
CpCo(III) Precatalysts for [2+2+2] Cycloadditions


Abstract

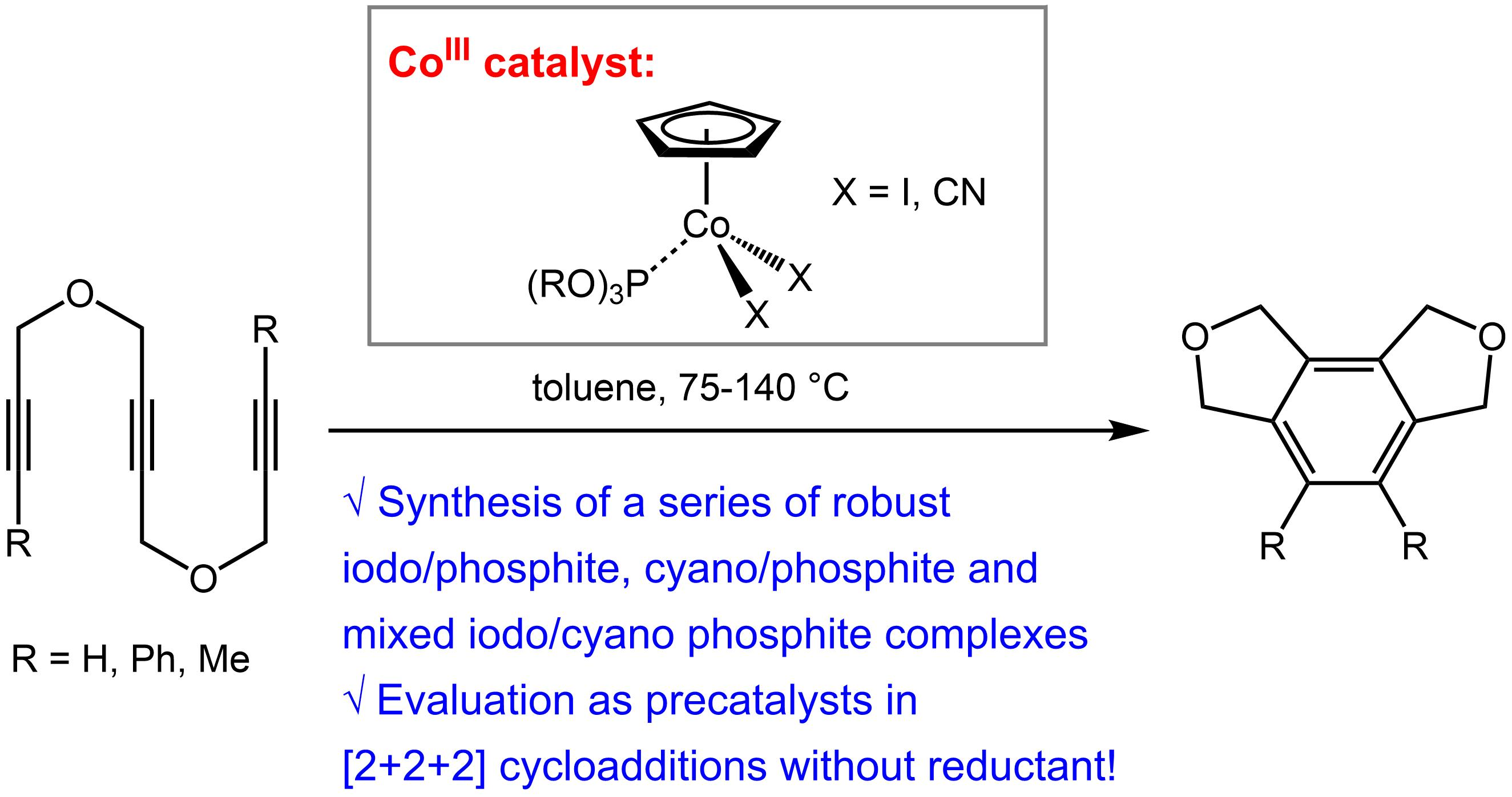
1. Introduction
2. Results and Discussion
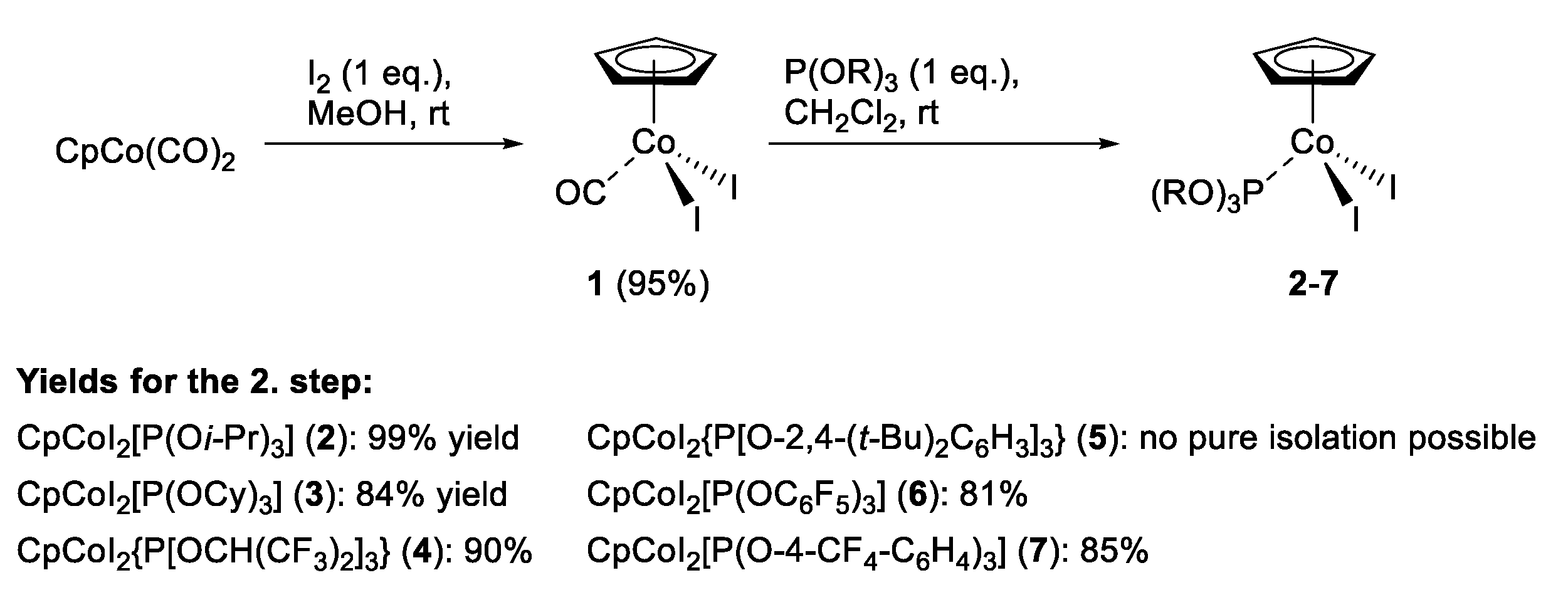
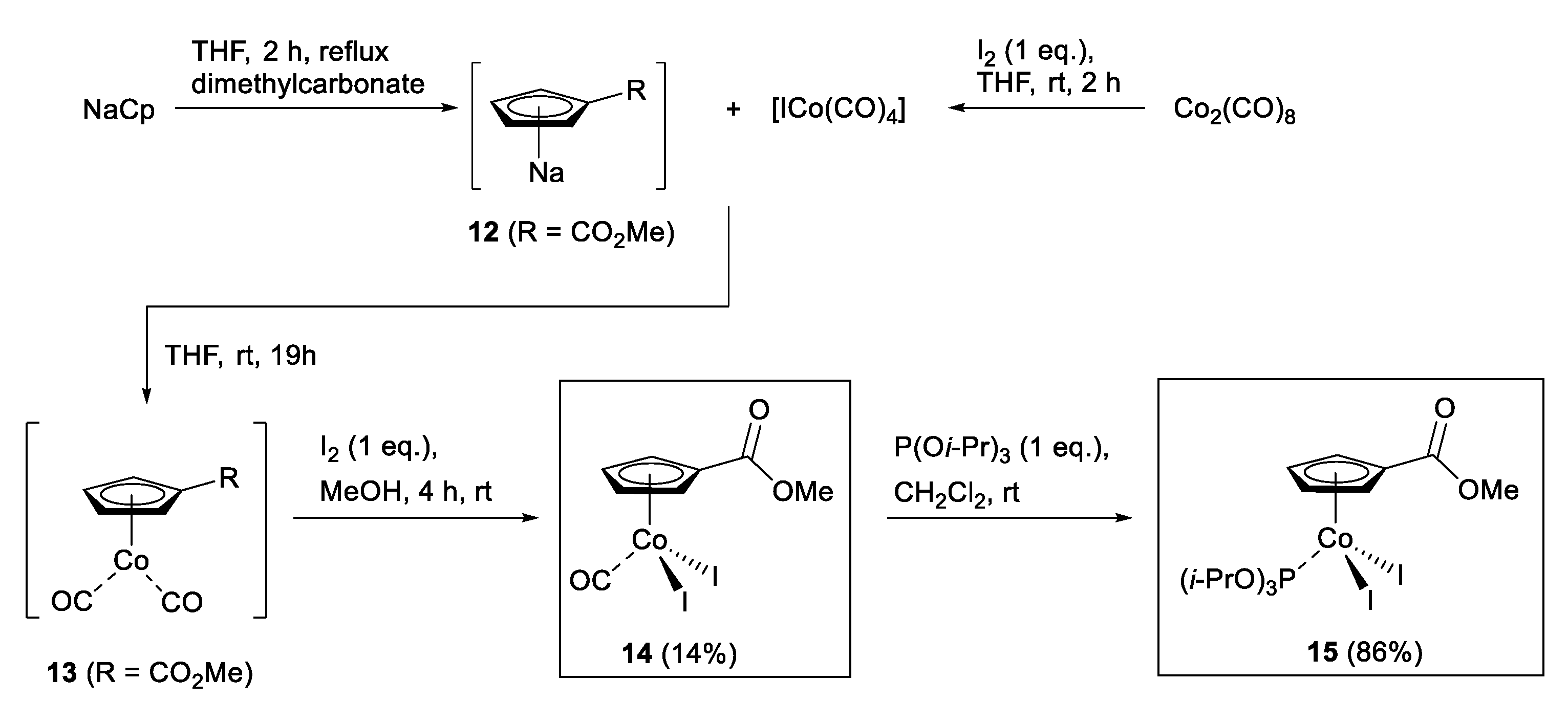
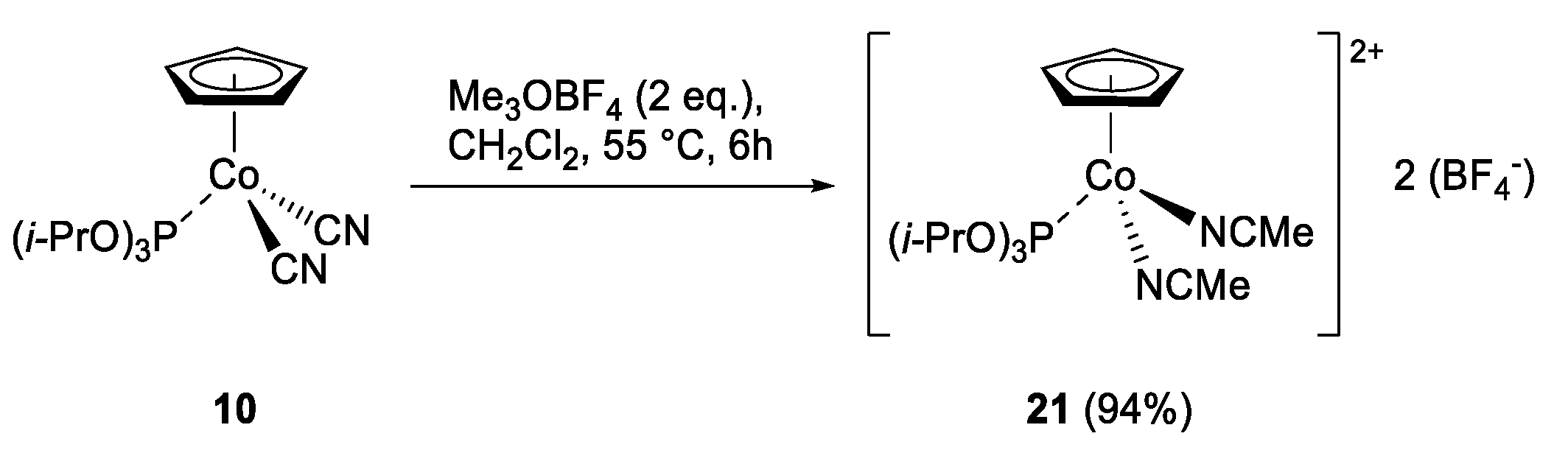
2.1. Synthesis of CpCo(III) Complexes
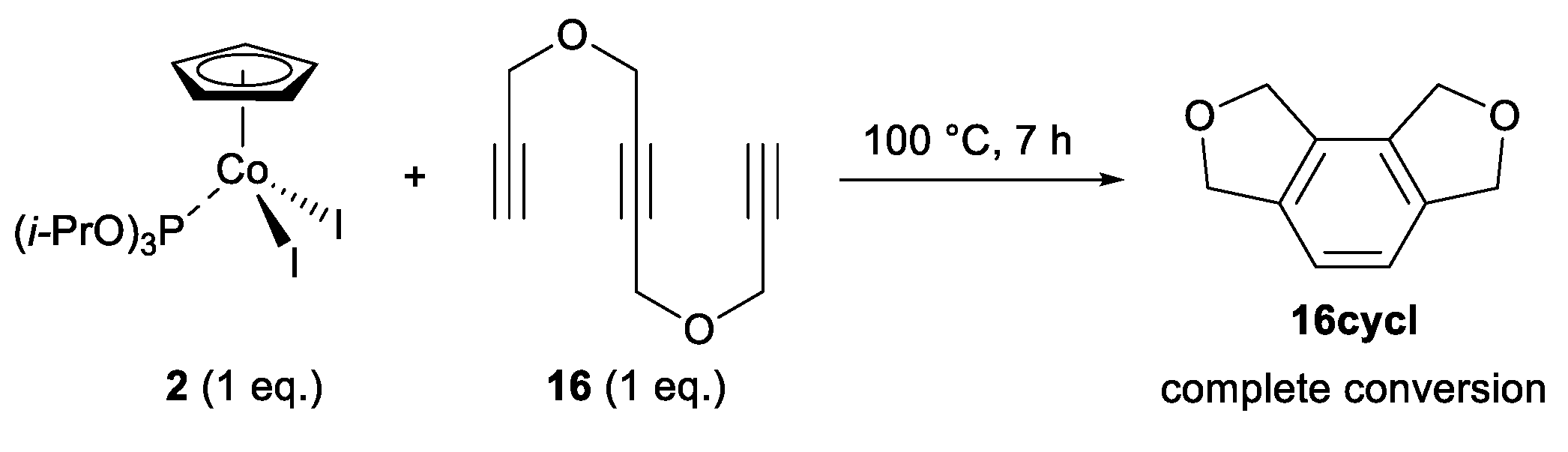
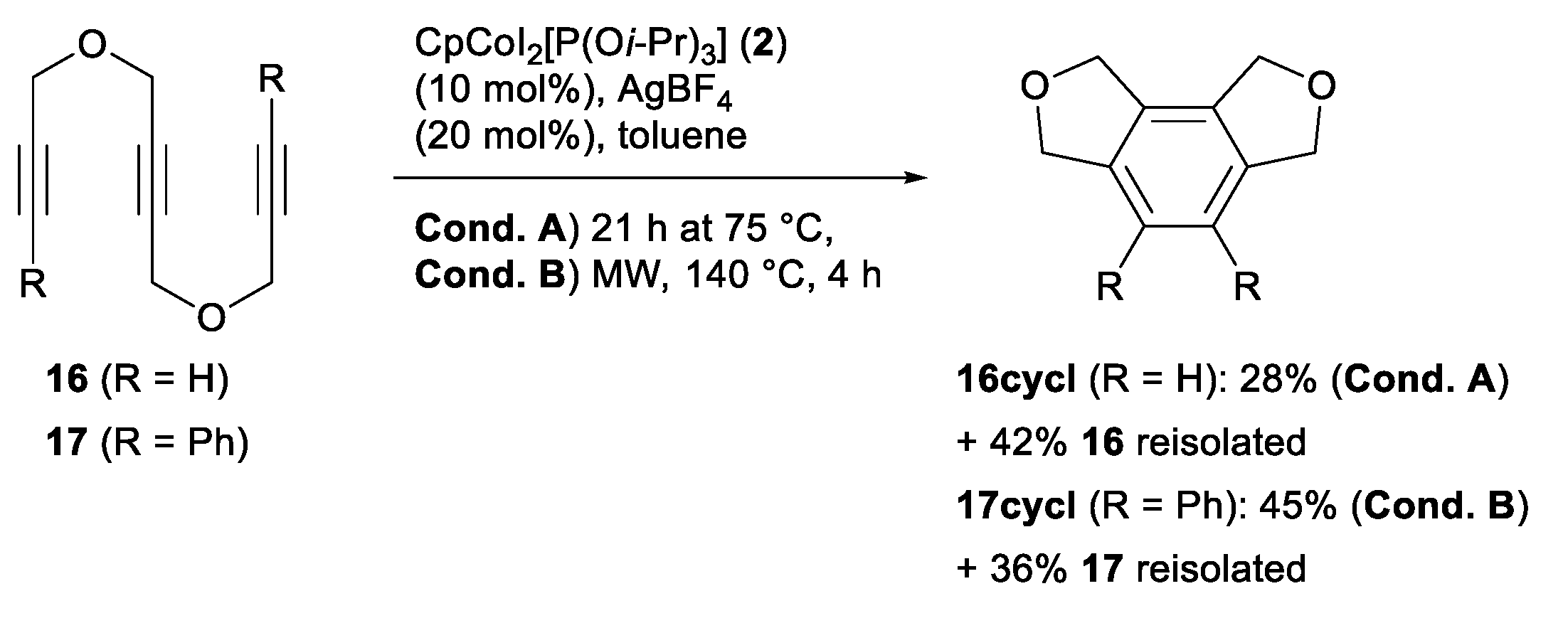
2.2. Screening on Catalytic Activity of the CpCo(III) Precatalysts
3. Experimental Section
3.1. Methods and Materials
3.2. Synthesis of Complexes
3.3. General Procedure 2 for Catalyst Screening Reactions with Triyne 16 (Table 1)
3.4. General Procedure 3 for Microwave Reactions with Precatalyst 2 (Table 2 and Scheme 4)
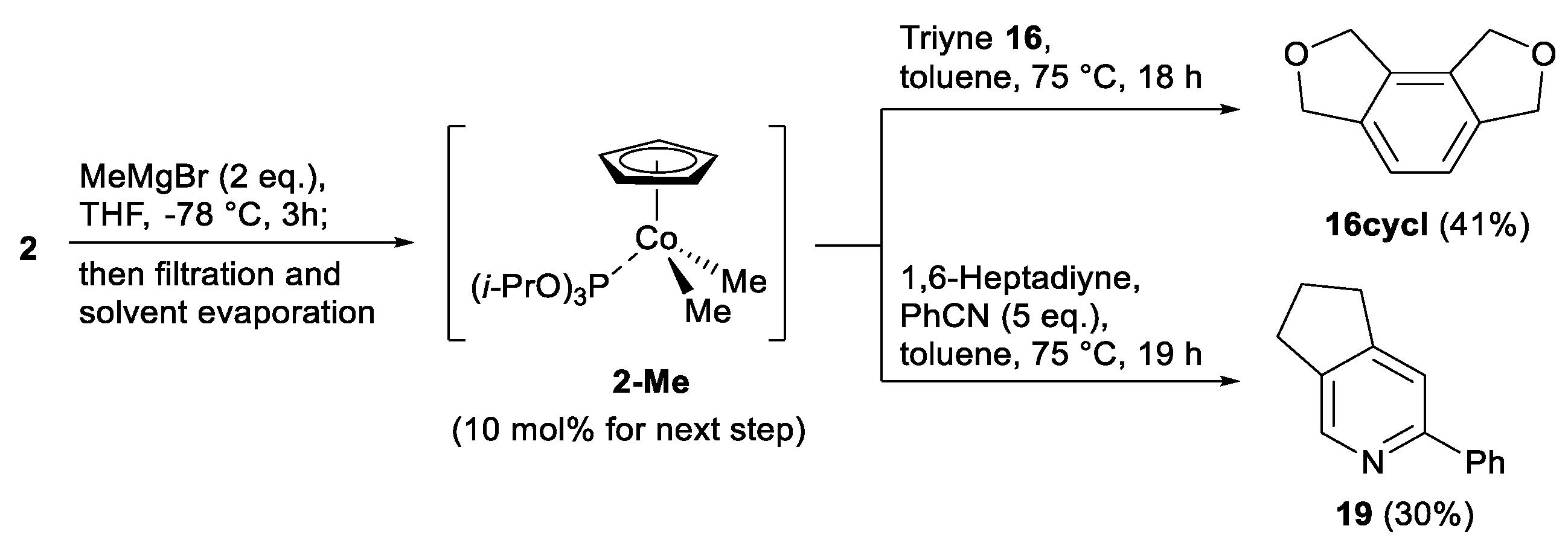
3.5. Synthesis and Reactions with Methylated Complexes 2 and 9 (Schemes 5 and 6)
3.6. Scavenger Experiment for Reductive Elimination Process (Scheme 7)
3.7. Reactivity of Precatalyst 2 in the Presence of AgBF4 (Scheme 9)
3.8. Reactivity of Isolated Precatalyst 21 (Scheme 11):
4. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yoshino, T.; Satake, S.; Matsunaga, S. Diverse Approaches for Enantioselective C−H Functionalization Reactions Using Group 9 CpxMIII Catalysts. Chem. Eur. J. 2020, 26, 7346–7357. [Google Scholar] [CrossRef] [PubMed]
- Loginov, D.A.; Shul’pina, L.S.; Muratov, D.V.; Shul’pin, G.B. Cyclopentadienyl cobalt(III) complexes: Synthetic and catalytic chemistry. Coord. Chem. Rev. 2019, 387, 1–31. [Google Scholar] [CrossRef]
- Pagano, J.K.; Stelmach, J.P.W.; Waterman, R. Cobalt-catalyzed ammonia borane dehydrocoupling and transfer hydrogenation under aerobic conditions. Dalton Trans. 2015, 44, 12074–12077. [Google Scholar] [CrossRef] [PubMed]
- Contakes, S.M.; Klausmeyer, K.K.; Rauchfuss, T.B. Cyanide compounds. Tricyanometalate building blocks and organometallic cyanide cages. Inorg. Synth. 2004, 34, 166–171. [Google Scholar]
- King, R.B. Organometallic Chemistry of the Transition Metals. XI. Some New Cyclopentadienyl Derivatives of Cobalt and Rhodium. Inorg. Chem. 1966, 5, 82–87. [Google Scholar] [CrossRef]
