
Comparison of Support Effects on Phillips and Metallocene Catalysts

Abstract
:
1. Introduction
2. Results
2.1. The Influence of Active Metal Ligation on the Polymer from Both Catalysts
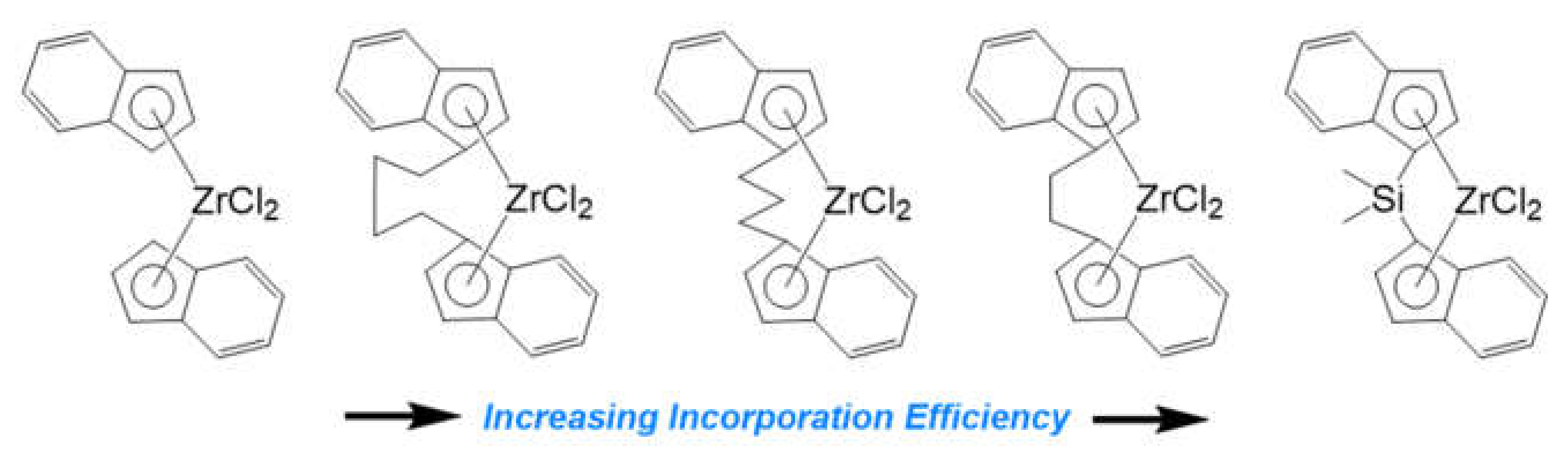
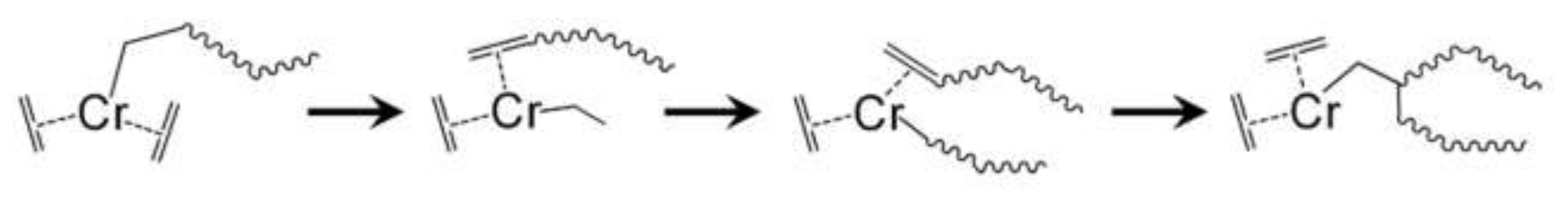
2.1.1. Shared Factors Influencing Comonomer Incorporation
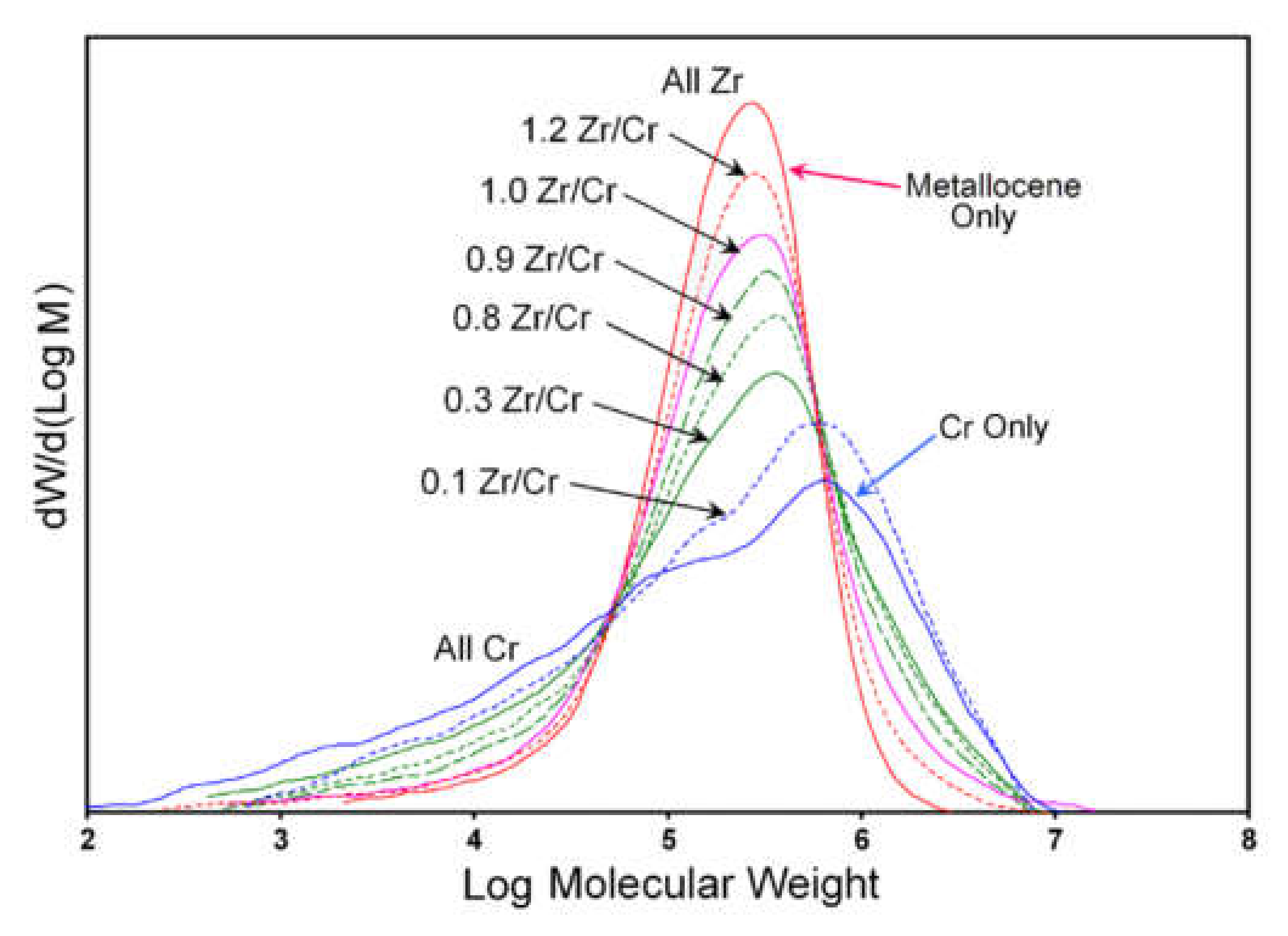
2.1.2. Shared Factors Influencing the Molecular Weight Distribution
