
Non-Covalent Interactions in Enantioselective Organocatalysis: Theoretical and Mechanistic Studies of Reactions Mediated by Dual H-Bond Donors, Bifunctional Squaramides, Thioureas and Related Catalysts




Abstract
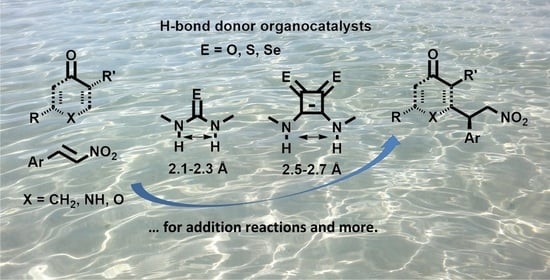
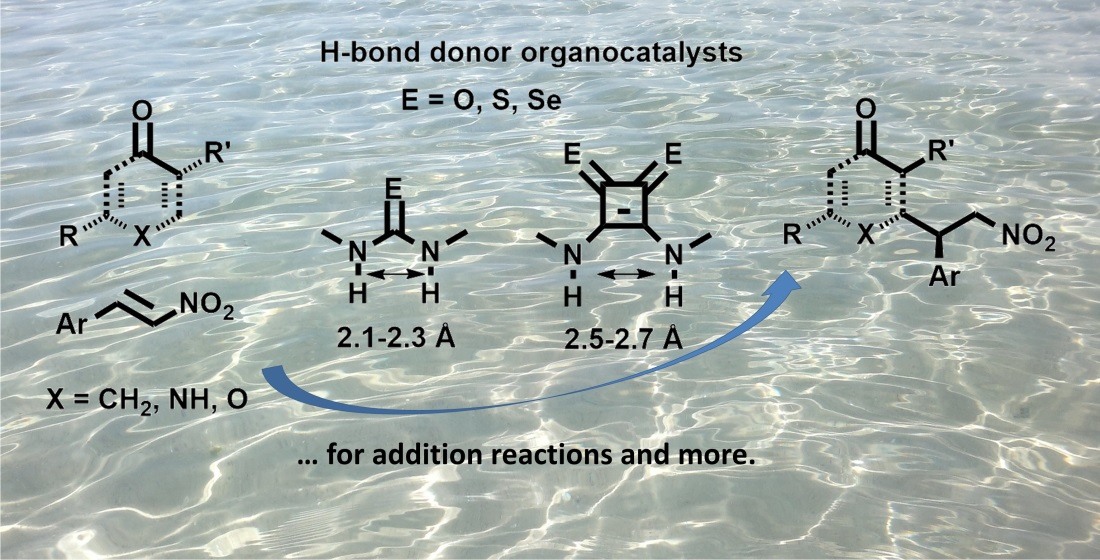
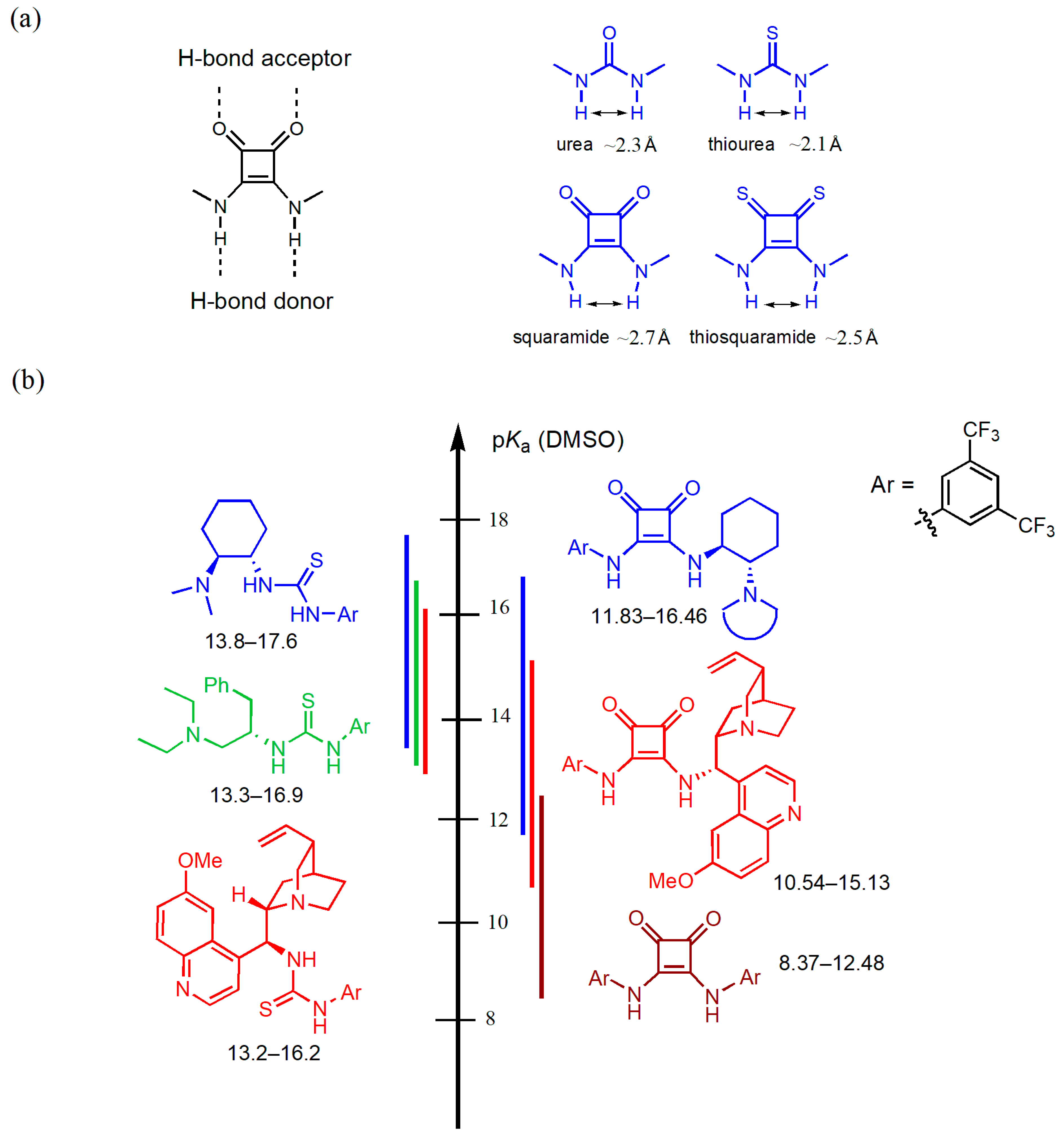
1. Introduction
- Introduction;
- Michael and other conjugate addition reactions;
- Cycloaddition reactions;
- Aldol and Henry reactions;
- Miscellaneous reactions involving anion-binding catalysis;
- Miscellaneous;
- Conclusions.
2. Michael and Other Conjugate Addition Reactions
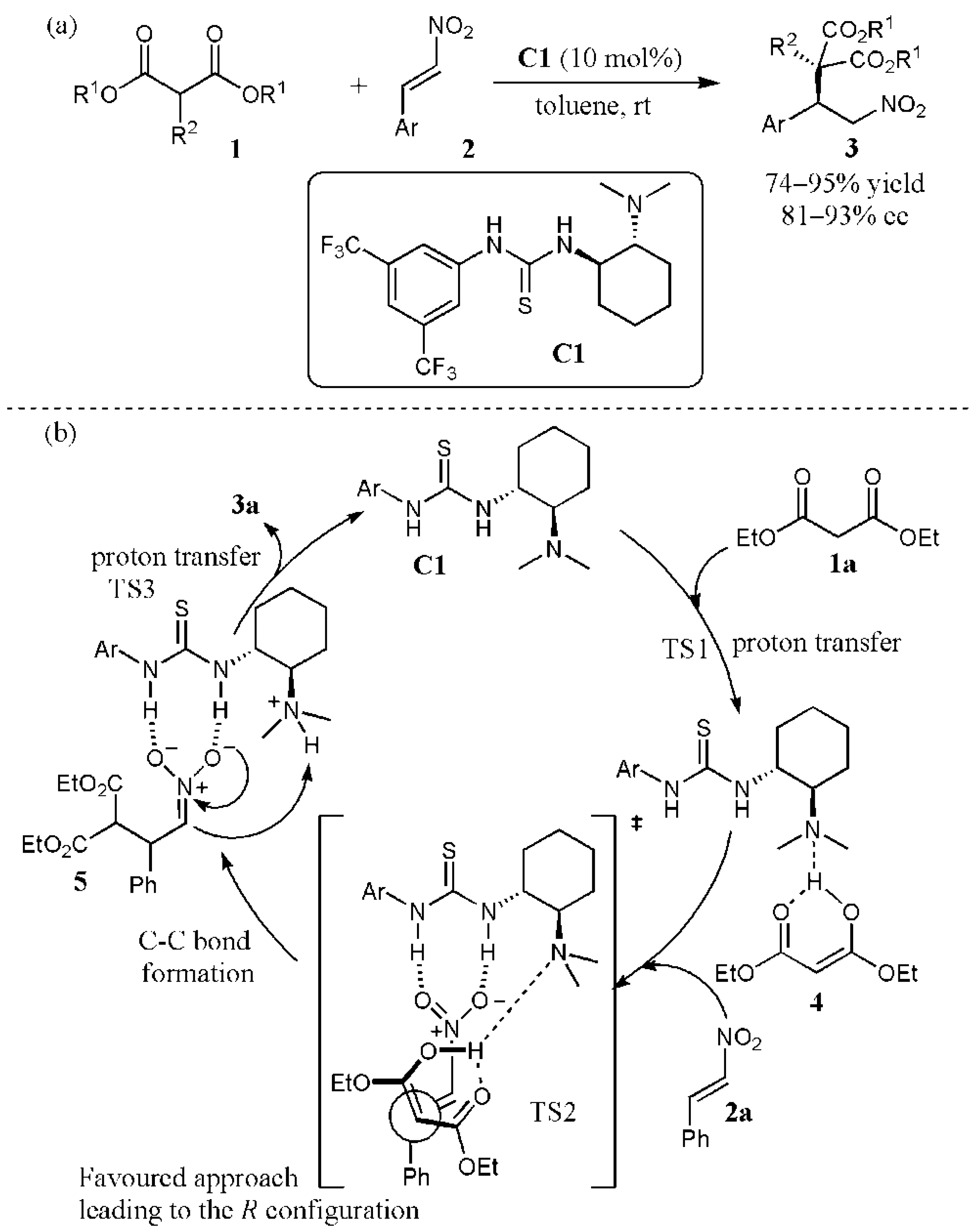
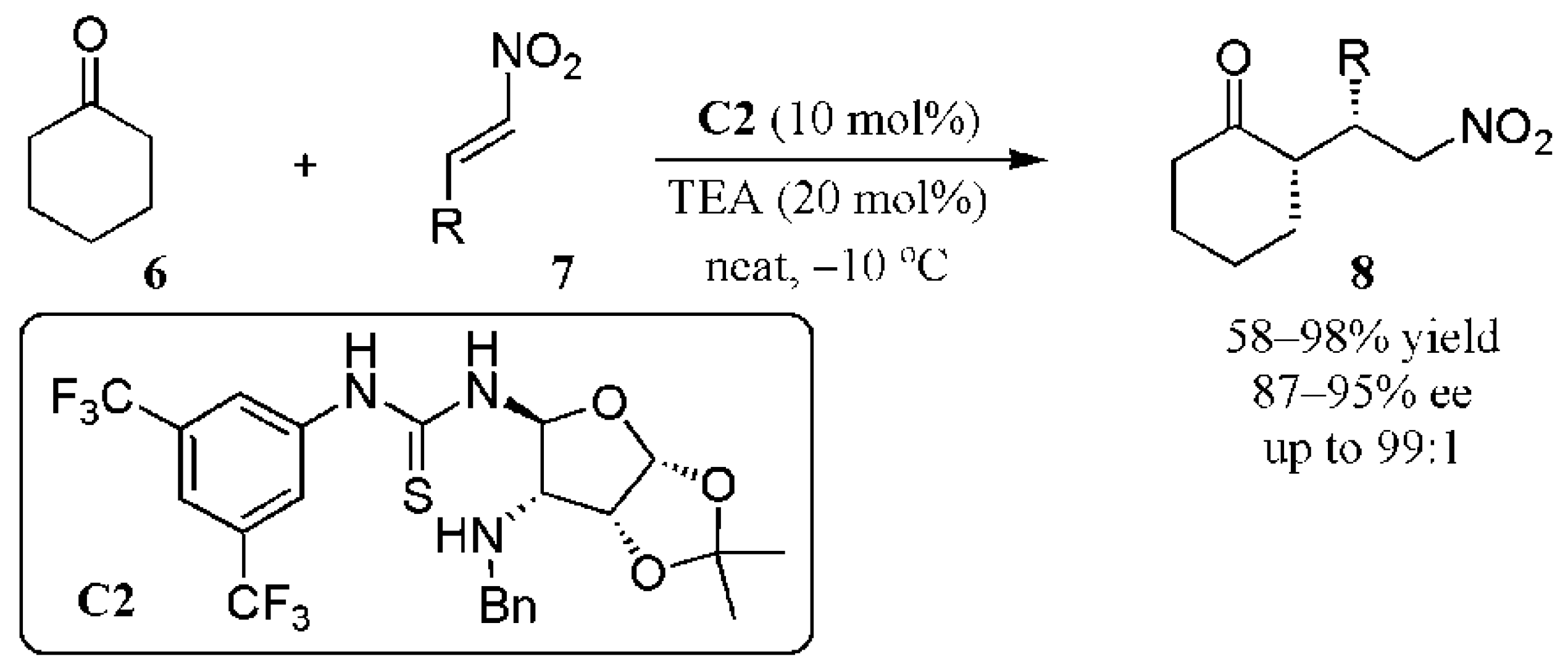
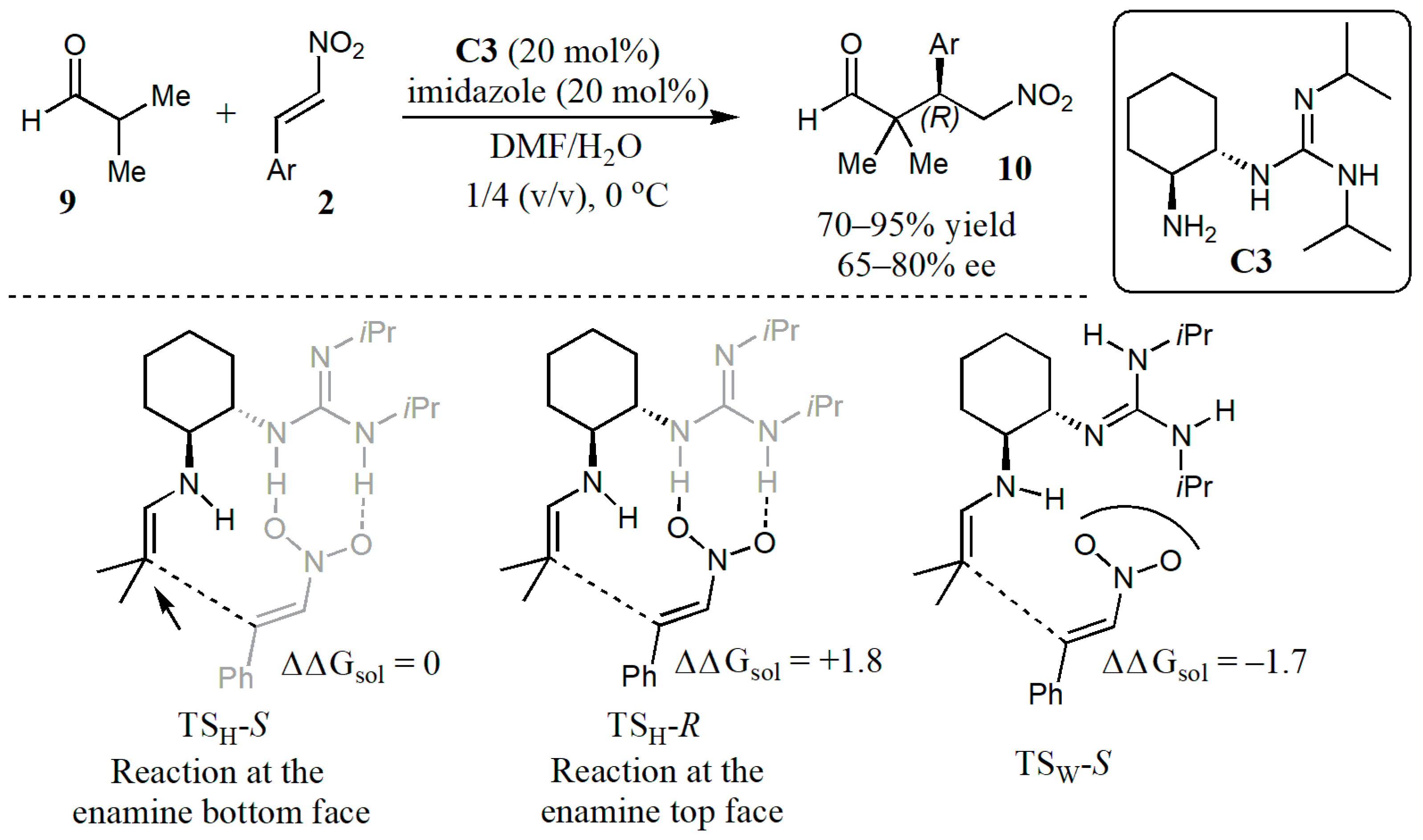
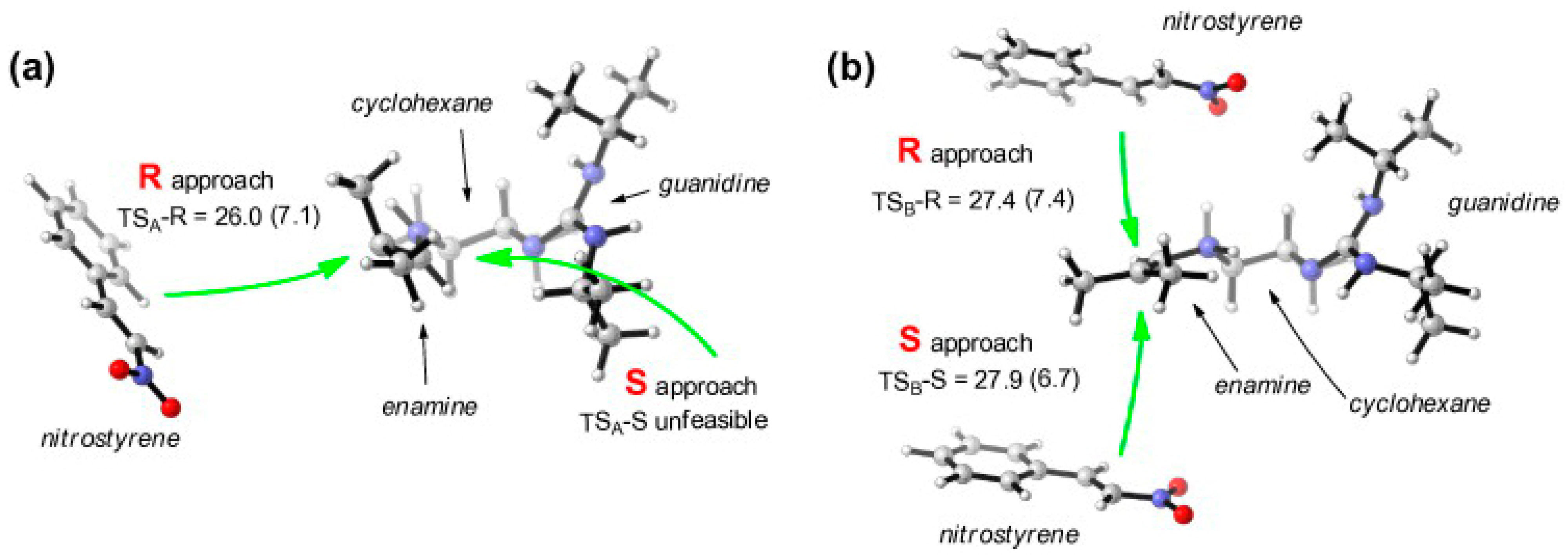
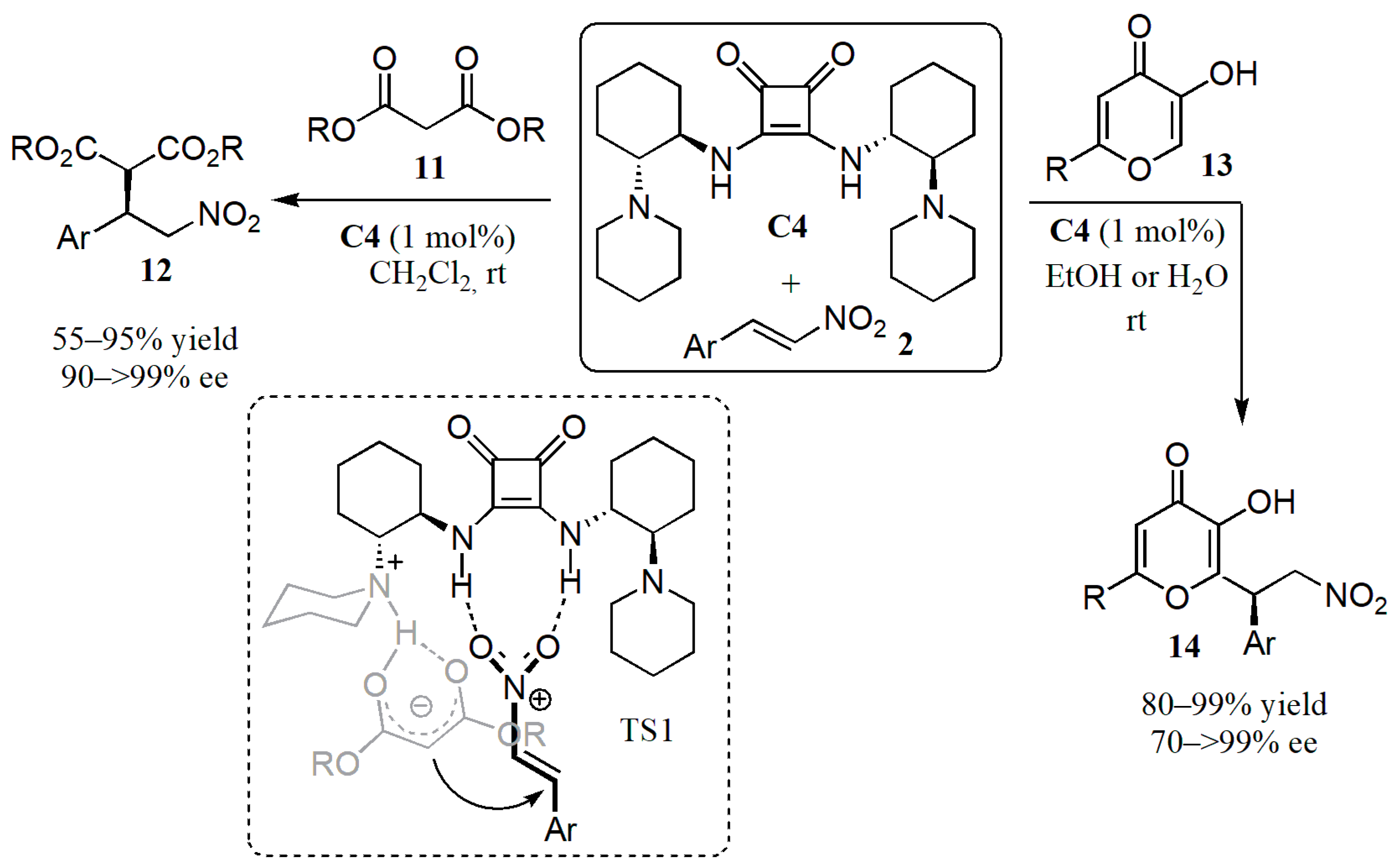
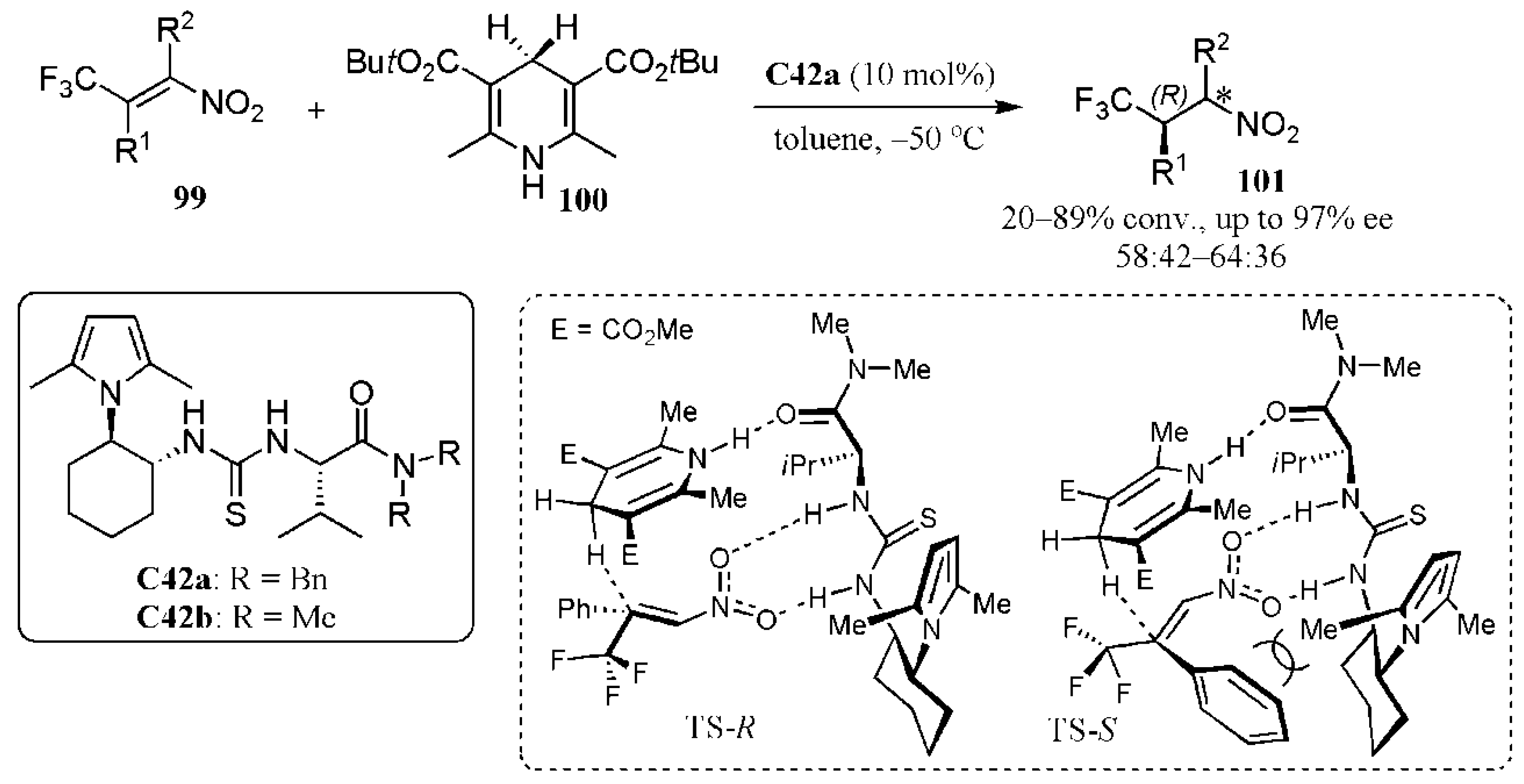
2.1. Additions to Nitroalkenes
2.2. Additions to Enones and Unsaturated Esters
2.3. Cascade Reactions Involving Conjugate Addition
3. Cycloaddition Reactions
4. Aldol and Henry Reactions
5. Miscellaneous Reactions Involving Anion-Binding Catalysis
6. Miscellaneous
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, B.-L.; Li, J.-H.; Du, D.-M. Squaramide-catalyzed asymmetric reactions. Chem. Rec. 2017, 17, 994–1018. [Google Scholar] [CrossRef]
- Parvin, T.; Yadava, R.; Choudhurym, L.H. Recent applications of thiourea-based organocatalysts in asymmetric multicomponent reactions (AMCRs). Org. Biomol. Chem. 2020, 18, 5513–5532. [Google Scholar] [CrossRef] [PubMed]
- Limnios, D.; Kokotos, C.G. Ureas and thioureas as asymmetric organocatalysts. In Sustainable Catalysis: Without Metals or Other Endangered Elements, Part 2; North, M., Ed.; The Royal Society of Chemistry: London, UK, 2016; pp. 196–255. [Google Scholar]
- Xiang, S.H.; Tan, B. Advances in asymmetric organocatalysis over the last 10 years. Nat. Commun. 2020, 11, 3786. [Google Scholar] [CrossRef]
- Ni, X.; Li, X.; Wang, Z.; Cheng, J.-P. Squaramide equilibrium acidities in DMSO. Org. Lett. 2014, 16, 1786–1789. [Google Scholar] [CrossRef] [PubMed]
- Storer, R.I.; Aciro, C.; Jones, L.H. Squaramides: Physical properties, synthesis and applications. Chem. Soc. Rev. 2011, 40, 2330–2346. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K.A. Squaramides: Bridging from molecular recognition to bifunctional organocatalysis. Chem. Eur. J. 2011, 17, 6890–6899. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.; Cheng, J.-P. Recent progress in equilibrium acidity studies of organocatalysts. Synlett 2019, 30, 1940–1949. [Google Scholar] [CrossRef]
- Li, X.; Deng, H.; Zhang, B.; Li, J.; Zhang, L.; Luo, S.; Cheng, J.-P. Physical organic study of structure–activity–enantioselectivity relationships in asymmetric bifunctional thiourea catalysis: Hints for the design of new organocatalysts. Chem. Eur. J. 2010, 16, 450–455. [Google Scholar] [CrossRef]
- Jakab, G.; Tancon, C.; Zhang, Z.; Lippert, K.M.; Schreiner, P.R. (Thio)urea organocatalyst equilibrium acidities in DMSO. Org. Lett. 2012, 14, 1724–1727. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Wheeler, S.E. Origin of the superior performance of (thio)squaramides over (thio)ureas in organocatalysis. Chem. Eur. J. 2013, 19, 15141–15147. [Google Scholar] [CrossRef]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral squaramide derivatives are excellent hydrogen bond donor catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef]
