
The Role of Redox Potential and Molecular Structure of Co(II)-Polypyridine Complexes on the Molecular Catalysis of CO2 Reduction

 , , and
, , and 
Abstract
:
1. Introduction
2. Results
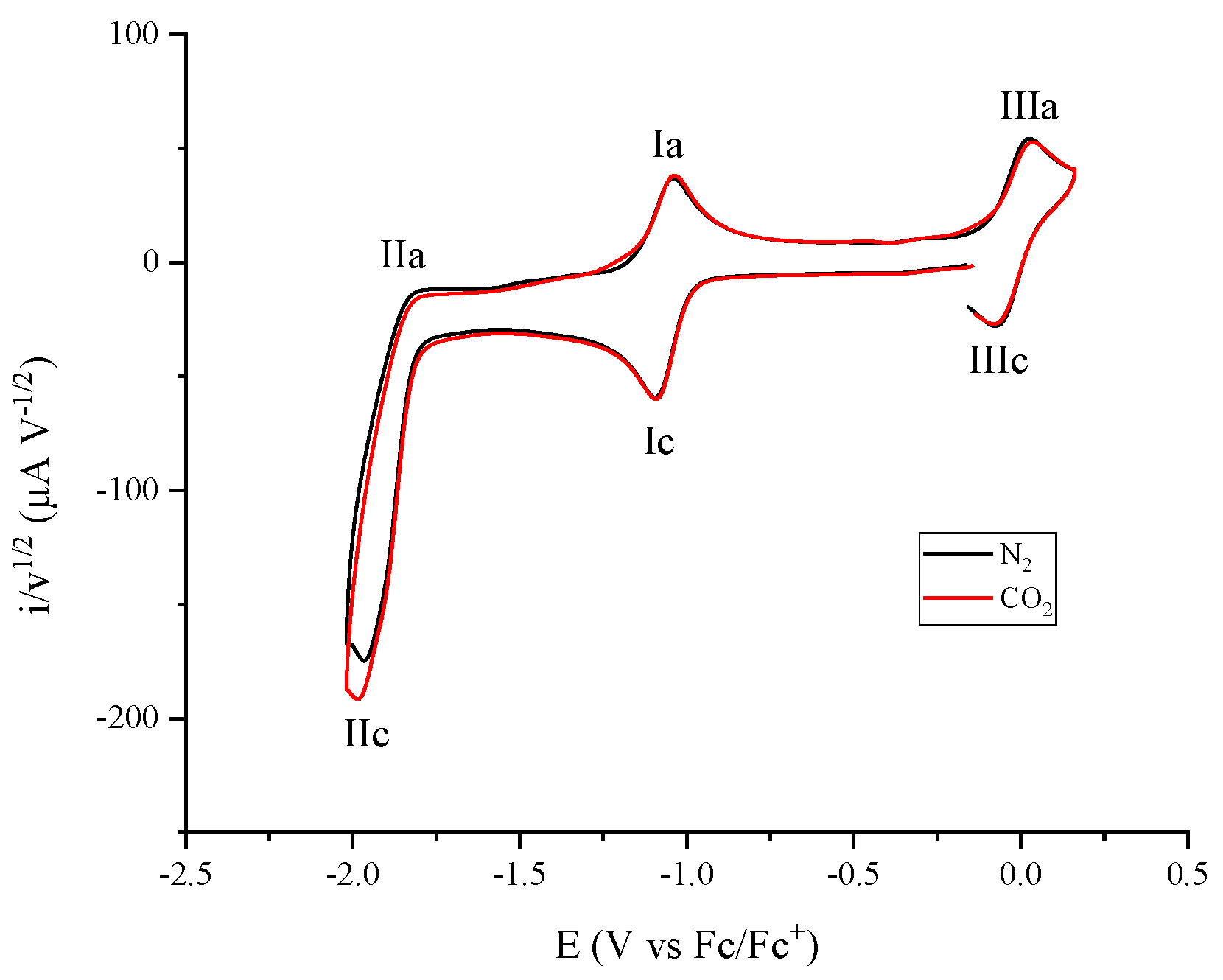
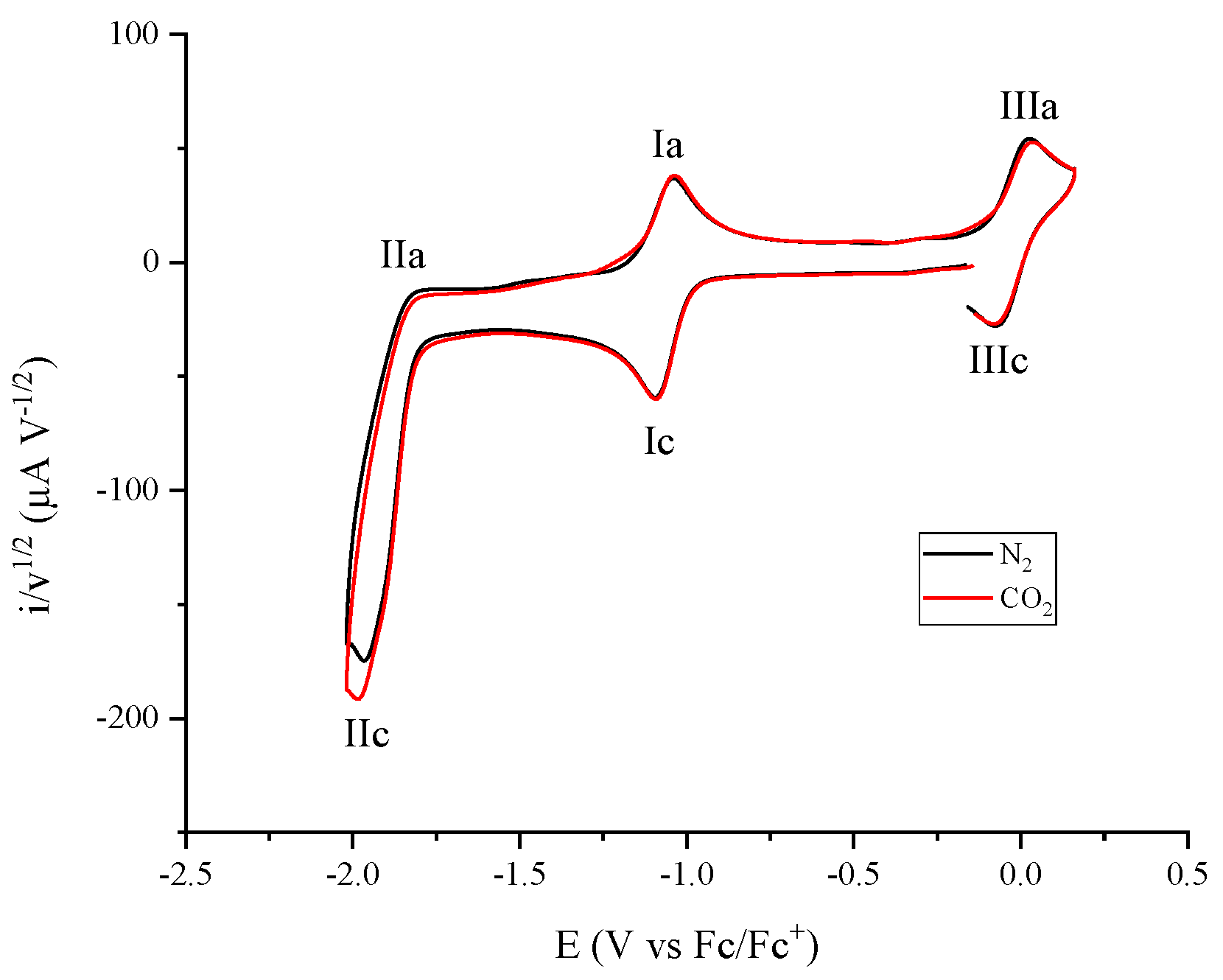
2.1. Electrochemical Response of Cobalt Coordination Compounds
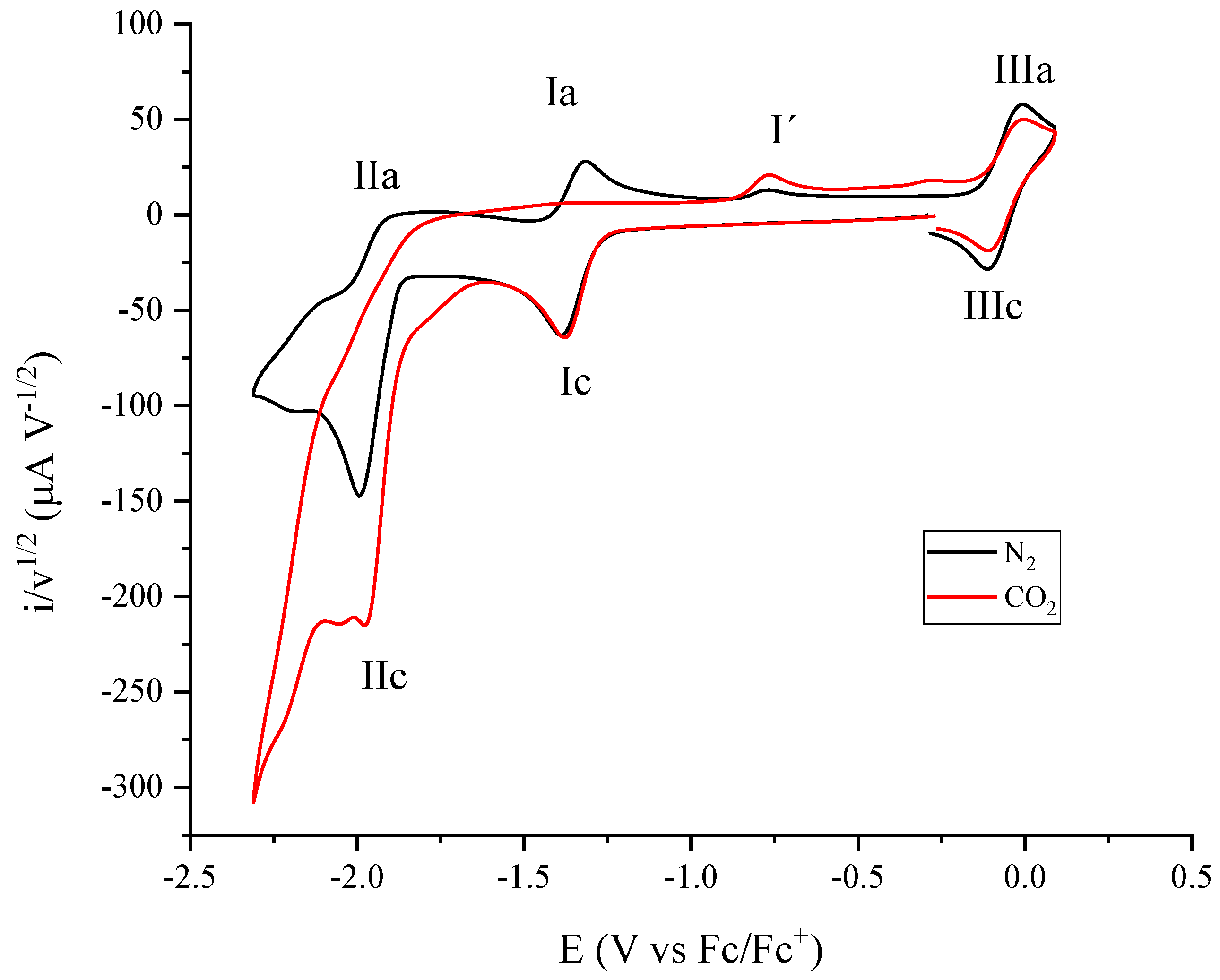
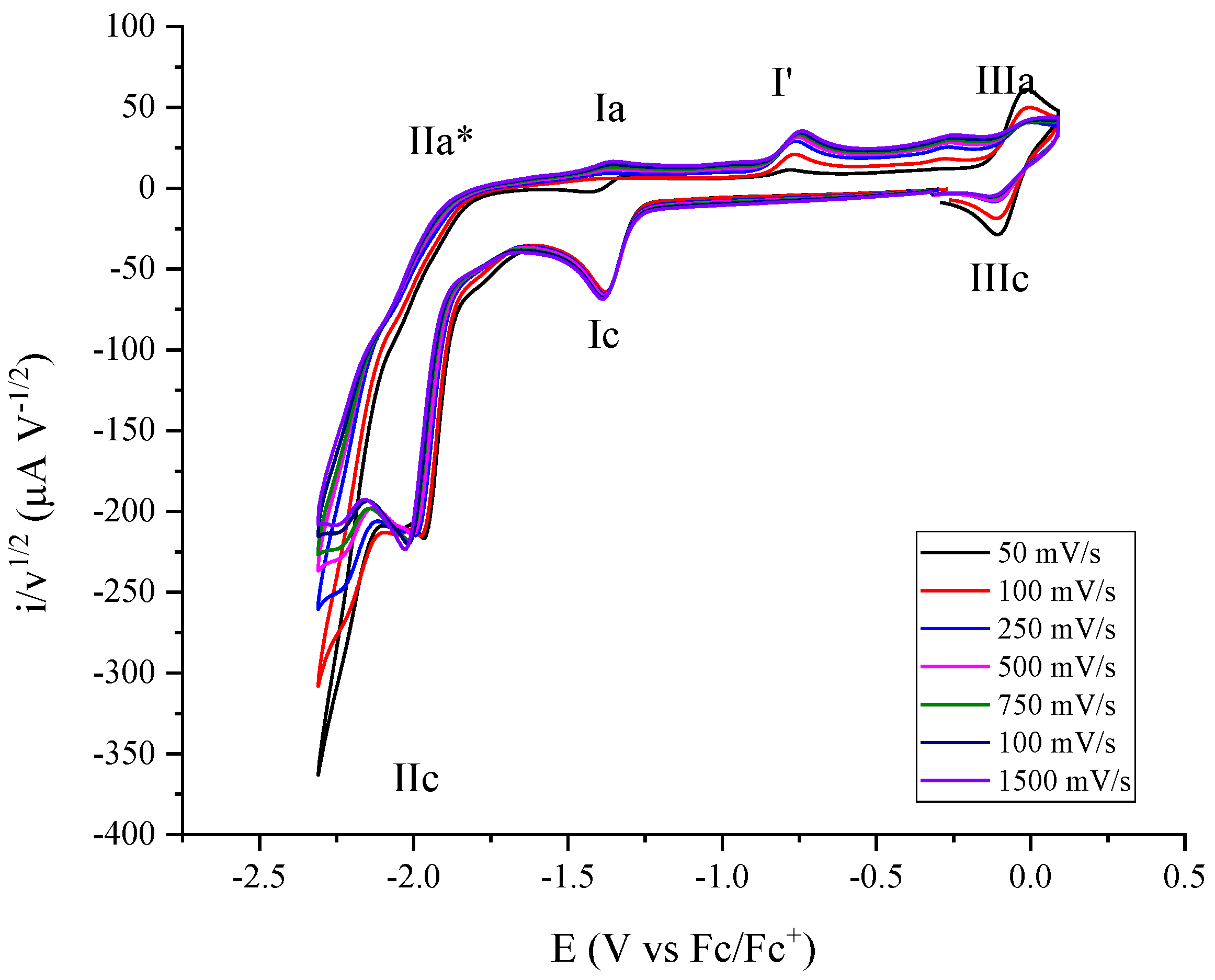
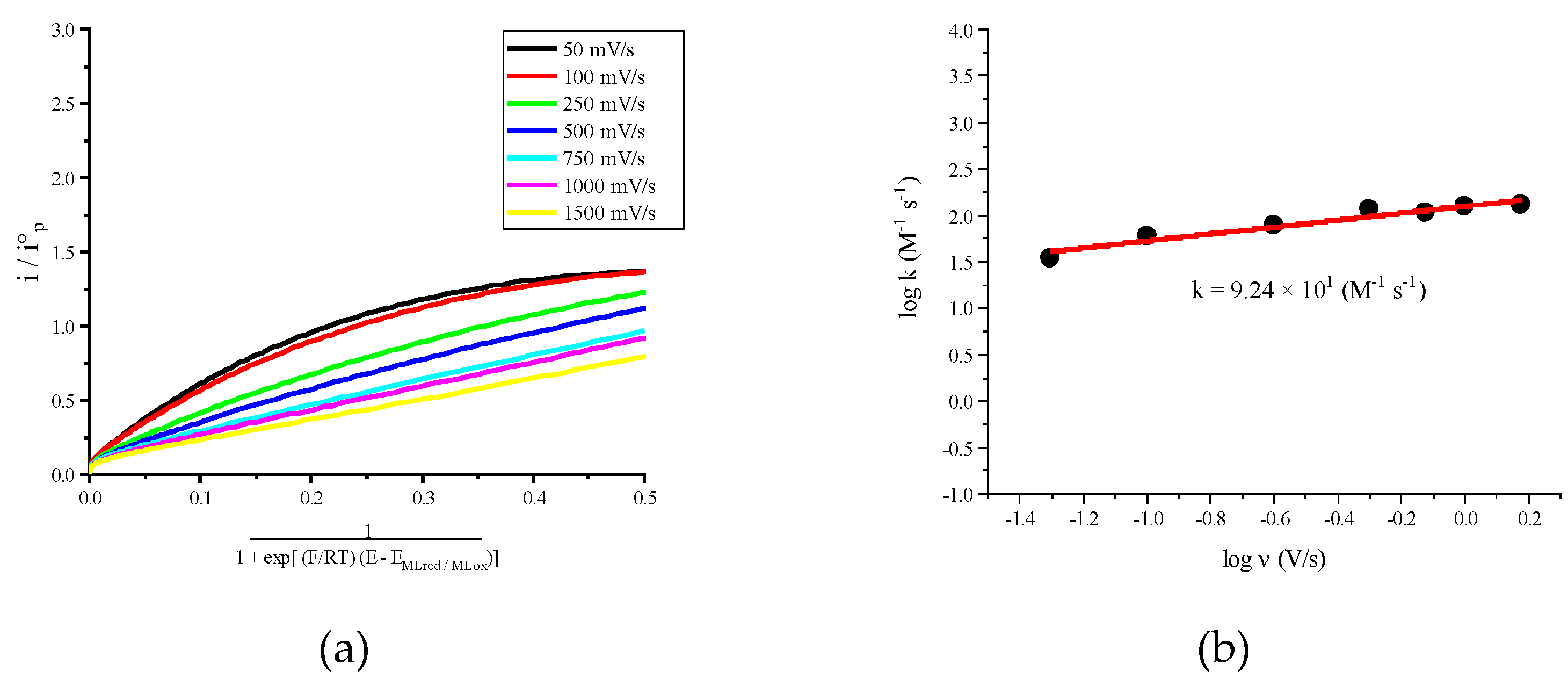
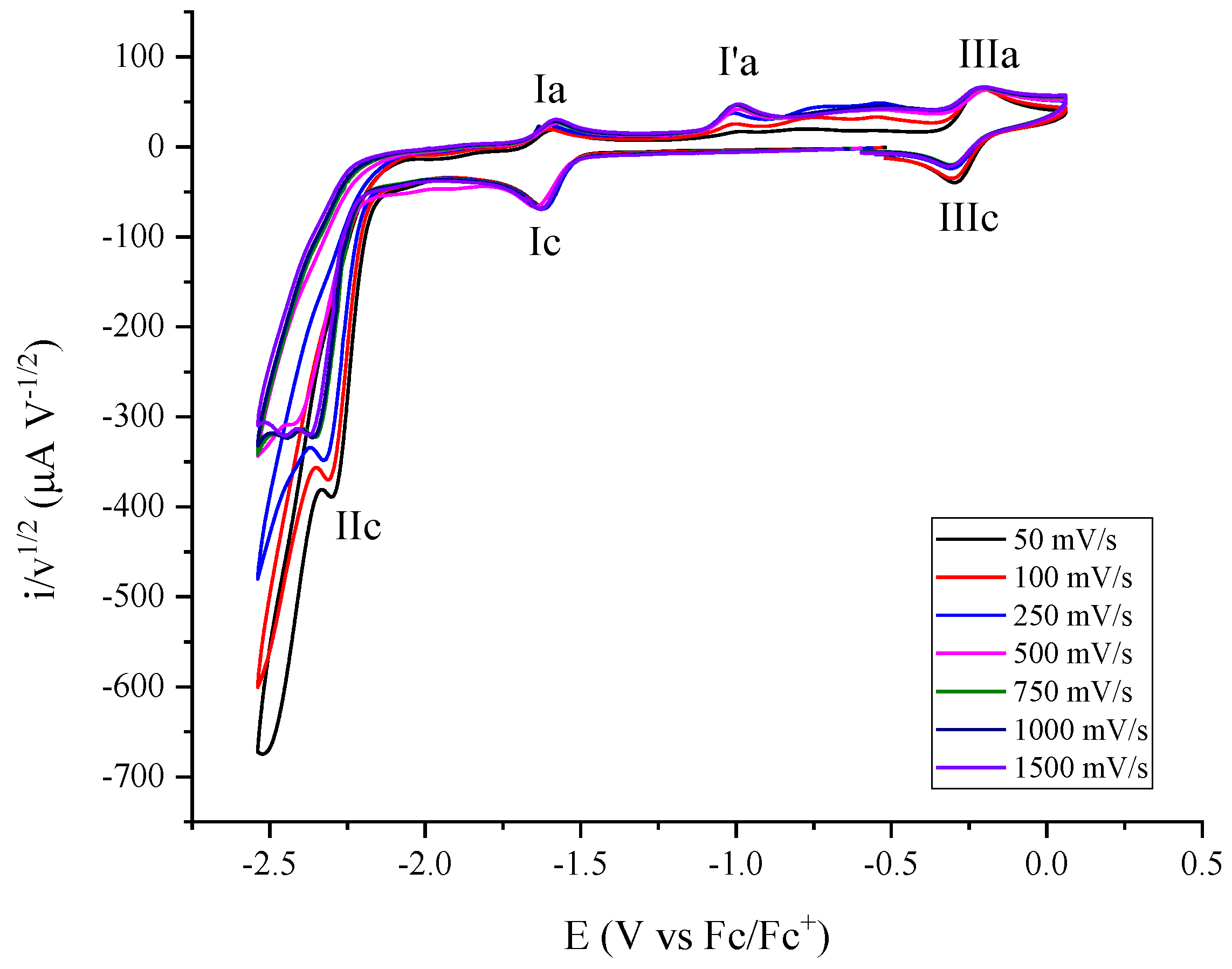
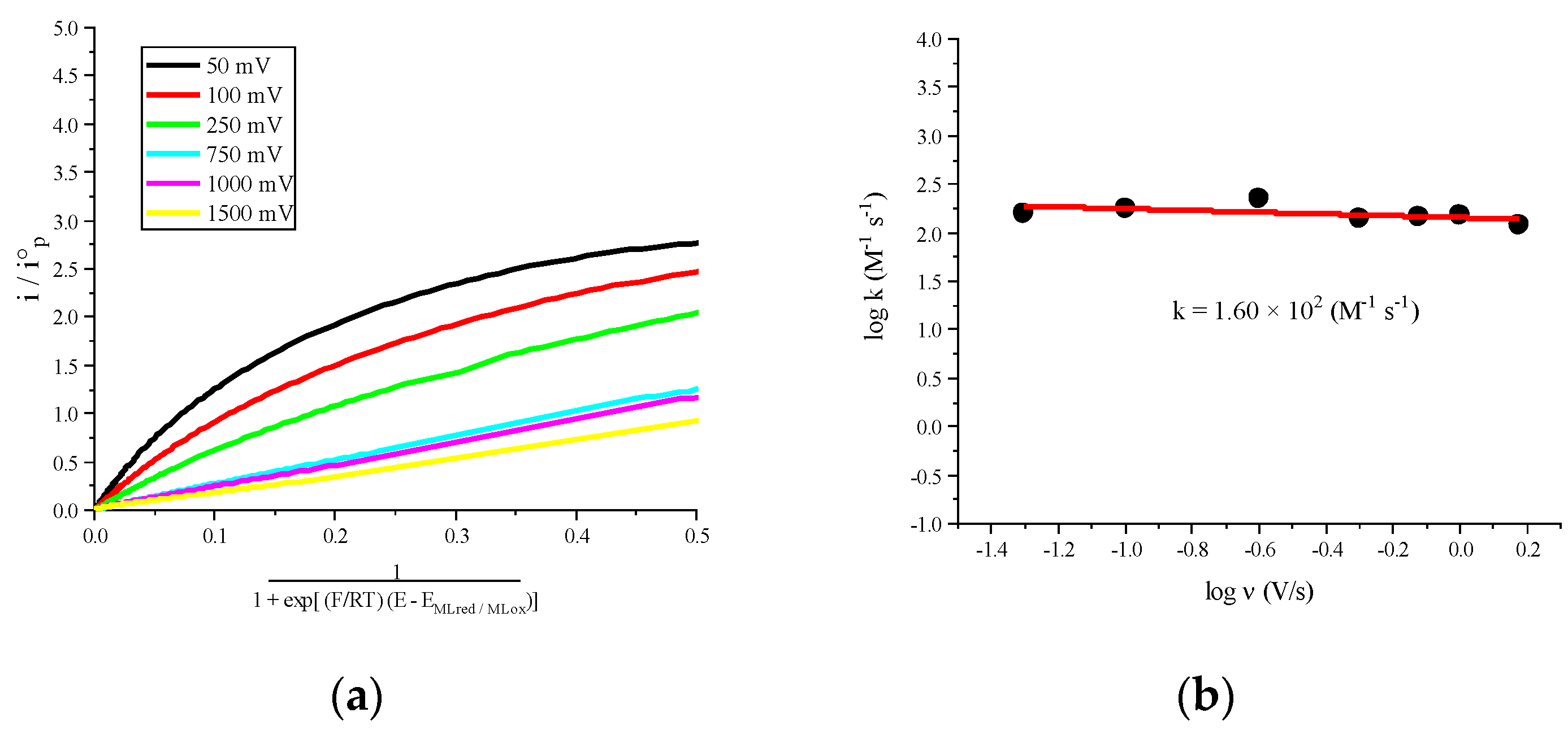
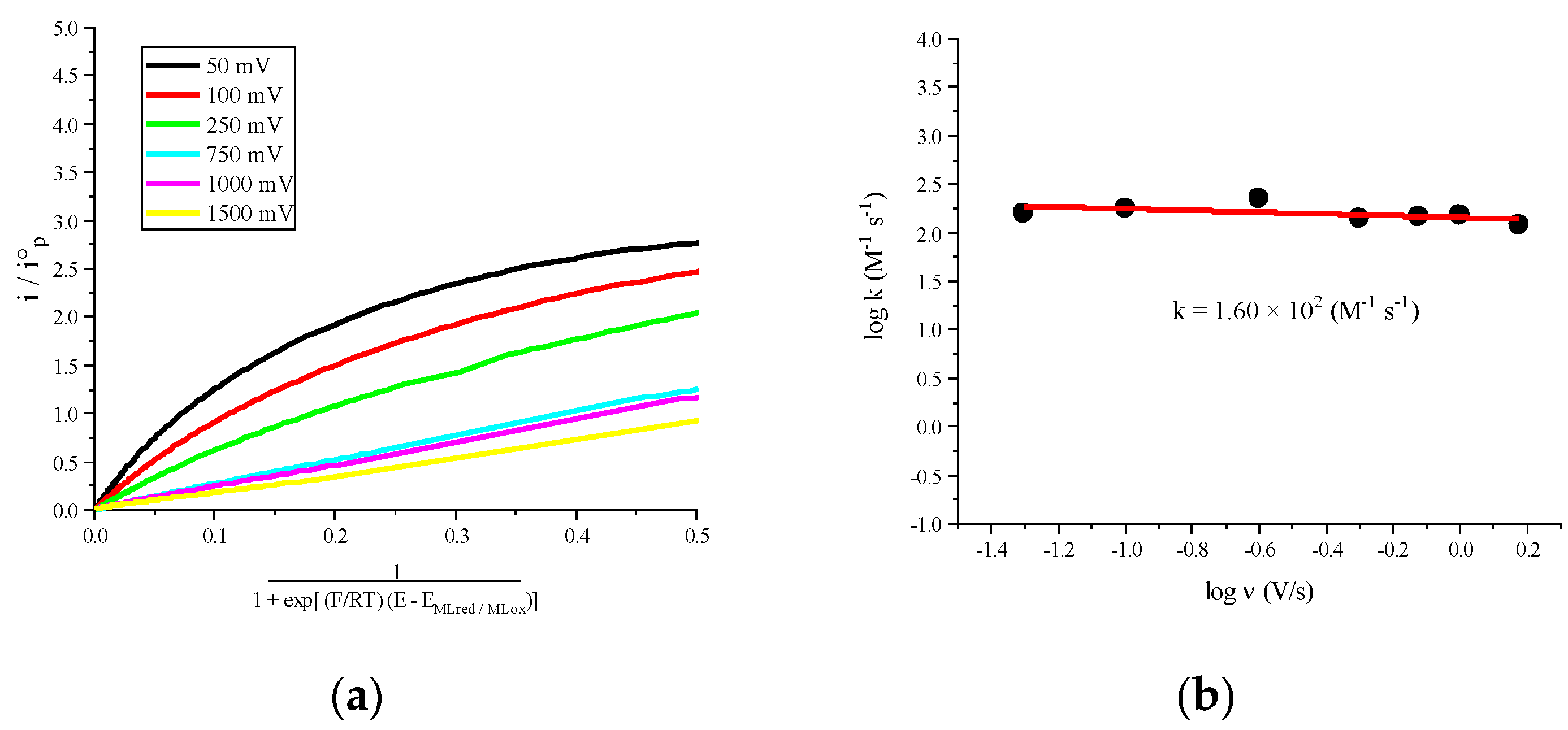
2.2. Molecular Catalysis of CO2
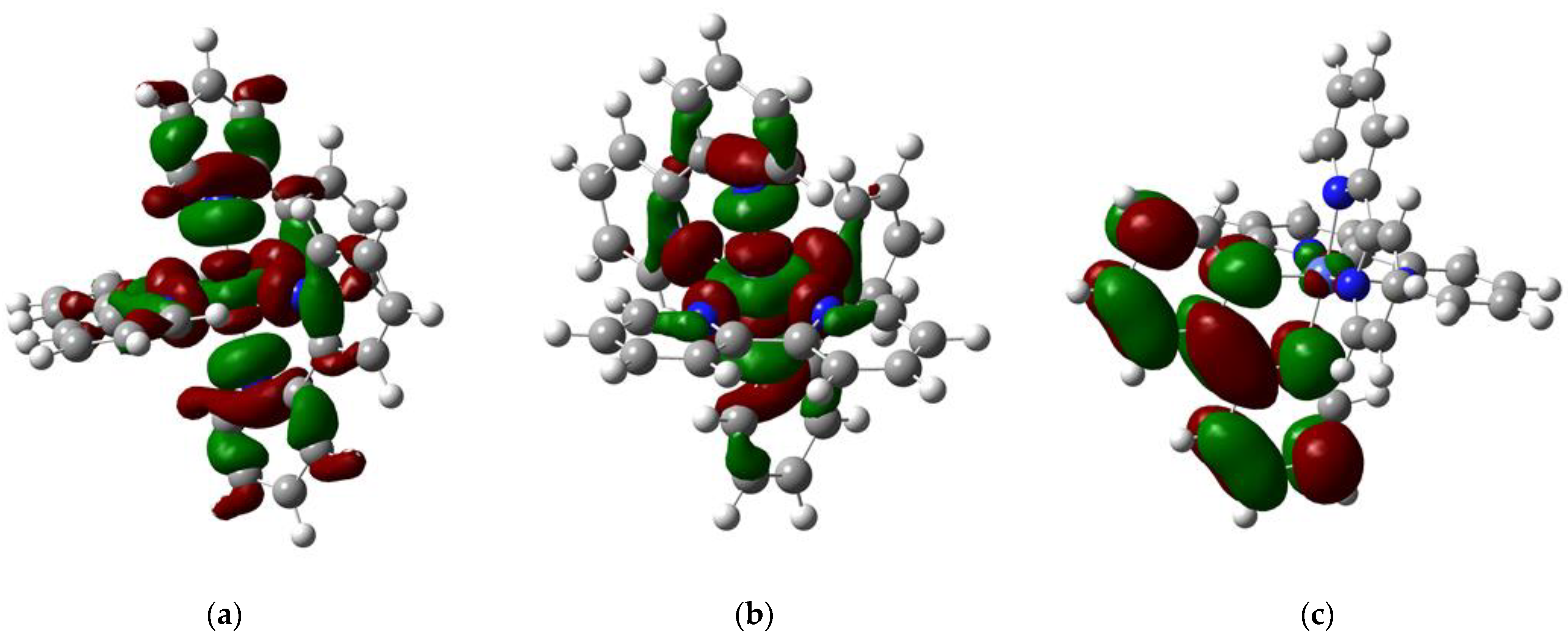
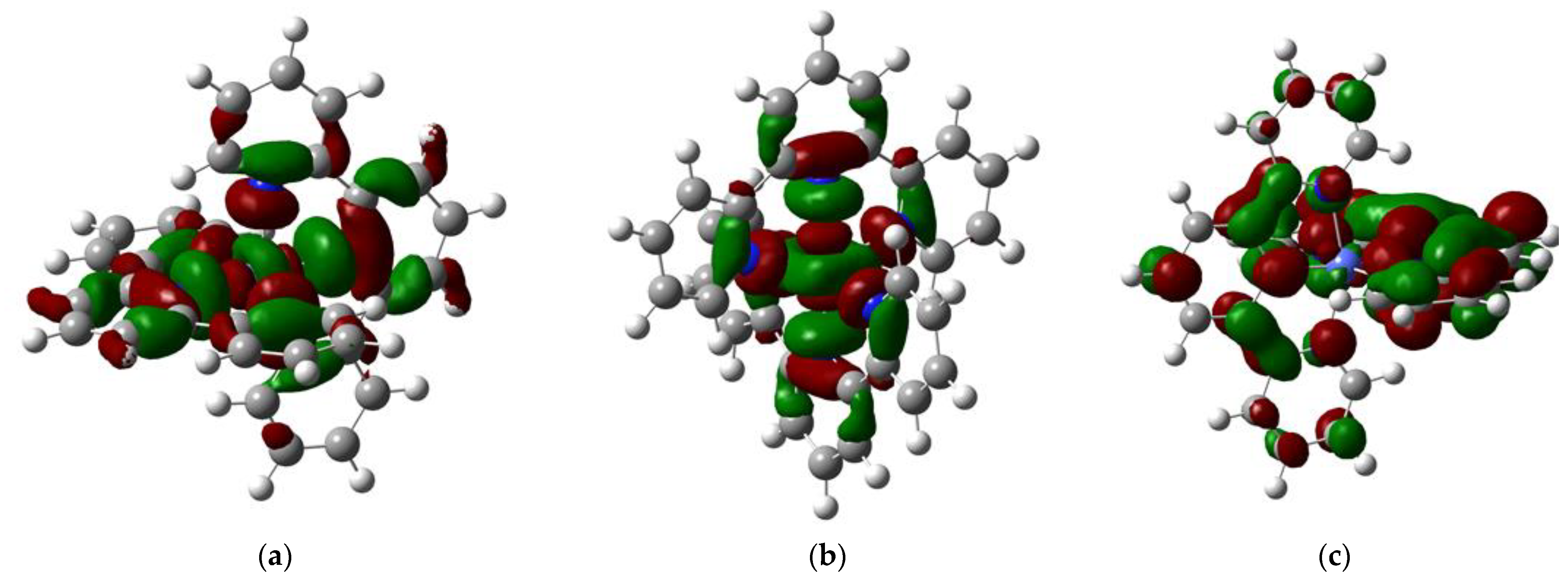
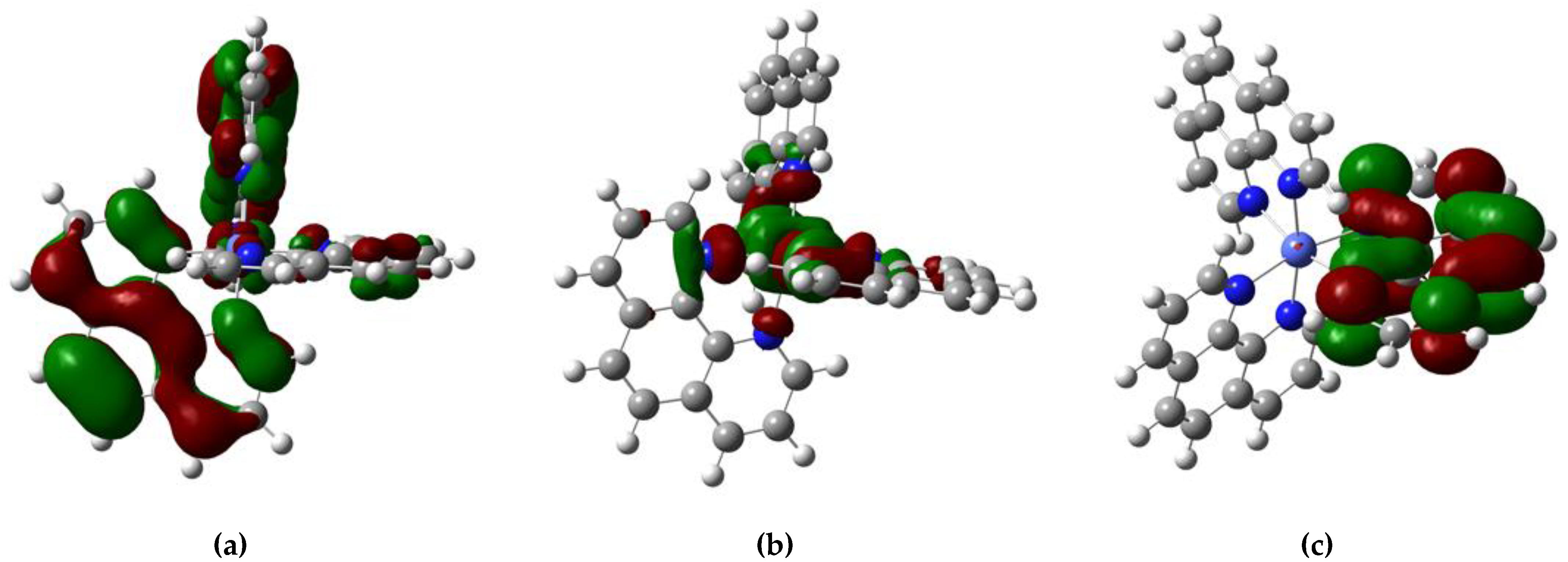
2.3. Molecular Orbitals Description
2.4. Identification of Products
3. Methodology
3.1. Synthesis and Characterization of Coordination Compounds
3.2. Electrochemical Characterization
3.3. DFT Calculations
3.4. Product Identification
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2017, 118, 434–504. [Google Scholar] [CrossRef]
- Sakakura, T.; Choi, J.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef]
- Reddy, M.V.V.S.; Lingam, K.V.; Gundu Rao, T.K. Studies of radicals in oxalate systems. Mol. Phys. 1980, 41, 1493–1500. [Google Scholar] [CrossRef]
- Schwarz, H.A.; Creutz, C.; Sutin, N. Homogeneous catalysis of the photoreduction of water by visible light. Cobalt(I) polypyridine complexes. Redox and substitutional kinetics and thermodynamics in the aqueous 2,2’-bipyridine and 4,4’-dimethyl-2,2’-bipyridine series studied by the pulse-r. Inorg. Chem. 1985, 24, 433–439. [Google Scholar] [CrossRef]
