
Catalytic Performance of Cycloalkyl-Fused Aryliminopyridyl Nickel Complexes toward Ethylene Polymerization by QSPR Modeling




Abstract
:
1. Introduction
2. Results and Discussions
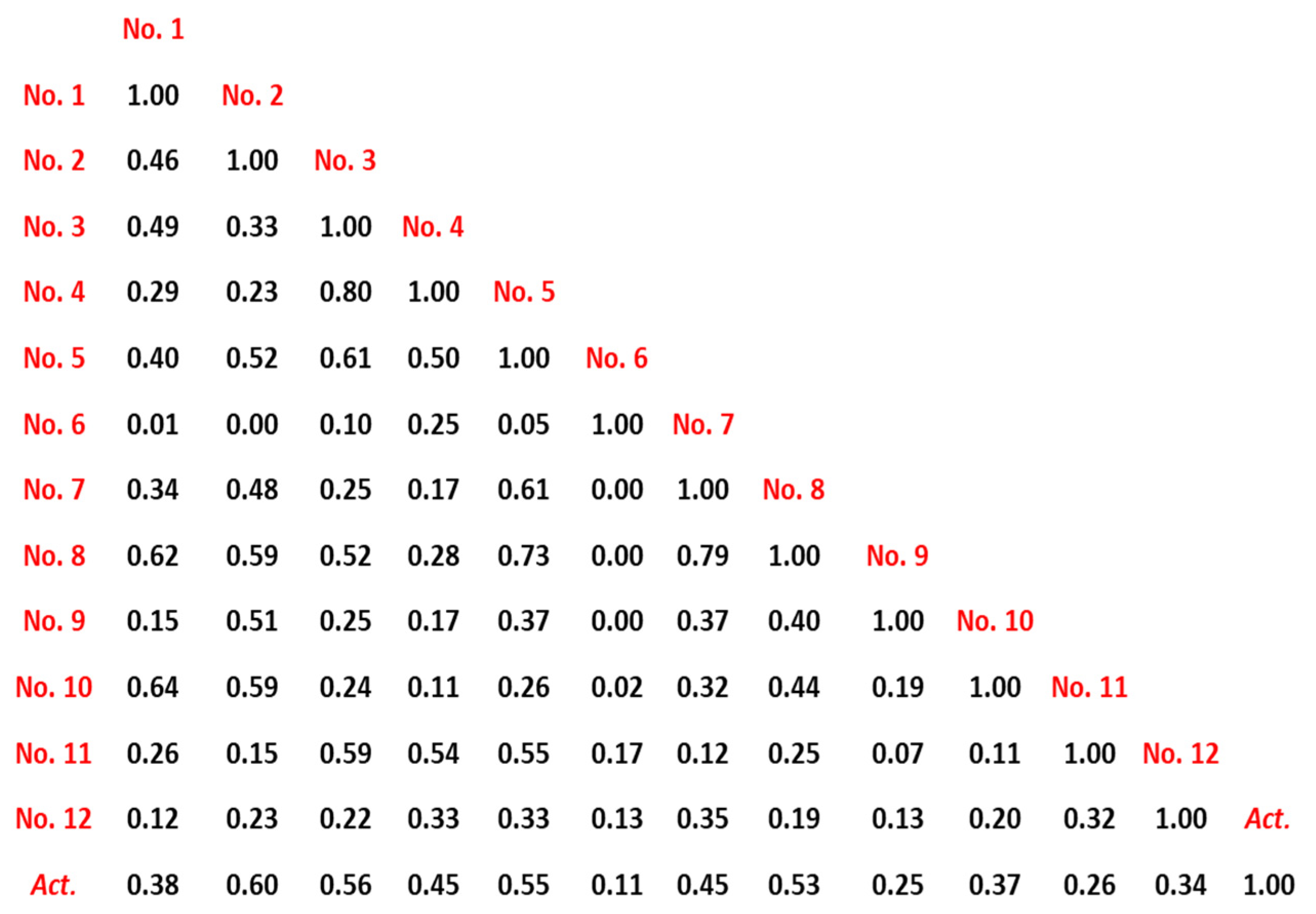
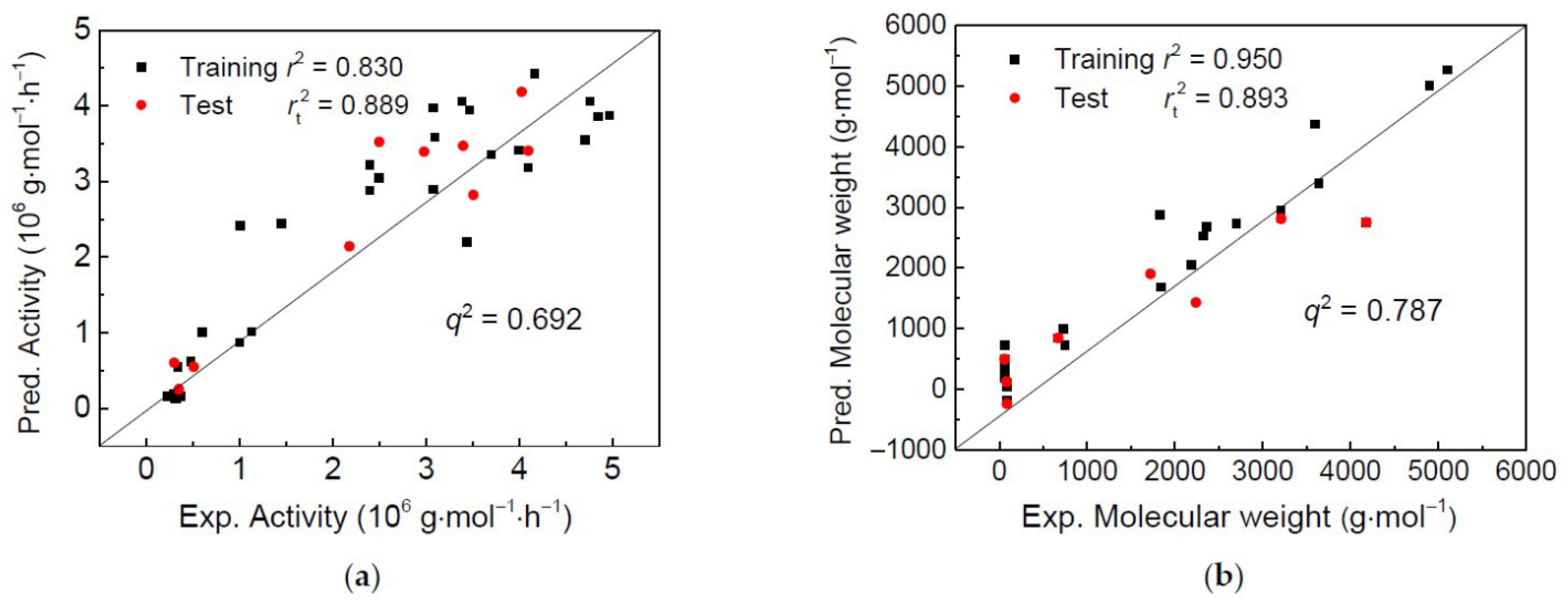
2.1. 2D-QSPR Modeling
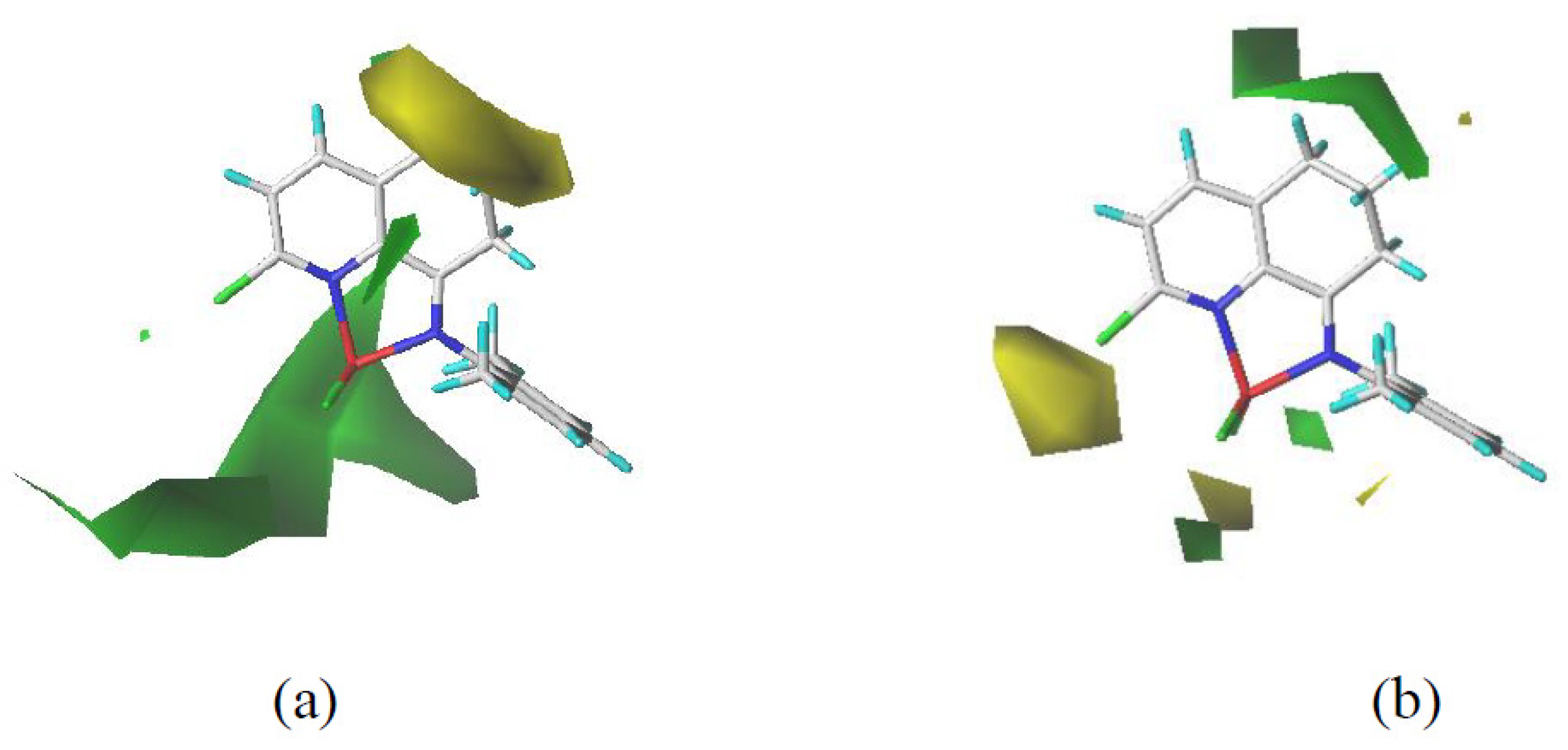
2.2. 3D-QSPR Modeling
3. Computational Methods
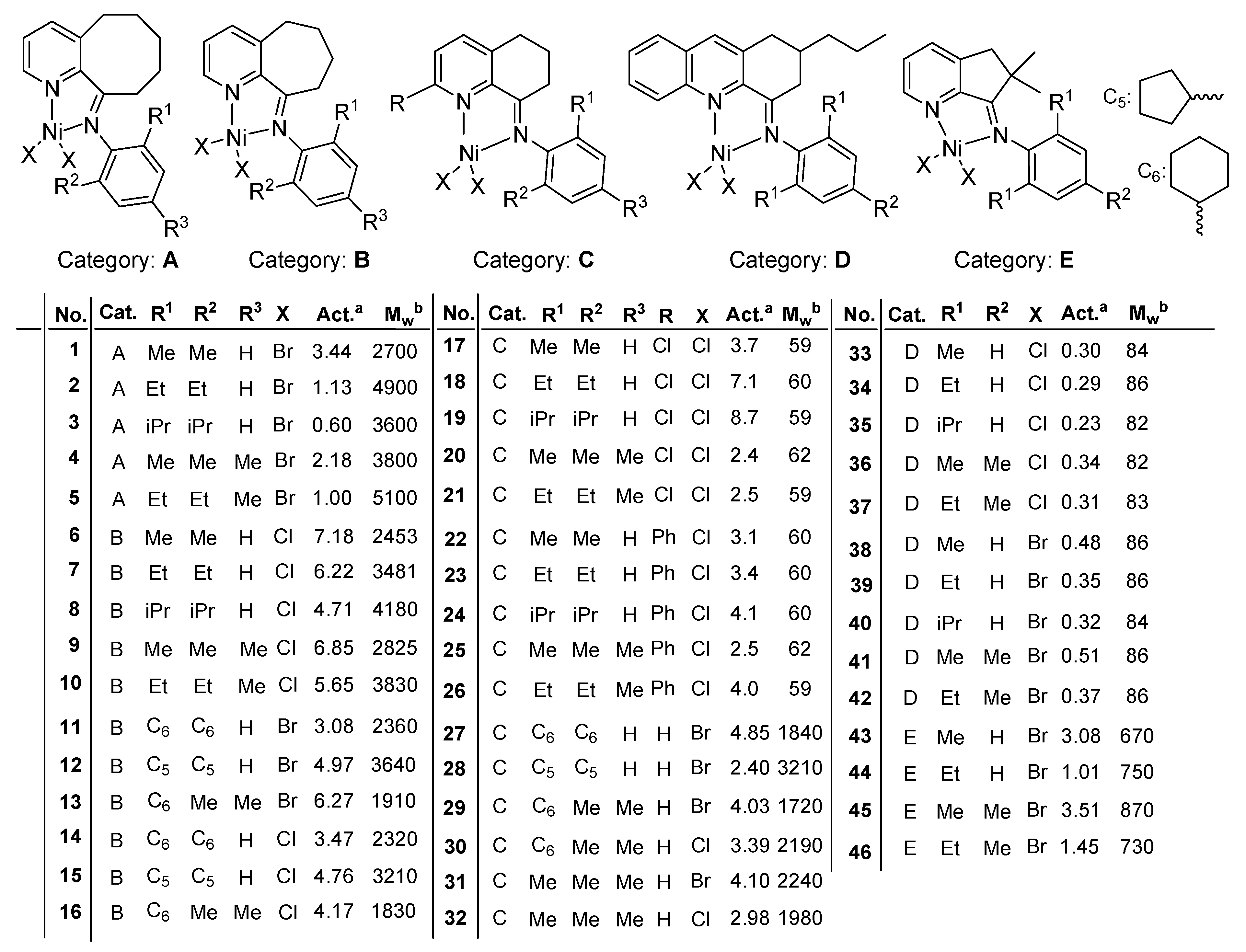
3.1. Data Set
3.2. QSPR Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Johnson, L.K.; Killian, C.M.; Brookhart, M. Pd(II)- and Ni(II)-Based Catalysts for Polymerization of Ethylene and alpha.-Olefins. J. Am. Chem. Soc. 1995, 23, 6414–6415. [Google Scholar] [CrossRef]
- Mecking, S. Cationic nickel and palladium complexes with bidentate ligands for the C–C linkage of olefins. Coord. Chem. Rev. 2000, 203, 325–351. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.H.; Ziller, J.W.; Guan, Z. Axial donating ligands: A new strategy for late transition metal olefin polymerization catalysis. J. Am. Chem. Soc. 2008, 24, 7538–7539. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, C.; Giambastiani, G.; Luconi, L.; Meli, A. Olefin oligomerization, homopolymerization and copolymerization by late transition metals supported by (imino) pyridine ligands. Coord. Chem. Rev. 2010, 254, 431–455. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Q.; Solan, G.A.; Sun, W.-H. Recent advances in Ni-mediated ethylene chain growth: Nimine-donor ligand effects on catalytic activity, thermal stability and oligo-/polymer structure. Coord. Chem. Rev. 2017, 350, 68–83. [Google Scholar] [CrossRef]
- Cruz, V.; Ramos, J.; Munoz-Escalona, A.; Lafuente, P.; Pena, B.; Martinez-Salazar, J. 3D-QSAR analysis of metallocene-based catalysts used in ethylene polymerisation. Polymer 2004, 45, 2061–2072. [Google Scholar] [CrossRef]