- Žabka, M.; Šebesta, R. Experimental and theoretical studies in hydrogen-bonding organocatalysis. Molecules 2015, 20, 15500–15524. [Google Scholar] [CrossRef]
- Faisca Phillips, A.M. Organocatalytic asymmetric nitro-Michael reactions. Curr. Org. Synth. 2016, 13, 687–725. [Google Scholar] [CrossRef]
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Gómez, C.; Guillena, G.; Pastor, I.M.; Ramón, D.J. Recent advances in asymmetric organocatalyzed conjugate additions to nitroalkenes. Molecules 2017, 22, 895. [Google Scholar] [CrossRef] [PubMed]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael reaction of malonates to nitroolefins catalyzed by bifunctional organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef] [PubMed]
- Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. Enantio- and diastereoselective Michael reaction of 1,3-dicarbonyl compounds to nitroolefins catalyzed by a bifunctional thiourea. J. Am. Chem. Soc. 2005, 127, 119–125. [Google Scholar] [CrossRef]
- Zhu, R.; Zhang, D.; Wu, J.; Liu, C. Theoretical study of the bifunctional-urea catalyzed Michael reaction of 1,3-dicarbonyl compounds and nitroolefins: Reaction mechanism and enantioselectivity. Tetrahedron Asymmetry 2006, 17, 1611–1616. [Google Scholar] [CrossRef]
- Hamza, A.; Schubert, G.; Soós, T.; Pápai, I. Theoretical studies on the bifunctionality of chiral thiourea-based organocatalysts: competing routes to C−C bond formation. J. Am. Chem. Soc. 2006, 128, 13151–13160. [Google Scholar] [CrossRef] [PubMed]
- Kreienborgm, N.M.; Pollok, C.H.; Merten, C. Towards an observation of active conformations in asymmetric catalysis: Interaction-induced conformational preferences of a chiral thiourea model compound. Chem. Eur. J. 2016, 22, 12455–12463. [Google Scholar] [CrossRef] [PubMed]
- Supady, A.; Hecht, S.; Baldauf, C. About underappreciated yet active conformations of thiourea organocatalysts. Org. Lett. 2017, 19, 4199–4202. [Google Scholar] [CrossRef]
- Luchini, G.; Ascough, D.M.H.; Alegre-Requena, J.V.; Gouverneur, V.; Paton, R.S. Data-mining the diaryl(thio)urea conformational landscape: Understanding the contrasting behavior of ureas and thioureas with quantum chemistry. Tetrahedron 2019, 75, 697–702. [Google Scholar] [CrossRef]
- Neuvonen, A.J.; Noutsias, D.; Topić, F.; Rissanen, K.; Földes, T.; Pápai, I.; Pihko, P.M. Dynamic refolding of ion-pair catalysts in response to different anions. J. Org. Chem. 2019, 84, 15009–15019. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-L.; Zhang, Y.; Liu, C.; Zheng, A.-M.; Wang, W. Mechanistic insights on organocatalytic enantioselective decarboxylative protonation by epicinchona-thiourea hybrid derivatives. J. Org. Chem. 2012, 77, 9813–9825. [Google Scholar] [CrossRef]
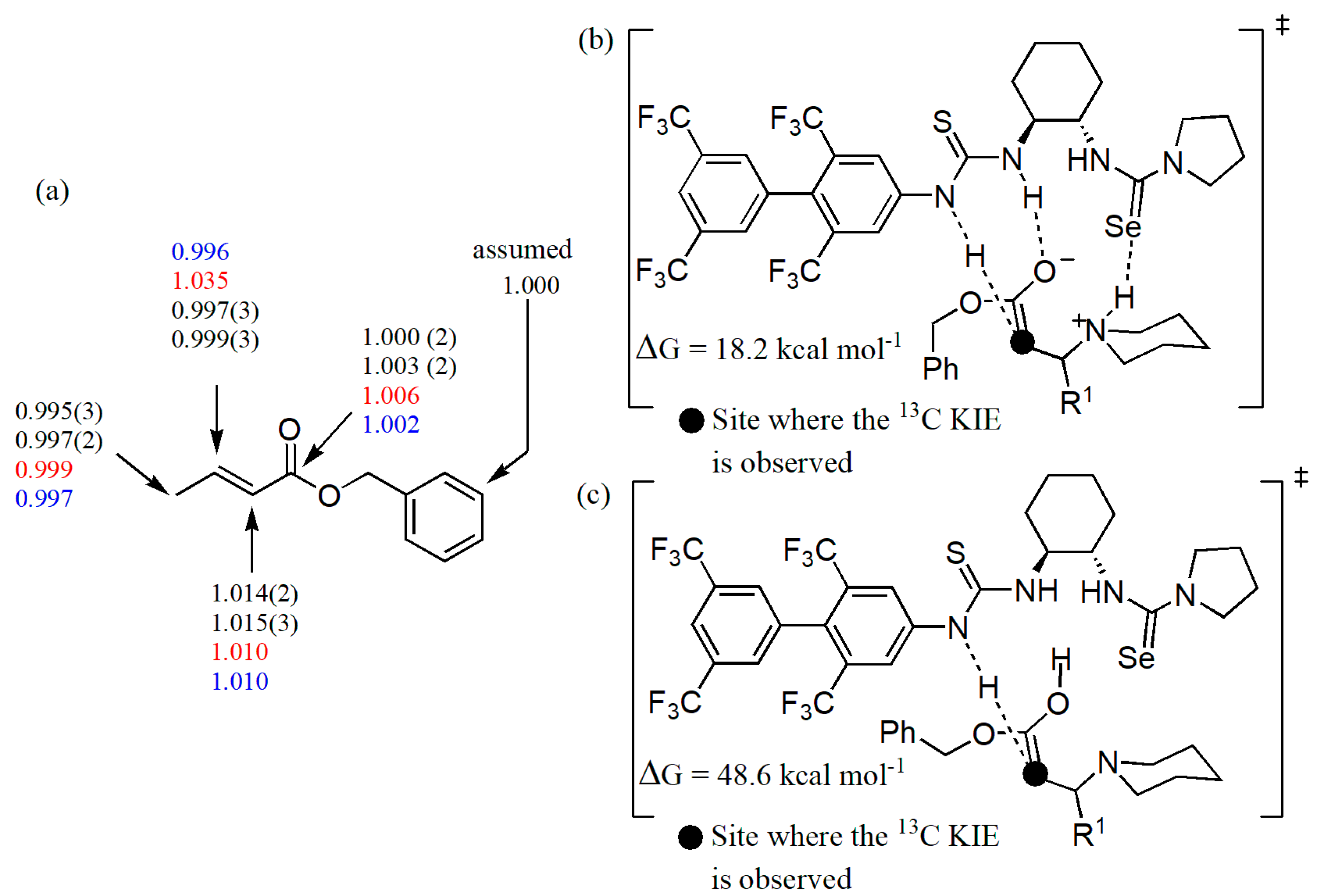
- Izzo, J.A.; Myshchuk, Y.; Hirschi, J.S.; Vetticatt, M.J. Transition state analysis of an enantioselective Michael addition by a bifunctional thiourea organocatalyst. Org. Biomol. Chem. 2019, 17, 3934–3939. [Google Scholar] [CrossRef] [PubMed]