- Amatore, C. Mechanism and Kinetic Characteristics of the Electrochemical Reduction of Carbon Dioxide in Media of Low Proton Availability. J. Am. Chem. Soc. 1981, 103, 5021–5023. [Google Scholar] [CrossRef]
- Chiericato, G.; Arana, C.; Casado, C.; Cuadrado, I.; Abruña, H. Electrocatalytic reduction of carbon dioxide mediated by transition metal complexes with terdentate ligands derived from diacetylpyridine. Inorg. Chim. Acta 2000, 300–302, 32–42. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.-M.; Tatin, A. Efficient and selective molecular catalyst for the CO2-to-CO electrochemical conversion in water. Proc. Natl. Acad. Sci. USA 2015, 112, 6882–6886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savéant, J.; Saveant, J.M. Molecular Catalysis of Electrochemical Reactions. Mechanistic Aspects. Chem. Rev. 2008, 108, 2348–2378. [Google Scholar] [CrossRef] [PubMed]
- Katsaounis, A.; Brosda, S.; Vayenas, C.G. Electrocatalysis. In Encyclopedia of Electrochemistry; Wiley: Hoboken, NJ, USA, 2007; Volume 5, ISBN 9783527303977. [Google Scholar]
- Bagotsky, V.S. Fundamentals of Electrochemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005; ISBN 9780471741992. [Google Scholar]
- Savéant, J.M. Elements of Molecular and Biomolecular Electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; ISBN 0471445738. [Google Scholar]