- Martínez, S.; Cruz, V.L.; Ramos, J.; Martínez-Salazar, J. Polymerization activity prediction of zirconocene single-site catalysts using 3D quantitative structure–activity relationship modeling. Organometallics 2012, 31, 1673–1679. [Google Scholar] [CrossRef]
- Cruz, V.L.; Martinez, J.; Martinez-Salazar, J.; Ramos, J.; Reyes, M.L.; Toro-Labbe, A.; Gutierrez-Oliva, S. QSAR model for ethylene polymerisation catalysed by supported bis (imino) pyridine iron complexes. Polymer 2007, 48, 7672–7678. [Google Scholar] [CrossRef]
- Yang, W.; Yi, J.; Ma, Z.; Sun, W.-H. 2D-QSAR modeling on the catalytic activities of 2-azacyclyl-6-aryliminopyridylmetal precatalysts in ethylene oligomerization. Catal. Commun. 2017, 101, 40–43. [Google Scholar] [CrossRef]
- Yang, W.; Ma, Z.; Yi, J.; Ahmed, S.; Sun, W.-H. Catalytic performance of bis (imino) pyridine Fe/Co complexes toward ethylene polymerization by 2D-/3D-QSPR modeling. J. Comput. Chem. 2019, 40, 1374–1386. [Google Scholar] [CrossRef] [PubMed]
- Manz, T.A. Deactivation of Ti and Zr half-metallocene complexes activated with B(C6F5)3: A case study in constructing DFT-based QSARs to predict unimolecular rate constants. RSC Adv. 2015, 5, 48246–48254. [Google Scholar] [CrossRef]
- Falivene, L.; Cavallo, L.; Talarico, G. Buried volume analysis for propene polymerization catalysis promoted by group 4 metals: A tool for molecular mass prediction. ACS Catal. 2015, 5, 6815–6822. [Google Scholar] [CrossRef] [Green Version]
- Ehm, C.; Vittoria, A.; Goryunov, G.P.; Izmer, V.V.; Kononovich, D.S.; Kulyabin, P.S.; Girolamo, R.D.; Budzelaar, P.H.M.; Voskoboynikov, A.Z.; Busico, V.; et al. A Systematic Study of the Temperature-Induced Performance Decline of ansa-Metallocenes for iPP. Macromolecules 2020, 53, 9325–9336. [Google Scholar] [CrossRef]