- Singleton, D.A.; Thomas, A.A. High-precision simultaneous determination of multiple small kinetic isotope effects at natural abundance. J. Am. Chem. Soc. 1995, 117, 9357–9358. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L.J. Effect of the damping function in dispersion corrected density functional theory. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Pliego, J.R., Jr. Theoretical free energy profile and benchmarking of functionals for amino-thiourea organocatalyzed nitro-Michael addition reaction. Phys. Chem. Chem. Phys. 2020, 22, 11529–11536. [Google Scholar] [CrossRef] [PubMed]
- Faisca Phillips, A.M. Applications of carbohydrate-based organocatalysts in enantioselective synthesis. Eur. J. Org. Chem. 2014, 7291–7303. [Google Scholar] [CrossRef]
- Azad, C.S.; Khan, I.A.; Narula, A.K. Organocatalyzed asymmetric Michael addition by an efficient bifunctional carbohydrate–thiourea hybrid with mechanistic DFT analysis. Org. Biomol. Chem. 2016, 14, 11454–11461. [Google Scholar] [CrossRef] [PubMed]
- Avila, A.; Chinchilla, R.; Fiser, B.; Gómez-Bengoa, E.; Nájera, C. Enantioselective Michael addition of isobutyraldehyde to nitroalkenes organocatalyzed by chiral primary amine-guanidines. Tetrahedron Asymmetry 2014, 25, 462–467. [Google Scholar] [CrossRef]
- Kucherenko, A.S.; Kostenko, A.A.; Komogortsev, A.N.; Lichitsky, B.V.; Fedotov, M.Y.; Zlotin, S.G. C2-Symmetric chiral squaramide, recyclable organocatalyst for asymmetric Michael reactions. J. Org. Chem. 2019, 84, 4304–4311. [Google Scholar] [CrossRef]
- Rombola, M.; Sumaria, C.S.; Montgomery, T.D.; Rawal, V.H. Development of chiral, bifunctional thiosquaramides: Enantioselective Michael additions of barbituric acids to nitroalkenes. J. Am. Chem. Soc. 2017, 139, 5297–5300. [Google Scholar] [CrossRef] [PubMed]
- Krautwald, S.; Carreira, E.M. Stereodivergence in asymmetric catalysis. J. Am. Chem. Soc. 2017, 139, 5627–5639. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.P.; Nájera, C.; Yus, M. Stereodivergent catalysis. Chem. Rev. 2018, 118, 5080–5200. [Google Scholar] [CrossRef]
- Ding, P.-G.; Zhou, F.; Wang, X.; Zhao, Q.-H.; Yu, J.-S.; Zhou, J. H-bond donor-directed switching of diastereoselectivity in the Michael addition of azido ketones to nitroolefins. Chem. Sci. 2020, 11, 3852–3861. [Google Scholar] [CrossRef]
- Herrera, R.P.; Sgarzani, V.; Bernardi, L.; Ricci, A. Catalytic enantioselective Friedel–Crafts alkylation of indoles by using a simple thiourea organocatalyst. Angew. Chem. Int. Ed. 2005, 44, 6576–6579. [Google Scholar] [CrossRef]
- Roca-López, D.; Marqués-López, E.; Alcaine, A.; Merino, P.; Herrera, R.P. A Friedel–Crafts alkylation mechanism using an aminoindanol-derived thiourea catalyst. Org. Biomol. Chem. 2014, 12, 4503–4510. [Google Scholar] [CrossRef]
- Pettersen, D.; Herrera, R.P.; Bernardi, L.; Fini, F.; Sgarzani, V.; Fernández, R.; Lassaletta, J.M.; Ricci, A. A broadened scope for the use of hydrazones as neutral nucleophiles in the presence of H-bonding organocatalysts. Synlett 2006, 239–242. [Google Scholar] [CrossRef]
- Etter, M.C.; Panunto, T.W. 1,3-Bis(m-nitrophenyl)urea: An exceptionally good complexing agent for proton acceptors. J. Am. Chem. Soc. 1988, 110, 5896–5897. [Google Scholar] [CrossRef]
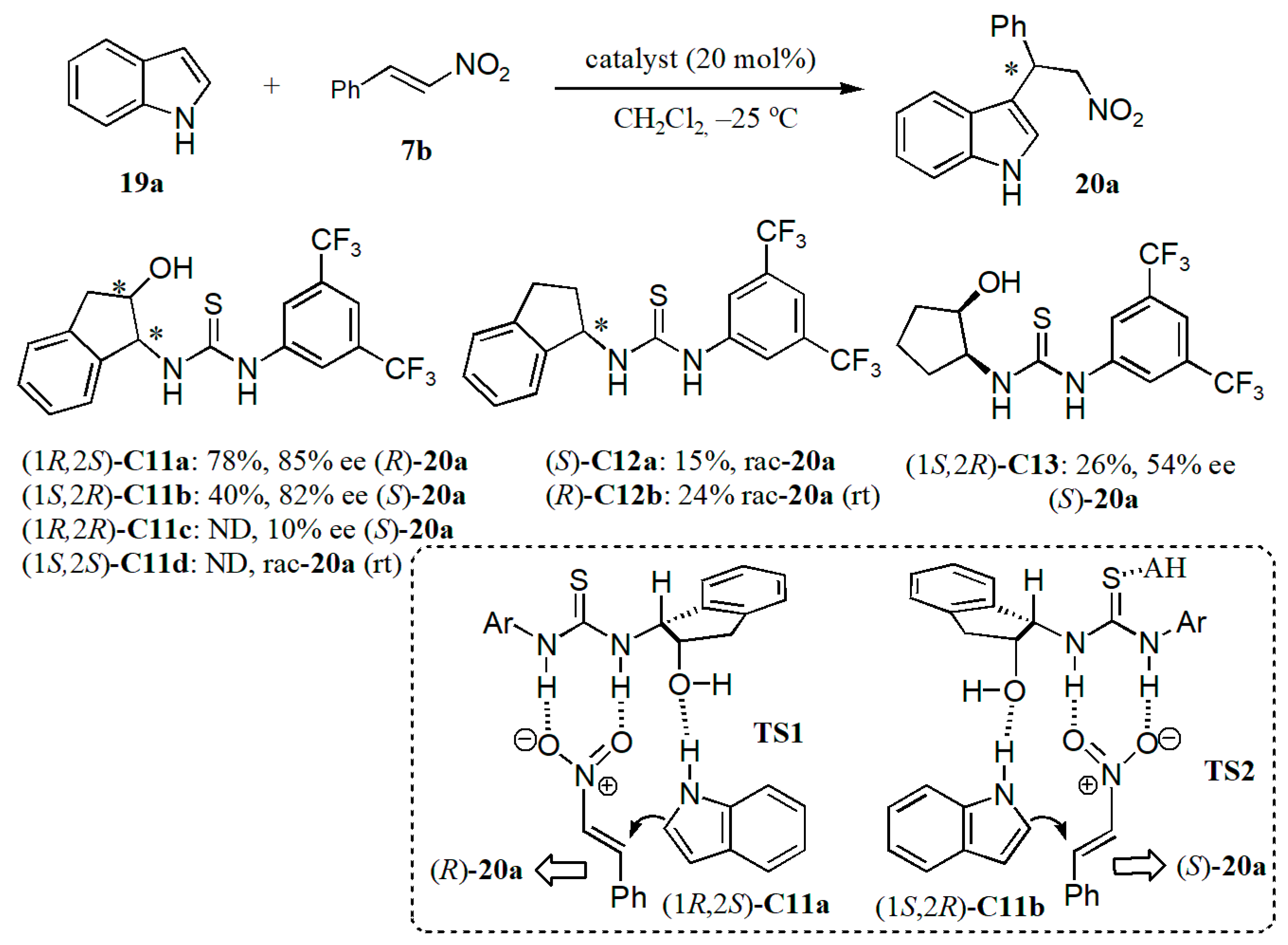
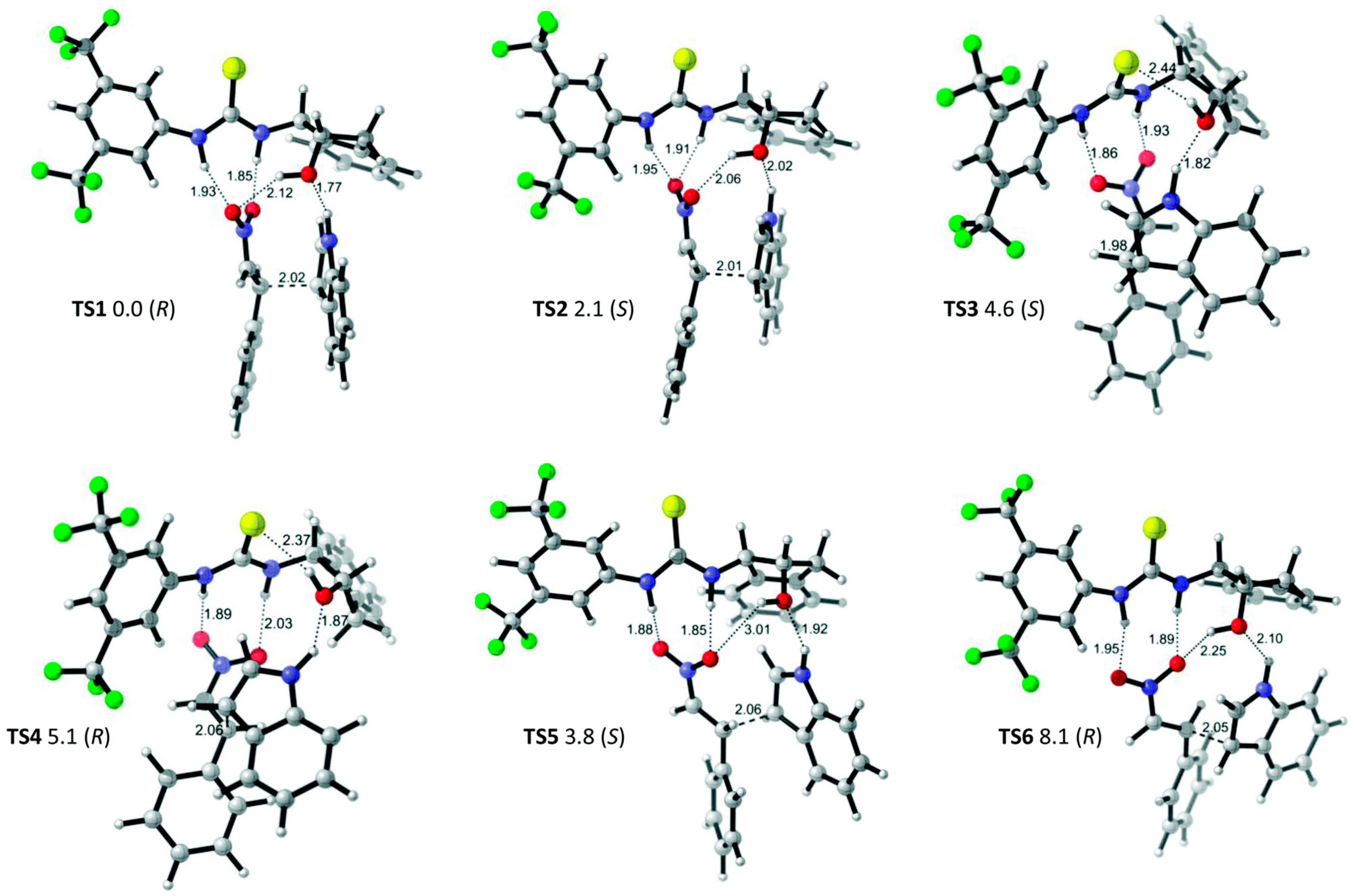
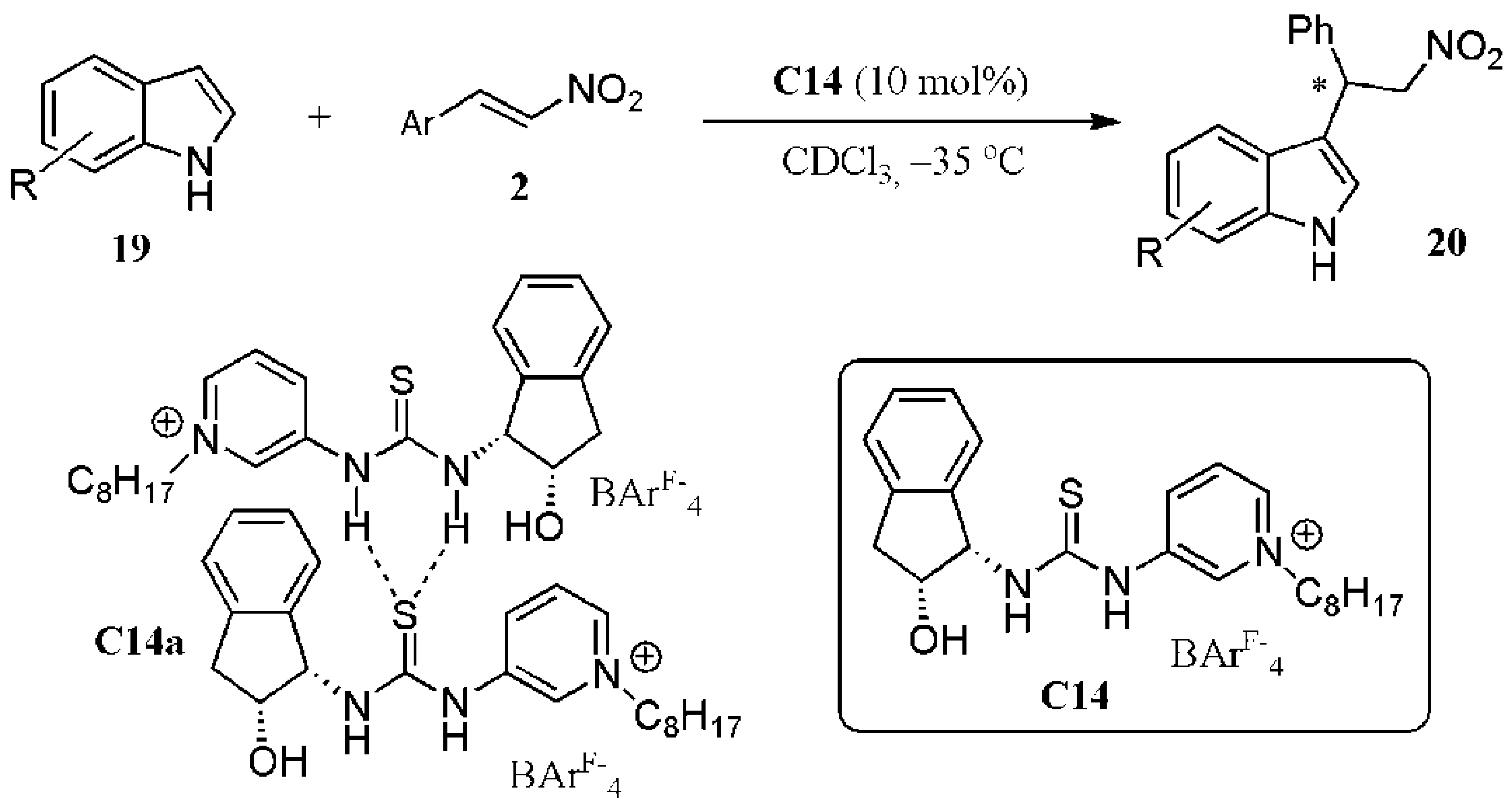
- Fan, Y.; Kass, S.R. Enantioselective Friedel–Crafts alkylation between nitroalkenes and indoles catalyzed by charge activated thiourea organocatalysts. J. Org. Chem. 2017, 82, 13288–13296. [Google Scholar] [CrossRef]
