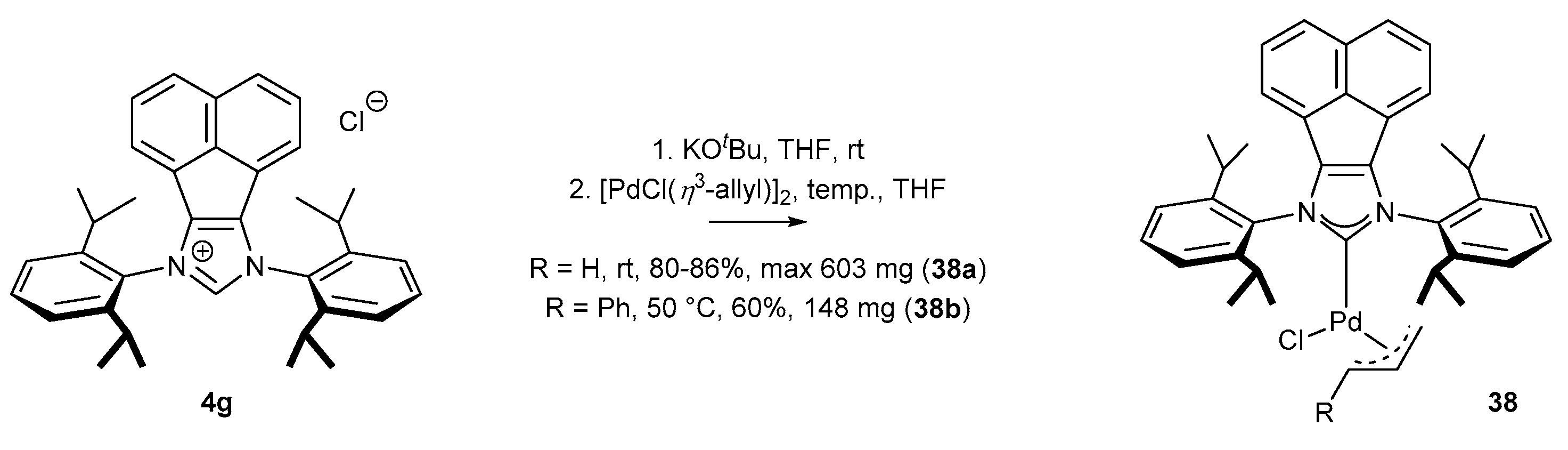
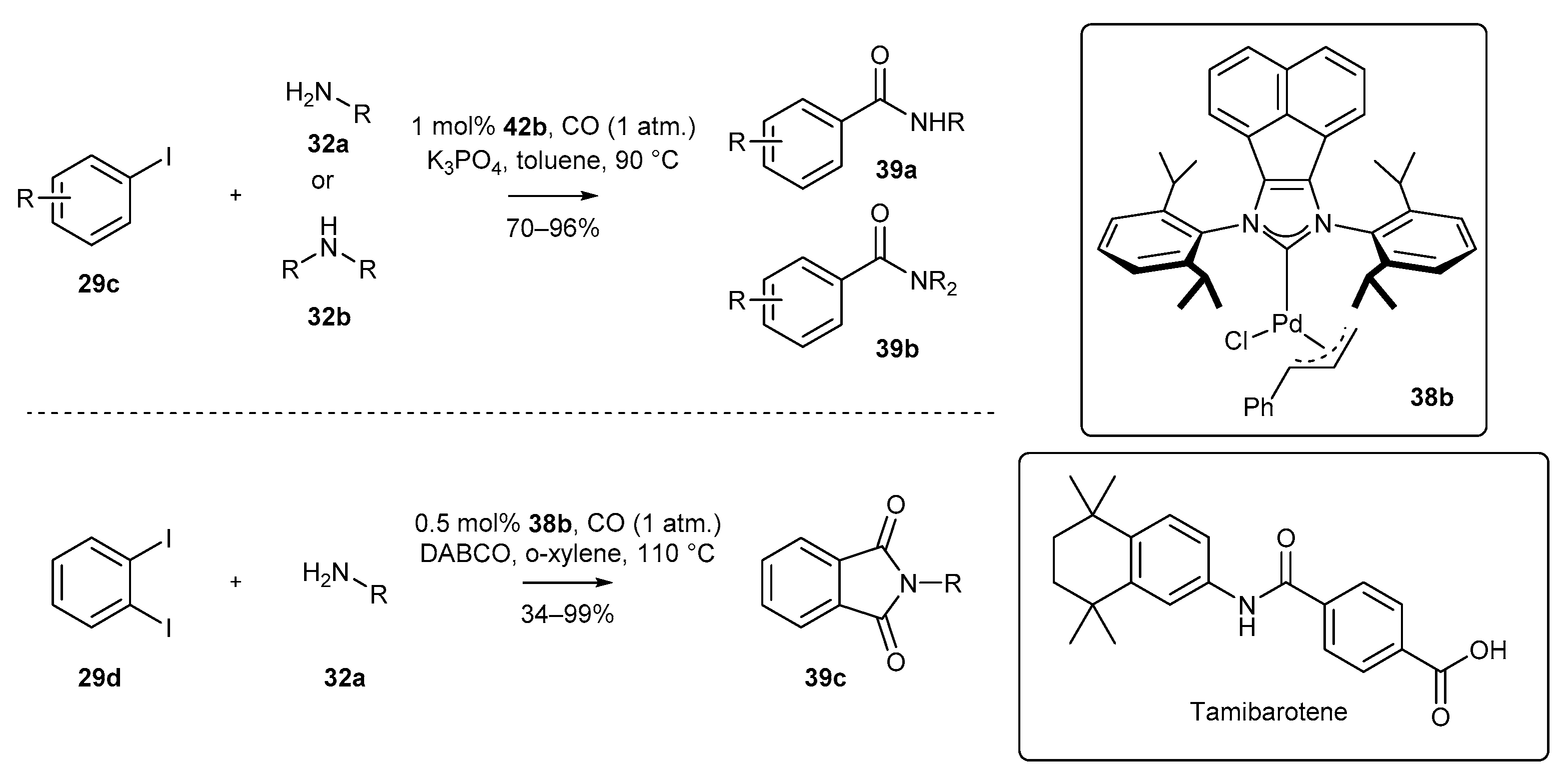
- Fang, W.; Deng, Q.; Xu, M.; Tu, T. Highly Efficient Aminocarbonylation of Iodoarenes at Atmospheric Pressure Catalyzed by a Robust Acenaphthoimidazolyidene Allylic Palladium Complex. Org. Lett. 2013, 15, 3678–3681. [Google Scholar] [CrossRef] [PubMed]
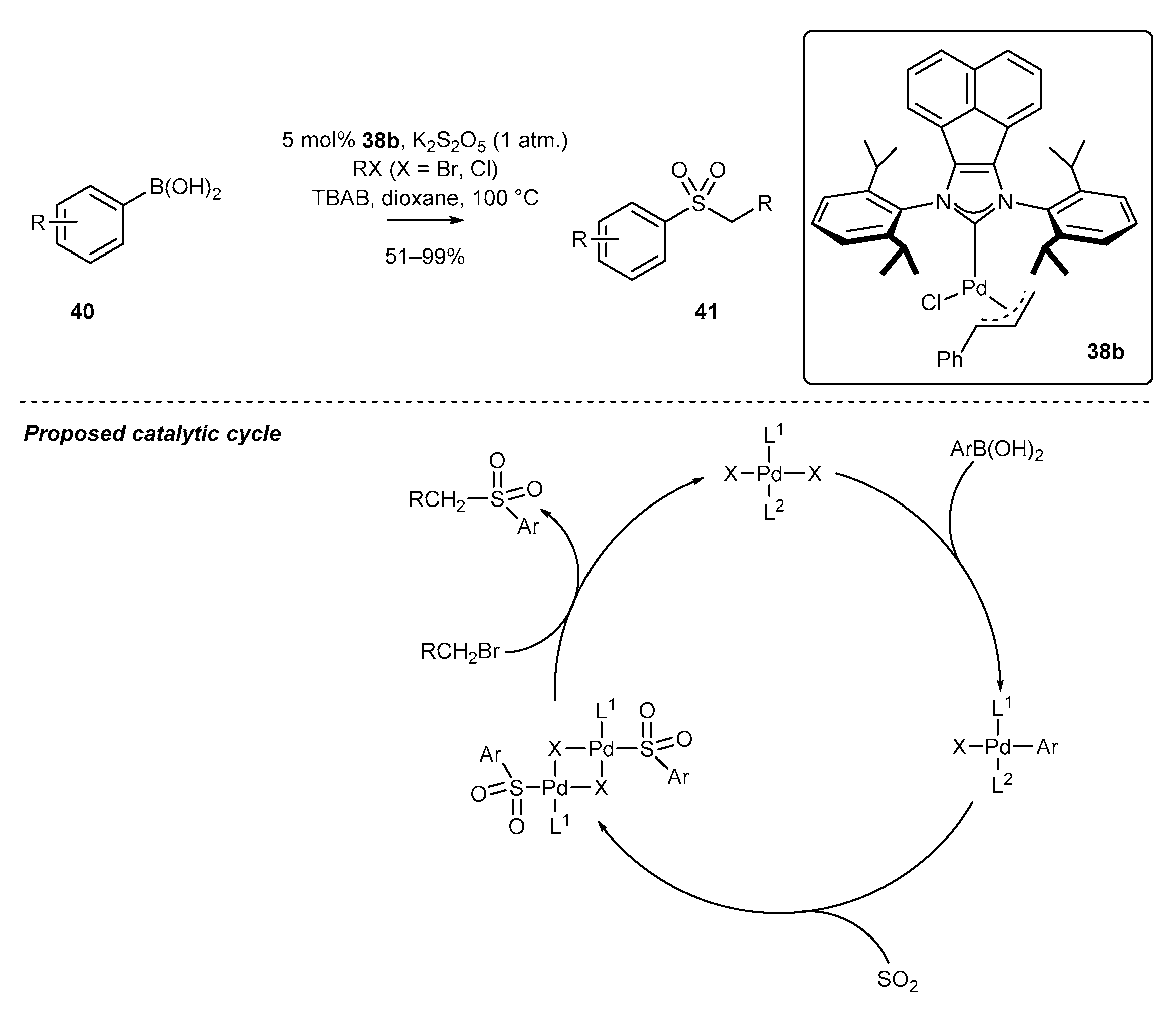
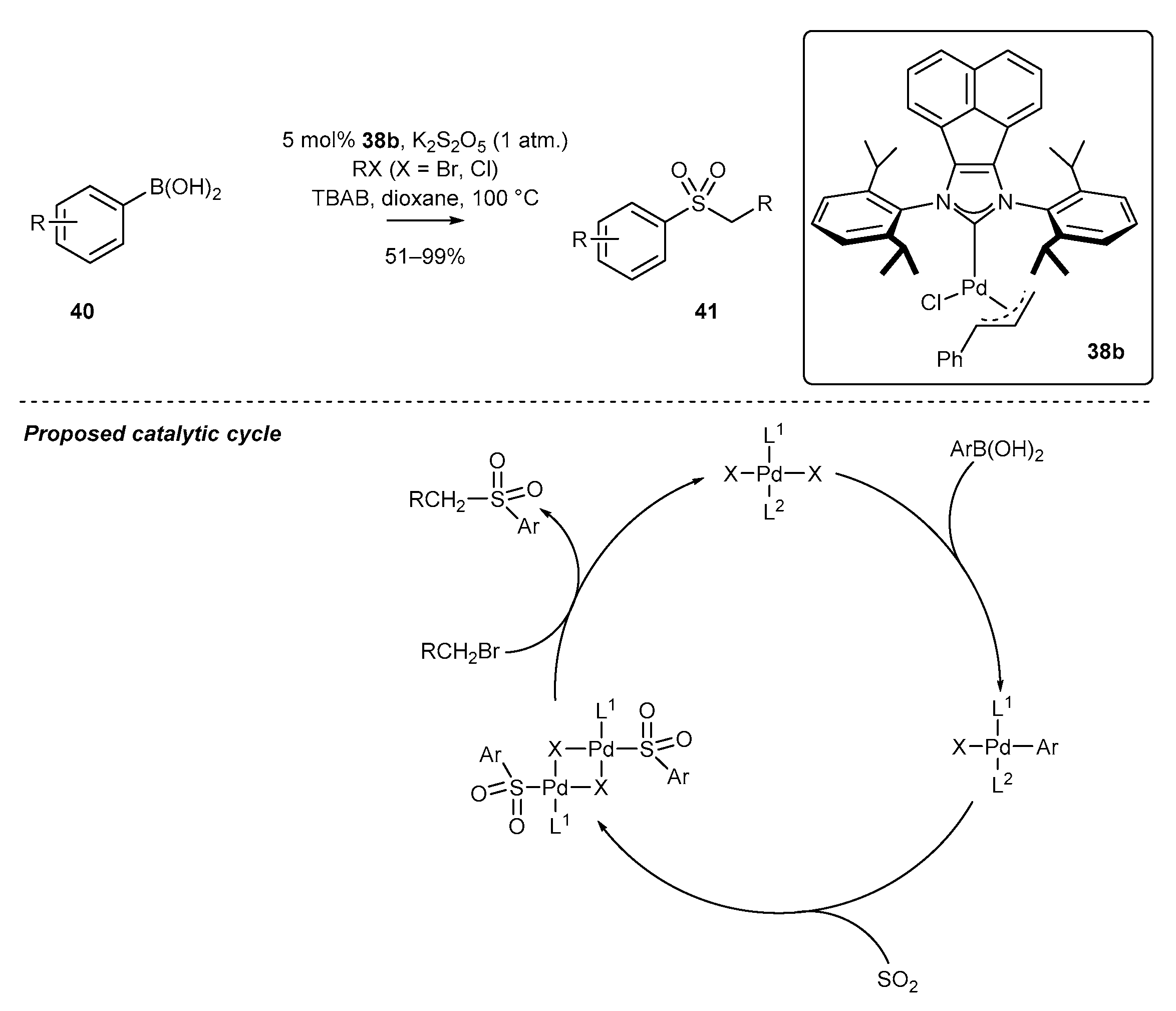
- Zhu, H.; Shen, Y.; Deng, Q.; Chen, J.; Tu, T. Pd(NHC)-catalyzed alkylsulfonylation of boronic acids: A general and efficient approach for sulfone synthesis. Chem. Commun. 2017, 53, 12473–12476. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Deng, Q.; Fang, W.; Gong, J.-F.; Song, M.-P.; Xu, M.; Tu, T. Efficient and scalable Pd-catalyzed double aminocarbonylations under atmospheric pressure at low catalyst loadings. Org. Chem. Front. 2014, 1, 1261–1265. [Google Scholar] [CrossRef]
- Deng, Q.; Zhang, Y.; Zhu, H.; Tu, T. Robust Acenaphthoimidazolylidene Palladacycles: Highly Efficient Catalysts for the Amination of N-Heteroaryl Chlorides. Chem. Asian. J. 2017, 12, 2364–2368. [Google Scholar] [CrossRef]