- Ehm, C.; Vittoria, A.; Goryunov, G.P.; Izmer, V.V.; Kononovich, D.S.; Samsonov, O.V.; Budzelaar, P.H.M.; Voskoboynikov, A.Z.; Busico, V.; Uborsky, D.V.; et al. On the limits of tuning comonomer affinity of ‘Spaleck-type’ansa-zirconocenes in ethene/1-hexene copolymerization: A high-throughput experimentation/QSAR approach. Dalton Trans. 2020, 49, 10162–10172. [Google Scholar] [CrossRef]
- Yang, W.; Ahmed, S.; Fidelis, T.T.; Sun, W.-H. Effect of cycloalkyl-fused ring on the catalytic performance of bis (imino) pyridine metal complexes by QSPR modeling. Catal. Commun. 2019, 132, 105820–105824. [Google Scholar] [CrossRef]
- Kennard, R.W.; Stone, L.A. Computer aided design of experiments. Technometrics 1969, 11, 137–148. [Google Scholar] [CrossRef]
- Stanton, D.T.; Jurs, P.C. Development and use of charged partial surface area structural descriptors in computer-assisted quantitative structure-property relationship studies. Anal. Chem. 1990, 62, 2323–2329. [Google Scholar] [CrossRef]
- Stanton, D.T.; Egolf, L.M.; Jurs, P.C.; Hicks, M.G. Computer-assisted prediction of normal boiling points of pyrans and pyrroles. J. Chem. Inf. Comput. Sci. 1992, 32, 306–316. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Z.; Flisak, Z.; Hao, X.; Liu, Q.; Sun, W.-H. Achieving branched polyethylene waxes by aryliminocycloocta [b] pyridylnickel precatalysts: Synthesis, characterization, and ethylene polymerization. J. Poly. Sci. Part. A Poly. Chem. 2017, 55, 2601–2610. [Google Scholar] [CrossRef]
- Sun, Z.; Huang, F.; Qu, M.; Yue, E.; Oleynik, I.V.; Oleynik, I.I.; Zeng, Y.; Liang, T.; Li, K.; Zhang, W.; et al. Targeting polyethylene waxes: 9-(2-cycloalkylphenylimino)-5, 6, 7, 8-tetrahydrocycloheptapyridylnickel halides and their use as catalysts for ethylene polymerization. RSC Adv. 2015, 5, 77913–77921. [Google Scholar] [CrossRef]