- Elgrishi, N.; Chambers, M.B.; Wang, X.; Fontecave, M. Molecular polypyridine-based metal complexes as catalysts for the reduction of CO2. Chem. Soc. Rev. 2017, 46, 761–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Electrocatalytic reduction of carbon dioxide mediated by Re(bipy)(CO)3Cl (bipy = 2,2?-bipyridine). J. Chem. Soc. Chem. Commun. 1984, 984, 328. [Google Scholar] [CrossRef]
- Ishida, H.; Tanaka, H.; Tanaka, K. Selective Formation of HCOO- in the electrochemical CO2 reduction catalysed by [Ru(byp)2(CO)2]2+ (byp = 2,2’ bipyridine). J. Chem. Soc. Chem. Commun. 1987, 2, 131–132. [Google Scholar] [CrossRef]
- Bourrez, M.; Molton, F.; Chardon-noblat, S.; Deronzier, A. [Mn (bipyridyl)(CO) 3 Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction **. Angew. Chem. Int. Ed. Engl. 2011, 50, 9903–9906. [Google Scholar] [CrossRef] [PubMed]
- Smieja, J.M.; Sampson, M.D.; Grice, K.A.; Benson, E.E.; Froehlich, J.D.; Kubiak, C.P. Manganese as a substitute for rhenium in CO2 reduction catalysts: The importance of acids. Inorg. Chem. 2003, 52, 2484–2491. [Google Scholar] [CrossRef]
- Agarwal, J.; Shaw, T.W.; Stanton, C.J.; Majetich, G.F.; Bocarsly, A.B.; Schaefer, H.F. NHC-containing manganese(I) electrocatalysts for the two-electron reduction of CO2. Angew. Chemie Int. Ed. 2014, 53, 5152–5155. [Google Scholar] [CrossRef]
- Rosas-Hernández, A.; Steinlechner, C.; Junge, H.; Beller, M. Photo- and Electrochemical Valorization of Carbon Dioxide Using Earth-Abundant Molecular Catalysts. Top. Curr. Chem. 2018, 376, 1. [Google Scholar] [CrossRef]
- Collin, J.P.; Sauvage, J.P. Electrochemical reduction of carbon dioxide mediated by molecular catalysts. Coord. Chem. Rev. 1989, 93, 245–268. [Google Scholar] [CrossRef]