- Deng, Q.; Shen, Y.; Zhu, H.; Tu, T. A magnetic nanoparticle-supported N-heterocyclic carbene-palladacycle: An efficient and recyclable solid molecular catalyst for Suzuki–Miyaura cross-coupling of 9-chloroacridine. Chem. Commun. 2017, 53, 13063–13066. [Google Scholar] [CrossRef]
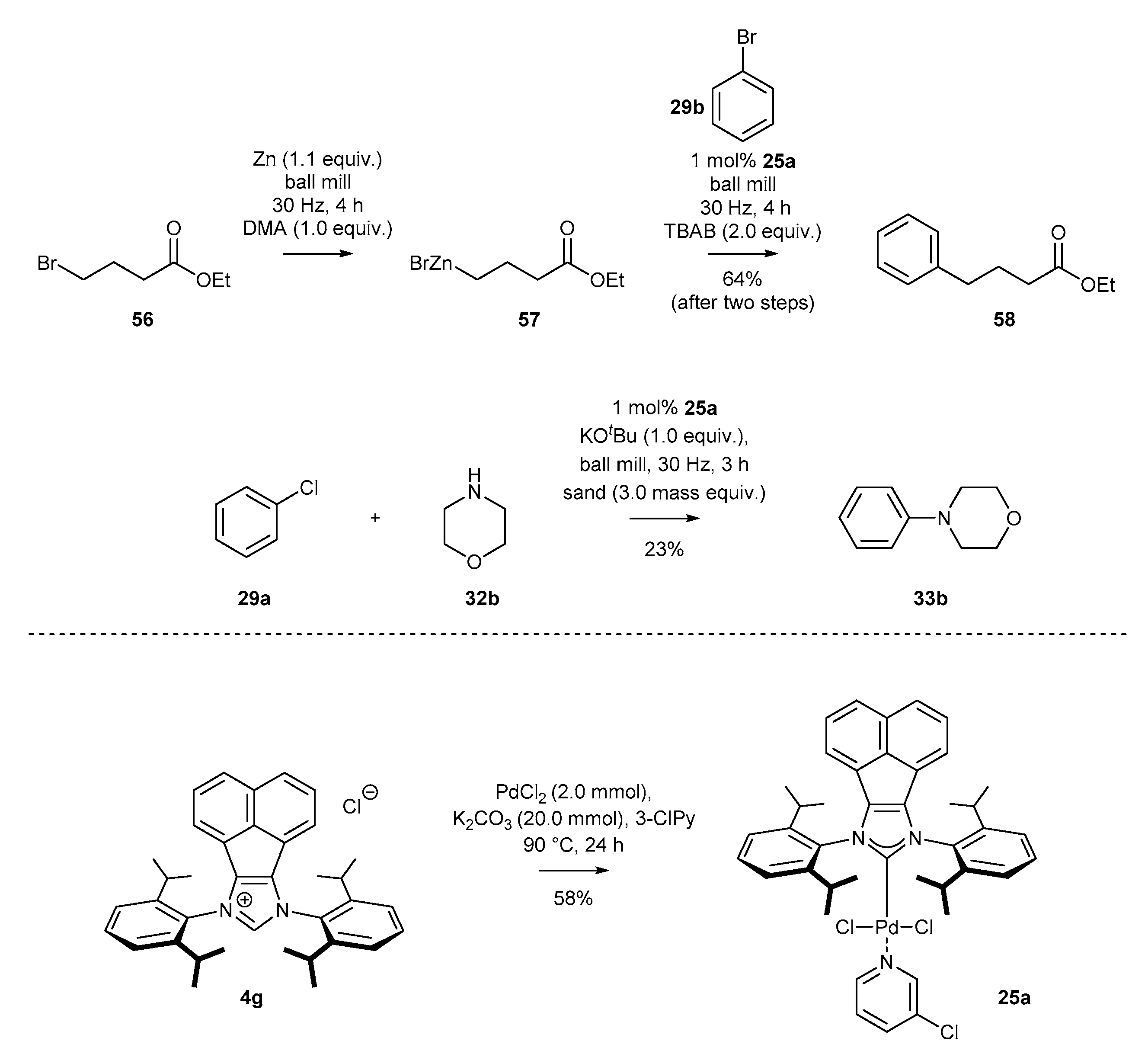
- Cao, Q.; Howard, J.L.; Wheatley, E.; Browne, D.L. Mechanochemical Activation of Zinc and Application to Negishi Cross-Coupling. Angew. Chem. Int. Ed. 2018, 57, 11339–11343. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Nicholson, W.; Jones, A.; Browne, D. Robust Buchwald–Hartwig amination enabled by ball-milling. Org. Biomol. Chem. 2018, 17, 1722–1726. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.R.; Makida, Y.; Meiries, S.; Slawin, A.M.Z.; Nolan, S.P. Enhanced Activity of [Ni(NHC)CpCl] Complexes in Arylamination Catalysis. Organometallics 2013, 32, 6265–6270. [Google Scholar] [CrossRef]
- Ben Halima, T.; Masson-Makdissi, J.; Newman, S.G. Nickel-Catalyzed Amide Bond Formation from Methyl Esters. Angew. Chem. Int. Ed. 2018, 57, 12925–12929. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.-W.; Li, R.; Li, J.-F.; Sun, J.; Ye, M. NHC ligand-enabled Ni-catalyzed reductive coupling of alkynes and imines using isopropanol as a reductant. Green Chem. 2019, 21, 2240–2244. [Google Scholar] [CrossRef]