- Yu, J.; Hu, X.; Zeng, Y.; Zhang, L.; Ni, C.; Hao, X.; Sun, W.-H. Synthesis, characterisation and ethylene oligomerization behaviour of N-(2-substituted-5, 6, 7-trihydroquinolin-8-ylidene) arylaminonickel dichlorides. New J. Chem. 2011, 35, 178–183. [Google Scholar] [CrossRef]
- Sun, Z.; Yue, E.; Qu, M.; Oleynik, I.V.; Oleynik, I.I.; Li, K.; Liang, T.; Zhang, W.; Sun, W.-H. 8-(2-Cycloalkylphenylimino)-5, 6, 7-trihydro-quinolylnickel halides: Polymerizing ethylene to highly branched and lower molecular weight polyethylenes. Inorg. Chem. Front. 2015, 2, 223–227. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, W.; Du, S.; Asuha, S.; Flisak, Z.; Sun, W.-H. Propyl substituted 4-arylimino-1, 2, 3-trihydroacridylnickel complexes: Their synthesis, characterization and catalytic behavior toward ethylene. J. Organomet. Chem. 2015, 789, 408–413. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Y.; Ma, Y.; Hu, X.; Solan, G.A.; Sun, Y.; Sun, W.-H. Molecular weight control of polyethylene waxes using a constrained imino-cyclopenta [b] pyridyl-nickel catalyst. J. Poly. Sci. Part. A Poly. Chem. 2017, 55, 3494–3505. [Google Scholar] [CrossRef]
- Huang, F.; Sun, Z.; Du, S.; Yue, E.; Ba, J.; Hu, X.; Liang, T.; Galland, G.B.; Sun, W.-H. Ring-tension adjusted ethylene polymerization by aryliminocycloheptapyridyl nickel complexes. Dalton Trans. 2015, 44, 14281–14292. [Google Scholar] [CrossRef]
- Winer, B.J.; Brown, D.R.; Michels, K.M. Single-Factor Experiments Having Repeated Measures on the Same Elements [Ch. 4]. In Statistical Principles in Experimental Design, 3rd ed.; McGraw-Hill: New York, NY, USA, 1991; pp. 220–281. [Google Scholar]
- Jolliffe, I.T. Principal Component Analysis Series. In Springer Series in Statistics, 2nd ed.; Springer: New York, NY, USA, 2002; p. 487. [Google Scholar]