- Thoi, V.S.; Chang, C.J. Nickel N-heterocyclic carbene-pyridine complexes that exhibit selectivity for electrocatalytic reduction of carbon dioxide over water. Chem. Commun. 2011, 47, 6578. [Google Scholar] [CrossRef] [Green Version]
- Daniele, S.; Ugo, P.; Bontempelli, G.; Fiorani, M. An electroanalytical investigation on the nickel-promoted electrochemical conversion of CO2 to CO. J. Electroanal. Chem. 1987, 219, 259–271. [Google Scholar] [CrossRef]
- Rebolledo-Chávez, J.P.F.; Cruz-Ramírez, M.; Patakfalvi, R.; Tenorio Rangel, F.J.; Ortiz-Frade, L. Insight on the mechanism of molecular catalysis of CO2 reduction with Fe(II)-polypyridine complexes. Electrochim. Acta 2017, 247, 241–251. [Google Scholar] [CrossRef]
- Lacy, D.C.; McCrory, C.C.L.; Peters, J.C. Studies of cobalt-mediated electrocatalytic CO2 reduction using a redox-active ligand. Inorg. Chem. 2014, 53, 4980–4988. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2001; ISBN 9780471043720. [Google Scholar]
- Huheey, J.E.; Keiter, E.A.; Keiter, R.L. Inorganic Chemistry: Principles of Structure and Reactivity, 4th ed.; Pearson: New York, NY, USA, 1993; ISBN 006042995X. [Google Scholar]
- Tokel-Takvoryan, N.E.; Hemingway, R.E.; Bard, A.J. Electrogenerated chemiluminescence. XIII. Electrochemical and electrogenerated chemiluminescence studies of ruthenium chelates. J. Am. Chem. Soc. 1973, 95, 6582–6589. [Google Scholar] [CrossRef]
- Tanaka, N.; Sato, Y. Polarographic Behavior of Tris(2,2′-bipyridine)cobalt(II) and Tris(2,2′-bipyridine)cobalt(III) in Acetonitrile Solutions. Bull. Chem. Soc. Jpn. 1968, 41, 2059–2064. [Google Scholar] [CrossRef]