- Li, B.-J.; Jiang, L.; Liu, M.; Chen, Y.-C.; Ding, L.-S.; Wu, Y. Asymmetric Michael addition of arylthiols to α,β-unsaturated carbonyl compounds catalyzed by bifunctional organocatalysts. Synlett 2005, 603–606. [Google Scholar] [CrossRef]
- Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional cinchona organocatalysts. Org. Lett. 2005, 7, 1967–1969. [Google Scholar] [CrossRef] [PubMed]
- McCooey, S.H.; Connon, S.J. Urea- and thiourea-substituted cinchona alkaloid derivatives as highly efficient bifunctional organocatalysts for the asymmetric addition of malonate to nitroalkenes: Inversion of configuration at C9 dramatically improves catalyst performance. Angew. Chem. Int. Ed. 2005, 44, 6367–6370. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Dixon, D.J.; Hynes, P.S. Enantioselective organocatalytic Michael addition of malonate esters to nitroolefins using bifunctional cinchonine derivatives. Chem. Commun. 2005, 4481–4483. [Google Scholar] [CrossRef]
- Grayson, M.N. Mechanism and origins of stereoselectivity in the cinchona thiourea- and squaramide-catalyzed asymmetric Michael addition of nitroalkanes to enones. J. Org. Chem. 2017, 82, 4396–4401. [Google Scholar] [CrossRef]
- Grayson, M.N.; Houk, K.N. Cinchona urea-catalyzed asymmetric sulfa-Michael reactions: The Brønsted acid−hydrogen bonding model. J. Am. Chem. Soc. 2016, 138, 9041–9044. [Google Scholar] [CrossRef]
- Zhang, G.; Zhu, C.; Liu, D.; Pan, J.; Zhang, J.; Hu, D.; Song, B. Solvent-free enantioselective conjugate addition and bioactivities of nitromethane to Chalcone containing pyridine. Tetrahedron 2017, 73, 129–136. [Google Scholar] [CrossRef]
- Duque, M.M.S.; Baslé, O.; Isambert, N.; Gaudel-Siri, A.; Genisson, Y.; Plaquevent, J.-C.; Rodriguez, J.; Constantieux, T. A cooperative participation of the amido group in the organocatalytic construction of all-carbon quaternary stereocenters by Michael addition with β-ketoamides. Org. Lett. 2011, 13, 3296–3299. [Google Scholar] [CrossRef] [PubMed]
- Quintard, A.; Cheshmedzhieva, D.; Duque, M.M.S.; Gaudel-Siri, A.; Naubron, J.-V.; Génisson, Y.; Plaquevent, J.-C.; Bugaut, X.; Rodriguez, J.; Constantieux, T. Origin of the enantioselectivity in organocatalytic Michael additions of β-ketoamides to α,β-unsaturated carbonyls: A combined experimental, spectroscopic and theoretical study. Chem. Eur. J. 2015, 21, 778–790. [Google Scholar] [CrossRef]
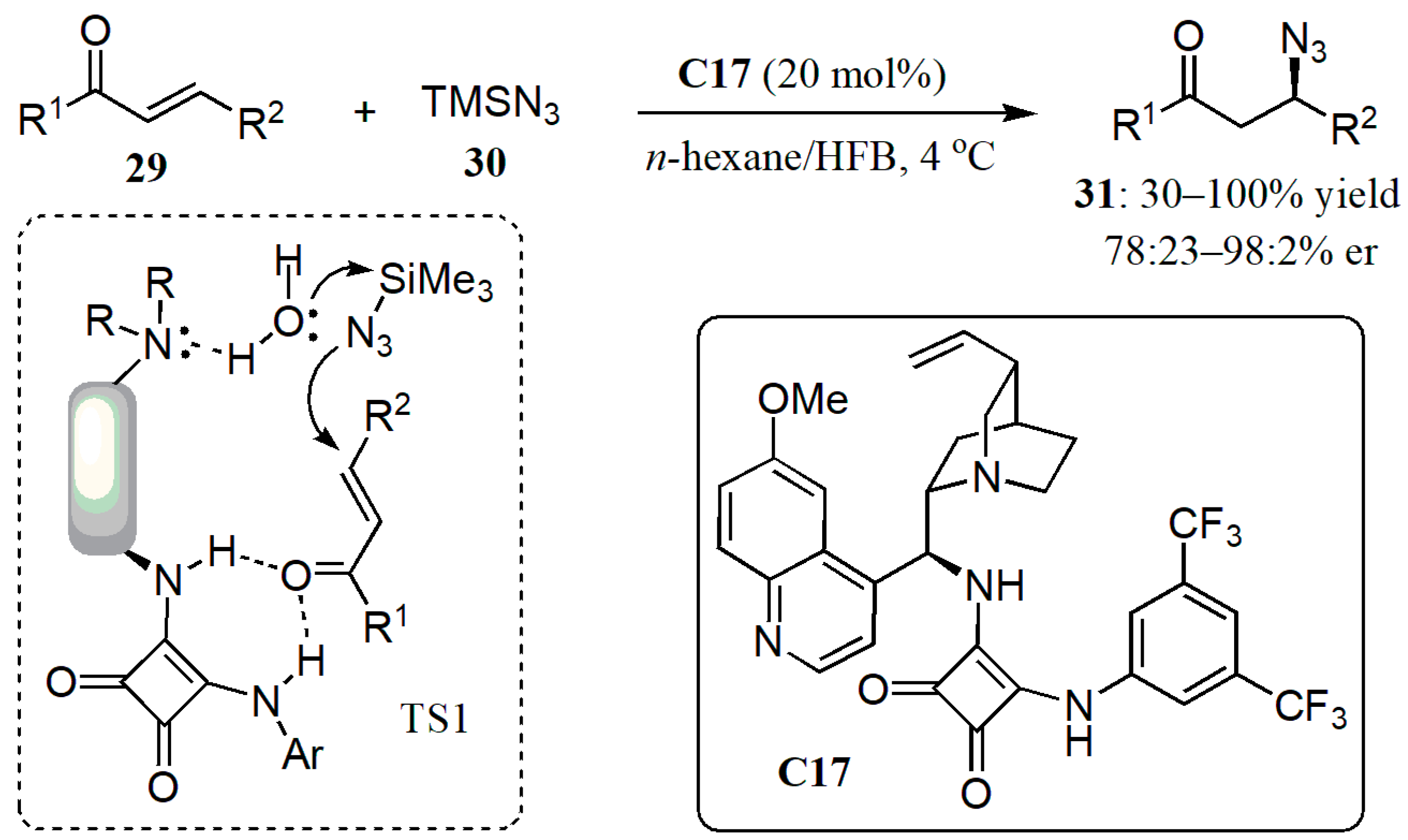
- Humbrías-Martín, J.; Pérez-Aguilar, M.C.; Mas-Ballesté, R.; Litta, A.D.; Lattanzi, A.; Sala, G.D.; Fernández-Salas, J.A.; Alemán, J. Enantioselective conjugate azidation of α,β-unsaturated ketones under bifunctional organocatalysis by direct activation of TMSN3. Adv. Synth. Catal. 2019, 361, 4790–4796. [Google Scholar] [CrossRef]
- Helder, R.; Arends, R.; Bolt, W.; Hiemstra, H.; Wynberg, H. Alkaloid catalyzed asymmetric synthesis III the addition of mercaptans to 2-cyclohexene-1-one; determination of enantiomeric excess using 13C NMR. Tetrahedron Lett. 1977, 18, 2181–2182. [Google Scholar] [CrossRef]
- Connon, S.J. Asymmetric catalysis with bifunctional cinchona alkaloid-based urea and thiourea organocatalysts. Chem. Commun. 2008, 2499–2510. [Google Scholar] [CrossRef] [PubMed]
- Melchiorre, P. Cinchona-based primary amine catalysis in the asymmetric functionalization of carbonyl compounds. Angew. Chem. Int. Ed. 2012, 51, 9748–9770. [Google Scholar] [CrossRef]