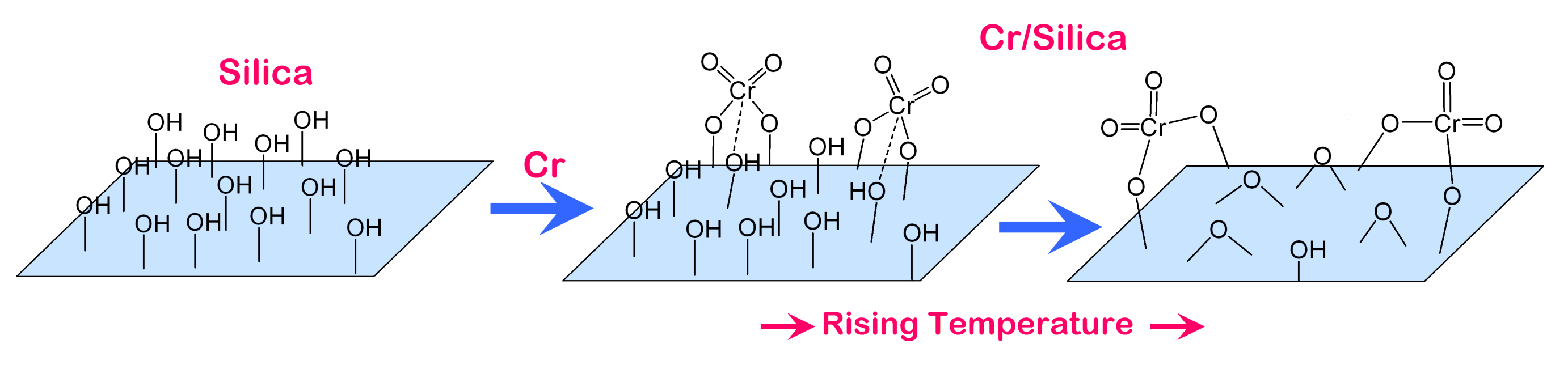
2.1.3. Attachment and Ligation on Silica and Alumina
2.1.4. Ligation on Mixed Oxide Supports
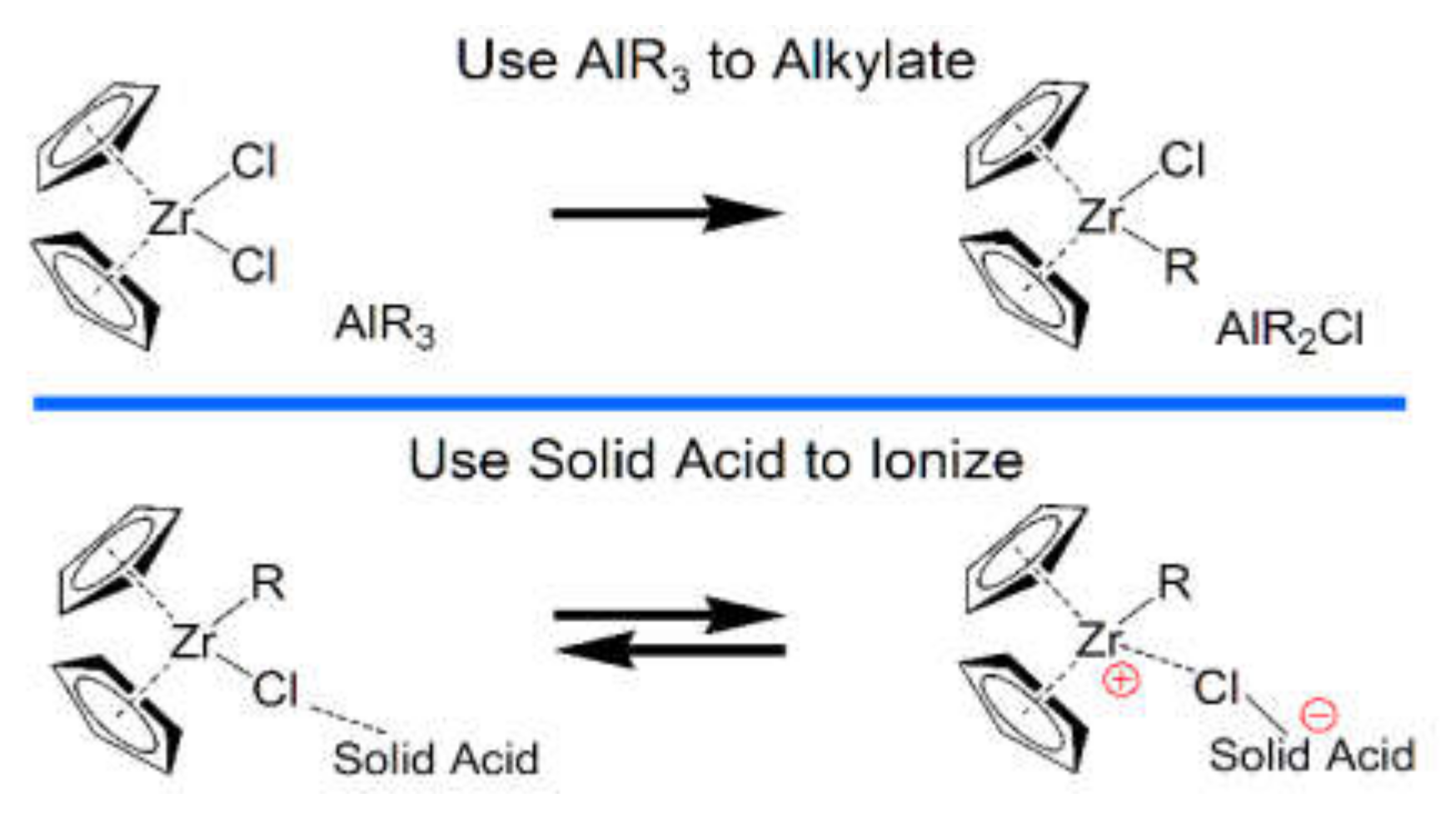
2.1.5. Similar Catalyst Formation and Behavior Allows Hybrid Catalysts
2.2. Influence by Physical Characteristics of the Support
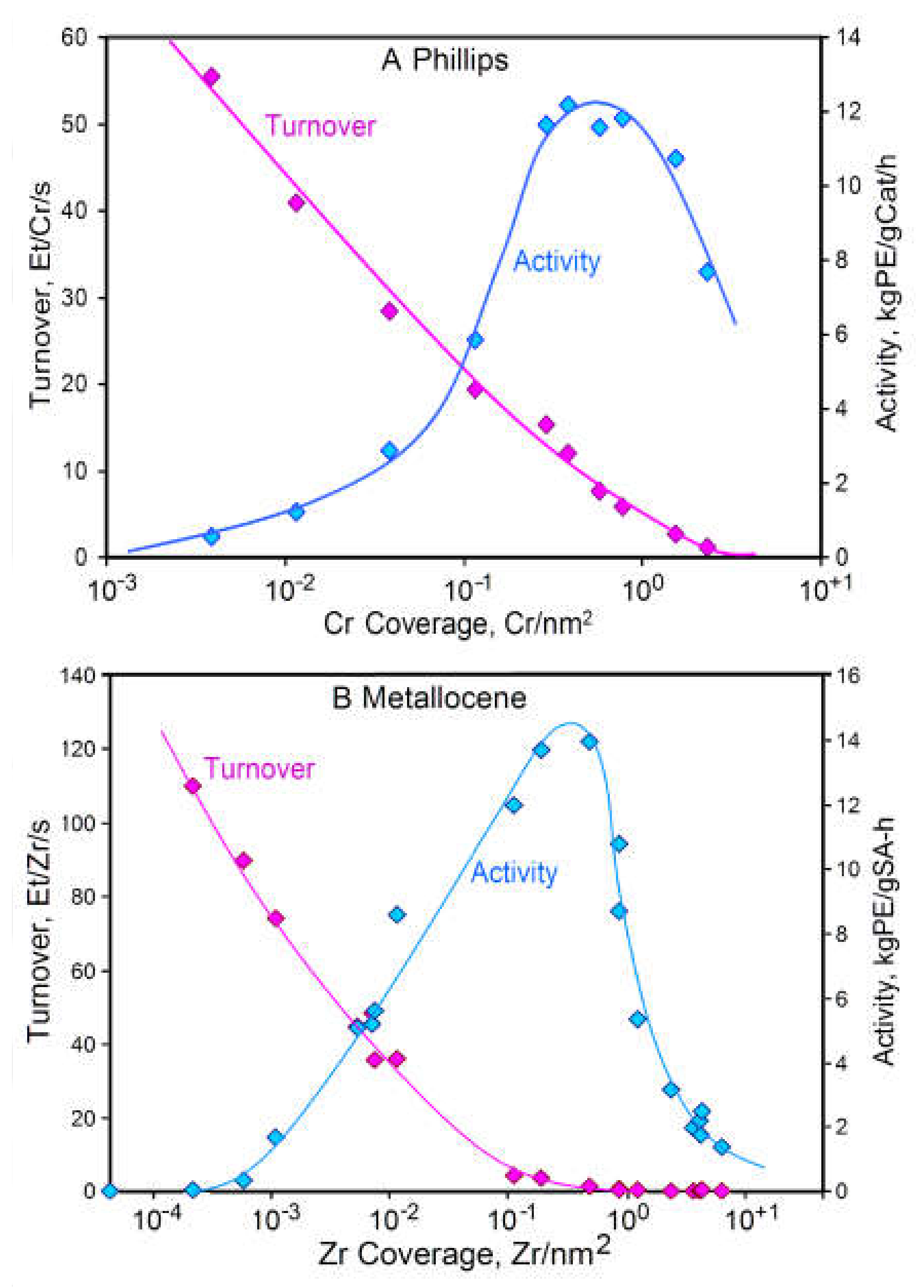
2.2.1. The Influence of Surface Area and Surface Coverage on Activity
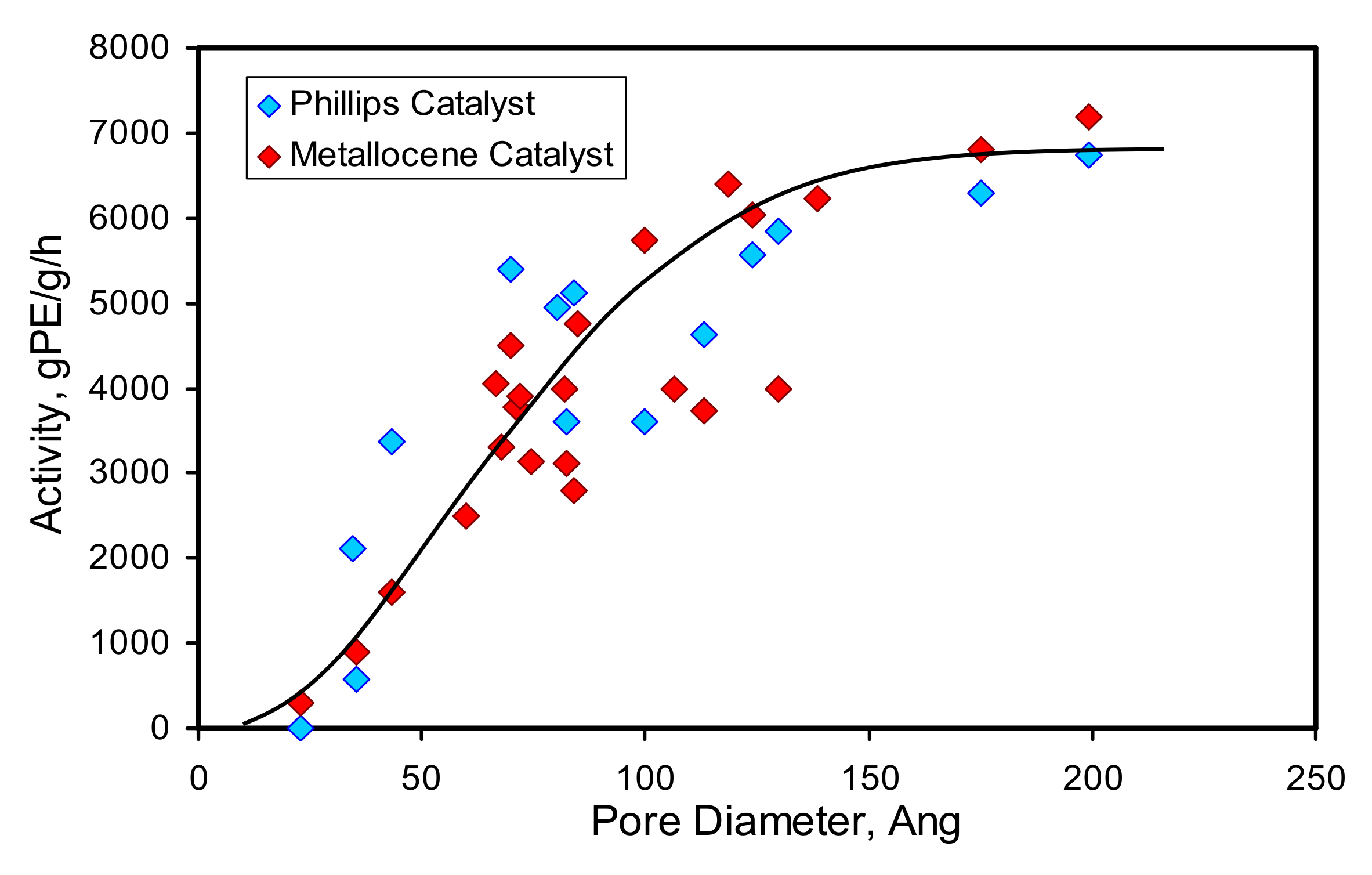