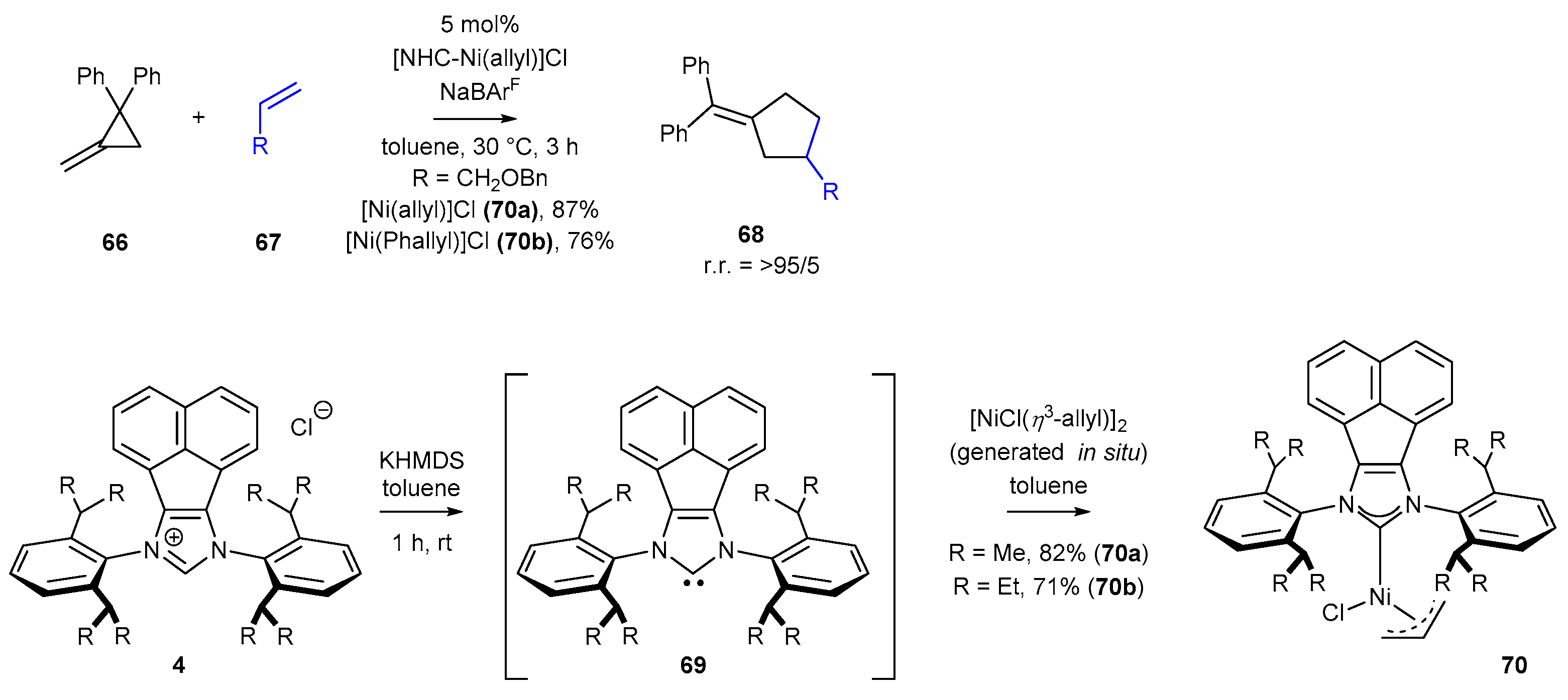
- Huang, J.-Q.; Ho, C.-Y. NHC/Nickel(II)-Catalyzed [3+2] Cross-Dimerization of Unactivated Olefins and Methylenecyclopropanes. Angew. Chem. Int. Ed. 2020, 59, 5288–5292. [Google Scholar] [CrossRef] [PubMed]
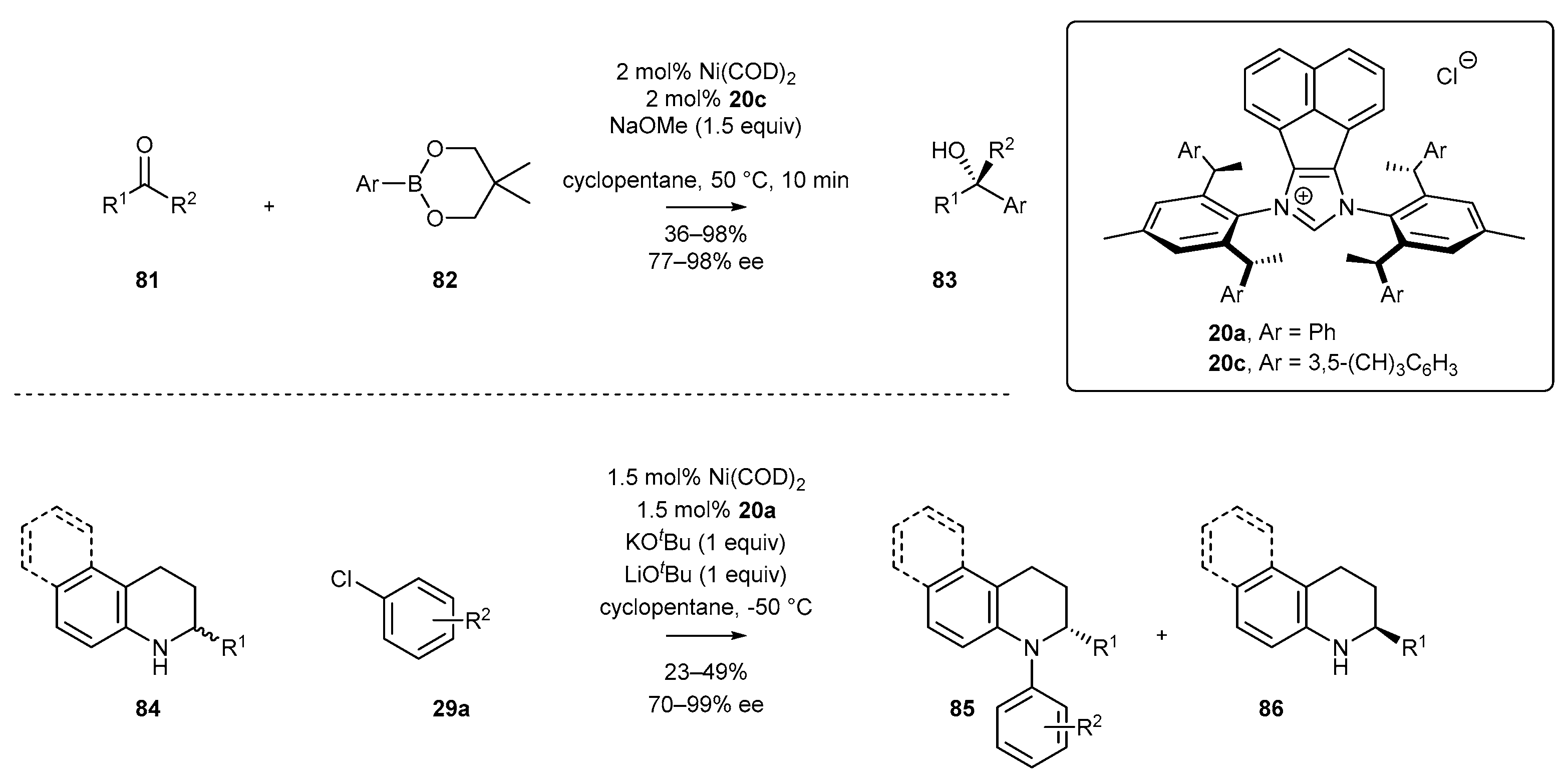
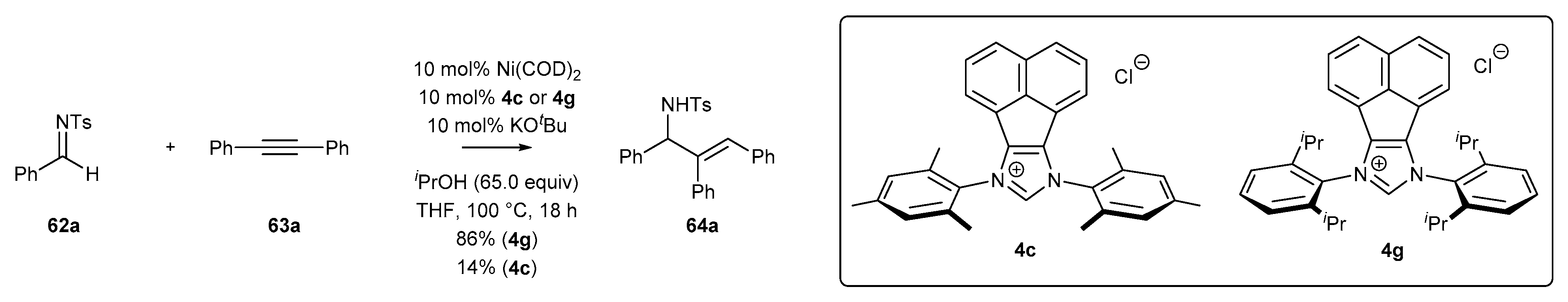
- Zhang, W.-B.; Yang, X.-T.; Ma, J.-B.; Su, Z.-M.; Shi, S.-L. Regio- and Enantioselective C–H Cyclization of Pyridines with Alkenes Enabled by a Nickel/N-Heterocyclic Carbene Catalysis. J. Am. Chem. Soc. 2019, 141, 5628–5634. [Google Scholar] [CrossRef] [PubMed]
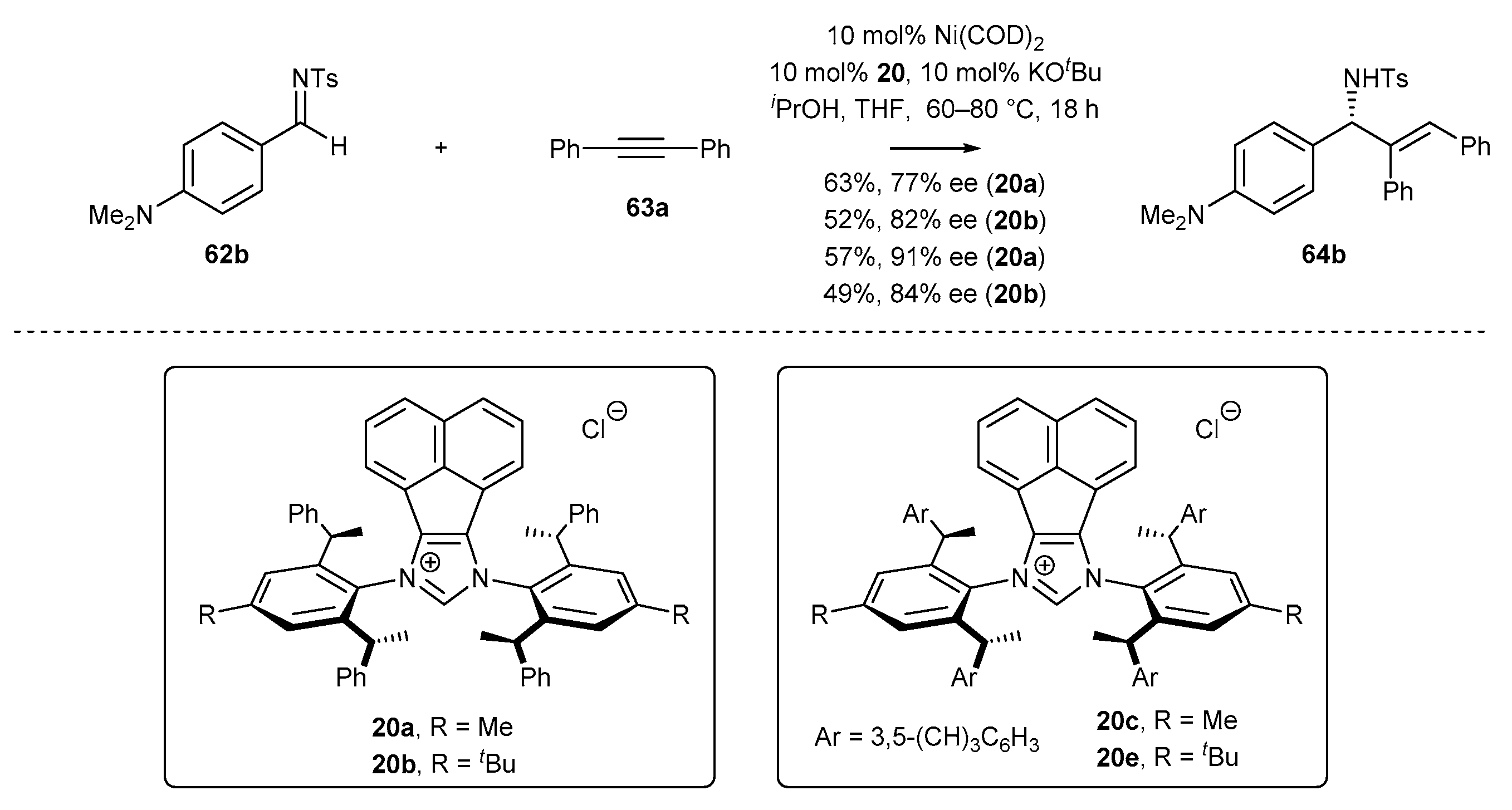
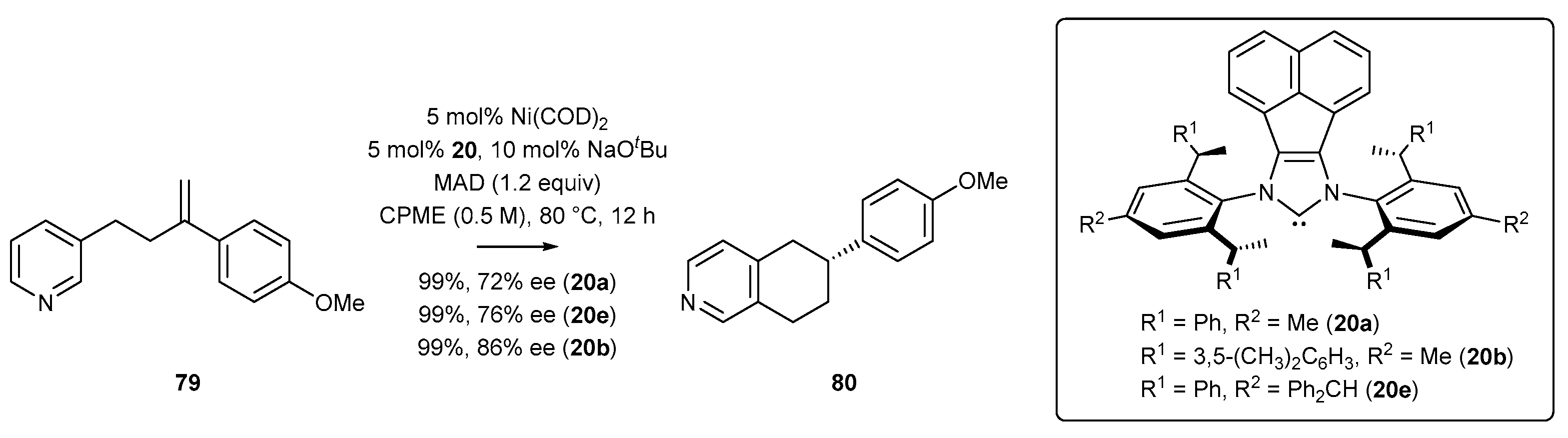
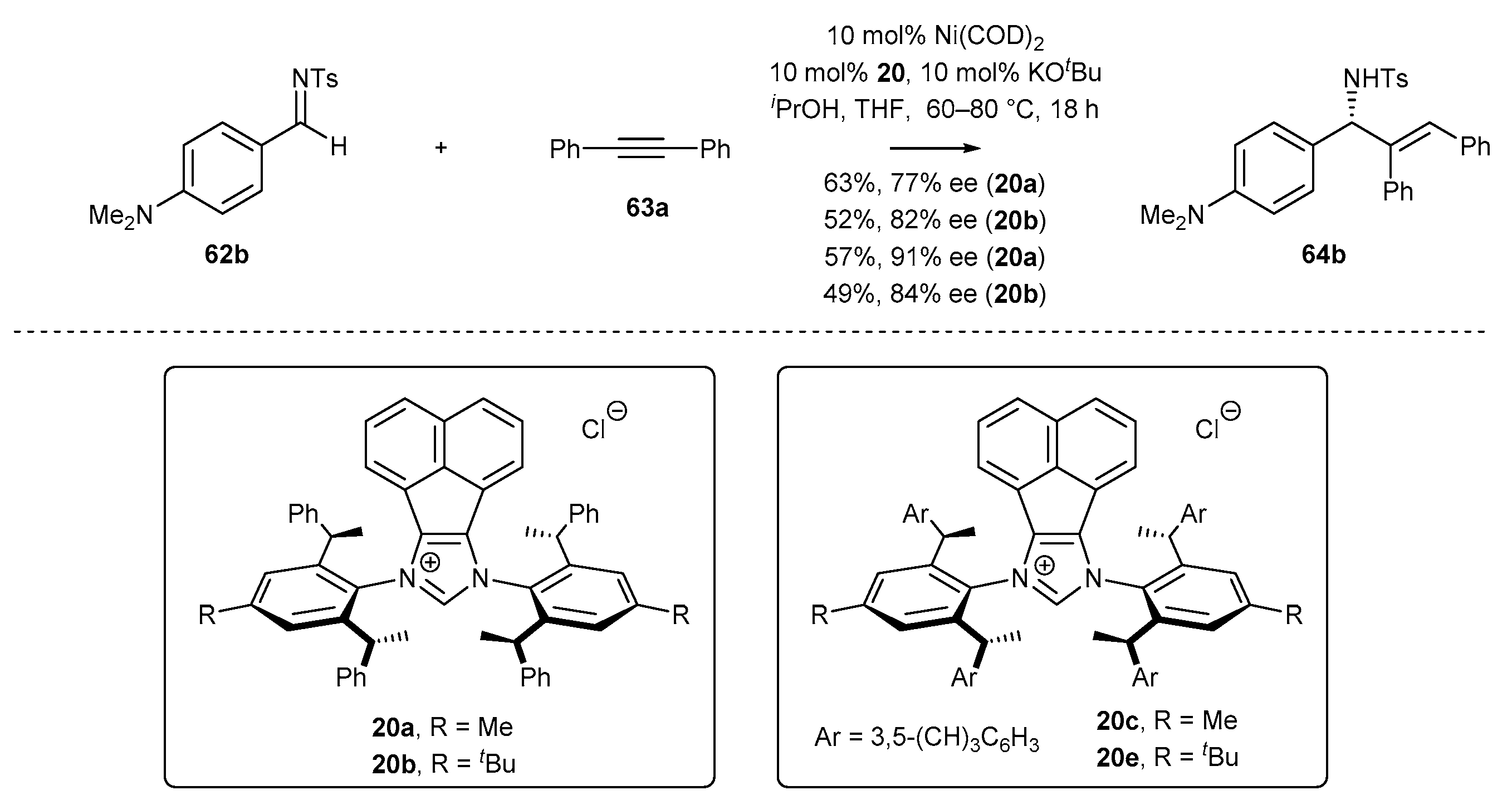
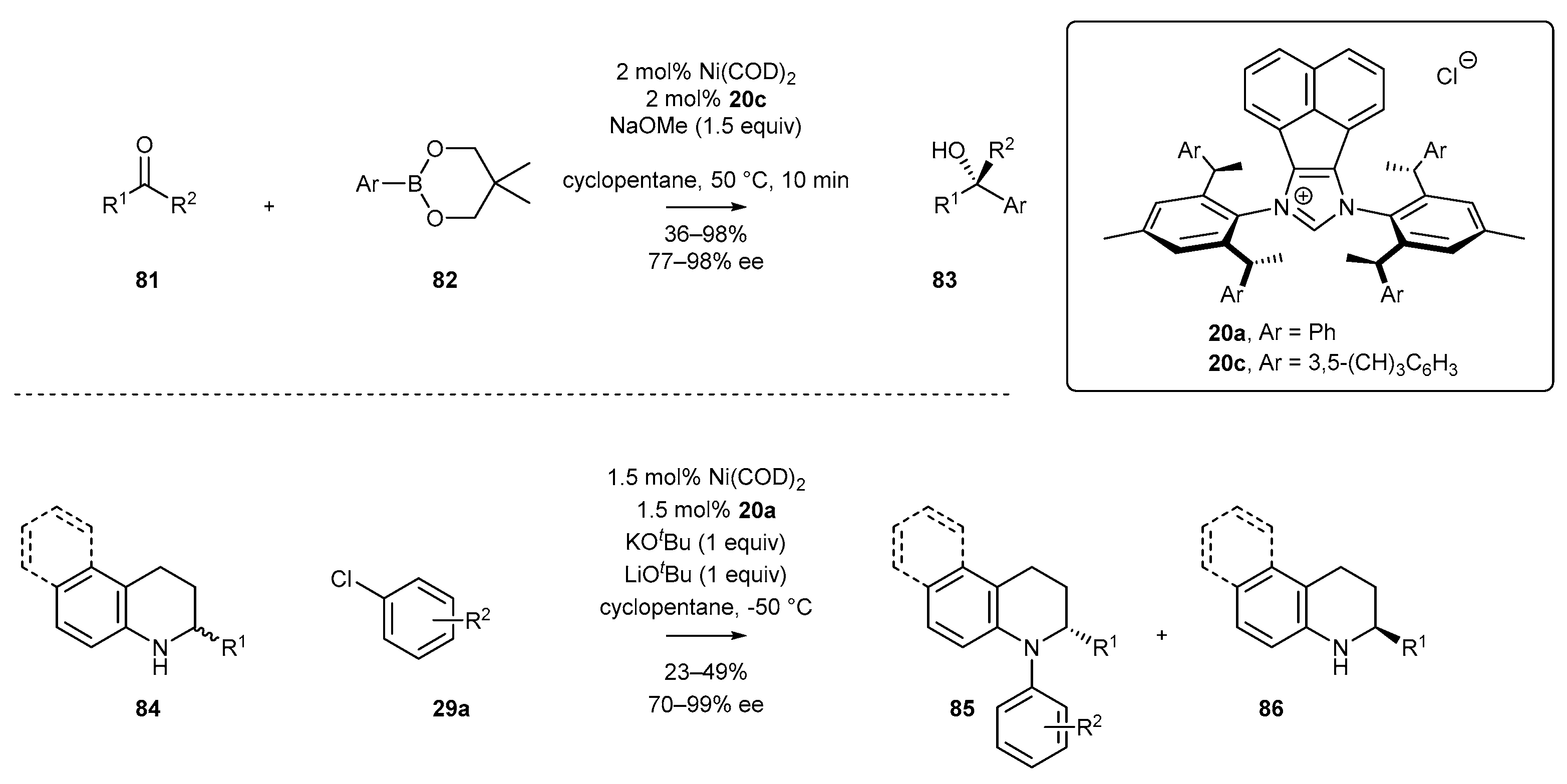
- Wang, Z.-C.; Xie, P.-P.; Xu, Y.; Hong, X.; Shi, S.-L. Low-Temperature Nickel-Catalyzed C−N Cross-Coupling via Kinetic Resolution Enabled by a Bulky and Flexible Chiral N-Heterocyclic Carbene Ligand. Angew. Chem. Int. Ed. 2021, 60, 16077–16084. [Google Scholar] [CrossRef] [PubMed]