- Sato, Y.; Tanaka, N. Polarographic Behavior of Tris(2,2′-bipyridine)manganese(II) in Acetonitrile Solutions. Bull. Chem. Soc. Jpn. 1968, 41, 2064–2066. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Sato, Y. Univalent tris(2,2′-bipyridine) niccolate anion in acetonitrile. Inorg. Nucl. Chem. Lett. 1968, 4, 487–490. [Google Scholar] [CrossRef]
- Tanaka, N.; Sato, Y. Univalent tris(2,2′-bipyridine)ferrate anion in acetonitrile. Inorg. Nucl. Chem. Lett. 1966, 2, 359–362. [Google Scholar] [CrossRef]
- Tanaka, N.; Sato, Y. Electrode reactions of tris(2,2′-bipyridine)-iron(II) and tris(2,2′-bipyridine)iron(III) complexes in acetonitrile solution. Electrochim. Acta 1968, 13, 335–346. [Google Scholar] [CrossRef]
- Azcarate, I.; Costentin, C.; Robert, M.; Savéant, J.-M. A Study of Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion. J. Am. Chem. Soc. 2016, 138, 16639–16644. [Google Scholar] [CrossRef]
- Costentin, C.; Drouet, S.; Passard, G.; Robert, M.; Savéant, J.-M. Proton-Coupled Electron Transfer Cleavage of Heavy-Atom Bonds in Electrocatalytic Processes. Cleavage of a C O Bond in the Catalyzed Electrochemical Reduction of CO2. J. Am. Chem. Soc. 2003, 135, 9023–9031. [Google Scholar] [CrossRef]
- Ramírez-Delgado, V.; Cruz-Ramirez, M.; Hernández-Ayala, L.F.; Reyes-Vidal, Y.; Patakfalvi, R.; García-Ramos, J.C.; Tenorio, F.J.; Ruiz-Azuara, L.; Ortiz-Frade, L. The Role of the π Acceptor Character of Polypyridine Ligands on the Electrochemical Response of Co (II) Complexes and its Effect on the Homogenous Electron Transfer Rate Constant with the Enzyme Glucose Oxidase. J. Mex. Chem. Soc. 2015, 59, 282–293. [Google Scholar] [CrossRef]