2.2.2. The Influence of Support Porosity on Catalyst Activity
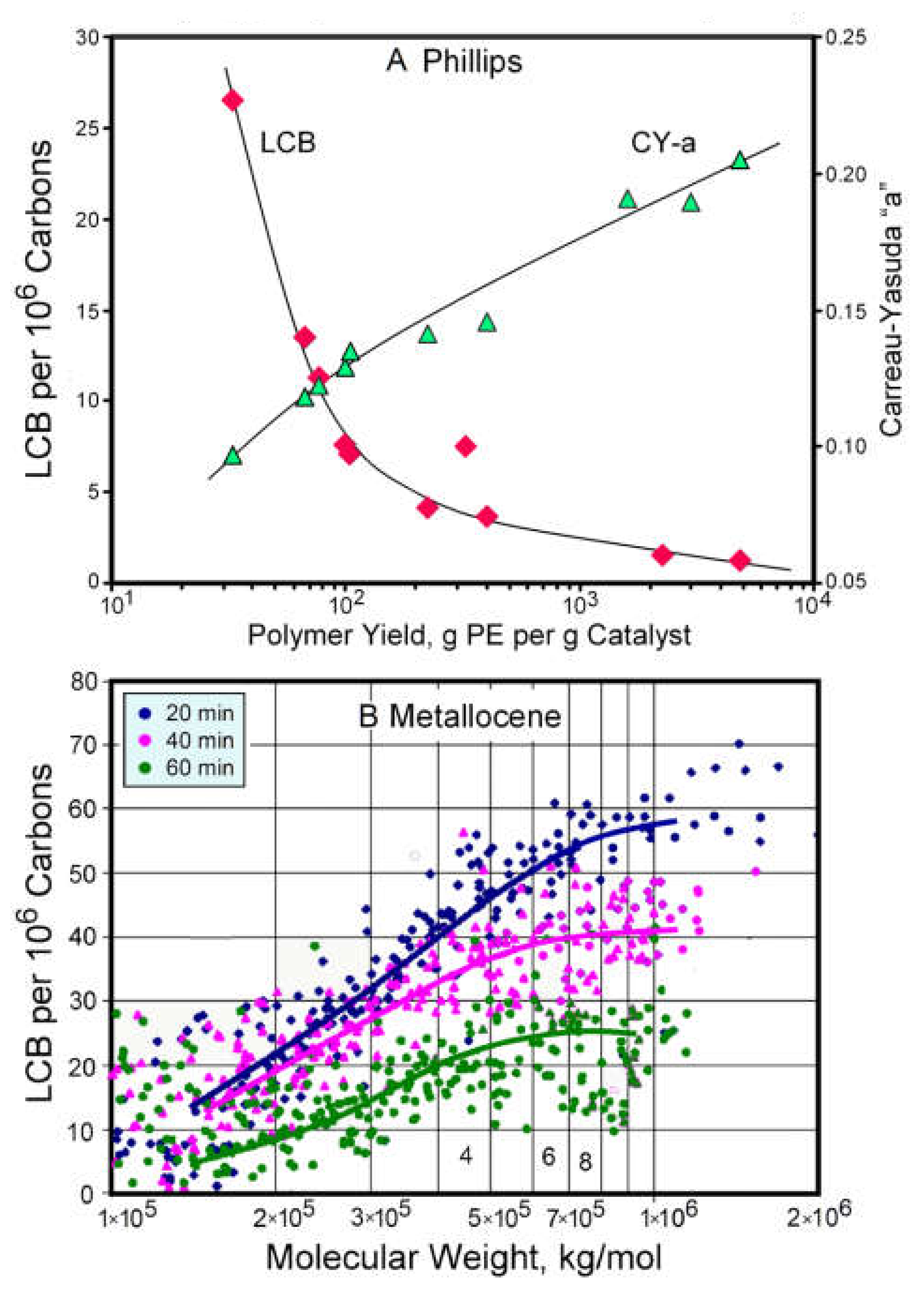
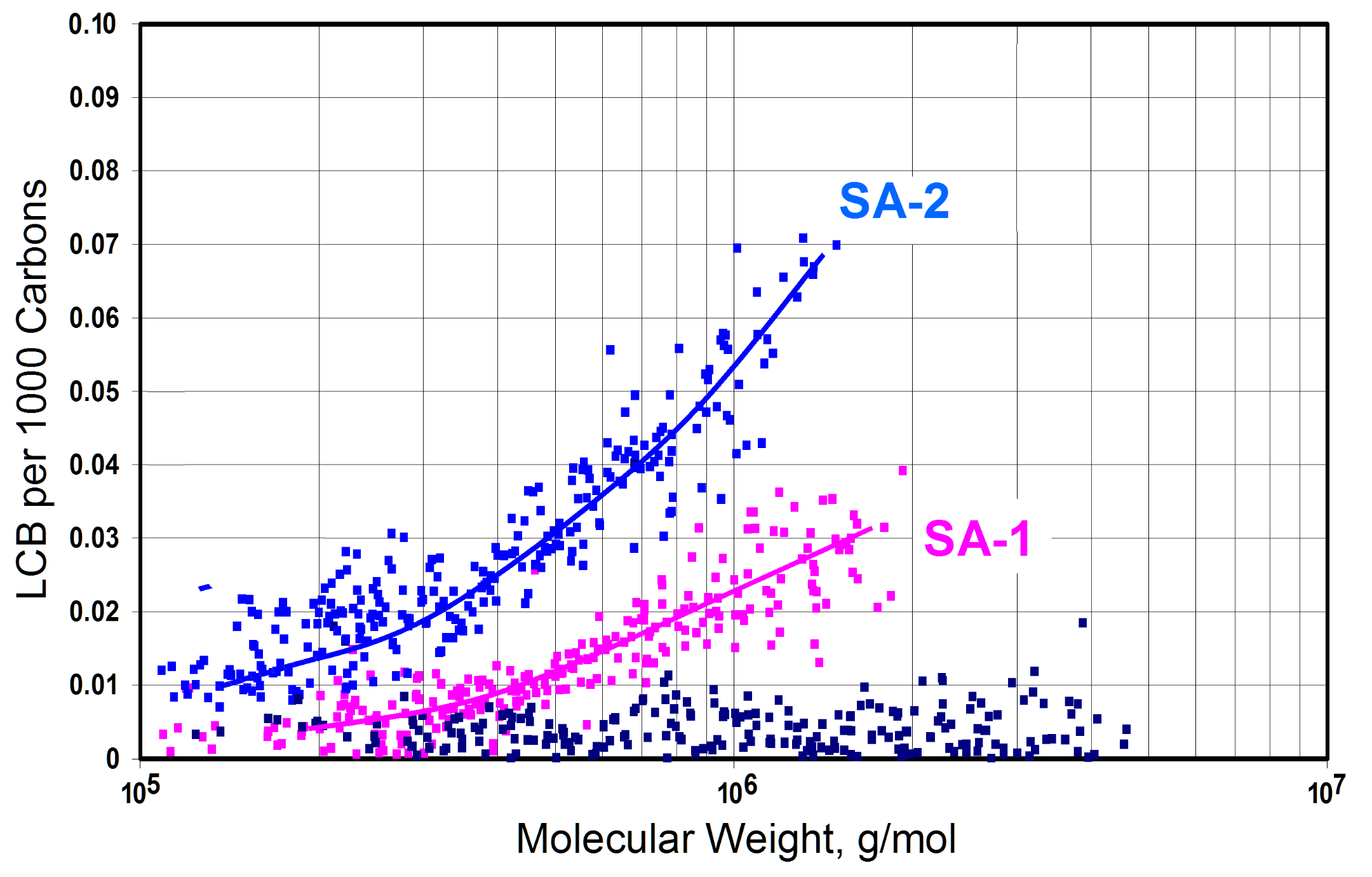
2.2.3. The Influence of Polymer Yield on Long-Chain Branching
2.3. Influence of Support Acidity on Activity and Long-Chain Branching
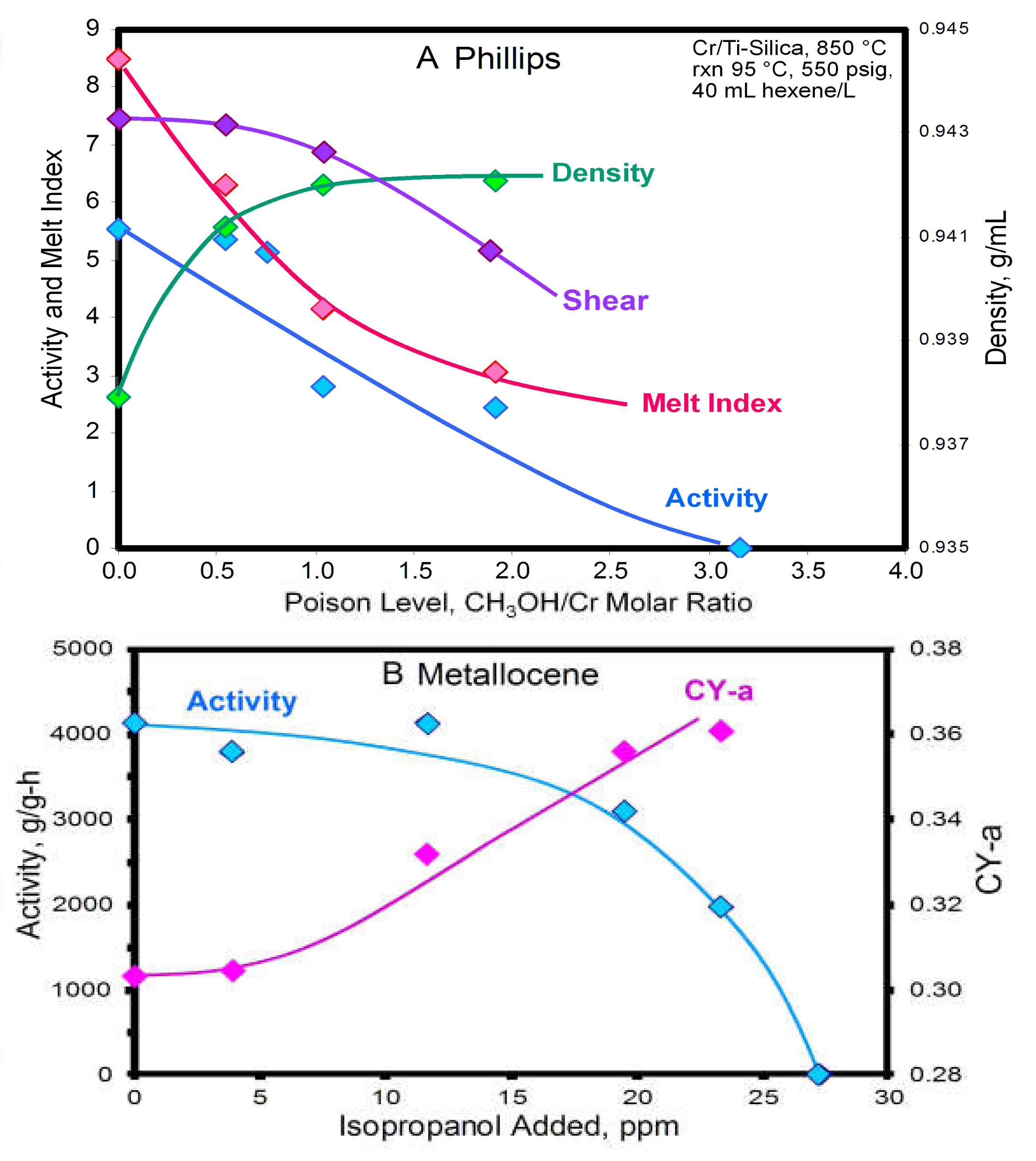
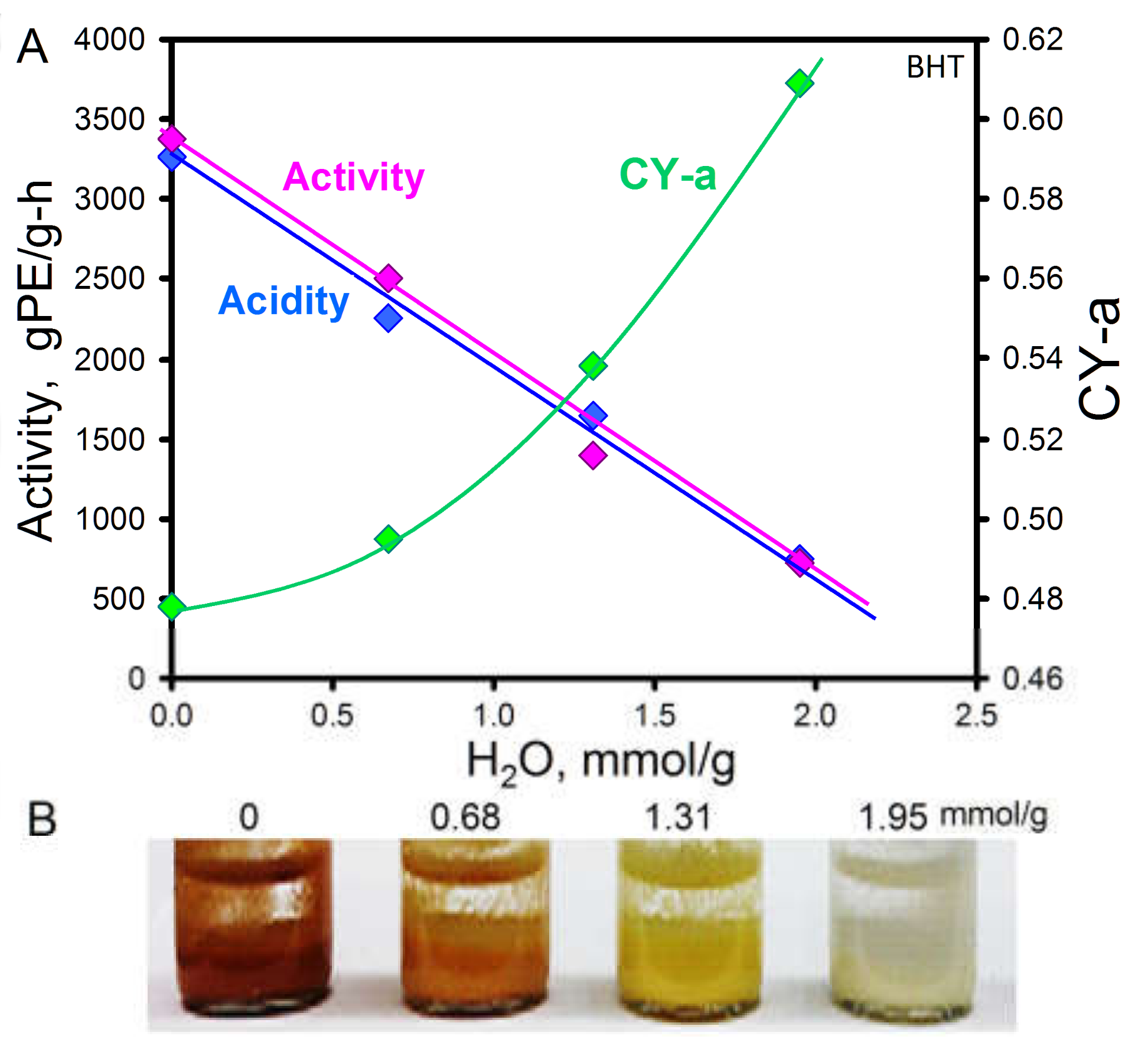
2.3.1. Macromer Retention and the Effect of Acidity
2.3.2. Measurement of Acidity