- Gardner, S.A.; Rausch, M.D. Reactions of π-cyclopentadienyltriphenylphosphinemetal diiodides and π-cyclopentadienylcarbonylmetal diiodides (M = Co, Rh, Ir) with 1,4-dilithio-1,2,3,4-tetraphenylbutadiene. J. Organomet. Chem. 1974, 78, 415–421. [Google Scholar] [CrossRef]
- Avilés, T.; Dinis, A.; Calhorda, M.J.; Pinto, P.; Félix, V.; Drew, M.G.B. Synthesis, X-ray structure, and theoretical studies of novel cationic mono-cyclopentadienyl complexes of Co(III): The orthometalation of trans-azobenzene. J. Organomet. Chem. 2001, 625, 186–194. [Google Scholar] [CrossRef]
- Kotha, S.; Lahiri, K.; Sreevani, G. Design and Synthesis of Aromatics through [2 + 2 + 2] Cyclotrimerization. Synlett 2018, 29, 2342–2361. [Google Scholar] [CrossRef]
- Babazadeh, M.; Soleimani-Amiri, S.; Vessally, E.; Hosseinian, A.; Edjlali, L. Transition metal-catalyzed [2 + 2 + 2] cycloaddition of nitrogen-linked 1,6-diynes: A straightforward route to fused pyrrolidine systems. RSC Adv. 2017, 7, 43716–43736. [Google Scholar] [CrossRef]
- Thakur, A.; Louie, J. Advances in Nickel-Catalyzed Cycloaddition Reactions to Construct Carbocycles and Heterocycles. Acc. Chem. Res. 2015, 48, 2354–2365. [Google Scholar] [CrossRef]