- Grela, K.; Harutyunyan, S.; Michrowska, A. A Highly Efficient Ruthenium Catalyst for Metathesis Reactions. Angew. Chem. Int. Ed. 2002, 41, 4038–4040. [Google Scholar] [CrossRef] [Green Version]
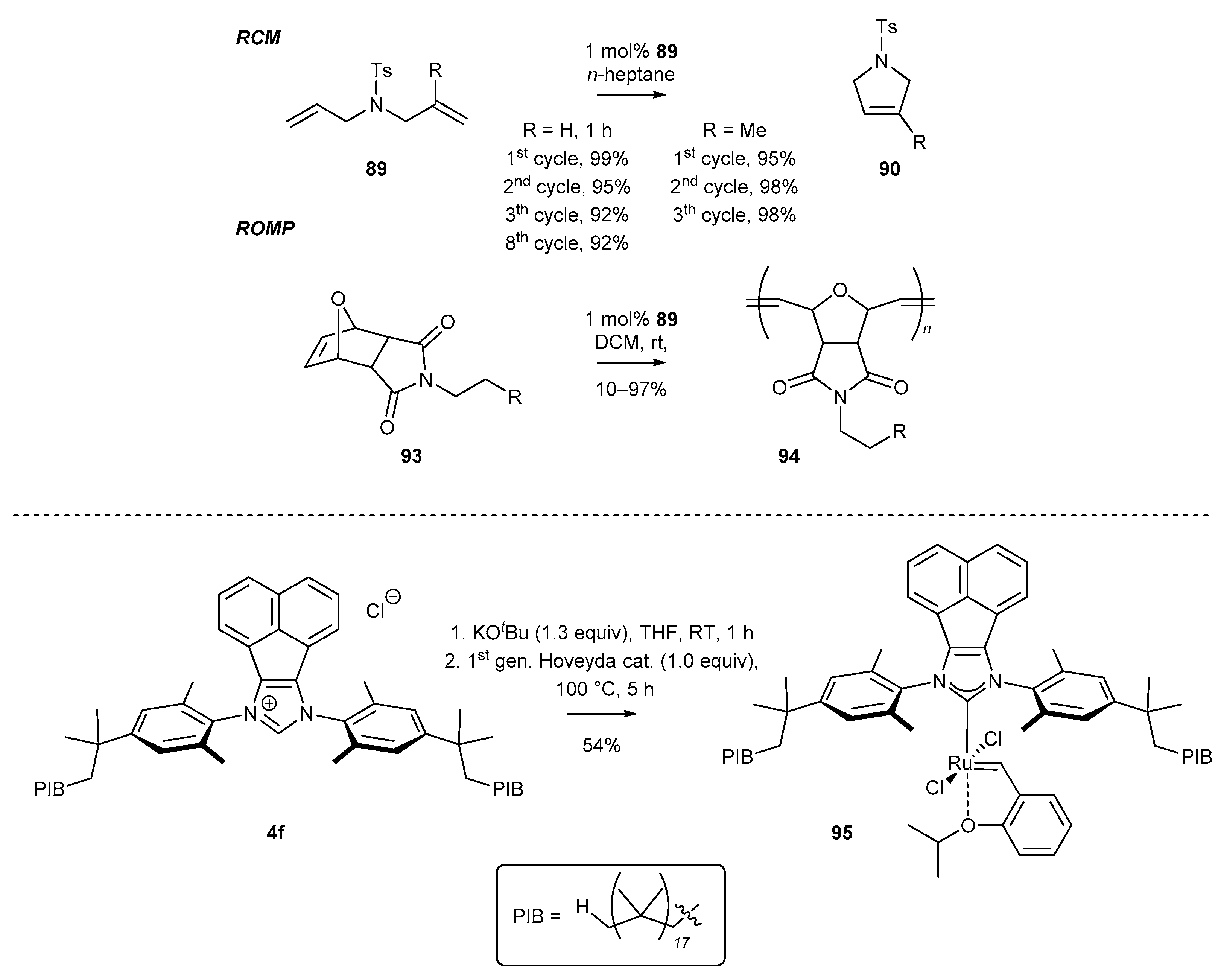
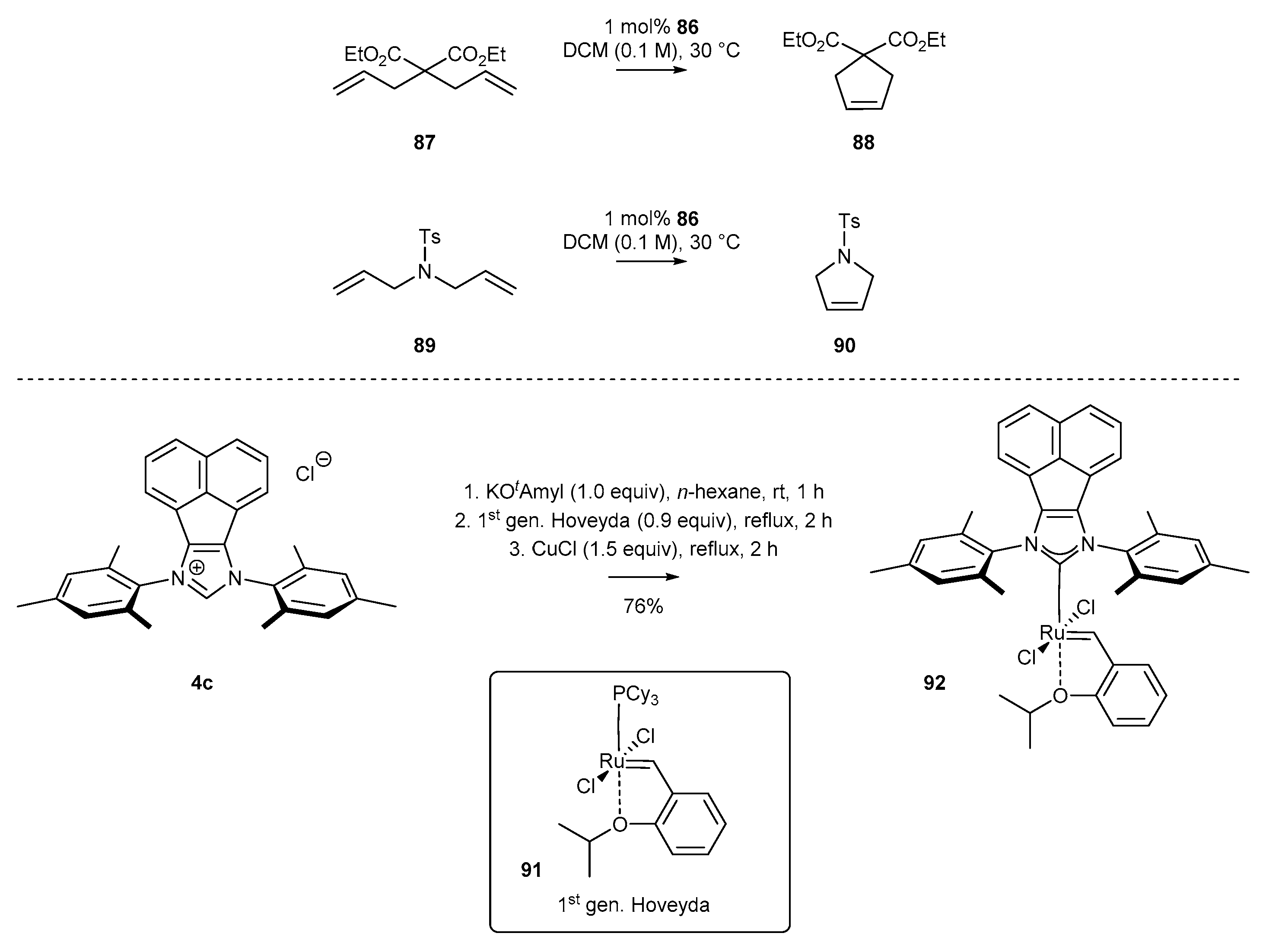
- Hlil, A.R.; Moncho, S.; Tuba, R.; Elsaid, K.; Szarka, G.; Brothers, E.N.; Grubbs, R.H.; Al-Hashimi, M.; Bazzi, H.S. Synthesis and catalytic activity of supported acenaphthoimidazolylidene N-heterocyclic carbene ruthenium complex for ring closing metathesis (RCM) and ring opening metathesis polymerization (ROMP). J. Catal. 2016, 344, 100–107. [Google Scholar] [CrossRef]
- Butorac, R.; Al-Deyab, S.; Cowley, A. Antimicrobial Properties of Some Bis(Iminoacenaphthene (BIAN)-Supported N-Heterocyclic Carbene Complexes of Silver and Gold. Molecules 2011, 16, 2285–2292. [Google Scholar] [CrossRef] [PubMed]
- Elzatahry, A.A.; Al-Enizi, A.M.; Elsayed, E.A.; Butorac, R.R.; Al-Deyab, S.S.; Wadaan, M.A.M.; Cowley, A.H. Nanofiber composites containing N-heterocyclic carbene complexes with antimicrobial activity. Int. J. Nanomed. 2012, 7, 2829–2832. [Google Scholar] [CrossRef] [Green Version]