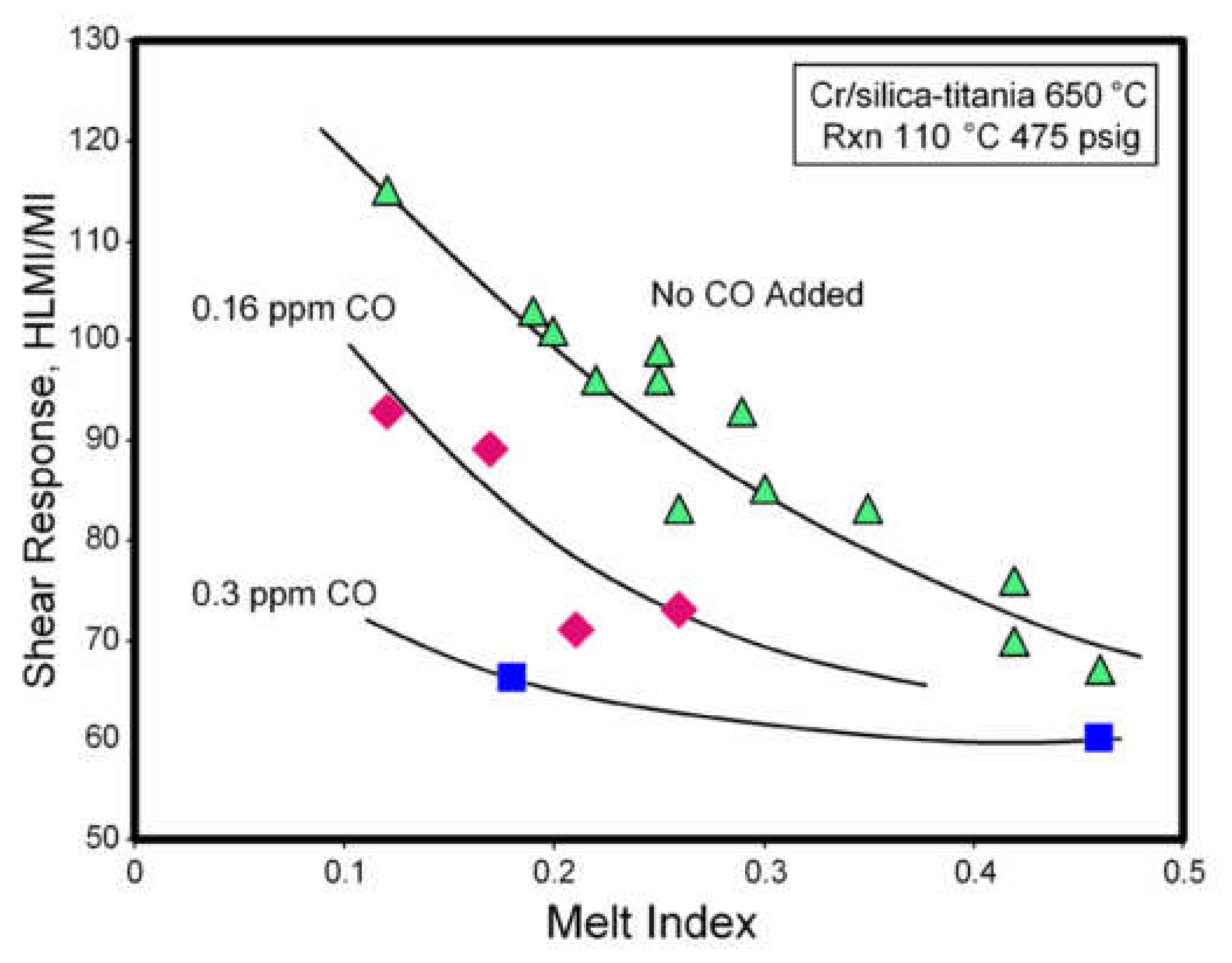
2.3.3. The Effect of Poisons on LCB Formation
2.3.4. The Influence of Fluoride Loading
2.3.5. The Influence of Sulfate Loading
2.3.6. The Influence of Adding Phosphate to Alumina
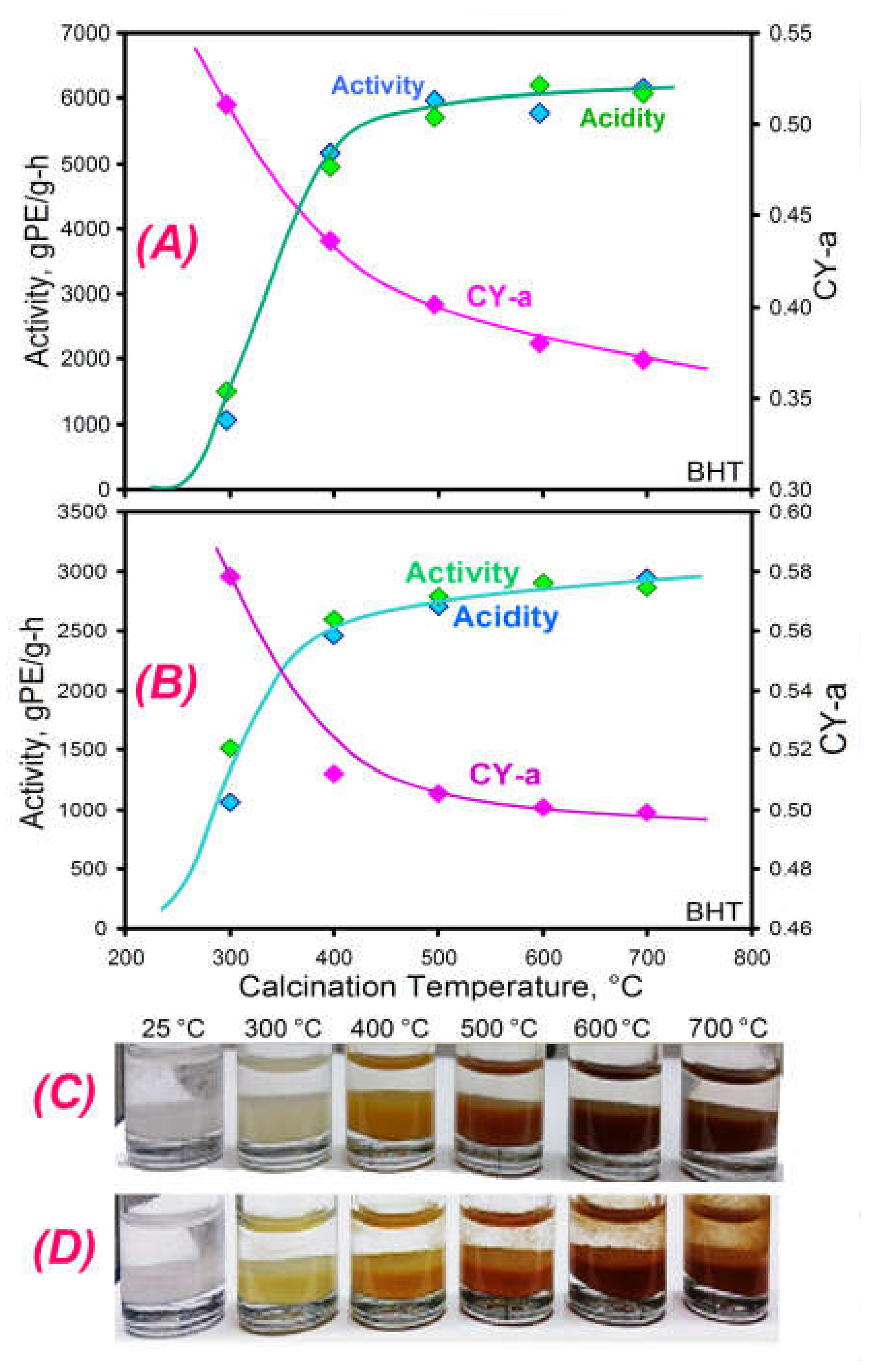
2.3.7. The Influence of Calcination Temperature on Activity and LCB Formation
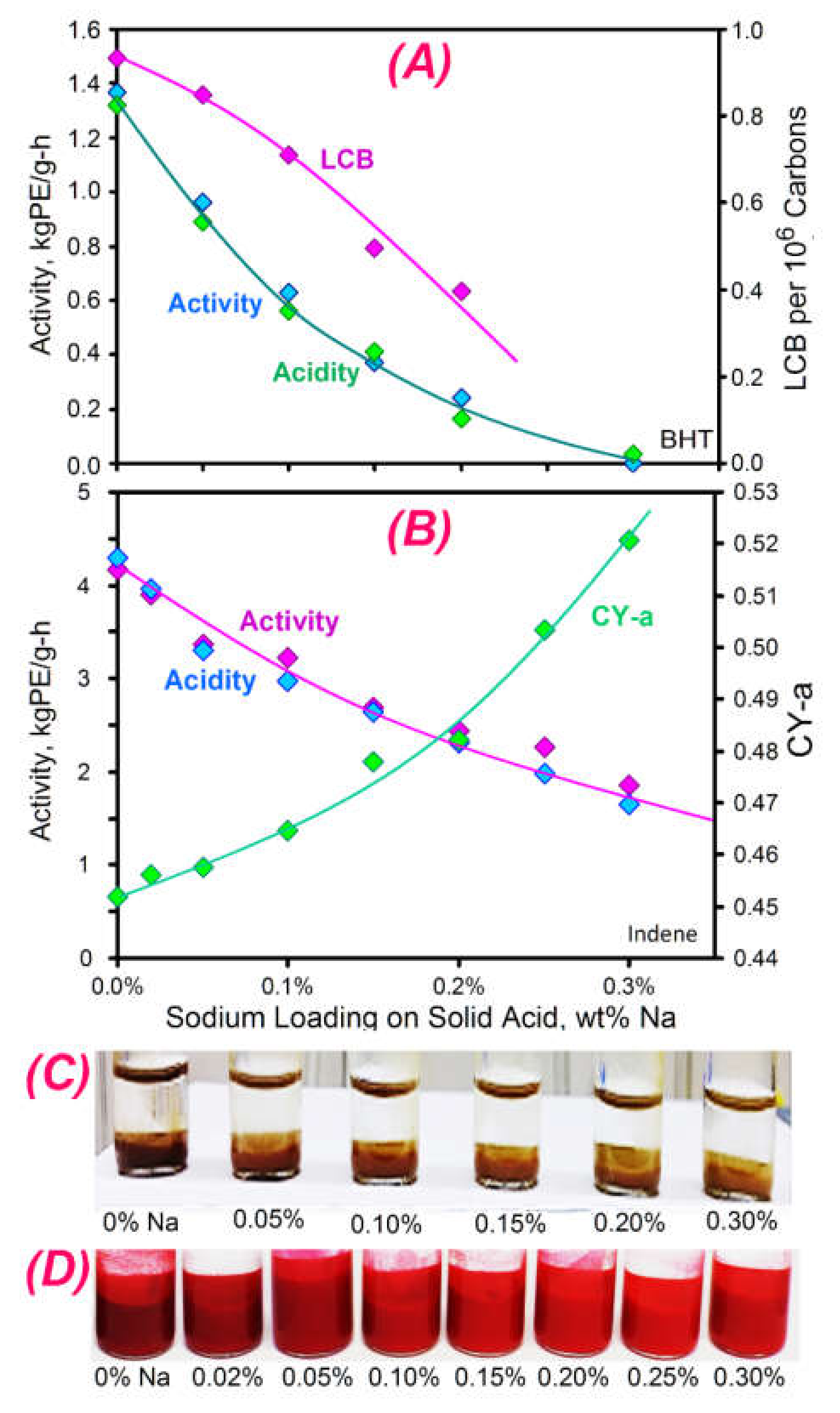
2.3.8. The Influence of Adding Sodium on Activity and LCB Formation
2.3.9. The Influence of Other Promoters on Activity and LCB Formation
3. Discussion
4. Materials and Methods
4.1. Catalyst Preparation
4.2. Calcination
4.3. Polymerization
4.4. Rheology
4.5. Molecular Weight
4.6. SEC-MALS
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hogan, J.P.; Banks, R.L. Polymers and Production Thereof, Belg. Pat. 530,617 (in French) and US 2,825,721 (in English), filed 26 March 1956, issued 4 March 1958.