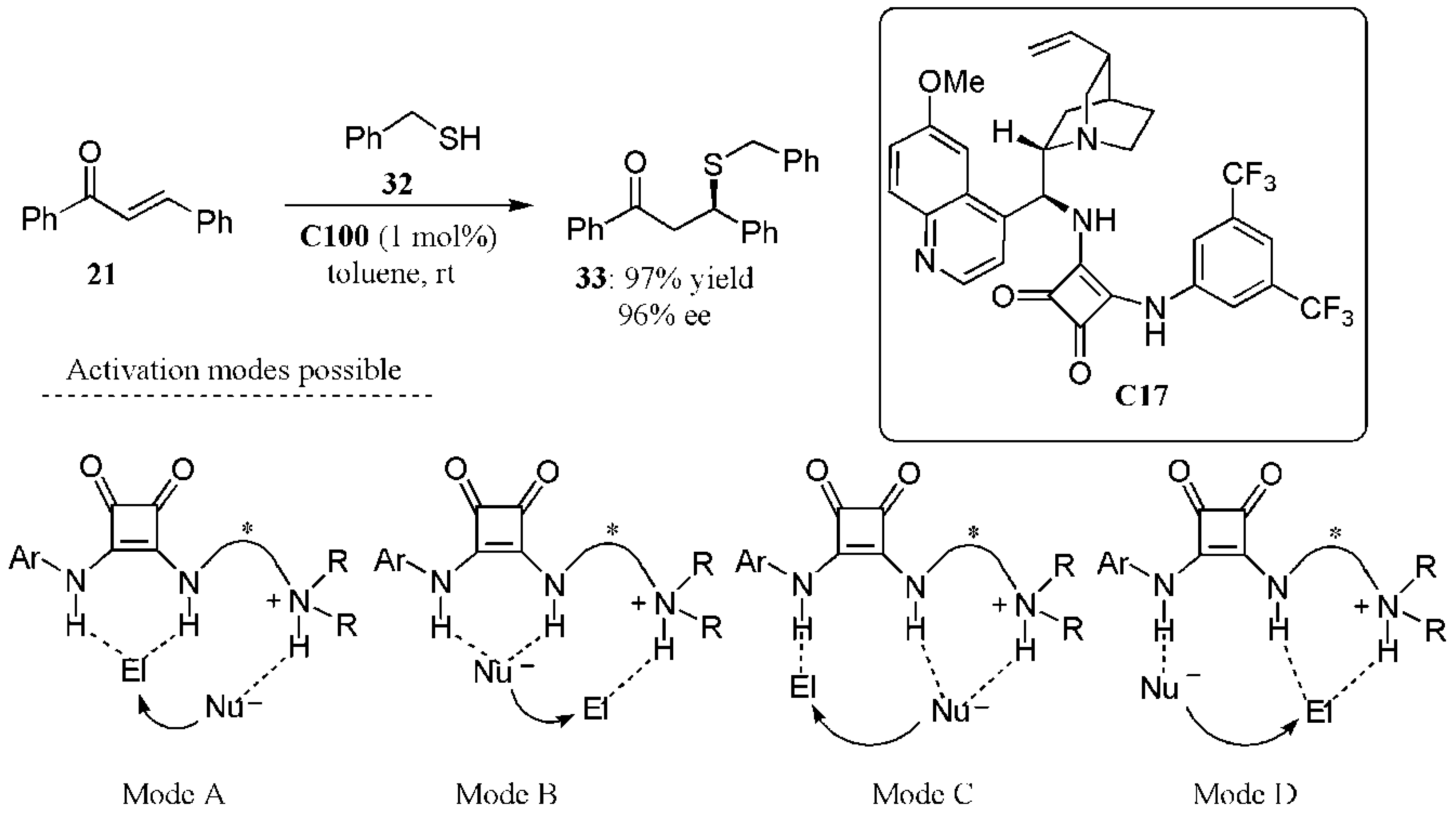
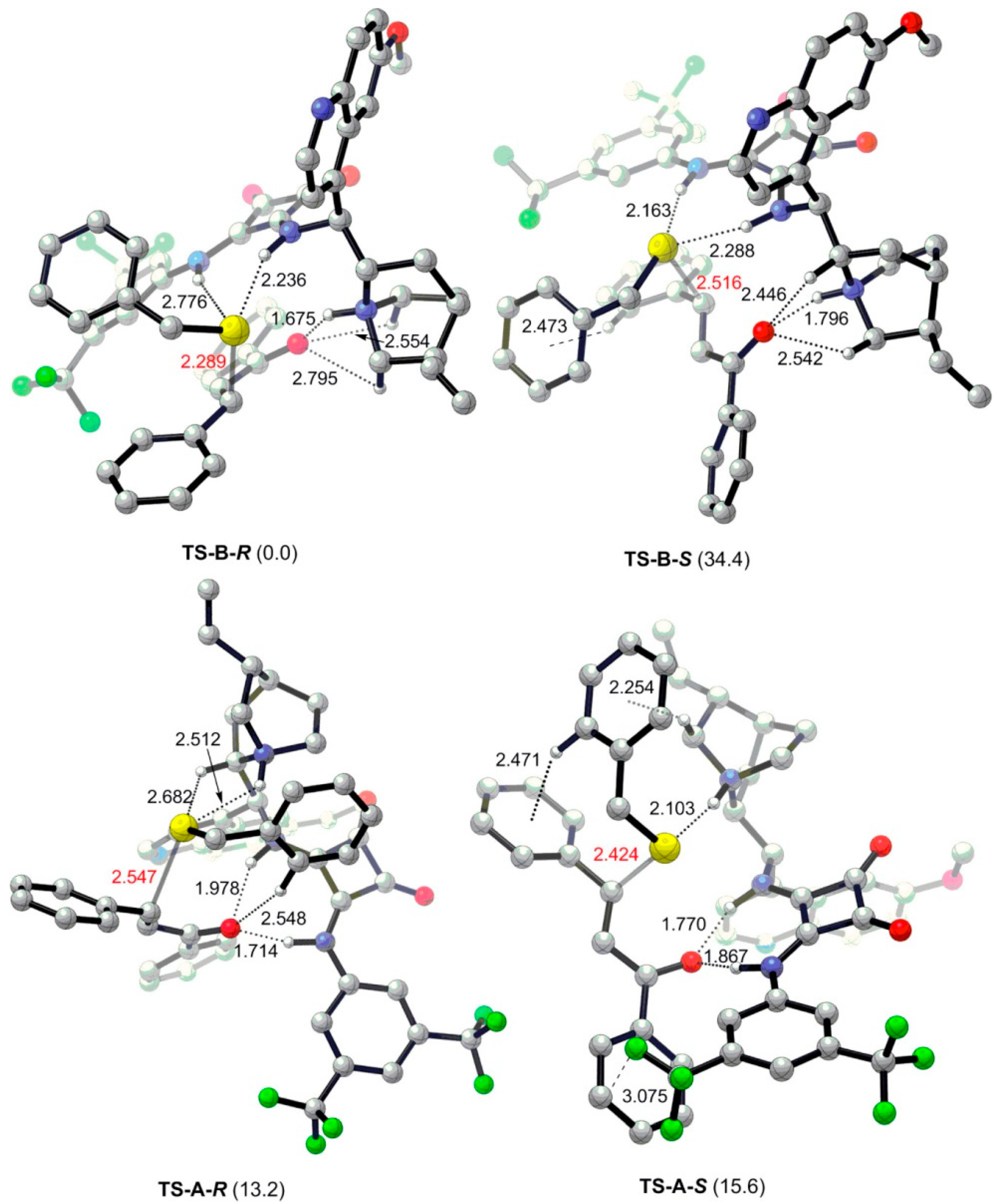
- Dai, L.; Wang, S.-X.; Chen, F.-E. A bifunctional cinchona alkaloid-squaramide catalyst for the highly enantioselective conjugate addition of thiols to trans-chalcones. Adv. Synth. Catal. 2010, 352, 2137–2141. [Google Scholar] [CrossRef]
- Guo, J.; Wong, M.W. Cinchona alkaloid-squaramide catalyzed sulfa-Michael addition reaction: Mode of bifunctional activation and origin of stereoinduction. J. Org. Chem. 2017, 82, 4362–4368. [Google Scholar] [CrossRef]
- Caner, H.; Biedermann, P.U.; Agranat, I. Conformational spaces of Cinchona alkaloids. Chirality 2003, 15, 637–645. [Google Scholar] [CrossRef]
- Dijkstra, G.D.H.; Kellogg, R.M.; Wynberg, H.; Svendsen, J.S.; Marko, I.; Sharpless, K.B. Conformational study of cinchona alkaloids. A combined NMR, molecular mechanics and x-ray approach. J. Am. Chem. Soc. 1989, 111, 8069–8076. [Google Scholar] [CrossRef]
- Grayson, M.N.; Houk, K.N. Cinchona alkaloid-catalyzed asymmetric conjugate additions: The bifunctional brønsted acid–hydrogen bonding model. J. Am. Chem. Soc. 2016, 138, 1170–1173. [Google Scholar] [CrossRef]
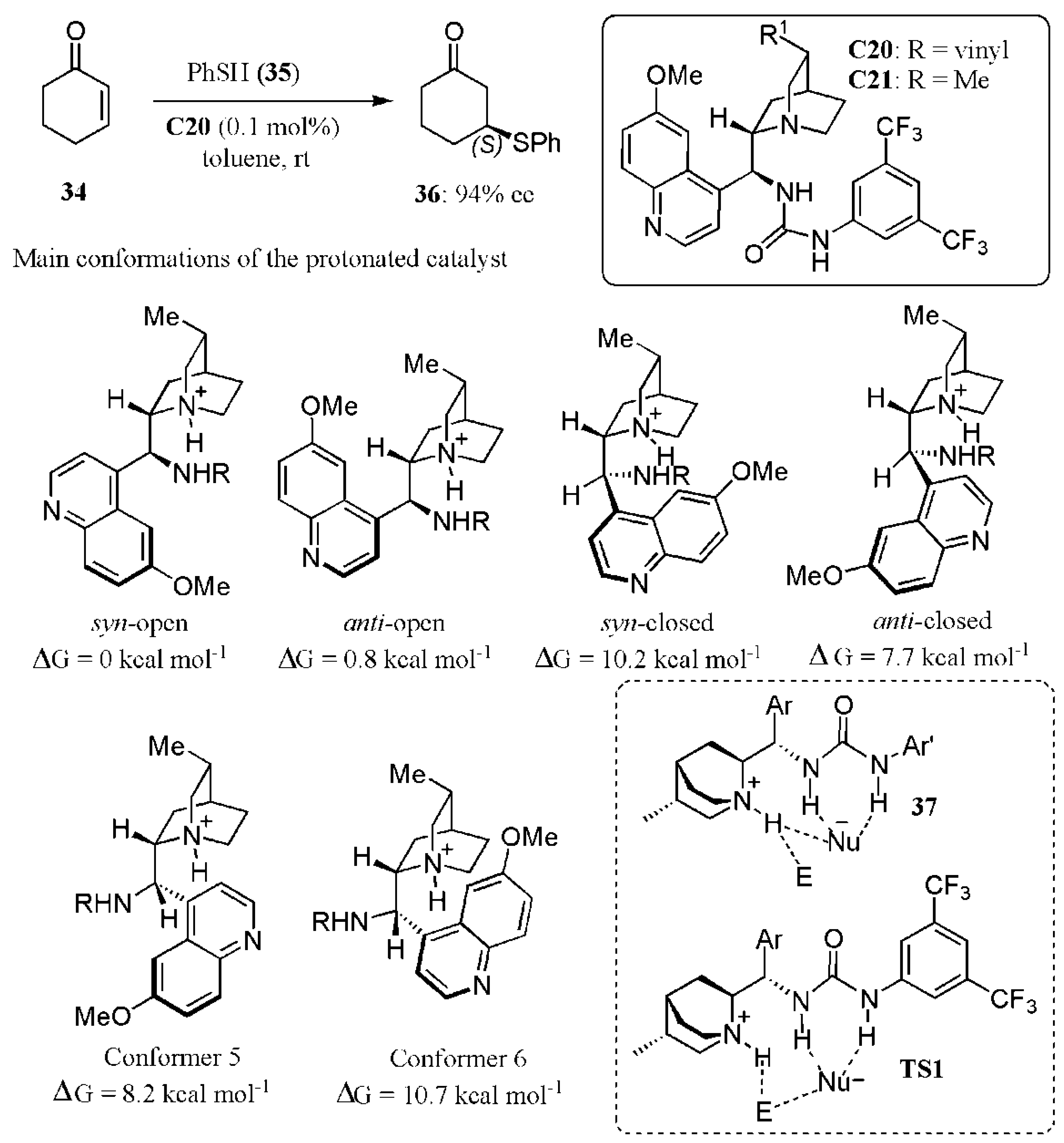
- Rana, N.K.; Selvakumar, S.; Singh, V.K. Highly enantioselective organocatalytic sulfa-Michael addition to α,β-unsaturated ketones. J. Org. Chem. 2010, 75, 2089–2091. [Google Scholar] [CrossRef]
- Bahlinger, A.; Fritz, S.P.; Wennemers, H. Stereoselective metal-free synthesis of β-amino thioesters with tertiary and quaternary stereogenic centers. Angew. Chem. Int. Ed. 2014, 53, 8779–8783. [Google Scholar] [CrossRef] [PubMed]
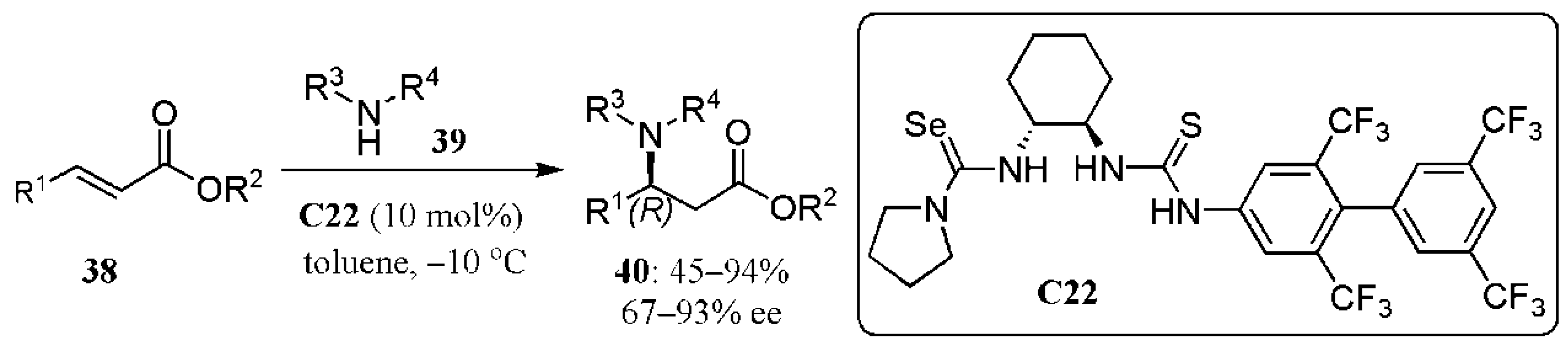
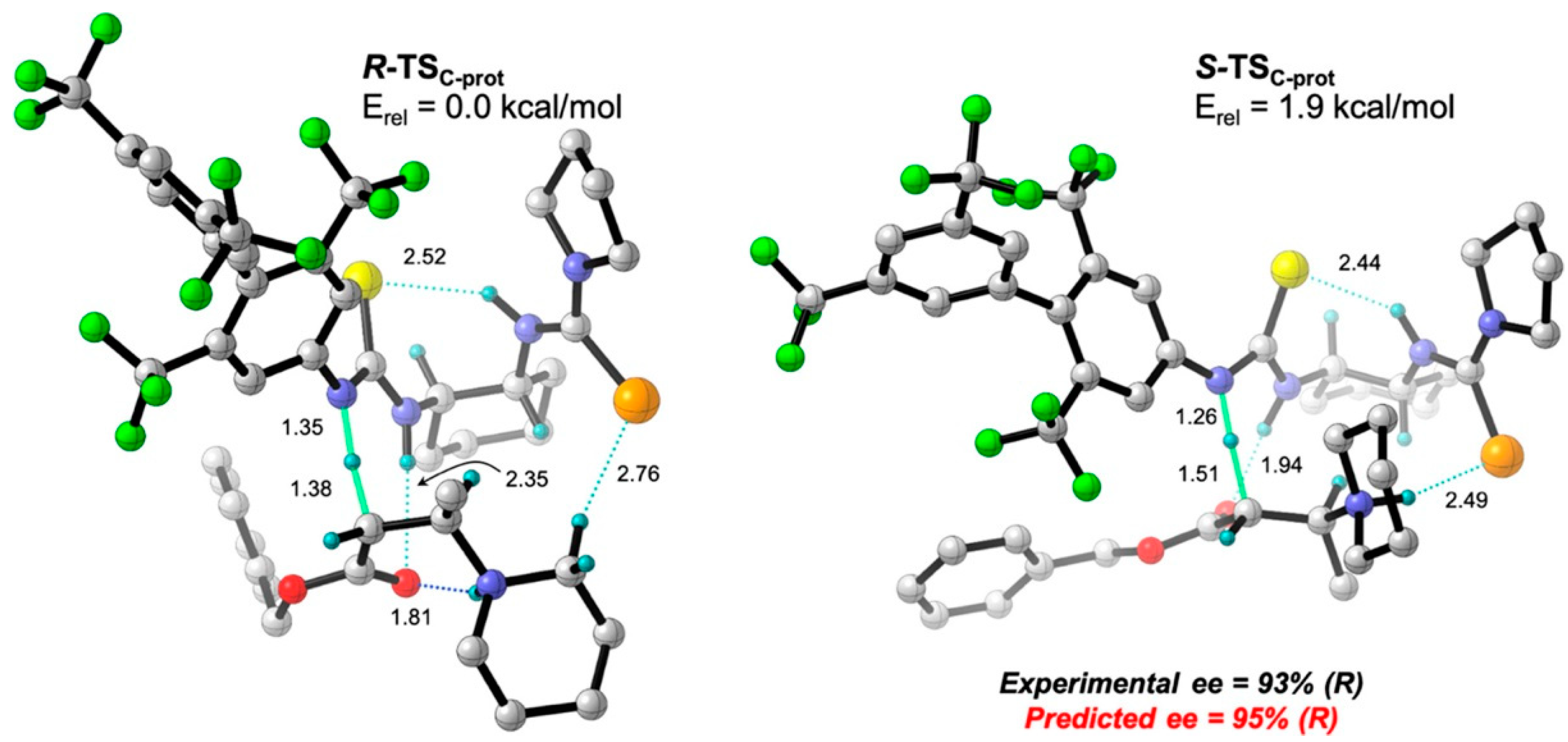
- Lin, Y.; Hirschi, W.J.; Kunadia, A.; Paul, A.; Ghiviriga, I.; Abboud, K.A.; Karugu, R.W.; Vetticatt, M.J.; Hirschi, J.S.; Seidel, D. A selenourea-thiourea Brønsted acid catalyst facilitates asymmetric conjugate additions of amines to α,β-unsaturated esters. J. Am. Chem. Soc. 2020, 142, 5627–5635. [Google Scholar] [CrossRef] [PubMed]
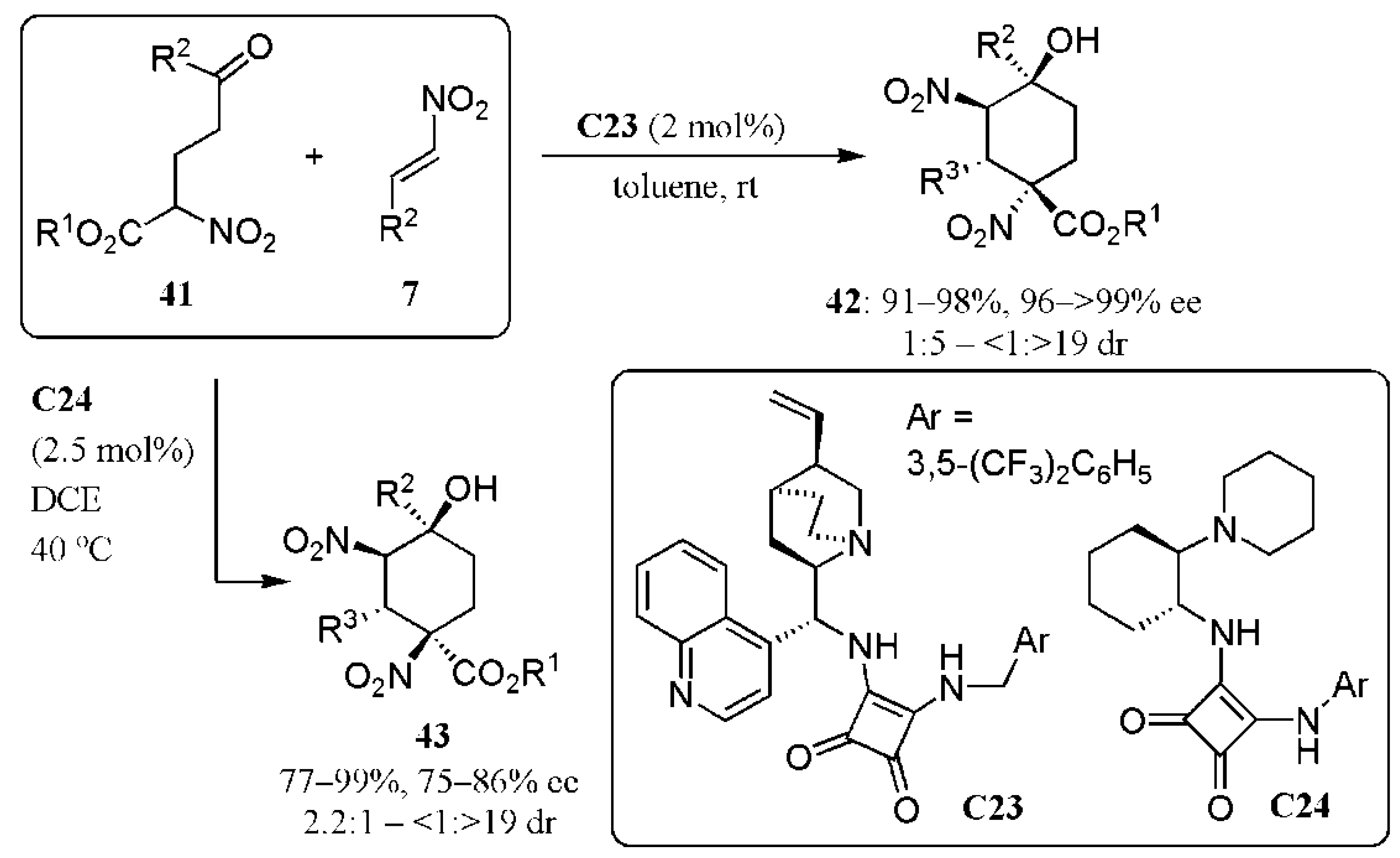
- Martínez, J.I.; Villar, L.; Uria, U.; Carrillo, L.; Reyes, E.; Vicario, J.L. Bifunctional squaramide catalysts with the same absolute chirality for the diastereodivergent access to densely functionalised cyclohexanes through enantioselective domino reactions. Synthesis and mechanistic studies. Adv. Synth. Catal. 2014, 356, 3627–3648. [Google Scholar] [CrossRef]
- Meninno, S.; Quaratesi, I.; Volpe, C.; Mazzanti, A.; Lattanzi, A. Catalytic enantioselective one-pot approach to cis- and trans-2,3-diaryl substituted 1,5-benzothiazepines. Org. Biomol. Chem. 2018, 16, 6923–6934. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wei, R.; Yi, X.; Gao, L.; Zhang, M.; Liu, H.; Li, Q.; Song, H.; Ban, S.J. Diastereo- and enantioselective synthesis of functionalized cyclopentenes containing a quaternary chiral center via a thiosquaramide-catalyzed cascade Michael−Henry reaction. Org. Chem. 2019, 84, 15655–15661. [Google Scholar] [CrossRef]
- Alishetty, S.; Shih, H.P.; Han, C.C. One-step, effective, and cascade syntheses of highly functionalized cyclopentenes with high diastereoselectivity. Org. Lett. 2018, 20, 2513–2516. [Google Scholar] [CrossRef]