- Farooq, M.; Taha, N.; Butorac, R.; Evans, D.; Elzatahry, A.; Elsayed, E.; Wadaan, M.; Al-Deyab, S.; Cowley, A. Biological Screening of Newly Synthesized BIAN N-Heterocyclic Gold Carbene Complexes in Zebrafish Embryos. Int. J. Mol. Sci. 2015, 16, 24718–24731. [Google Scholar] [CrossRef] [Green Version]
- Ciancaleoni, G.; Biasiolo, L.; Bistoni, G.; Macchioni, A.; Tarantelli, F.; Zuccaccia, D.; Belpassi, L. NHC-Gold-Alkyne Complexes: Influence of the Carbene Backbone on the Ion Pair Structure. Organometallics 2013, 32, 4444–4447. [Google Scholar] [CrossRef]
- Visbal, R.; Laguna, A.; Gimeno, M.C. Simple and efficient synthesis of [MCI(NHC)] (M = Au, Ag) complexes. Chem. Commun. 2013, 49, 5642–5644. [Google Scholar] [CrossRef]
- Collado, A.; Gomez-Suarez, A.; Martin, A.R.; Slawin, A.M.Z.; Nolan, S.P. Straightforward synthesis of [Au(NHC)X] (NHC = N-heterocyclic carbene, X = Cl, Br, I) complexes. Chem. Commun. 2013, 49, 5541–5543. [Google Scholar] [CrossRef] [PubMed]
- Plenio, H.; Heidrich, M.; Bergmann, M.; Müller-Borges, D. Bispentiptycenyl-N-Heterocyclic Carbene (NHC) Gold Complexes: Highly Active Catalysts for the Room Temperature Hydration of Alkynes. Adv. Synth. Catal. 2018, 360, 3572–3578. [Google Scholar] [CrossRef]