- Cossee, P.; Catalysis, Z.-N.I. Mechanism of Polymerization of α-olefins with Ziegler–Natta Catalyst. J. Catal. 1964, 3, 80–88. [Google Scholar] [CrossRef]
- McDaniel, M.P. Review of the Phillips Polymerization Catalyst for Ethylene Polymerization. Adv. Catal. 2010, 53, 123–606. [Google Scholar]
- Groppo, E.; Lamberti, C.; Bordiga, S.; Spoto, A.G.; Zecchina, A. The Structure of Active Centers and the Ethylene Polymerization Mechanism on the Cr/SiO2 Catalyst: A Frontier for the Characterization Methods. Chem. Rev. 2005, 105, 115–184. [Google Scholar] [CrossRef]
- Hogan, J.P. Ethylene polymerization catalysis over chromium oxide. J. Polym. Sci. Part A-1 Polym. Chem. 1970, 8, 2637–2652. [Google Scholar] [CrossRef]
- Fubini, B.; Ghiotti, G.; Stradella, L.; Garonne, E.; Morterra, C. The Chemistry of Silica-Supported Cr Ions: Reduced and Oxidized Forms of Chromia/Silica Catalyst by Calorimetry and Ultraviolet-Visible Spectroscopy. Support J. Catal. 1980, 66, 200–213. [Google Scholar] [CrossRef]
- Iannibello, A.; Marengo, S.; Tittarelli, P.; Morelli, G.; Zecchina, A. Spectroscopic study of the structure of chromium(VI), molybdenum(VI) and tungsten(VI) oxospecies anchored on aluminium oxide. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1984, 80, 2209–2223. [Google Scholar] [CrossRef]
- Groppo, E.; Lamberti, C.; Spoto, G.; Bordiga, S.; Magnacca, G.; Zecchina, A. Tuning the structure, distribution and reactivity of polymerization centres of Cr(II)/SiO2 Phillips catalyst by controlled annealing. J. Catal. 2005, 236, 233–244. [Google Scholar] [CrossRef]
- Martino, G.A.; Barzan, C.; Piovano, A.; Budnyk, A.; Groppo, E. Tracking the reasons for the peculiarity of Cr/Al2O3 catalyst in ethylene polymerization. J. Catal. 2018, 357, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Peek, N.M.; Jeffcoat, D.B.; Moisii, C.L.; van de Burgt, S.; Profeta, S.L., Jr.; Stiegman, S.A.E. Do Mono-oxo Sites Exist in Sili-ca-Supported Cr(VI) Materials? Reassessment of the Resonance Raman Spectra. J. Phys. Chem. C 2018, 122, 4349–4358. [Google Scholar] [CrossRef]
- Weckhuysen, B.M.; Wachs, I.E. In Situ Raman Spectroscopy of Supported Chromium Oxide Catalysts: 18O2-16O2 Isotopic La-beling Studies. J. Phys. Chem. B 1997, 101, 2793–2796. [Google Scholar] [CrossRef] [Green Version]
- Weckhuysen, B.M.; De Ridder, L.M.; Schoonheydt, R.A. A quantitative diffuse reflectance spectroscopy study of supported chromium catalysts. J. Phys. Chem. 1993, 97, 4756–4763. [Google Scholar] [CrossRef] [Green Version]
- Weckhuysen, B.M.; Verberckmoes, A.A.; Baets, A.R.; Schoonheydt, R.A. Diffuse Reflectance Spectroscopy of Supported Chromium Oxide Catalysts: A Self-Modeling Mixture Analysis. J. Catal. 1997, 166, 160–171. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Liu, Z.; Cheng, R.; Tang, S.; Qiu, P.; He, X.; Terano, M.; Liu, B. Effect of F-modification over Phillips Cr/SiO2 catalyst for ethylene polymerization. ChemCatChem 2012, 4, 872–881. [Google Scholar] [CrossRef]
- Groppo, E.; Martino, G.A.; Piovano, A.; Barzan, C. The Active Sites in the Phillips Catalysts: Origins of a Lively Debate and a Vision for the Future. ACS Catal. 2018, 8, 10846–10863. [Google Scholar] [CrossRef]
- Morra, E.; Martino, G.A.; Piovano, A.; Barzan, C.; Groppo, E.; Chiesa, M. In SituX- and Q-Band EPR Investigation of Ethylene Polymerization on Cr/SiO2Phillips Catalyst. J. Phys. Chem. C 2018, 122, 21531–21536. [Google Scholar] [CrossRef]
- Potter, K.C.; Beckerle, C.W.; Jentoft, F.C.; Schwerdtfeger, E.; McDaniel, M.P. Reduction of the Phillips catalyst by various olefins: Stoichiometry, thermochemistry, reaction products and polymerization activity. J. Catal. 2016, 344, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.; Potter, K.C.; Wulfers, M.J.; Schwerdtfeger, E.; McDaniel, M.P.; Jentoft, F.C. Products of the initial reduction of the Phillips catalyst by olefins. J. Catal. 2019, 377, 550–564. [Google Scholar] [CrossRef]
- Weckhuysen, B.M.; Schoonheydt, R.A. Olefin polymerization over supported chromium oxide catalysts. Catal. Today 1999, 51, 215–221. [Google Scholar] [CrossRef] [Green Version]
- Jongkind, M.K.; Meirer, F.; Bossers, K.W.; Have, I.T.; Ohldag, H.; Watts, B.; van Kessel, T.; Friederichs, N.; Weckhuysen, B.M. Influence of Metal-Alkyls on Early-Stage Ethylene Polymerization over a Cr/SiO2 Phillips Catalyst: A Bulk Characterization and X-ray Chemical Imaging Study. Chem. A Eur. J. 2021, 27, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; Garrone, E.; Spoto, G.; Ugliengo, P.; Zecchina, A. Reactions of silica strained rings: An experimental and ab-initio study. Surf. Sci. 1995, 323, 151–162. [Google Scholar] [CrossRef]
- McDaniel, M.P.; Welch, M.B. The Activation of the Phillips Polymerization Catalyst, 1. Influence of the OH Population. J. Catal. 1983, 82, 98–109. [Google Scholar] [CrossRef]
- Groppo, E.; Damin, A.; Bonino, F.; Zecchina, A.; Bordiga, S.; Lamberti, C. New Strategies in the Raman Study of the Cr/SiO2Phillips Catalyst: Observation of Molecular Adducts on Cr(II) Sites. Chem. Mater. 2005, 17, 2019–2027. [Google Scholar] [CrossRef]
- Demmelmaier, C.A.; White, R.E.; Van Bokhoven, J.A.; Scott, S.L. Evidence for a chromasiloxane ring size effect in Phillips (Cr/SiO2) polymerization catalysts. J. Catal. 2009, 262, 44–56. [Google Scholar] [CrossRef]
- Welch, M.; McDaniel, M. The activation of the Phillips polymerization catalyst II. Activation by reduction/reoxidation. J. Catal. 1983, 82, 110–117. [Google Scholar] [CrossRef]
- McDaniel, M.P.; Johnson, M.M. A comparison of Cr/SiO2 and Cr/AlPO4 polymerization catalysts. 2. Chain transfer. Macromolecules 1987, 20, 773–778. [Google Scholar] [CrossRef]
- McDaniel, M.P.; Leigh, C.H.; Wharry, S.M. Ethylene Polymerization from Supported Organotransition Metal Complexes, III. Support Interactions. J. Catal. 1989, 120, 170–181. [Google Scholar] [CrossRef]
- Zecchina, A.; Groppo, E. Surface chromium single sites: Open problems and recent advances. Proc. R. Soc. A Math. Phys. Eng. Sci. 2012, 468, 2087–2098. [Google Scholar] [CrossRef]
- Smith, P.D.; McDaniel, M.P. Ethylene Polymerization from Supported Organotransition Metal Complexes, II Chromium Al-kyls. J. Polym. Sci. Part A Polymer Chem. 1990, 28, 3587–3601. [Google Scholar] [CrossRef]
- Bordiga, S.; Barzan, C.; Damin, A.A.; Budnyk, A.; Zecchina, A.; Groppo, E. Pre-reduction of the Phillips CrVI/SiO2 catalyst by cyclohexene: A model for the induction period of ethylene polymerization. J. Catal. 2017, 337, 45–51. [Google Scholar]
- Smith, P.D.; McDaniel, M.P. Ethylene Polymerization from Supported Organotransition Metal Complexes, I. Pentadienyl De-rivatives of Ti, V and Cr. J. Polym. Sci. Part A Polym. Chem. 1989, 27, 2695–2710. [Google Scholar] [CrossRef]
- Zheng, X.X.; Smit, M.M.; Chadwick, J.C.; Loos, J.J. Fragmentation Behavior of Silica-Supported Metallocene/MAO Catalyst in the Early Stages of Olefin Polymerization. Macromolecules 2005, 38, 4673–4678. [Google Scholar] [CrossRef]
- Kaminsky, W. The discovery of metallocene catalysts and their present state of the art. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 3911–3921. [Google Scholar] [CrossRef]
- Charoenchaidet, S.; Chavadej, S.; Gulari, E. Borane-functionalized silica supports: In situ activated heterogeneous zirconocene catalysts for MAO-free ethylene polymerization. J. Mol. Catal. A: Chem. 2002, 185, 167–177. [Google Scholar] [CrossRef]
- Welch, M.B.; Palackal, S.J.; Hauger, B.E.; Dockter, D.W.; Koppl, A.; Alt, H.G. Metallocenes, polymerization catalyst systems, their preparation, and use. U.S. Patent 6,992,035, 31 January 2006. [Google Scholar]
- Welborn, H.C.; Ewen, J.A., Jr. Metallocene Catalysts using MAO. U.S. Patent 5,324,800, 28 June 1994. [Google Scholar]
- McDaniel, M.P.; Jensen, M.D.; Jayaratne, K.; Collins, K.S.; Benham, E.A.; McDaniel, N.D.; Das, P.K.; Martin, J.L.; Yang, Q.; Thorn, M.G.; et al. Metallocene Activation by Solid Acids. Tailor-Made Polym. 2008, 171–210. [Google Scholar] [CrossRef]
- Jenxen, M.D.; Hain, J.; Myers, A. What is driving change in polyolefin catalyst technology. Hydrocarb. Process. 2008, 1–3. [Google Scholar]
- Dietz, R.E. Silica-Titania Cogel Catalyst. U.S. Patent 3,119,569, 3 June 1975. [Google Scholar]
- McDaniel, M. The state of Cr(VI) on the Phillips polymerization catalyst IV. Saturation coverage. J. Catal. 1982, 76, 37–47. [Google Scholar] [CrossRef]