- Delley, B. Ground-state enthalpies: Evaluation of electronic structure approaches with emphasis on the density functional method. J. Phys. Chem. A 2006, 110, 13632–13639. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B.J. From molecules to solids with the DMol3 approach. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B. 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A multicenter numerical integration scheme for polyatomic molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Bergner, A.; Dolg, M.; Küchle, W.; Stoll, H.; Preuß, H. Ab initio energy-adjusted pseudopotentials for elements of groups 13–17. Mol. Phys. 1993, 80, 1431–1441. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Lobanov, V.S.; Karelson, M. Comprehensive Descriptors for Structural and Statistical Analysis (Codessa), Referance Manual; Semichem, Inc.: Shawnee Mission, KS, USA; Florida University: Gainesville, FL, USA, 2004. [Google Scholar]
- Frisch, M.J.E.A.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Gill, P.M.; Johnson, B.G.; Pople, J.A.; Frisch, M.J. An investigation of the performance of a hybrid of Hartree-Fock and density functional theory. Int. J. Quantum Chem. 1992, 26, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.; Yang, W.; Sun, W.-H. Quantitative Investigation of the Electronic and Steric Influences on Ethylene Oligo/Polymerization by 2-Azacyclyl-6-aryliminopyridylmetal (Fe, Co, and Cr) Complexes. Macromol. Chem. Phys. 2016, 217, 757–764. [Google Scholar] [CrossRef]
- Yang, W.; Ma, Z.; Sun, W.-H. Modeling study on the catalytic activities of 2-imino-1, 10-phenanthrolinylmetal (Fe, Co, and Ni) precatalysts in ethylene oligomerization. RSC Adv. 2016, 6, 79335–79342. [Google Scholar] [CrossRef]
- Ahmed, S.; Yang, W.; Ma, Z.; Sun, W.-H. Catalytic activities of bis (pentamethylene) pyridyl (Fe/Co) complex analogues in ethylene polymerization by modeling method. J. Phys. Chem. A. 2018, 122, 9637–9644. [Google Scholar] [CrossRef]