- Amatore, M.; Aubert, C. Recent Advances in Stereoselective [2 + 2 + 2] Cycloadditions. Eur. J. Org. Chem. 2015, 2015, 265–286. [Google Scholar] [CrossRef]
- Okamoto, S.; Sugiyama, Y. From the Development of Catalysts for Alkyne and Alkyne-Nitrile [2 + 2 + 2] Cycloaddition Reactions to Their Use in Polymerization Reactions. Synlett 2013, 24, 1044–1060. [Google Scholar] [CrossRef]
- Shibata, Y.; Tanaka, K. Rhodium-Catalyzed [2 + 2 + 2] Cycloaddition of Alkynes for the Synthesis of Substituted Benzenes: Catalysts, Reaction Scope, and Synthetic Applications. Synthesis 2012, 44, 323–350. [Google Scholar] [CrossRef]
- Okamoto, S. Synthesis of 2,2′-Bipyridines by Transition Metal-Catalyzed Alkyne/Nitrile [2 + 2 + 2] Cycloaddition Reactions. Heterocycles 2012, 85, 1579–1602. [Google Scholar] [CrossRef]
- Broere, D.L.J.; Ruijter, E. Recent Advances in Transition-Metal-Catalyzed [2 + 2 + 2]-Cyclo(co)trimerization Reactions. Synthesis 2012, 44, 2639–2672. [Google Scholar] [CrossRef]