- Held, F.E.; Tsogoeva, S.B. Asymmetric cycloaddition reactions catalyzed by bifunctional thiourea and squaramide organocatalysts: Recent advances. Catal. Sci. Technol. 2016, 6, 645–667. [Google Scholar] [CrossRef]
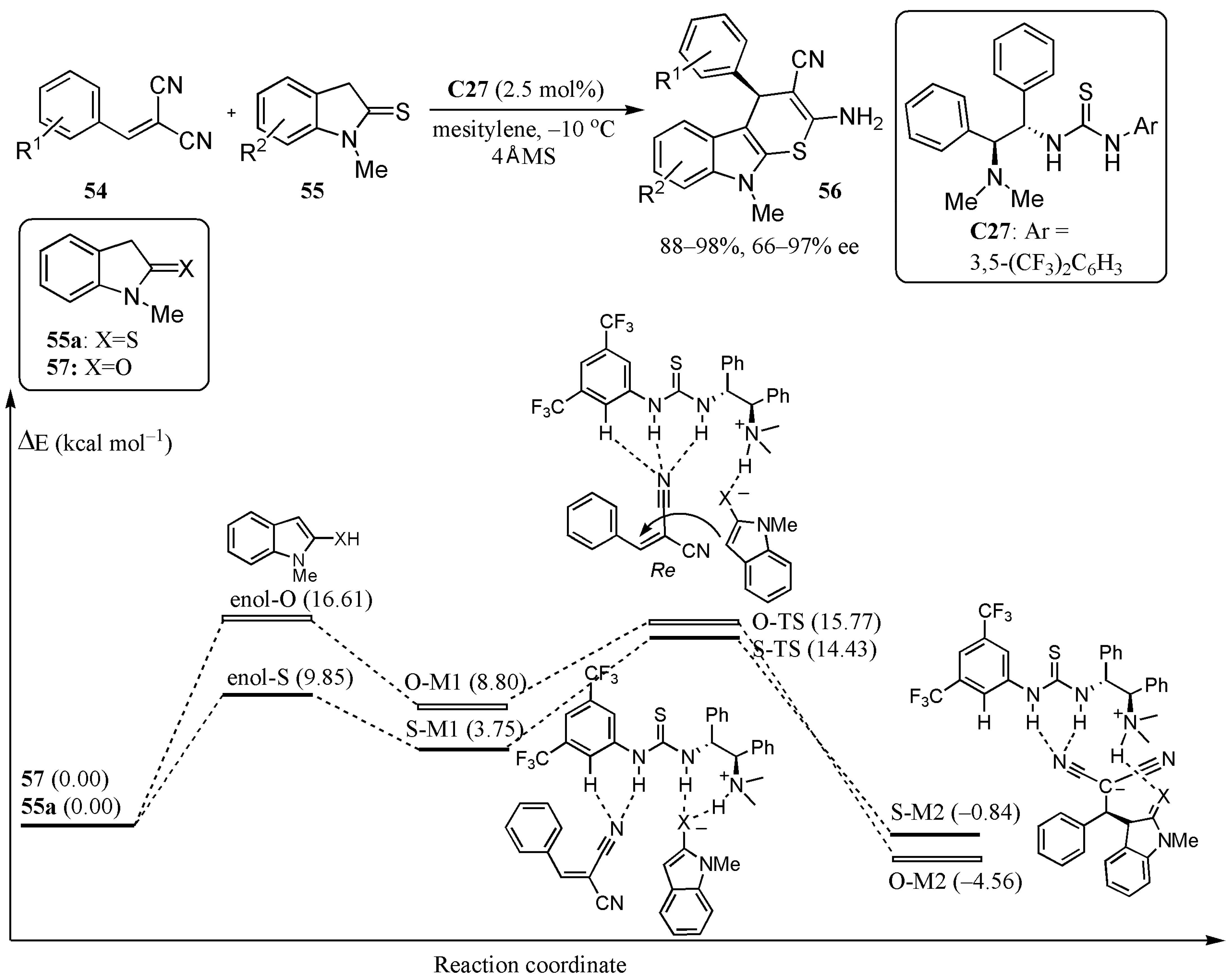
- Chen, X.; Qi, Z.-H.; Zhang, S.-Y.; Kong, L.-P.; Wang, Y.; Wang, X.-W. Enantioselective construction of functionalized thiopyrano-indole annulated heterocycles via a formal thio [3 + 3]-cyclization. Org. Lett. 2015, 17, 42–45. [Google Scholar] [CrossRef]
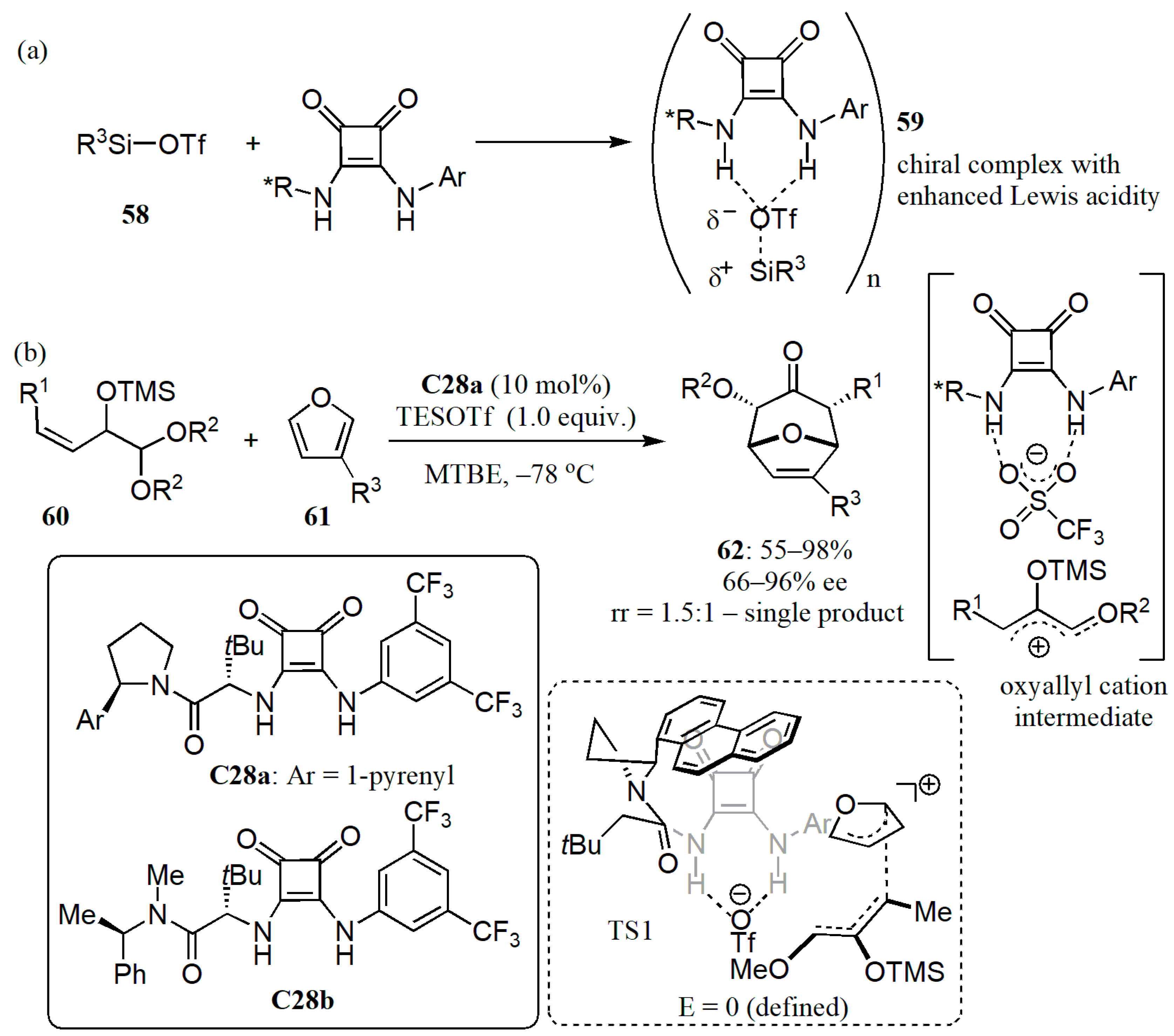
- Banik, S.M.; Levina, A.; Hyde, A.M.; Jacobsen, E.N. Lewis acid enhancement by hydrogen-bond donors for asymmetric catalysis. Science 2017, 358, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, B.; Ghosez, L. Trimethylsilyl bis(trifluoromethanesulfonyl)imide as a tolerant and environmentally benign Lewis acid catalyst of the Diels–Alder reaction. Tetrahedron 2002, 58, 8219–8226. [Google Scholar] [CrossRef]
- James, T.; van Gemmeren, M.; List, B. Development and applications of disulfonimides in enantioselective organocatalysis. Chem. Rev. 2015, 115, 9388–9409. [Google Scholar] [CrossRef]
- Burés, J. A simple graphical method to determine the order in catalyst. Angew. Chem. Int. Ed. 2016, 55, 2028–2031. [Google Scholar] [CrossRef]
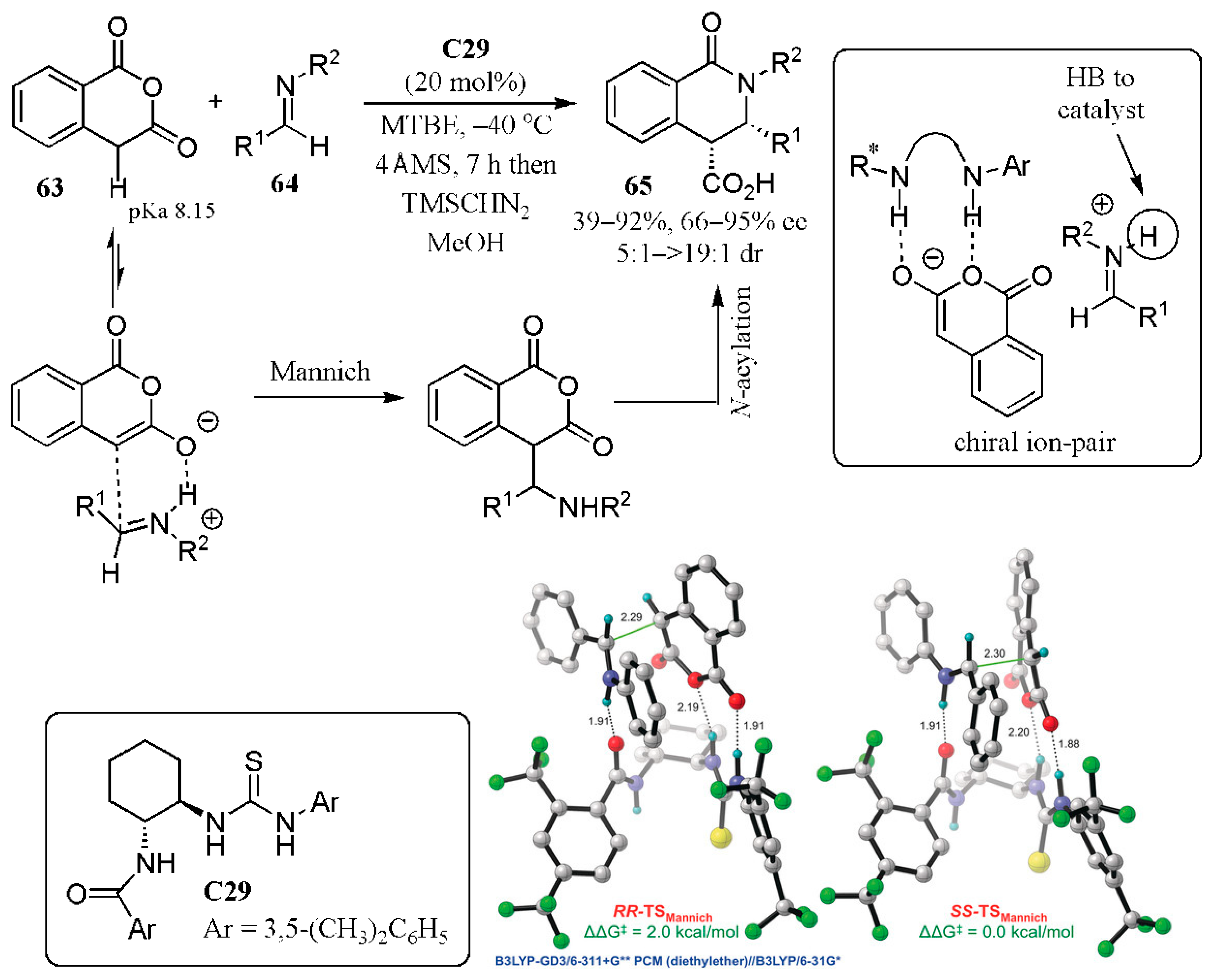
- Jarvis, C.L.; Hirschi, J.S.; Vetticatt, M.J.; Seidel, D. Catalytic enantioselective synthesis of lactams through formal [4 + 2] cycloaddition of imines with homophthalic anhydride. Angew. Chem. Int. Ed. 2017, 56, 2670–2674. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, N. The condensation of succinic anhydride with benzylidinemethylamine. A stereoselective synthesis of trans- and cis-1-methyl-4-carbosy-5-phenyl-2-pyrrolidinone. J. Org. Chem. 1969, 34, 3187–3189. [Google Scholar] [CrossRef]
- Castagnoli, N., Jr.; Cushman, M. Condensation of succinic anhydrides with Schiff bases. Scope and mechanism. J. Org. Chem. 1971, 36, 3404–3406. [Google Scholar] [CrossRef] [PubMed]
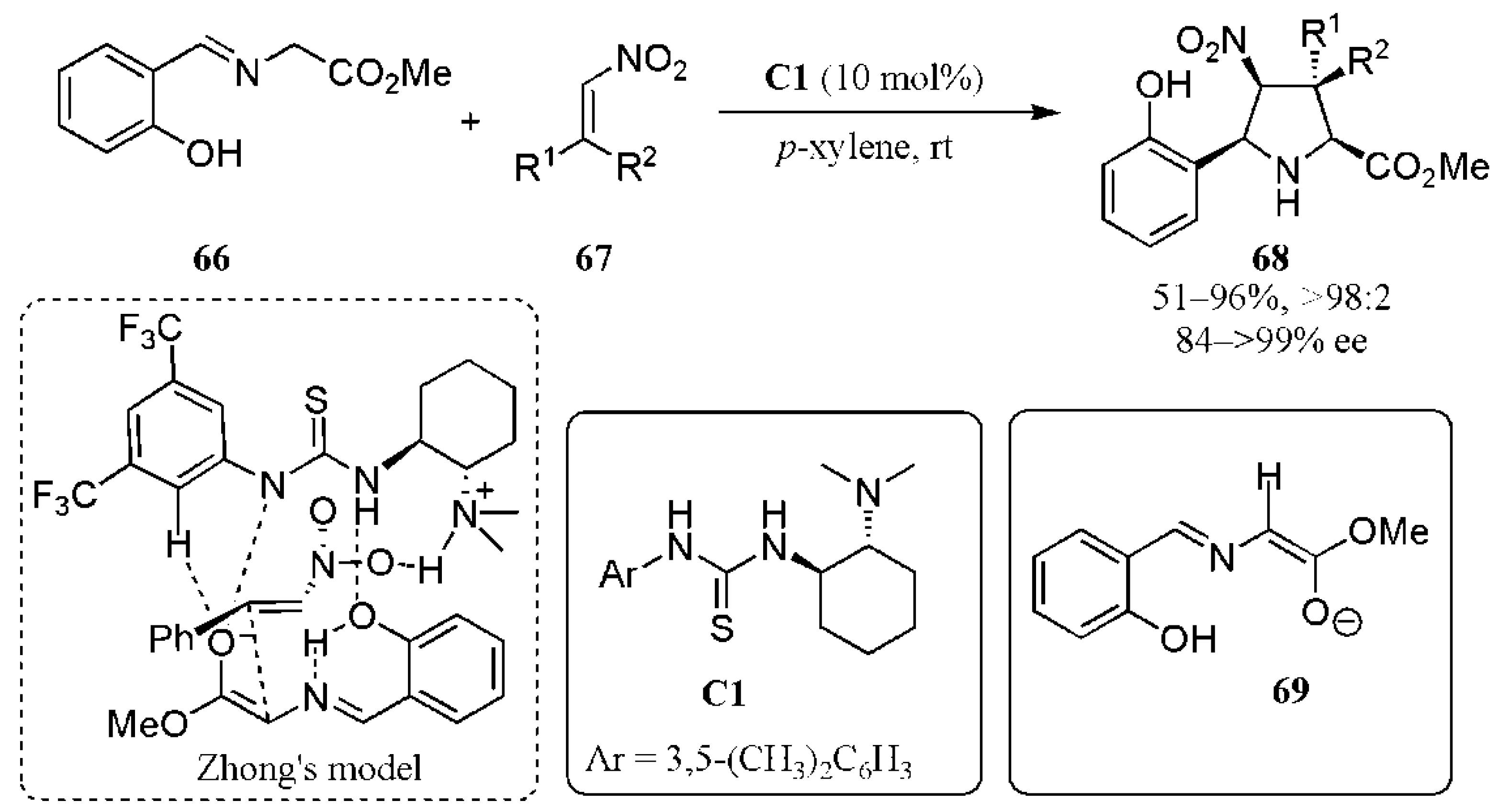
- Esteban, F.; Cieślik, W.; Arpa, E.M.; Guerrero-Corella, A.; Díaz-Tendero, S.; Perles, J.; Fernández-Salas, J.A.; Fraile, A.; Alemán, J. Intramolecular hydrogen bond activation: Thiourea-organocatalyzed enantioselective 1,3-dipolar cycloaddition of salicylaldehyde-derived azomethine ylides with nitroalkenes. ACS Catal. 2018, 8, 1884–1890. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, X.; Li, S.; Xue, L.; Shan, C.; Zhao, Z.; Yan, H. Organocatalytic atroposelective intramolecular [4 + 2] cycloaddition: Synthesis of axially chiral heterobiaryls. Angew. Chem. Int. Ed. 2018, 57, 6491–6495. [Google Scholar] [CrossRef]
- Shan, C.; Zhang, T.; Xiong, Q.; Yan, H.; Bai, R.; Lan, Y. Hydrogen-bond-induced chiral axis construction: Theoretical study of cinchonine–thiourea-catalyzed enantioselective intramolecular cycloaddition. Chem. Asian J. 2019, 14, 2731–2736. [Google Scholar] [CrossRef]
- Manoni, F.; Farid, U.; Trujillo, C.; Connon, S.J. Catalytic asymmetric Tamura cycloadditions involving nitroalkenes. Org. Biomol. Chem. 2017, 15, 1463–1474. [Google Scholar] [CrossRef]
- Cornaggia, C.; Manoni, F.; Torrente, E.; Tallon, S.; Connon, S.J. A catalytic asymmetric reaction involving enolizable anhydrides. Org. Lett. 2012, 14, 1850–1853. [Google Scholar] [CrossRef]