- Zhu, H.; Shen, Y.; Deng, Q.; Chen, J.; Tu, T. Acenaphthoimidazolylidene Gold Complex-Catalyzed Alkylsulfonylation of Boronic Acids by Potassium Metabisulfite and Alkyl Halides: A Direct and Robust Protocol to Access Sulfones. ACS Catal. 2017, 7, 4655–4659. [Google Scholar] [CrossRef]
- Gatto, M.; Del Zotto, A.; Segato, J.; Zuccaccia, D. Hydration of Alkynes Catalyzed by L–Au–X under Solvent- and Acid-Free Conditions: New Insights into an Efficient, General, and Green Methodology. Organometallics 2018, 37, 4685–4691. [Google Scholar] [CrossRef]
- Zhu, H.; Shen, Y.; Wen, D.; Le, Z.-G.; Tu, T. Selective Synthesis of ortho-Substituted Diarylsulfones by Using NHC-Au Catalysts under Mild Conditions. Org. Lett. 2019, 21, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, R.; Gong, X.; Li, Z.; Huang, Q.; Wang, H.; Song, G. Synthesis and Herbicidal Activity of N,N-Diethyl-3-(arylselenonyl)-1H-1,2,4-triazole-1-carboxamide. J. Agric. Food Chem. 2006, 54, 7724–7728. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Tobisu, M.; Chatani, N. C–H Borylation by Platinum Catalysis. Bull. Chem. Soc. Jpn. 2017, 90, 332–342. [Google Scholar] [CrossRef]