- Thiel, I.; Jiao, H.; Spannenberg, A.; Hapke, M. Fine-Tuning the Reactivity and Stability by Systematic Ligand Variations in CpCoI Complexes as Catalysts for [2 + 2 + 2] Cycloaddition Reactions. Chem. Eur. J. 2013, 19, 2548–2554. [Google Scholar] [CrossRef]
- Towle, D.K.; Landon, S.J.; Brill, T.B.; Tulip, T.H. A double Michaelis-Arbusov rearrangement involving (η5-cyclopentadienyl)(carbonyl)diiodocobalt and trimethyl phosphite. Formation of the cobalt “supersandwich” complex. Organometallics 1982, 1, 295–301. [Google Scholar] [CrossRef]
- Dineen, J.A.; Pauson, P.L. Cyanide and isocyanide metal complexes: III. Further studies of the alkylation of cyano groups in cyclopentadienyl-cobalt and -iron complexes. J. Organomet. Chem. 1974, 71, 77–85. [Google Scholar] [CrossRef]
- Fischer, F.; Pientka, T.; Jiao, H.; Spannenberg, A.; Hapke, M. CpCo(I) precatalysts for [2 + 2 + 2] cycloaddition reactions: Synthesis and reactivity. Catal. Sci. Technol. 2020, 10, 8005–8014. [Google Scholar] [CrossRef]
- Morán, M. Reactions of (η5-C5H5)Co(CO)2 and (η5-EtMe4C5)Co(CO)2 with Iodine and Cyanogen Halides, XCN (X = Br OR I). Zeit. Naturfosch. B 1981, 36, 431–433. [Google Scholar] [CrossRef]