- Trasatti, S. International Union of Pure and Applied Chemistry Commission on Electrochemistry * the Absolute Electrode Potential: An Explanatory Note. Pure Appl. Chem. 1986, 58, 955–966. [Google Scholar] [CrossRef]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.M. Turnover numbers, turnover frequencies, and overpotential in molecular catalysis of electrochemical reactions. Cyclic voltammetry and preparative-scale electrolysis. J. Am. Chem. Soc. 2012, 134, 11235–11242. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, A.K.; Callaway, J. Inhomogeneous electron gas. Phys. Rev. B 1973, 7, 1912–1919. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.B.M.; Petersson, G.A.; Nakatsuji, M.H.; et al. Gaussian 09, Revision D; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Hehre, W.J.; Stewart, R.F.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. I. Use of Gaussian Expansions of Slater-Type Atomic Orbitals. J. Chem. Phys. 1969, 51, 2657–2664. [Google Scholar] [CrossRef]
- Collins, J.B.; von, R.; Schleyer, P.; Binkley, J.S.; Pople, J.A. Self-consistent molecular orbital methods. XVII. Geometries and binding energies of second-row molecules. A comparison of three basis sets. J. Chem. Phys. 1976, 64, 5142–5151. [Google Scholar] [CrossRef]
- Maurya, R.C.; Malik, B.A.; Mir, J.M.; Vishwakarma, P.K.; Rajak, D.K.; Jain, N. Mixed-ligand cobalt(II) complexes of bioinorganic and medicinal relevance, involving dehydroacetic acid and β-diketones: Their synthesis, hyphenated experimental-DFT, thermal and bactericidal facets. J. Mol. Struct. 2015, 1099, 266–285. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | E°(I) | E°(II) | E°(III) | k M−1 s−1 |