- Trujillo, C.; Rozas, I.; Botte, A.; Connon, S.J. A DFT mechanistic study of the organocatalytic asymmetric reaction of aldehydes and homophthalic anhydride. Chem. Commun. 2017, 53, 8874–8877. [Google Scholar] [CrossRef]
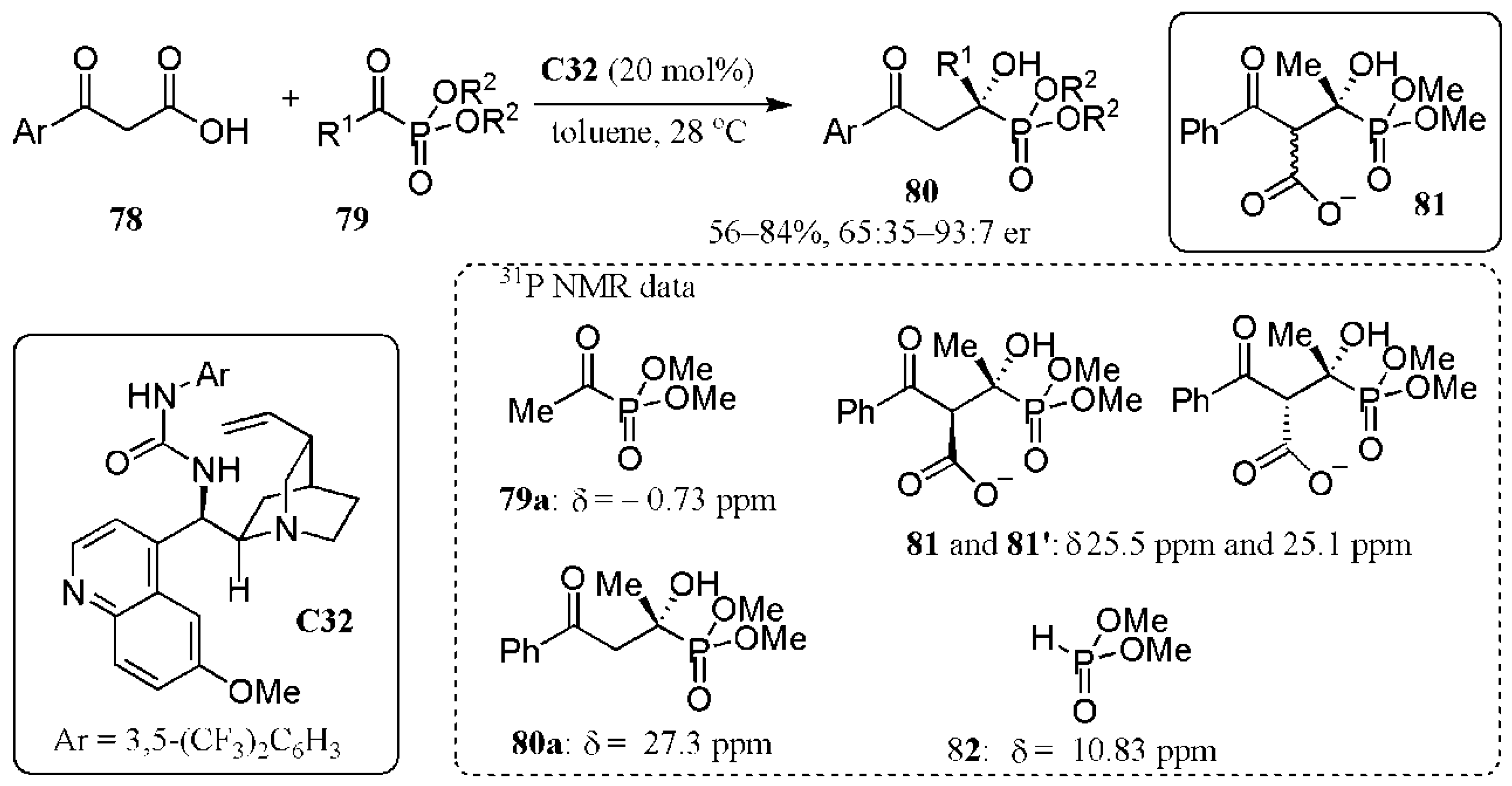
- Vamisetti, G.B.; Chowdhury, R.; Ghosha, S.K. Organocatalytic decarboxylative aldol reaction of β-ketoacids with α-ketophosphonates en route to the enantioselective synthesis of tertiary α-hydroxyphosphonates. Org. Biomol. Chem. 2017, 15, 3869–3873. [Google Scholar] [CrossRef]
- Barros, M.T.; Faísca Phillips, A.M. Organocatalyzed synthesis of tertiary α-hydroxyphosphonates by a highly regioselective modified Pudovik reaction. Eur. J. Org. Chem. 2011, 4028–4036. [Google Scholar] [CrossRef]
- Faísca Phillips, A.M.; Barros, M.T.; Pacheco, M.; Dias, R. Synthesis and biological evaluation of α-hydroxyalkylphosphonates as new antimicrobial agents. Bioorg. Med. Chem. Lett. 2014, 24, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Faisca Phillips, A.M.M.M. Synthesis and applications of pharmacologically relevant phosphonates and phosphinates. In Organic and Medicinal Chemistry; Banik, B.K., Ed.; Nova Science Publishers: New York, NY, USA, 2018; Volume 2, pp. 249–319. [Google Scholar]
- Ni, X.; Li, X.; Cheng, J.-P. Equilibrium acidities of cinchona alkaloid organocatalysts bearing 6′-hydrogen bonding donors in DMSO. Org. Chem. Front. 2016, 3, 170–176. [Google Scholar] [CrossRef]
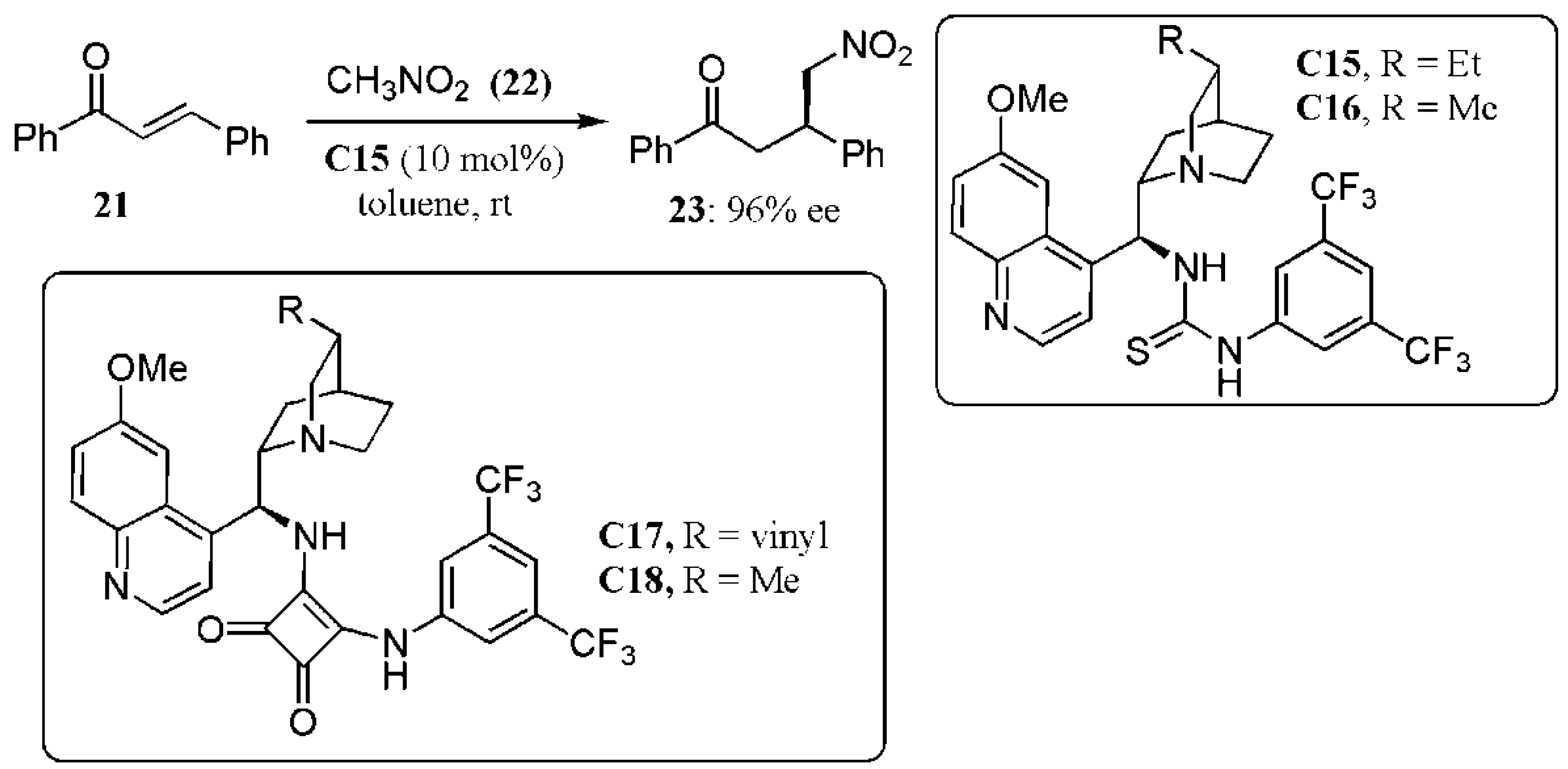
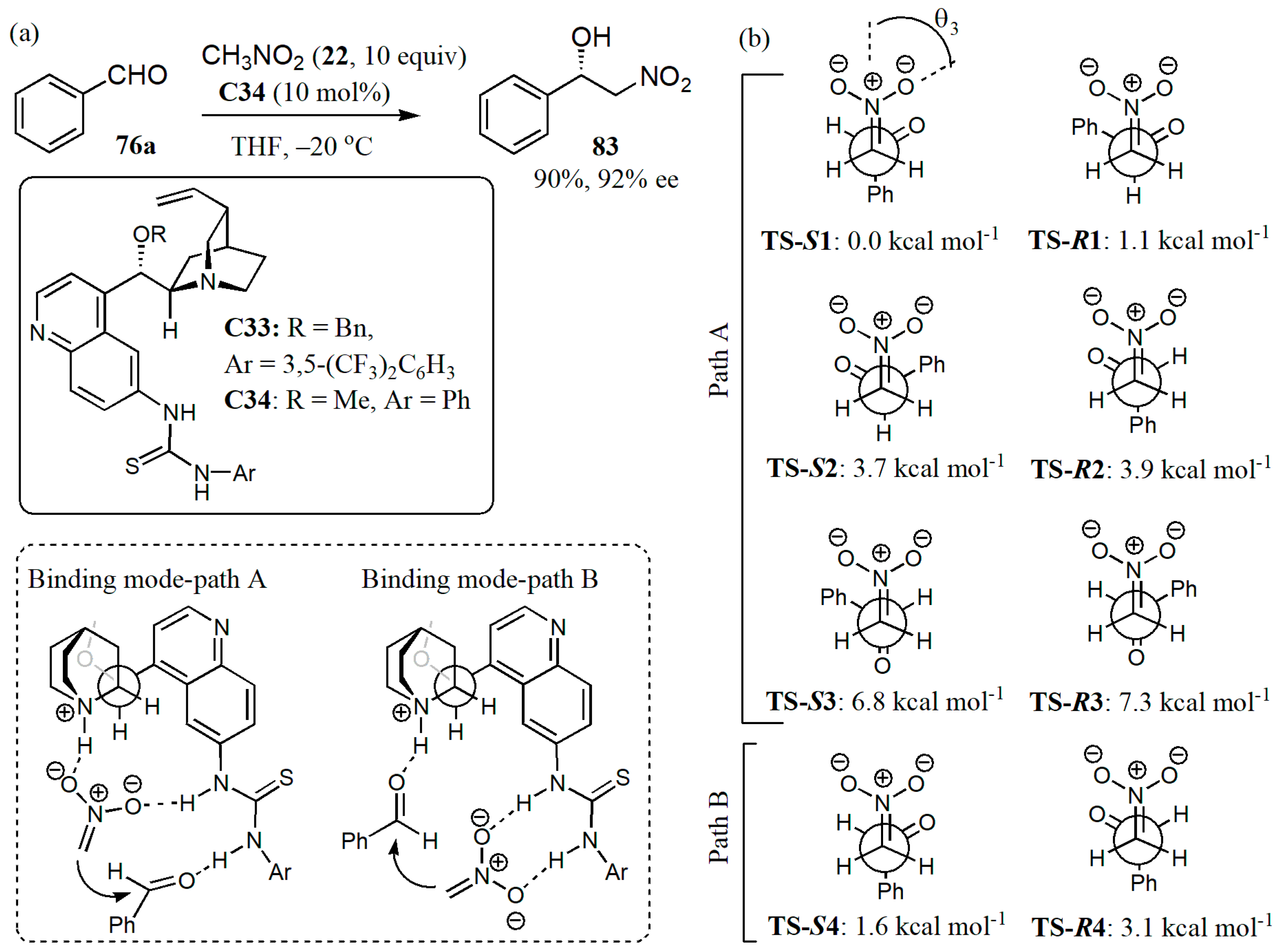
- Hammar, P.; Marcelli, T.; Hiemstra, H.; Himoa, F. Density functional theory study of the Cinchona thiourea- catalyzed Henry reaction: Mechanism and enantioselectivity. Adv. Synth. Catal. 2007, 349, 2537–2548. [Google Scholar] [CrossRef]
- Otevrel, J.; Bobal, P. Diamine-tethered bis(thiourea) organocatalyst for asymmetric Henry reaction. J. Org. Chem. 2017, 82, 8342–8358. [Google Scholar] [CrossRef]
- Alegre-Requena, J.V.; Marqués-López, E.; Herrera, R.P. Optimizing the accuracy and computational cost in theoretical squaramide catalysis: The Henry reaction. Chem. Eur. J. 2017, 23, 15336–15347. [Google Scholar] [CrossRef]
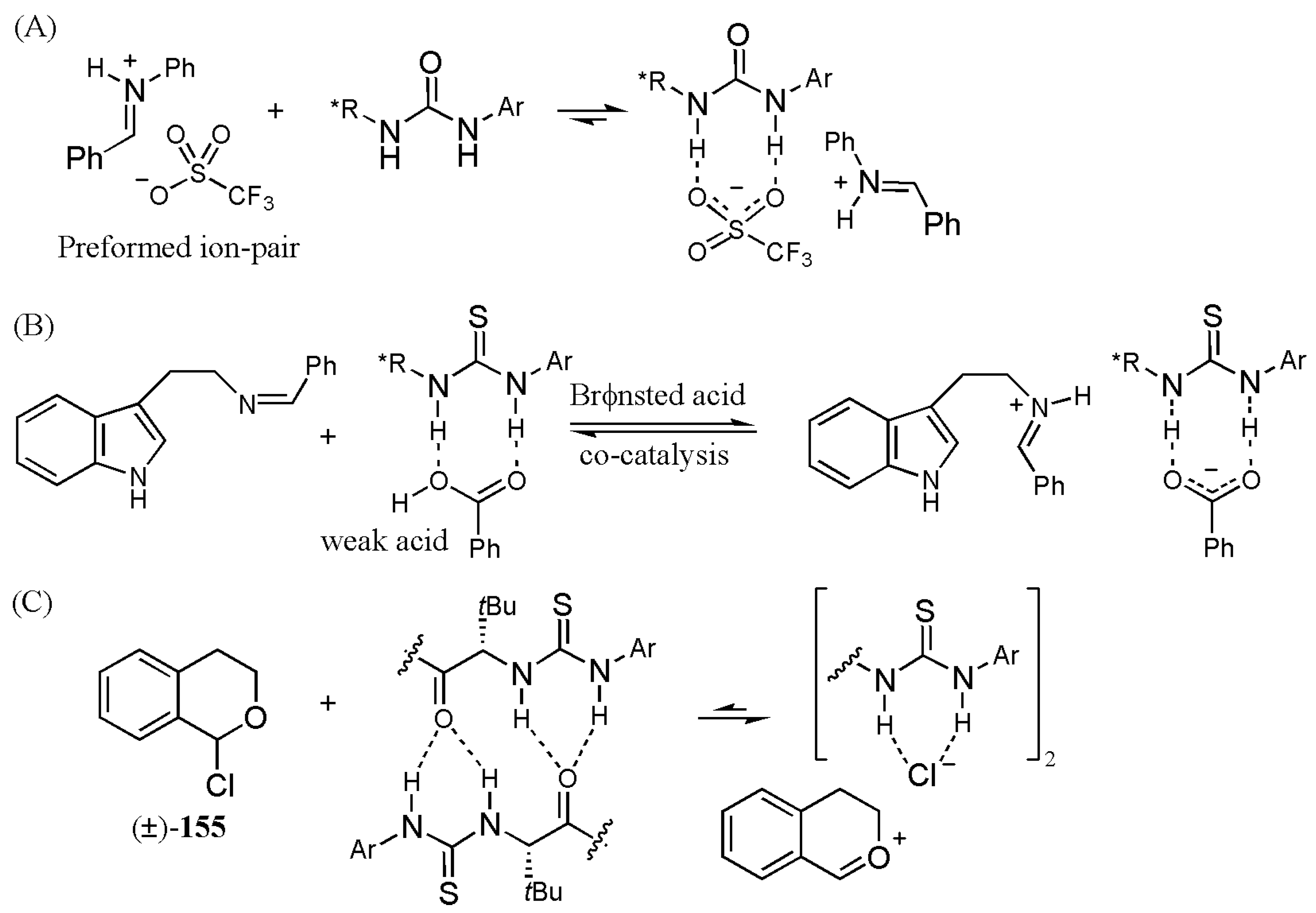
- Ford, D.D.; Lehnherr, D.; Kennedy, C.R.; Jacobsen, E.N. Anion-abstraction catalysis: The cooperative mechanism of α-chloroether activation by dual hydrogen-bond donors. ACS Catal. 2016, 6, 4616–4620. [Google Scholar] [CrossRef]
- Ford, D.D.; Lehnherr, D.; Kennedy, C.R.; Jacobsen, E.N. On- and off-cycle catalyst cooperativity in anion-binding catalysis. J. Am. Chem. Soc. 2016, 138, 7860–7863. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.R.; Lehnherr, D.; Rajapaksa, N.S.; Ford, D.D.; Park, Y.; Jacobsen, E.N. Mechanism-guided development of a highly active bis-thiourea catalyst for anion-abstraction catalysis. J. Am. Chem. Soc. 2016, 138, 13525–13528. [Google Scholar] [CrossRef] [PubMed]
- Lehnherr, D.; Ford, D.D.; Bendelsmith, A.J.; Kennedy, C.R.; Jacobsen, E.N. Conformational control of chiral amido-thiourea catalysts enables improved activity and enantioselectivity. Org. Lett. 2016, 18, 3214–3217. [Google Scholar] [CrossRef] [PubMed]
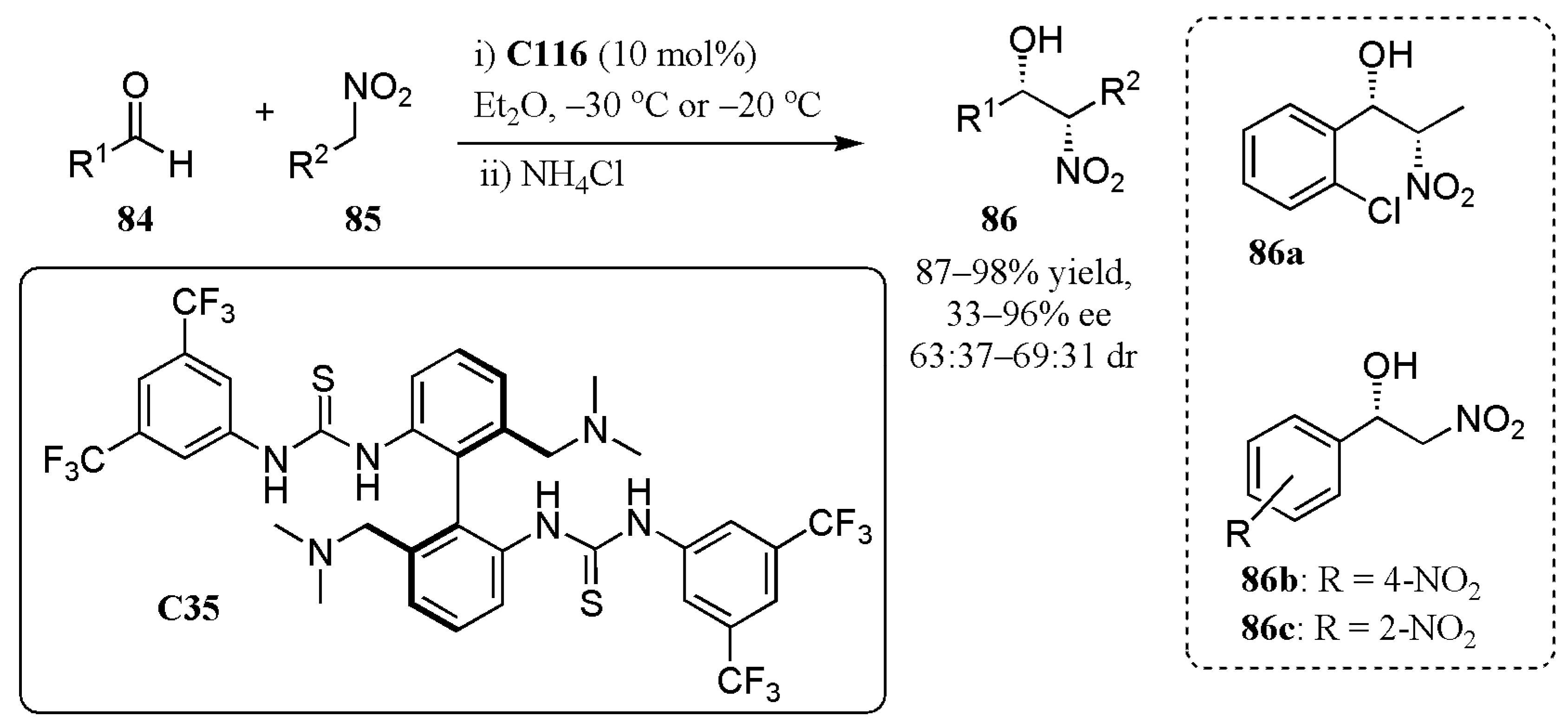
- Mittal, N.; Lippert, K.M.; De, C.K.; Klauber, E.G.; Emge, T.J.; Schreiner, P.R.; Seidel, D.J. A dual-catalysis anion-binding approach to the kinetic resolution of amines: Insights into the mechanism via a combined experimental and computational study. Am. Chem. Soc. 2015, 137, 5748–5758. [Google Scholar] [CrossRef]