- Dineen, J.A.; Pauson, P.L. Some Cyclopentadienylcobalt cyanides and isocyanides. J. Organomet. Chem. 1972, 43, 203–207. [Google Scholar] [CrossRef]
- Fiebig, L.; Kuttner, J.; Hilt, G.; Schwarzer, M.C.; Frenking, G.; Schmalz, H.-G.; Schäfer, M. Cobalt Catalysis in the Gas Phase: Experimental Characterization of Cobalt(I) Complexes as Intermediates in Regioselective Diels–Alder Reactions. J. Org. Chem. 2013, 78, 10485–10493. [Google Scholar] [CrossRef] [PubMed]
- Chirila, P.G.; Whiteoak, C.J. Recent advances using [Cp*Co(CO)I2] catalysts as a powerful tool for C-H functionalisation. Dalton Trans. 2017, 46, 9721–9739. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Khan, B.A.; Roy, S.K.; Zain ul, A.; Mahmood, R.; Khan, J.; Ashraf, H. Theoretical insights on the C-C bond reductive elimination from Co(III) center. Comp. Theor. Chem. 2018, 1130, 140–147. [Google Scholar] [CrossRef]
- Yamazaki, H.; Hagihara, N. Preparations and reactions of alkylcobalt complexes and diphenylacetylenecobalt complexes having a π-cyclopentadienyl group as a ligand. J. Organomet. Chem. 1970, 21, 431–443. [Google Scholar] [CrossRef]