|---|---|---|---|---|
| [Co(bipy)3]2+ | −1.351 | −1.962 | −0.059 | 9.24 × 101 |
| [Co(phen)3]2+ | −1.580 | −2.286 | −0.245 | 1.60 × 102 |
| [Co(3,4,7,8-tm-phen)3]2+ | −1.514 | −2.197 | −0.204 | 2.40 × 103 |
| [Co(5,6-dm-phen)3]2+ | −1.364 | −2.074 | −0.115 | 5.19 × 101 |
| [Co(4,7-dph-phen)3]2+ | −1.252 | −1.915 | −0.090 | 1.64 × 102 |
| [Co(terpy)3]2+ | −1.404 | −2.283 | −0.341 | 5.78 × 101 |
| [Co(4-Cl-terpy)3]2+ | −1.067 | −1.964 a | −0.002 | n.o. |
| Chemical Species | SOMO Energy (kcal/mol) | k M−1 s−1 | |
|---|---|---|---|
| [CoII(bipy)3]2+ [CoI(bipy)3]+ [CoI(bipy)(bipy −)2]− | −250.2 −117.6 64.1 | 9.24 × 101 | 2.29 |
| [CoII(phen)3]2+ [CoI(phen)3]+ [CoII(phen)(phen−)2]− | −244.0 −116.1 65.7 | 1.60 × 102 | 3.50 |
| [CoII(3,4,7,8-tm-phen)3]2+ [CoI(3,4,7,8-tm-phen)3]+ [CoI(3,4,7,8-tm-phen)(3,4,7,8-tm-phen−)2]− | −227.1 −103.4 65.5 | 2.40 × 103 | 4.56 |
| [CoII (5,6-dm-phen)3]2+ [CoI(5,6-dm-phen)3]+ [CoI(5,6-dm-phen)(5,6-dm-phen−)2]− | −230.0 −110.7 65.9 | 5.19 × 101 | 3.3 |
| [CoII(4,7-dphen-phen)3]2+ [CoI(4,7-dphen-phen)3]+ [CoI(4,7-dphen-phen)(4,7-dphen-phen−)2]− | −205.1 −106.2 46.4 | 1.64 × 102 | 4.4 |
| [CoII(terpy)3]2+ [CoI(terpy)3]+ [CoI(terpy)(terpy−)] | −247.7 −117.7 15.8 | 5.78 × 101 | 1.1 |
| [CoII(4-Cl-terpy)3]2+ [CoI(4-Cl-terpy)3]+ [CoI(4-Cl-terpy)(4-Cl-terpy−)] | −259.2 −131.7 −19.4 | n.o. | n.o. |
| Chemical Species | HOMO Energy (kcal/mol) | SUMO Energy (kcal/mol) | LUMO Energy (kcal/mol) |