- Malik, A.A.; Yang, W.; Ma, Z.; Sun, W.-H. The Catalytic Activities of Carbocyclic Fused Pyridineimine Nickel Complexes Analogues in Ethylene Polymerization by Modeling Study. Catalysts 2019, 9, 520. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.N. On a heuristic method of test construction and its use in multivariate analysis. Ann. Math. Stat. 1953, 24, 220–239. [Google Scholar] [CrossRef]
- Shao, J.J. Linear model selection by cross-validation. J. Am. Stat. Assoc. 1993, 88, 486–494. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Chong, I.G.; Jun, C.H. Performance of some variable selection methods when multicollinearity is present. Chemom. Intell. Lab. Syst. 2005, 78, 103–112. [Google Scholar] [CrossRef]
- Gosselin, R.; Rodrigue, D.; Duchesne, C. A Bootstrap-VIP approach for selecting wavelength intervals in spectral imaging applications. Chemom. Intell. Lab. Syst. 2010, 100, 12–21. [Google Scholar] [CrossRef]
- Li, H.-D.; Xu, Q.-S.; Liang, Y.-Z. libPLS: An Integrated Library for Partial Least Squares Regression and Discriminant Analysis. Chemom. Intell. Lab. Systems 2018, 176, 34–43. Available online: http://www.libpls.net (accessed on 15 March 2020). [CrossRef]
- Martin, Y.C.; Kim, K.H.; Liu, C.T. Comparative Molecular Field Analysis: CoMFA. In Advances in Quant. Str.-Property Relationships; JAI Press: Greenwich, CT, USA, 1996; Volume 1, pp. 1–52. [Google Scholar]
- Cramer, R.D.; Bunce, J.D.; Patterson, D.E.; Frank, I.E. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | A | R2 | Q2 (k = 5) | RMSEF | RMSEV |
|---|---|---|---|---|---|
| 63 | 6 | 0.9211 | 0.5810 | 0.6390 | 0.2772 |
| 22 | 5 | 0.8770 | 0.6892 | 0.5503 | 0.3461 |
| 12 | 5 | 0.8755 | 0.7328 | 0.5103 | 0.3483 |
| 5 | 2 | 0.7363 | 0.6731 | 0.5644 | 0.5069 |
| No. | Molecular Descriptor | Coeff. | Contr.(%) |