- Evitt, E.R.; Bergman, R.G. Substitution, alkyl-transfer, and thermal decomposition reactions of η5-cyclopentadienyl(triphenylphosphine)dimethylcobalt(III). J. Am. Chem. Soc. 1980, 102, 7003–7011. [Google Scholar] [CrossRef]
- Roglans, A.; Pla-Quintana, A.; Sola, M. Mechanistic Studies of Transition-Metal-Catalyzed [2 + 2 + 2] Cycloaddition Reactions. Chem. Rev. 2021, 121, 1894–1979. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| # | Precatalyst | Time (h) | Temperature (°C) | Yield (%) a |
|---|---|---|---|---|
| Entry 1 | 1 | 19 | 75 | 38 |
| Entry 2 | 2 | 22 | 75 | 55 |
| Entry 3 | 3 | 20 | 75 | 22 |
| Entry 4 | 4 | 21 | 75 | 27 |
| Entry 5 | 6 | 21 | 75 | 25 |
| Entry 6 | 7 | 21 | 75 | 8 |
| Entry 7 | 8 | 14 | 75 | - b,c |
| Entry 8 | 9 | 21 | 75 | - b,c |
| Entry 9 | 9 | 23 | 100 | - b,c |
| Entry 10 | 10 | 21 | 75 | - b,d |
| Entry 11 | 10 | 21 | 105 | 84 |
| Entry 12 | 14 | 16 | 75 | 48 |
| Entry 13 | 15 | 20 | 75 | 44 |
| Entry 14 | CpCo(CO)2 | 19 | 75 | - b,e |