- Savka, R.; Plenio, H. Facile synthesis of [(NHC)MX(cod)] and [(NHC)MCl(CO)2] (M = Rh, Ir; X = Cl, I) complexes. Dalton Trans. 2015, 44, 891–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dastgir, S.; Coleman, K.S.; Cowley, A.R.; Green, M.L.H. Stable crystalline annulated diaminocarbenes: Coordination with rhodium(i), iridium(i) and catalytic hydroformylation studies. Dalton Trans. 2009, 35, 7203–7214. [Google Scholar] [CrossRef]
- Gómez-Suárez, A.; Nelson, D.J.; Nolan, S.P. Quantifying and understanding the steric properties of N-heterocyclic carbenes. Chem. Commun. 2017, 53, 2650–2660. [Google Scholar] [CrossRef] [Green Version]
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef] [Green Version]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Ar | R | Conditions | 19 (%) | 20 (%) |
|---|---|---|---|---|---|
| 1 | Ph | Me | A | 70 (19a) | 40 (20a) |
| 2 | Ph | tBu | A | 60 (19b) | 17 (20b) |
| 3 | 3,5−(CH3)2C6H3 | Me | A | 54 (19c) | 52 (20c) |
| 4 | 3,5−(tBu)2C6H3 | Me | B | 23 (19d) | 10 (20d) |