- Weckhuysen, B.M.; Wachs, A.I.E.; Schoonheydt, R.A. Surface Chemistry and Spectroscopy of Chromium in Inorganic Oxides. Chem. Rev. 1996, 96, 3327–3350. [Google Scholar] [CrossRef] [Green Version]
- Bird, R.B.; Armstrong, R.C.; Hassager, O. Dynamics of Polymeric Liquids Fluid Mechanics; John Wiley & Sons: New York, NY, USA, 1987; Volume 1. [Google Scholar]
- Janzen, J.; Colby, R. Diagnosing long-chain branching in polyethylenes. J. Mol. Struct. 1999, 485–486, 569–583. [Google Scholar] [CrossRef]
- Wyatt, P.J. Light scattering and the absolute characterization of macromolecules. Anal. Chim. Acta 1993, 272, 1–40. [Google Scholar] [CrossRef]
- Yu, Y.; DesLauriers, P.J.; Rohlfing, D.C. SEC-MALS method for the determination of long-chain branching and long-chain branching distribution in polyethylene. Polymer 2005, 46, 5165–5182. [Google Scholar] [CrossRef]
- Cavallo, L.; Fait, A.; Piemontesi, F. Selectivity in Propene Polymerization with Metallocene Catalysts. Chem. Rev. 2000, 100, 1253–1346. [Google Scholar]
- Zaccaria, F.; Ehm, C.; Budzelaar, P.H.M.; Busico, V. Accurate Prediction of Copolymerization Statistics in Molecular Olefin Polymerization Catalysis: The Role of Entropic, Electronic, and Steric Effects in Catalyst Comonomer Affinity. ACS Catal. 2017, 7, 1512–1519. [Google Scholar] [CrossRef]
- Ehm, C.; Vittoria, A.; Goryunov, G.P.; Kulyabin, P.S.; Budzelaar, P.H.M.; Voskoboynikov, A.Z.; Busico, V.; Uborsky, D.V.; Cipullo, R. Connection of Stereoselectivity, Regioselectivity, and Molecular Weight Capability in rac-R′2Si(2-Me-4-R-indenyl)2ZrCl2 Type Catalysts. Macromolecules 2018, 51, 8073–8083. [Google Scholar] [CrossRef]
- Kulyabin, P.S.; Goryunov, G.P.; Sharikov, M.I.; Izmer, V.V.; Vittoria, A.; Budzelaar, P.H.; Busico, M.V.; Voskoboynikov, A.Z.; Ehm, C.; Cipullo, R.; et al. Ansa-Zirconocene Catalysts for Isotactic-Selective Propene Polymerization at High Temperature: A Long Story Finds a Happy Ending. J. Am. Chem. Soc. 2021, 143, 7641–7647. [Google Scholar] [CrossRef]
- Brintzinger, H.H.; Fischer, D.R.; Rieger, M.B.; Waymouth, R.M. Stereospecific Olefin Polymerization with Chiral Metallocene Catalysts Angew. Chem. Int. Ed. 1995, 34, 1143–1170. [Google Scholar] [CrossRef] [Green Version]
- Röll, W.; Brintzinger, H.H.; Rieger, B.; Stero, R.Z. Regioselectivity of Chiral, Alkyl-substituted ansa-Zirconece Catalysts in Methylalumoxane-activator Propene Polymerization Angew. Chem. Int. Ed. 1990, 3, 279–280. [Google Scholar] [CrossRef]
- Uborsky, D.V.; Mladentsev, D.Y.; Guzeev, B.A.; Borisov, I.S.; Vittoria, A.; Ehm, C.; Cipullo, R.; Hendriksen, C.; Busico, N.F.V.; Voskoboynikov, A.Z. C1-Symmetric Si-bridged (2- indenyl)(1-indenyl) ansa-metallocenes as efficient eth-ene/1-hexene copolymerization catalysts. Dalton Trans. 2020, 49, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Ehm, C.; Vittoria, A.; Goryunov, G.P.; Izmer, V.V.; Kononovich, D.S.; Samsonov, O.V.; Budzelaar, P.H.M.; Voskoboynikov, A.Z.; Busico, V.; Uborsky, D.V.; et al. On the limits of tuning comonomer affinity of ‘Spaleck-type’ ansa-zirconocenes in ethene/1-hexene copolymerization: A high-throughput experimentation/QSAR approach. Dalton Trans. 2020, 49, 10162–10172. [Google Scholar] [CrossRef]
- Yang, Q.; Jensen, M.D.; McDaniel, M.P. Alternative View of Long Chain Branch Formation by Metallocene Catalysts. Macromolecules 2010, 43, 8836–8852. [Google Scholar] [CrossRef]
- DesLauriers, P.J.; McDaniel, M.P. Short chain branching profiles in polyethylene from the Phillips Cr/silica catalyst. J. Polym. Sci. Part A: Polym. Chem. 2007, 45, 3135–3149. [Google Scholar] [CrossRef]
- McDaniel, M.P.; Martin, S.J. Poisoning Studies on Cr/Silica, Carbon Monoxide. J. Phys. Chem. 1991, 95, 3289–3293. [Google Scholar] [CrossRef]
- Zecchina, A.; Garroñe, E.; Ghiotti, G.; Coluccia, S. On the Chemistry of Silica-Supported Chromium Ions, One-Ligand Complexes, Adsorption of Carbon Monoxide, Carbon Dioxide and Pyridine. J. Phys. Chem. 1975, 79, 972–978. [Google Scholar] [CrossRef]
- Cotes, M.P.; Jordan, R.E. Cationic Aluminum Alkyl Complexes Incorporating Amidinate Ligands. J. Am. Chem. Soc. 1997, 119, 8125–8216. [Google Scholar]
- Crowther, D.J.; Borkowsky, S.L.; Swenson, D.; Meyer, T.Y.; Jordan, R.F. Coordination of CB11H12 to cationic zirconium(IV) complexes. Organometallics 1993, 12, 2897–2903. [Google Scholar] [CrossRef]
- Baldwin, S.M.; Bercaw, J.E.; Brintzinger, H.H. Cationic Alkylaluminum-Complexed Zirconocene Hydrides as Participants in Olefin Polymerization Catalysis. J. Am. Chem. Soc. 2010, 132, 13969–13971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herwig, J.; Kaminsky, W. Halogen-Free Soluble Ziegler Catalysts with MAO as Catalyst. Polym. Bull. 1983, 9, 464–469. [Google Scholar]
- Ballard, D.G.H.; Jones, E.; Padget, J.C.; Pioli, A.J.P.; Walker, A.R.J.; Wyatt, R.J. Polymerization Process. U.S. Patent 4,056,669, 1 November 1977. [Google Scholar]
- Ahn, H.; Nicholas, C.P.; Marks, T.J. Surface Organozirconium Electrophiles Activated by Chemisorption on “Super Acidic” Sulfated Zirconia as Hydrogenation and Polymerization Catalysts. A Synthetic, Structural, and Mechanistic Catalytic Study. Organometallics 2002, 21, 1788–1806. [Google Scholar] [CrossRef]
- McDaniel, M.P.; Short, J.N. Catalyst Comprising Phosphate and Chromocene. U.S. Patent 4,424,139, 3 January 1984. [Google Scholar]
- Freeman, J.W.; Wilson, D.R.; Ernst, R.D.; Smith, P.D.; Klendworth, D.D.; McDaniel, M.P. Ethylene polymerization over organochromium catalysts: A comparison between closed and open pentadienyl ligands. J. Polym. Sci. Part A Polym. Chem. 1987, 25, 2063–2075. [Google Scholar] [CrossRef]
- Martino, G.A.; Piovano, A.; Barzan, C.; Liao, Y.K.; Morra, E.; Hirokane, K.; Chiesa, M.; Monoi, T.; Groppo, E. Cr[CH(SiMe3)2]3/SiO2 catalysts for ethene polymerization: The correlation at a molecular level between the chromium loading and the microstructure of the produced polymer. J. Catal. 2021, 394, 131–141. [Google Scholar] [CrossRef]
- Smith, P.D. Polychromium Compounds. U.S. Patent 4,668,808, 26 May 1987. [Google Scholar]
- Walker, D.W.; Czenkusch, E.L. Polymerization Process using Supported Diarene Catalysts. U.S. Patent 3,157,712, 17 November 1964. [Google Scholar]
- McDaniel, M.P.; Johnson, M.M. Phosphate Containing Zerovalent Chromium. U.S. Patent 4,364,841, 21 December 1982. [Google Scholar]
- McDaniel, M.P.; Smith, P.D. Polymerization Catalyst, Method of Making and use Therefor. U.S. Patent 4,619,980, 28 October 1986. [Google Scholar]
- McDaniel, M.P.; Johnson, M.M. Chromium on Mixed Metal Phosphate base with Organoboron Cocatalyst. U.S. Patent 4,481,302, 6 November 1984. [Google Scholar]
- McDaniel, M.P.; Smith, P.D.; Norwood, D.D. Silicon and Fluorine-Treated Alumina Containing a Chromium Catalyst and method of Producing Same. U.S. Patent 4,806,513, 21 February 1989. [Google Scholar]
- Vuurman, M.A.; Hardcastle, F.D.; Wachs, I.E. Characterization of CrO3/Al2O3 Catalysts Under Ambient Conditions. J. Molec. Catal. 1993, 84, 193–205. [Google Scholar] [CrossRef]
- McDaniel, M.P.; Benham, E.A.; Martin, S.J.; Collins, K.S.; Smith, J.L.; Hawley, G.R.; Wittner, C.E.; Jensen, M.D. Compositions That Can Produce Polymers. U.S. Patent 6,300,271, 9 October 2001. [Google Scholar]
- McDaniel, M.; Welch, M.; Dreiling, M. The activation of the Phillips polymerization catalyst III. Promotion by titania. J. Catal. 1983, 82, 118–126. [Google Scholar] [CrossRef]
- McDaniel, M.P.; Collins, K.S.; Smith, J.L.; Benham, E.A.; Johnson, M.M.; Eaton, A.P.; Jensen, M.D.; Martin, J.L.; Hawley, G.R. Organometal catalyst compositions. U.S. Patent 6,395,666, 28 May 2002. [Google Scholar]
- McDaniel, M.P.; Collins, K.S. Methods of Preparing a Polymerization Catalyst. U.S. Patent 7,638,456, 29 December 2009. [Google Scholar]
- McDaniel, M.P.; Johnson, M.M. Polymerization process using phosphate supported chromium catalyst. U.S. Patent 4,444,964, 24 April 1984. [Google Scholar]
- McDaniel, M.P.; Rohlfing, D.C.; Benham, E.A. Long Chain Branching in Polyethylene from the Phillips Chromium Catalyst. Polym. React. Eng. 2003, 11, 101–132. [Google Scholar] [CrossRef]
- Yu, Y.; Schwerdtfeger, E.D.; McDaniel, M.P. Size-Exclusion Chromatography Coupled to Multiangle Light Scattering Detec-tion of Long-Chain Branching in Polyethylene Made with Phillips Catalyst. J. Polym. Sci., A. Polym. Chem. 2012, 50, 1166–1173. [Google Scholar] [CrossRef]
- DesLauriers, P.J.; Tso, C.; Yu, Y.; Rohlfing, D.L.; McDaniel, M.P. Long-chain branching in PE from Cr/aluminophosphate cata-lysts. Appl. Catal. A: Gen. 2010, 388, 102–112. [Google Scholar] [CrossRef]