| # | 2 (mol%) | Time (h) | Temperature (°C) | Yield (%) a |
|---|---|---|---|---|
| Entry 1 | 10 | 4 | 100 (MW) b | 25 |
| Entry 2 | 10 | 12 | 100 (MW) | 41 |
| Entry 3 | 20 | 4 | 100 (MW) | 56 |
| Entry 4 | 10 | 18 | 75 | 20 (58) c,f |
| Entry 5 | 10 | 18 | 75 | 5 (74) d,f |
| Entry 6 | 20 (2 × 10) | 24 (2 × 12 h) | 100 | 43 (15) f |
| Entry 7 | 10 | 48 | 75 | 53 (8) e,f |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, F.; Eder, M.; Hapke, M. CpCo(III) Precatalysts for [2+2+2] Cycloadditions. Catalysts 2021, 11, 596. https://doi.org/10.3390/catal11050596
Fischer F, Eder M, Hapke M. CpCo(III) Precatalysts for [2+2+2] Cycloadditions. Catalysts. 2021; 11(5):596. https://doi.org/10.3390/catal11050596
Chicago/Turabian StyleFischer, Fabian, Michael Eder, and Marko Hapke. 2021. "CpCo(III) Precatalysts for [2+2+2] Cycloadditions" Catalysts 11, no. 5: 596. https://doi.org/10.3390/catal11050596
APA StyleFischer, F., Eder, M., & Hapke, M. (2021). CpCo(III) Precatalysts for [2+2+2] Cycloadditions. Catalysts, 11(5), 596. https://doi.org/10.3390/catal11050596