|---|---|---|---|
| 2 | Min partial charge for a C atom [Zefirov’s PC] | 0.7854 | 29.17 |
| 3 | WNSA-3 Weighted PNSA (PNSA3*TMSA/1000) [Zefirov’s PC] | 0.6168 | 17.38 |
| 9 | HA dependent HDCA-2/TMSA [Quantum-Chemical PC] | −0.3804 | 12.74 |
| 12 | Min valency of a H atom | −0.2534 | 9.21 |
| 11 | Avg valency of a N atom | 0.4056 | 9.15 |
| 10 | Max SIGMA-SIGMA bond order | 0.1834 | 5.47 |
| 8 | WNSA-3 Weighted PNSA (PNSA3*TMSA/1000)[Quantum-Chemical PC] | 0.1593 | 5.32 |
| 1 | Average Information content (order 2) | 0.1381 | 3.85 |
| 6 | Max 1-electron react. index for a C atom | 0.1501 | 3.59 |
| 5 | Max electroph. react. index for a C atom | 0.0899 | 3.29 |
| 7 | FNSA-2 Fractional PNSA(PNSA-2/TMSA)[Quantum-Chemical PC] | −0.0123 | 0.44 |
| 4 | HOMO–LUMO energy gap | −0.0141 | 0.40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meraz, M.M.; Malik, A.A.; Yang, W.; Sun, W.-H. Catalytic Performance of Cycloalkyl-Fused Aryliminopyridyl Nickel Complexes toward Ethylene Polymerization by QSPR Modeling. Catalysts 2021, 11, 920. https://doi.org/10.3390/catal11080920
Meraz MM, Malik AA, Yang W, Sun W-H. Catalytic Performance of Cycloalkyl-Fused Aryliminopyridyl Nickel Complexes toward Ethylene Polymerization by QSPR Modeling. Catalysts. 2021; 11(8):920. https://doi.org/10.3390/catal11080920
Chicago/Turabian StyleMeraz, Md Mostakim, Arfa Abrar Malik, Wenhong Yang, and Wen-Hua Sun. 2021. "Catalytic Performance of Cycloalkyl-Fused Aryliminopyridyl Nickel Complexes toward Ethylene Polymerization by QSPR Modeling" Catalysts 11, no. 8: 920. https://doi.org/10.3390/catal11080920
APA StyleMeraz, M. M., Malik, A. A., Yang, W., & Sun, W.-H. (2021). Catalytic Performance of Cycloalkyl-Fused Aryliminopyridyl Nickel Complexes toward Ethylene Polymerization by QSPR Modeling. Catalysts, 11(8), 920. https://doi.org/10.3390/catal11080920