| Entry | Solvent | Base | 34 (%) |
|---|---|---|---|
| 1 | DMSO | K3PO4 | 65 |
| 2 | DMSO | CsF | 26 |
| 3 | DMSO | Na2CO3 | 42 |
| 4 | DMSO | K2CO3 | 68 |
| 5 | DMSO | Cs2CO3 | 20 |
| 6 | THF | K2CO3 | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baczewska, P.; Śniady, K.; Kośnik, W.; Michalak, M. Acenaphthene-Based N-Heterocyclic Carbene Metal Complexes: Synthesis and Application in Catalysis. Catalysts 2021, 11, 972. https://doi.org/10.3390/catal11080972
Baczewska P, Śniady K, Kośnik W, Michalak M. Acenaphthene-Based N-Heterocyclic Carbene Metal Complexes: Synthesis and Application in Catalysis. Catalysts. 2021; 11(8):972. https://doi.org/10.3390/catal11080972
Chicago/Turabian StyleBaczewska, Paulina, Katarzyna Śniady, Wioletta Kośnik, and Michał Michalak. 2021. "Acenaphthene-Based N-Heterocyclic Carbene Metal Complexes: Synthesis and Application in Catalysis" Catalysts 11, no. 8: 972. https://doi.org/10.3390/catal11080972
APA StyleBaczewska, P., Śniady, K., Kośnik, W., & Michalak, M. (2021). Acenaphthene-Based N-Heterocyclic Carbene Metal Complexes: Synthesis and Application in Catalysis. Catalysts, 11(8), 972. https://doi.org/10.3390/catal11080972