|---|---|---|---|
| CO2 | −145.22 | − | 66.52 |
| CO2•− | − | 197.64 | 320.88 |
| CO22− | 459.64 | − | 571.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rebolledo-Chávez, J.P.F.; Toral, G.T.; Ramírez-Delgado, V.; Reyes-Vidal, Y.; Jiménez-González, M.L.; Cruz-Ramírez, M.; Mendoza, A.; Ortiz-Frade, L. The Role of Redox Potential and Molecular Structure of Co(II)-Polypyridine Complexes on the Molecular Catalysis of CO2 Reduction. Catalysts 2021, 11, 948. https://doi.org/10.3390/catal11080948
Rebolledo-Chávez JPF, Toral GT, Ramírez-Delgado V, Reyes-Vidal Y, Jiménez-González ML, Cruz-Ramírez M, Mendoza A, Ortiz-Frade L. The Role of Redox Potential and Molecular Structure of Co(II)-Polypyridine Complexes on the Molecular Catalysis of CO2 Reduction. Catalysts. 2021; 11(8):948. https://doi.org/10.3390/catal11080948
Chicago/Turabian StyleRebolledo-Chávez, Juan Pablo F., Gionnany Teodoro Toral, Vanesa Ramírez-Delgado, Yolanda Reyes-Vidal, Martha L. Jiménez-González, Marisela Cruz-Ramírez, Angel Mendoza, and Luis Ortiz-Frade. 2021. "The Role of Redox Potential and Molecular Structure of Co(II)-Polypyridine Complexes on the Molecular Catalysis of CO2 Reduction" Catalysts 11, no. 8: 948. https://doi.org/10.3390/catal11080948
APA StyleRebolledo-Chávez, J. P. F., Toral, G. T., Ramírez-Delgado, V., Reyes-Vidal, Y., Jiménez-González, M. L., Cruz-Ramírez, M., Mendoza, A., & Ortiz-Frade, L. (2021). The Role of Redox Potential and Molecular Structure of Co(II)-Polypyridine Complexes on the Molecular Catalysis of CO2 Reduction. Catalysts, 11(8), 948. https://doi.org/10.3390/catal11080948