- Schwerdtfeger, E.D.; Jensen, M.D.; Yang, Q.; McDaniel, M.P. Controlling Long-Chain Branch Formation in Polyethylene. Curr. Topics Catal. 2016, 12, 1–27. [Google Scholar]
- McDaniel, M.P.; Benham, E.A.; Collins, K.S.; Smith, J.L.; Hawlely, G.R.; Wittner, C.E.; Jensen, M.D. Process for Producing Polymers. U.S. Patent 6,831,141, 9 October 2001. [Google Scholar]
- McDaniel, M.P.; Collins, K.S. Methods of Preparing a Polymerization Catalyst. U.S. Patent 9,068,027, 30 June 2015. [Google Scholar]
- McDaniel, M.P.; Collins, K.S. Ethylene polymerization by Cr(III) salts on acidic carriers. Appl. Catal. A Gen. 2016, 527, 116–126. [Google Scholar] [CrossRef]
- McDaniel, M.P. Influence of Catalyst Porosity on Ethylene Polymerization. ACS Catal. 2011, 1, 1394–1407. [Google Scholar] [CrossRef]
- McDaniel, M.P. Influence of porosity on PE molecular weight from the Phillips Cr/silica catalyst. J. Catal. 2009, 261, 34–49. [Google Scholar] [CrossRef]
- McDaniel, M.P.; Collins, K.S. The influence of porosity on the Phillips Cr/silica catalyst 2. Polyethylene elasticity. J. Polym. Sci. Part A Polym. Chem. 2008, 47, 845–865. [Google Scholar] [CrossRef]
- McDaniel, M.P.; Kelly, S.L. Reinforcement of Cr/silica catalysts by secondary deposition of silicate oligomers. Appl. Catal. A Gen. 2018, 554, 88–94. [Google Scholar] [CrossRef]
- Alt, H.G. The heterogenization of homogeneous metallocene catalysts for olefin polymerization. J. Chem. Soc. Dalton Trans. 1999, 1703–1710. [Google Scholar] [CrossRef]
- Jensen, M.D.; Yang, Q.; Yu, Y.; McDaniel, M.P. Kinetics of Long-Chain Branch Formation in Polyethylene. ACS Catal. 2017, 8, 725–737. [Google Scholar] [CrossRef]
- Monwar, M.M.; Cruz, C.A.; Barr, J.L.; McDaniel, M.P. Ethylene polymerization by hydrocarbon-reduced Cr/silica catalyst. J. Catal. 2021, 394, 451–464. [Google Scholar] [CrossRef]
- Groppo, E. Acidic Properties of γ-Al2O3 and Sulfated Al2O3 Investigated by FT-IR Spectroscopy of Molecular Probes. 2016; in press, unpublished. [Google Scholar]
- Hammett, L.P.; Deyrup, A.J. A Series of Simple Basic Indicators. I. the Acidity Functions of Mixtures of Sulfuric and Perchloric Acids with Water1. J. Am. Chem. Soc. 1932, 54, 2721–2739. [Google Scholar] [CrossRef]
- Walling, C. The Acid Strength of Surfaces. J. Am. Chem. Soc. 1950, 72, 1164–1168. [Google Scholar] [CrossRef]
- Benesi, H.A. Acidity of Catalyst Surfaces. II. Amine Titration Using Hammett Indicators. J. Phys. Chem. 1957, 61, 970–973. [Google Scholar] [CrossRef]
- Johnson, O. Acidity and Polymerization Activity of Solid Acid Catalysts. J. Phys. Chem. 1955, 59, 827–831. [Google Scholar] [CrossRef]
- Kladnig, W.F. Use of Hammcatt Indicators for Acidity Measurements in Zeolites. J. Phys. Chem. 1979, 83, 765–766. [Google Scholar] [CrossRef]
- Alemdaroğlu, T. Determination methods for the acidity of solid surfaces. Commun. Fac. Sci. Univ. Ank. Ser. B Chem. Chem. Eng. 2001, 47, 027–035. [Google Scholar] [CrossRef]
- Gold, V.; Hawes, B.W.V. The Ionization of Triarylcarbinols in Strong Acids and the Definition of a New Acidity Function. J. Chem. Soc. 1951, 465, 2102–2111. [Google Scholar] [CrossRef]
- Deno, N.C.; Jaruzelski, J.J.; Schriesheim, A. Carbonium Ions. I. An Acidity Function (C0) Derived from Arylcarbonium Ion Equilibria1. J. Am. Chem. Soc. 1955, 77, 3044–3051. [Google Scholar] [CrossRef]
- Liu, X.; Iu, K.-K.; Thomas, J.K.; He, H.; Klinowski, J. Spectroscopic Studies of Protonated Aromatic Species and Radical Cations in H+-Zeolites. J. Am. Chem. Soc. 1994, 116, 11811–11818. [Google Scholar] [CrossRef]
- Hogan, J.P. Ethylene Homopolymer and Copolymer Polymerization Process and the Products Produced Therefrom. U.S. Patent 5,155,186, 13 October 1992. [Google Scholar]
- Amercan Society for Testing and Materials (ASTM). Procedure D1238-E, condition 190/2, at 190 C. with a 2,160 gram weight.
- Grayson, M.; McDaniel, M. Sulfide poisoning of ethylene polymerization over Phillips Cr/silica catalyst. J. Mol. Catal. 1991, 65, 139–144. [Google Scholar] [CrossRef]
- McDaniel, M.P.; Collins, K.S.; Benham, E.A.; Jensen, M.D.; Martin, J.L.; Hawley, G.R. Organometal Catalyst Compositions. U.S. Patent 6,613,852, 2 September 2003. [Google Scholar]
- McDaniel, M.P.; Collins, K.S.; Benham, E.A.; Deslauriers, P.J. Methods of Preparing Active Chromium/Alumina Catalysts via Treatment with Sulfate. U.S. Patent 7,214,642, 8 May 2007. [Google Scholar]
- McDaniel, M.; Johnson, M. A comparison of Cr/SiO2 and Cr/AlPO4 polymerization catalysts I. Kinetics. J. Catal. 1986, 101, 446–457. [Google Scholar] [CrossRef]
- Cheng, R.; Xu, C.; Liu, Z.; Dong, Q.; He, X.; Fang, Y.; Terano, M.; Hu, Y.; Pullukat, T.J.; Liu, B. High-resolution spectroscopy (XPS, 1H MAS solid-state NMR) and DFT investigations into Ti-modified Phillips CrOx/SiO2 catalysts. J. Catal. 2010, 273, 103–115. [Google Scholar] [CrossRef]
- McDaniel, M. The state of Cr(VI) on the Cr/Silica polymerization catalyst. J. Catal. 1981, 67, 71–76. [Google Scholar] [CrossRef]
- Qiu, P.; Li, X.; Zhang, S.; Cheng, R.; Dong, Q.; Liu, B.; Li, L.; Yu, Y.; Tang, Y.; Xie, J.; et al. Supporting mechanism of non-toxic chromium (III) acetate on silica for preparation of Phillips ethylene polymerization catalysts. Asia-Pacific J. Chem. Eng. 2009, 4, 660–665. [Google Scholar] [CrossRef]
- Sun, Q.; Cheng, R.; Liu, Z.; He, X.; Zhao, N.; Liu, B. Effect of Fluoride-Modification on the Phillips Cr/SiO2 Catalyst for Ethylene Polymerization. ChemCatChem 2017, 9, 3364–3373. [Google Scholar] [CrossRef]
- Cheng, R.; Liu, X.; Fang, Y.; Terano, M.; Liu, B. High-resolution 29Si CP/MAS solid state NMR spectroscopy and DFT in-vestigation on the role of geminal and single silanols in grafting chromium species over Phillips Cr/silica catalyst. Appl. Catal. A: Gen. 2017, 543, 26–33. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Titania Ti/nm2 | Activity kgPE/gCat/h | MW kg/mol | MW Breadth MW/MN | LCB per 106 C |
|---|---|---|---|---|
| 0.00 | 1.9 | 158 | 7.9 | 2.8 |
| 0.25 | 3.4 | 152 | 9.3 | 3.4 |
| 0.50 | 3.8 | 143 | 12.3 | 5.1 |
| 0.80 | 4.5 | 134 | 16.2 | 7.0 |
| 1.10 | 4.7 | 128 | 17.5 | 7.3 |
| 1.40 | 4.7 | 120 | 18.8 | 7.7 |
| 2.20 | 4.2 | 112 | 21.3 | 10.3 |
| Silica | A | B |
|---|---|---|
| Pore volume, mL/g | 2.6 | 1.0 |
| Surface area, m2/g | 350 | 600 |
| Avg. pore diam., Ang | 297 | 67 |
| Activity, kgPE/h per mol Cr or Zr | ||
| Catalyst type 1 (Cr) | 16.6 | 4.2 |
| Catalyst type 2 (met) | 103 | 7.7 |
| Catalyst type 3 (met) | 29.7 | 3.7 |
| Catalyst type 4 (met) | 47.2 | 4.6 |
| SO4/nm2 | 0 | 2.01 | 4.01 | 8.03 |
| HLMI | 4.84 | 3.12 | 2.28 | 0.27 |
| Shear Response | >200 | 46.8 | 42.7 | 31.7 |
| MW, kg/mol | 473 | 339 | 311 | 379 |
| MZ, kg/mol | 3708 | 1402 | 1303 | 1595 |
| MW / MN | 99.8 | 13.1 | 10.8 | 10.9 |
| MZ / MW | 7.6 | 4.0 | 4.1 | 4.0 |
| Eta(0), Pa-s | 8.1 × 106 | 4.9 × 105 | 5.7 × 105 | 1.4 × 106 |
| Tau-eta, s | 141 | 2.04 | 2.33 | 5.17 |
| CY-a | 0.2749 | 0.2962 | 0.3014 | 0.3846 |
| Metallocene | Phillips | |||||
|---|---|---|---|---|---|---|
| Promoter | Amount | Support Type | Calcination Temp. | Met. type | Activity gPE/g/h | Activity gPE/g/h |
| None | 0 mmol/g | Alumina | 600 °C | C | 92 | 274 |
| Boria | 3.0 mmol/g | Alumina | 600 °C | C | 760 | 5495 |
| Chloride | 0.10 mol/g | Alumina | 600 °C | C | 339 | 282 |
| Silica | 10 wt% | Alumina | 600 °C | C | 2820 | 984 |
| Zinc | 1.5 mmol/g | Alumina | 600 °C | C | 3294 | 466 |
| None | 0 mmol/g | Si-Alumina | 600 °C | C | 591 | 1882 |
| Boria | 3.0 mmol/g | Si-Alumina | 600 °C | C | 1320 | 2020 |
| None | 0 mmol/g | Cl-Alumina | 600 °C | A | 2800 | 282 |
| Tin | 2.0 mmol/g | Cl-Alumina | 600 °C | A | 6859 | 533 |
| Zinc | 1.5 mmol/g | Cl-Alumina | 600 °C | A | 14,000 | 466 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; McDaniel, M.P. Comparison of Support Effects on Phillips and Metallocene Catalysts. Catalysts 2021, 11, 842. https://doi.org/10.3390/catal11070842
Yang Q, McDaniel MP. Comparison of Support Effects on Phillips and Metallocene Catalysts. Catalysts. 2021; 11(7):842. https://doi.org/10.3390/catal11070842
Chicago/Turabian StyleYang, Qing, and Max Paul McDaniel. 2021. "Comparison of Support Effects on Phillips and Metallocene Catalysts" Catalysts 11, no. 7: 842. https://doi.org/10.3390/catal11070842
APA StyleYang, Q., & McDaniel, M. P. (2021). Comparison of Support Effects on Phillips and Metallocene Catalysts. Catalysts, 11(7), 842. https://doi.org/10.3390/catal11070842