- Taylor, J.E.; Bull, S.D.; Williams, J.M.J. Amidines, isothioureas, and guanidines as nucleophilic catalysts. Chem. Soc. Rev. 2012, 41, 2109–2121. [Google Scholar] [CrossRef]
- Seidel, D. The anion-binding approach to catalytic enantioselective acyl transfer. Synlett 2014, 25, 783–794. [Google Scholar] [CrossRef]
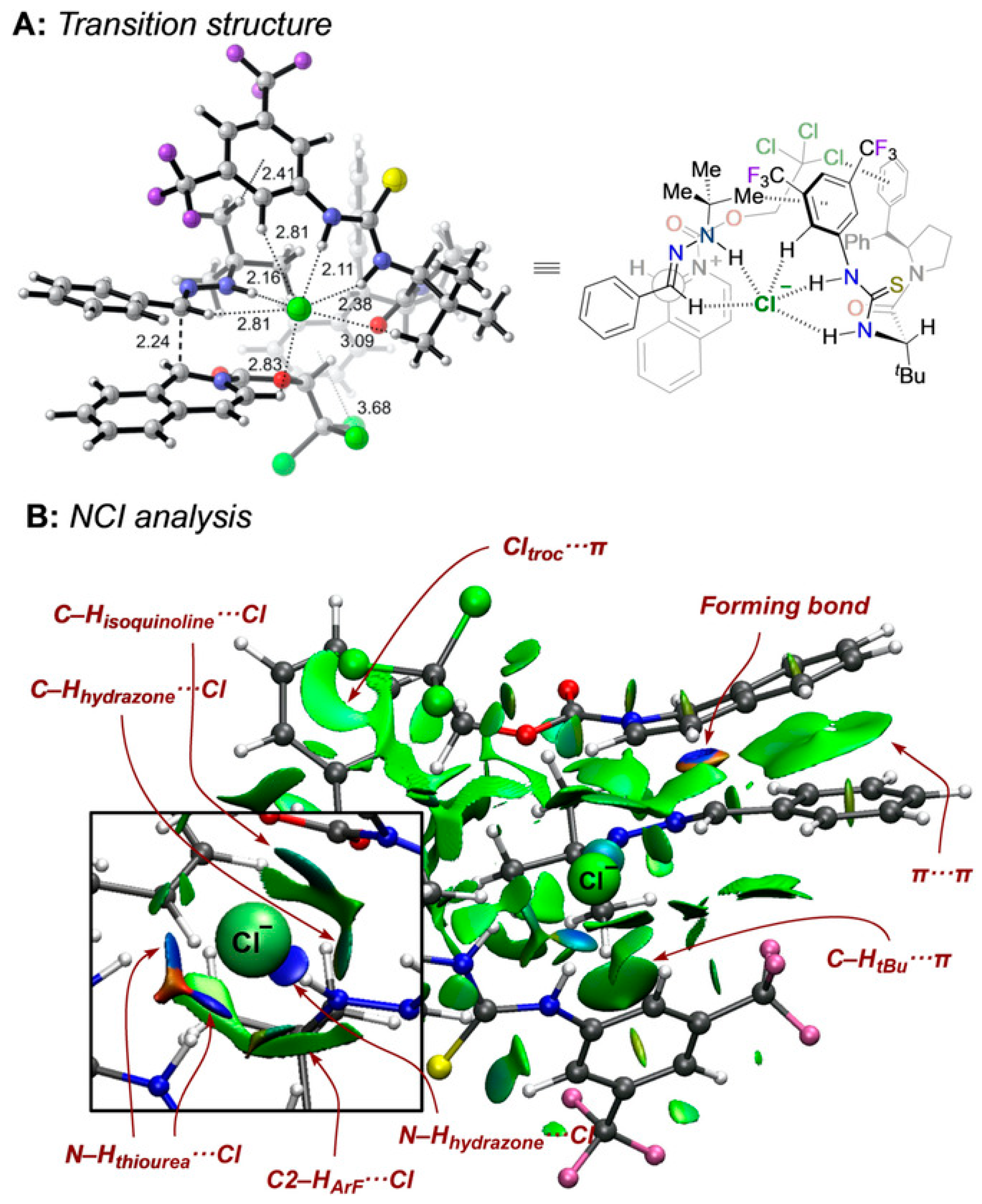
- Matador, E.; Iglesias-Sigüenza, J.; Monge, D.; Merino, P.; Fernández, R.; Lassaletta, J.M. Enantio- and diastereoselective nucleophilic addition of N-tert-butylhydrazones to isoquinolinium ions through anion-binding catalysis. Angew. Chem. Int. Ed. 2021, 60, 5096–5101. [Google Scholar] [CrossRef]
- Boto, R.A.; Peccati, F.; Laplaza, R.; Quan, C.; Carbone, A.; Piquemal, J.-P.; Maday, Y.; Contreras-García, J.J. NCIPLOT4: Fast, robust, and quantitative analysis of noncovalent interactions. Chem. Theory Comput. 2020, 16, 4150–4158. [Google Scholar] [CrossRef]
- Faisca Phillips, A.M.; Pombeiro, A.J.L. Recent advances in organocatalytic enantioselective transfer hydrogenation. Org. Biomol. Chem. 2017, 15, 2307–2340. [Google Scholar] [CrossRef]
- Massolo, E.; Benaglia, M.; Orlandi, M.; Rossi, S.; Celentano, G. Enantioselective Organocatalytic Reduction of b-Trifluoromethyl Nitroalkenes: An Efficient Strategy for the Synthesis of Chiral β-Trifluoromethyl Amines. Chem. Eur. J. 2015, 21, 3589–3595. [Google Scholar] [CrossRef]
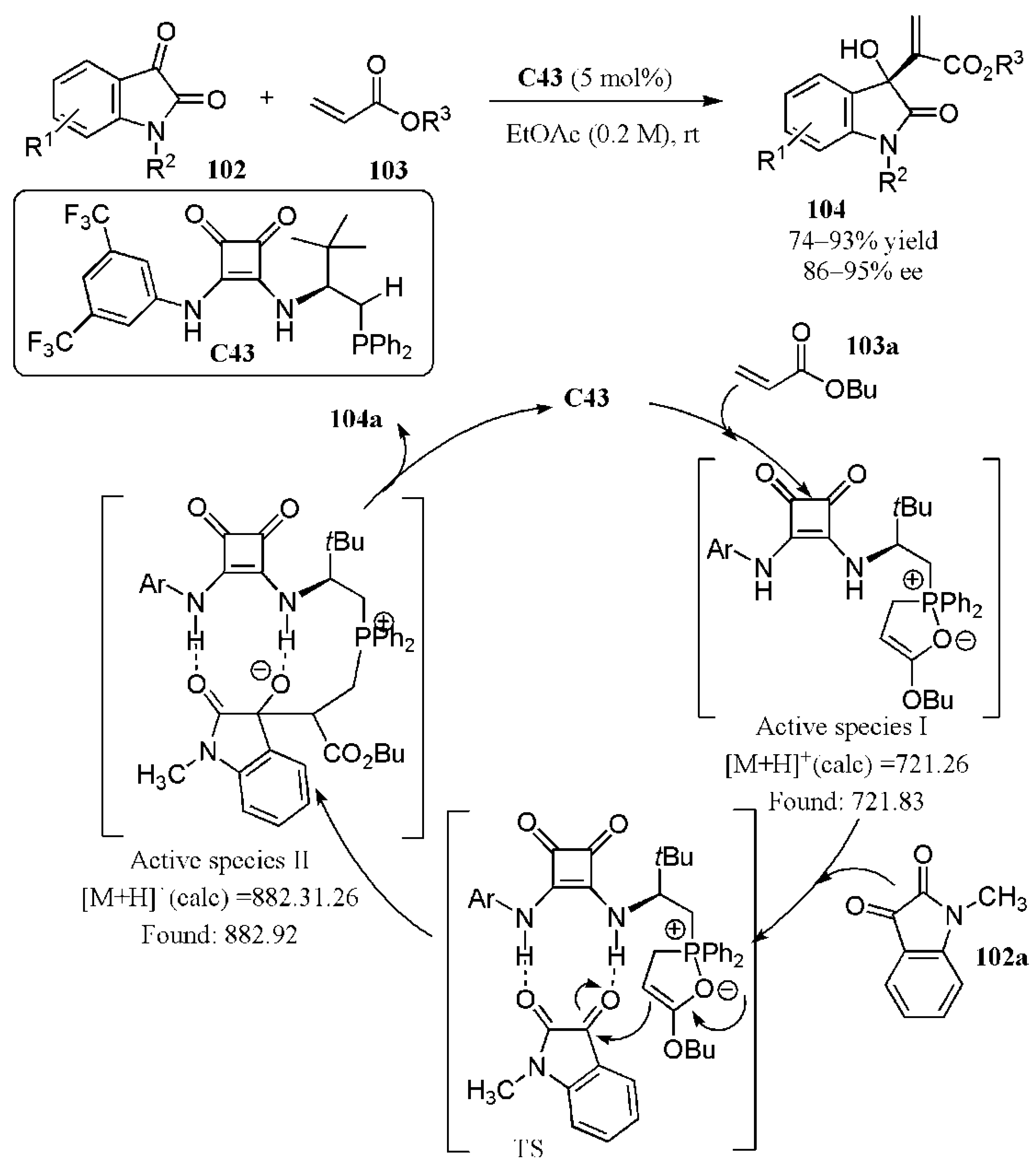
- Dong, Z.; Yan, C.; Gao, Y.; Dong, C.; Qiu, G.; Zhou, H.-B. Tunable bifunctional phosphine–squaramide promoted Morita–Baylis–Hillman reaction of N-alkyl isatins with acrylates. Adv. Synth. Catal. 2015, 357, 2132–2142. [Google Scholar] [CrossRef]
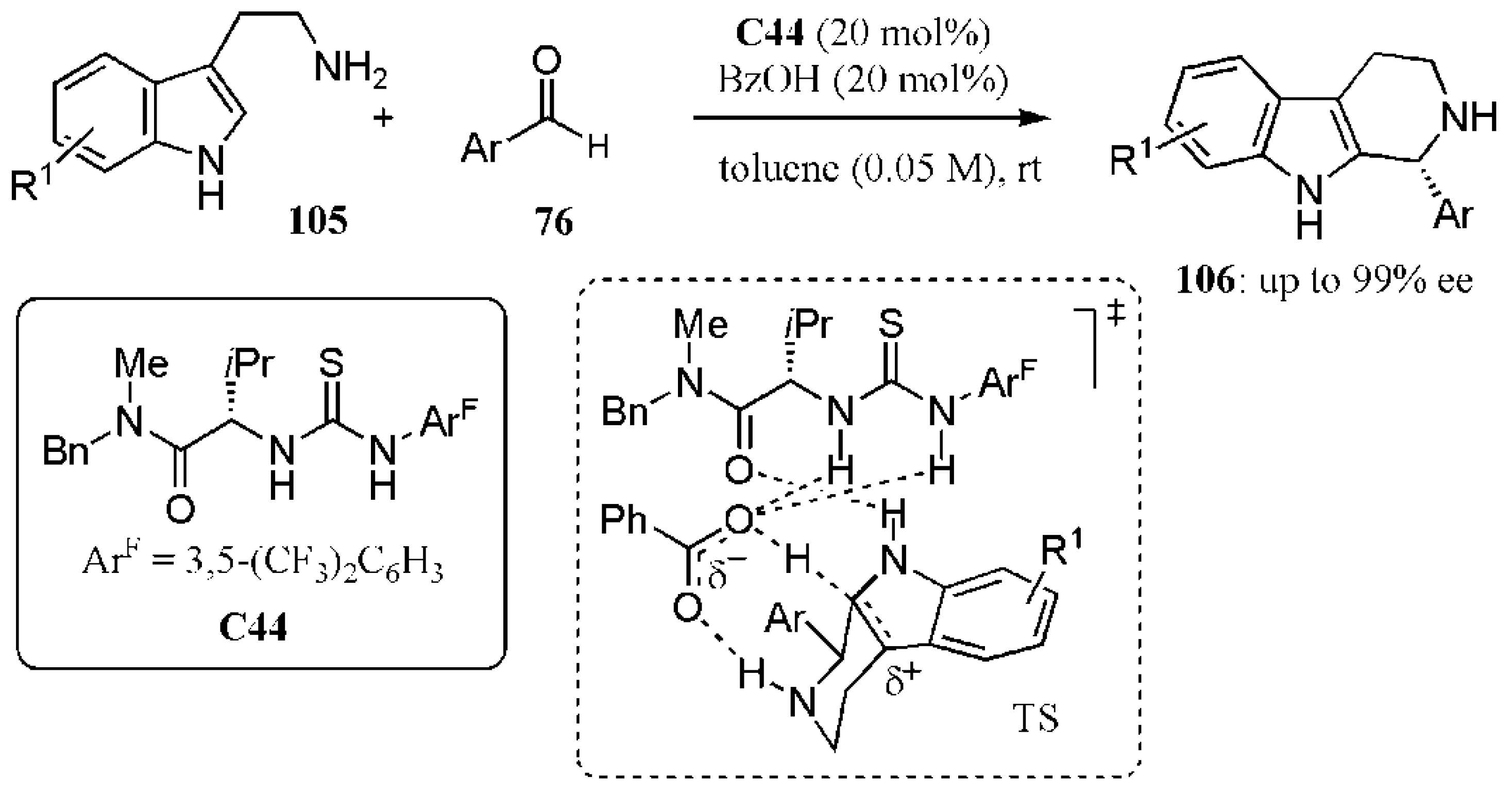
- Klausen, R.S.; Kennedy, C.R.; Hyde, A.M.; Jacobsen, E.N. Chiral thioureas promote enantioselective Pictet−Spengler cyclization by stabilizing every intermediate and transition state in the carboxylic acid-catalyzed reaction. J. Am. Chem. Soc. 2017, 139, 12299–12309. [Google Scholar] [CrossRef]
- Klausen, R.S.; Jacobsen, E.N. Weak Brønsted acid−thiourea co-catalysis: Enantioselective, catalytic protio-Pictet−Spengler reactions. Org. Lett. 2009, 11, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Hou, H.; Ling, F.; Zhong, W. The cinchona alkaloid squaramide catalyzed asymmetric Pictet–Spengler reaction and related theoretical studies. Org. Biomol. Chem. 2018, 16, 566–574. [Google Scholar] [CrossRef] [PubMed]
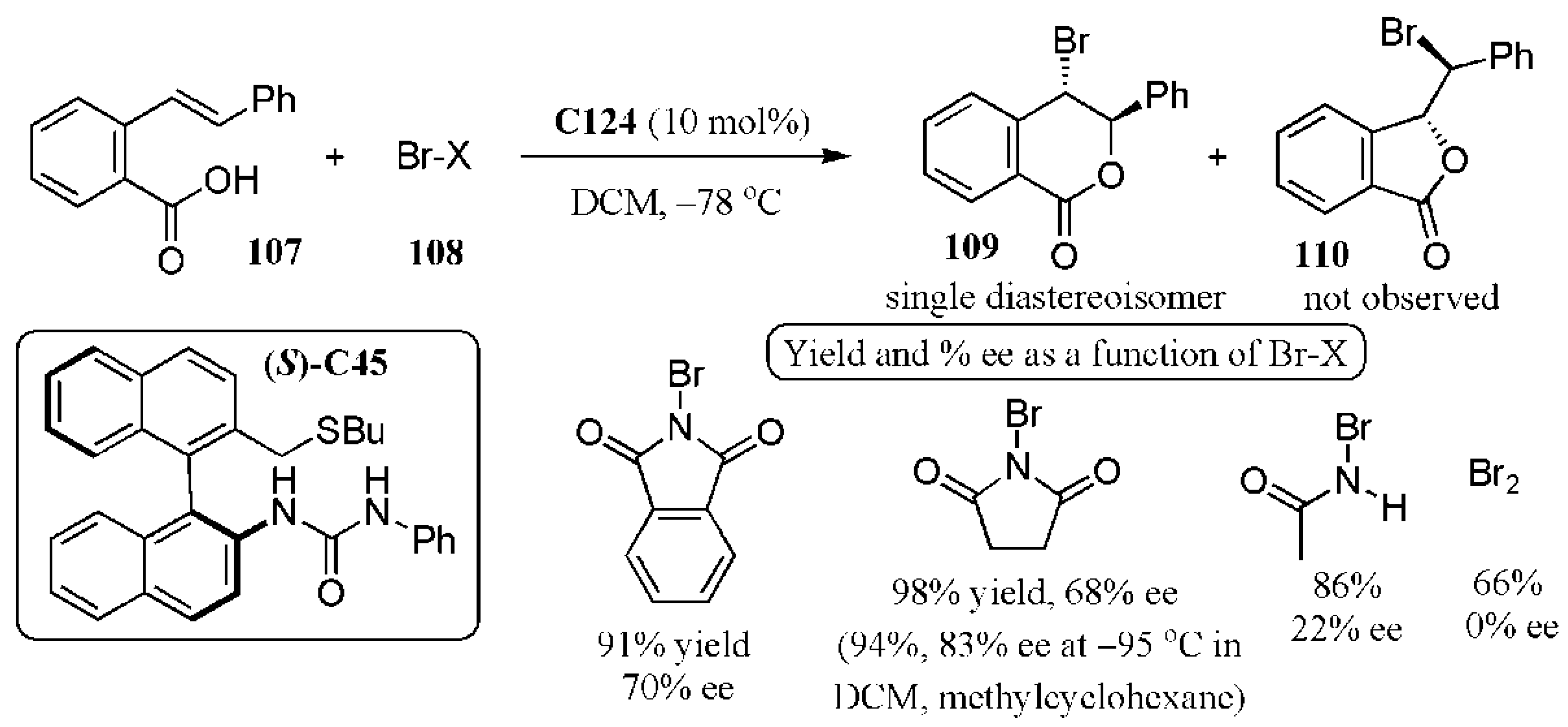
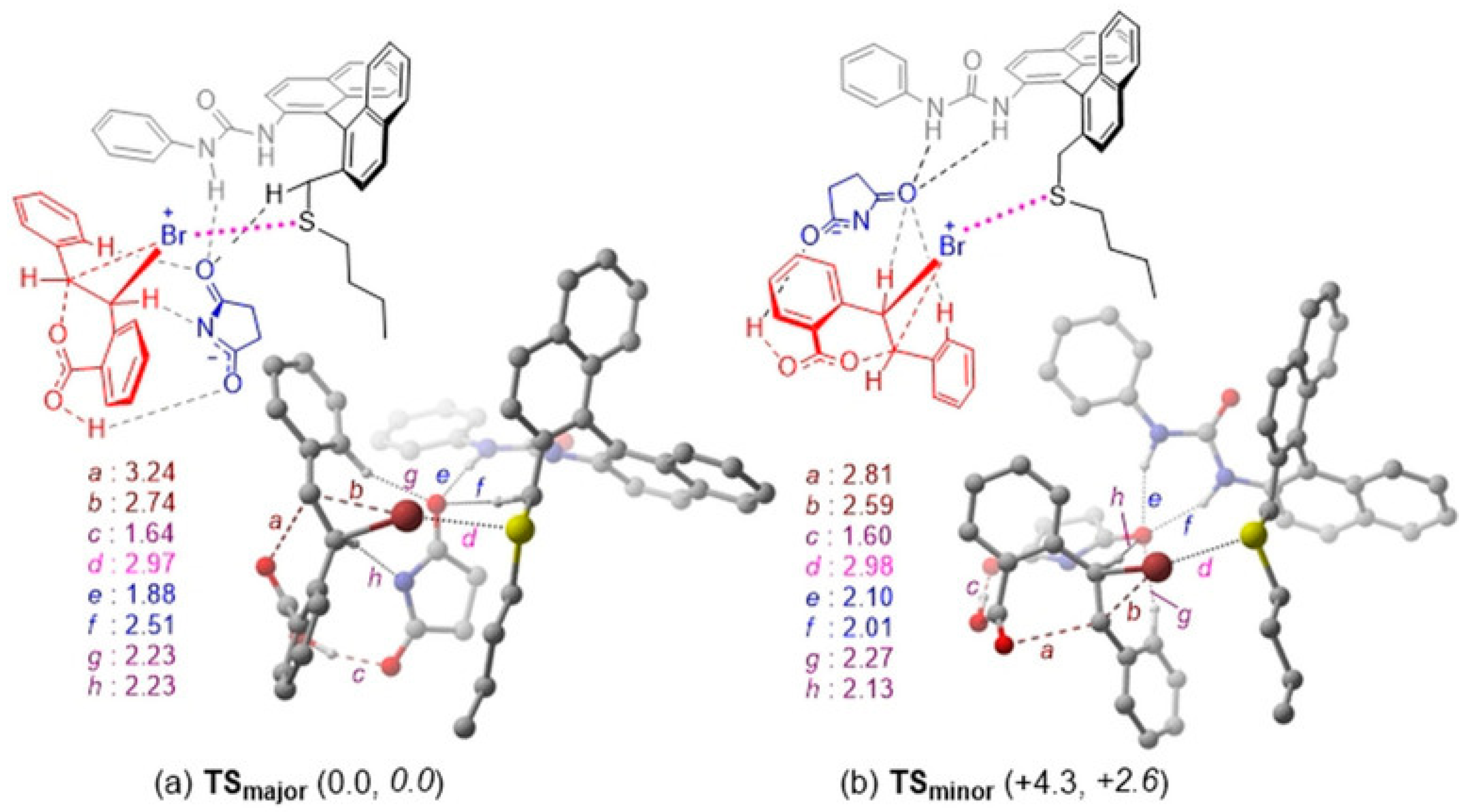
- Nishiyori, R.; Tsuchihashi, A.; Mochizuki, A.; Kaneko, K.; Yamanaka, M.; Shirakawa, S. Design of chiral bifunctional dialkyl sulfide catalysts for regio-, diastereo-, and enantioselective bromolactonization. Chem. Eur. J. 2018, 24, 16747–16752. [Google Scholar] [CrossRef]
- Werth, J.; Sigman, M.S. Connecting and analyzing enantioselective bifunctional hydrogen bond donor catalysis using data science tools. J. Am. Chem. Soc. 2020, 142, 16382–16391. [Google Scholar] [CrossRef] [PubMed]






















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
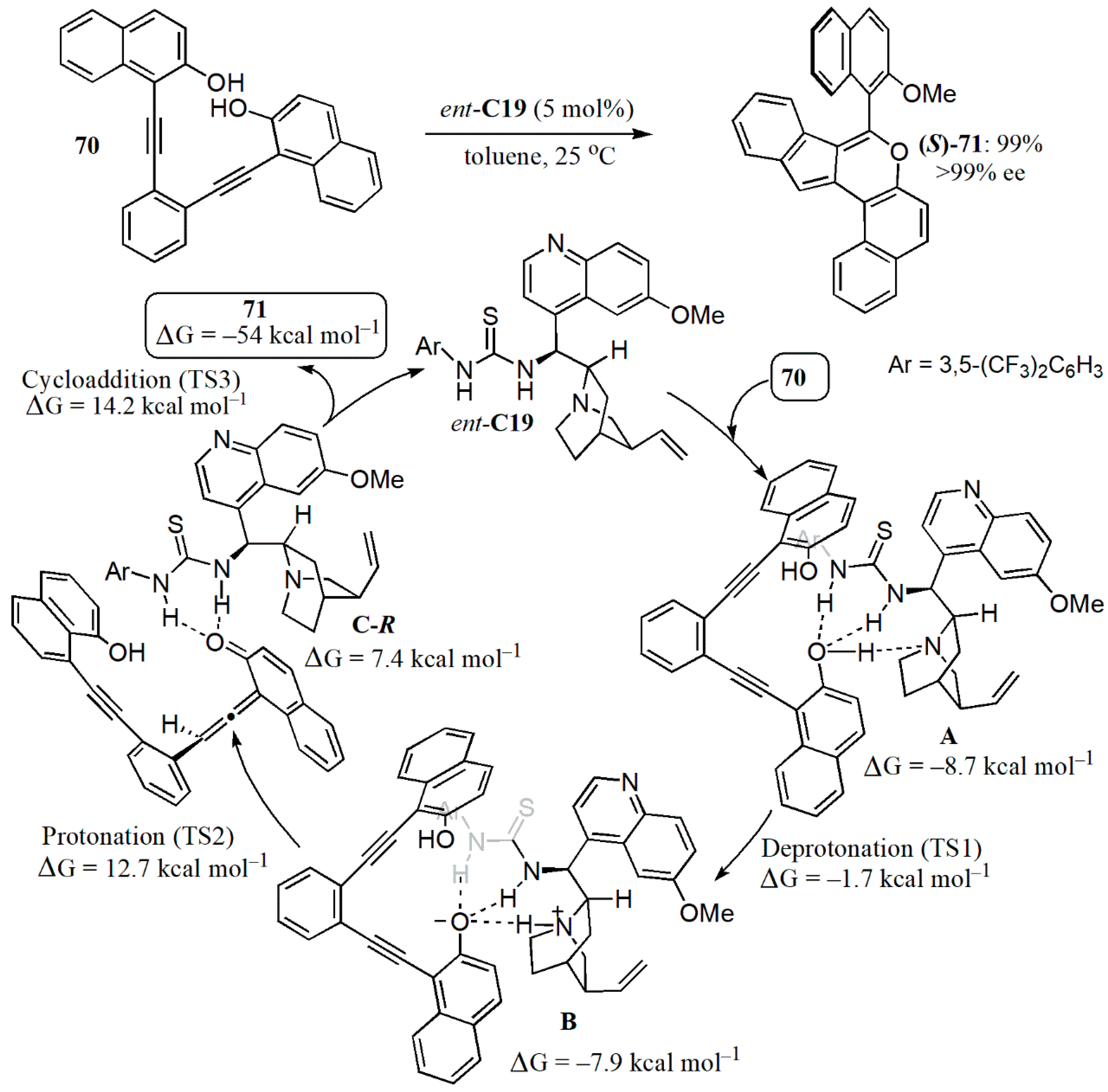{kind=link}
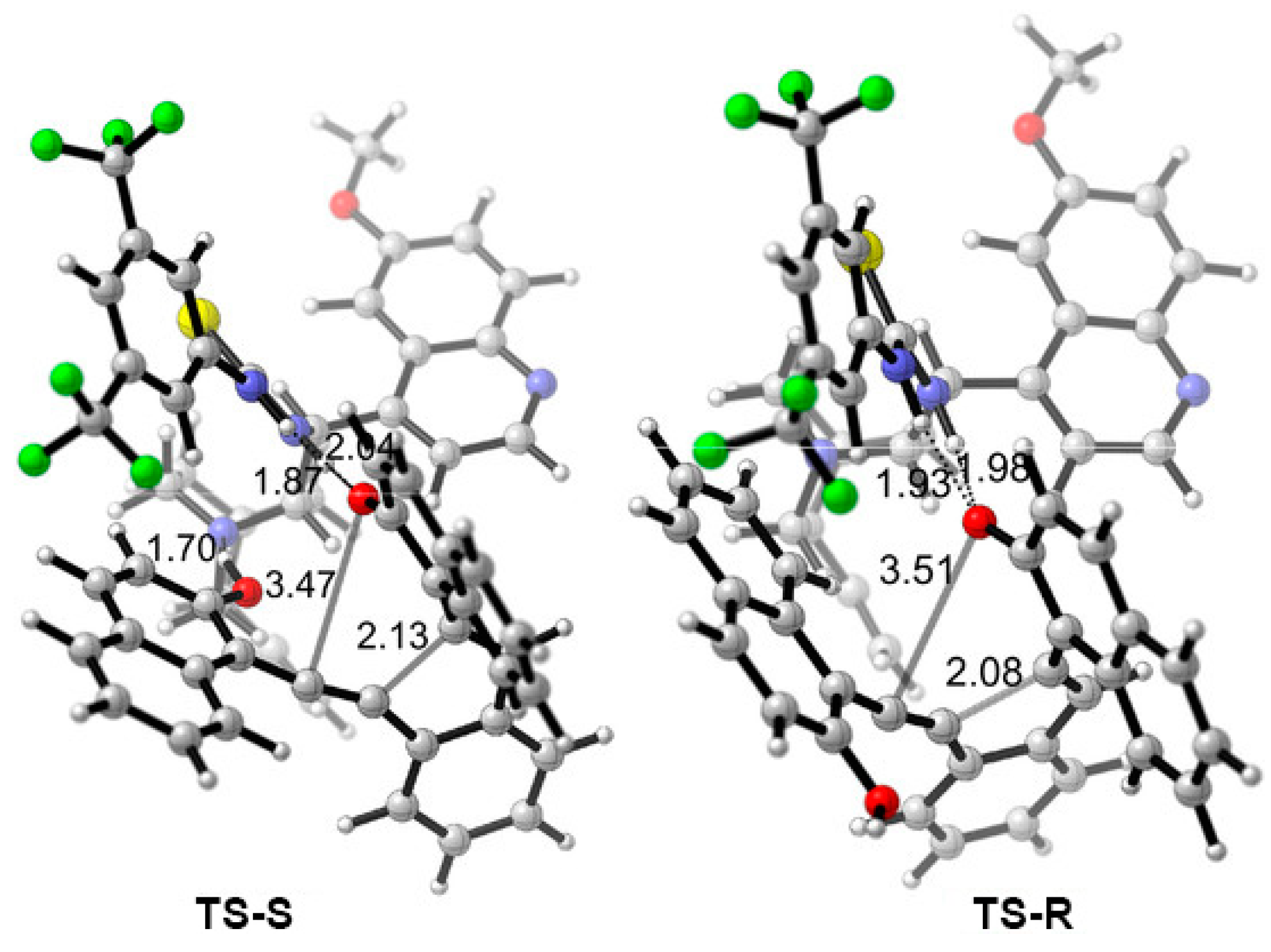{kind=link}
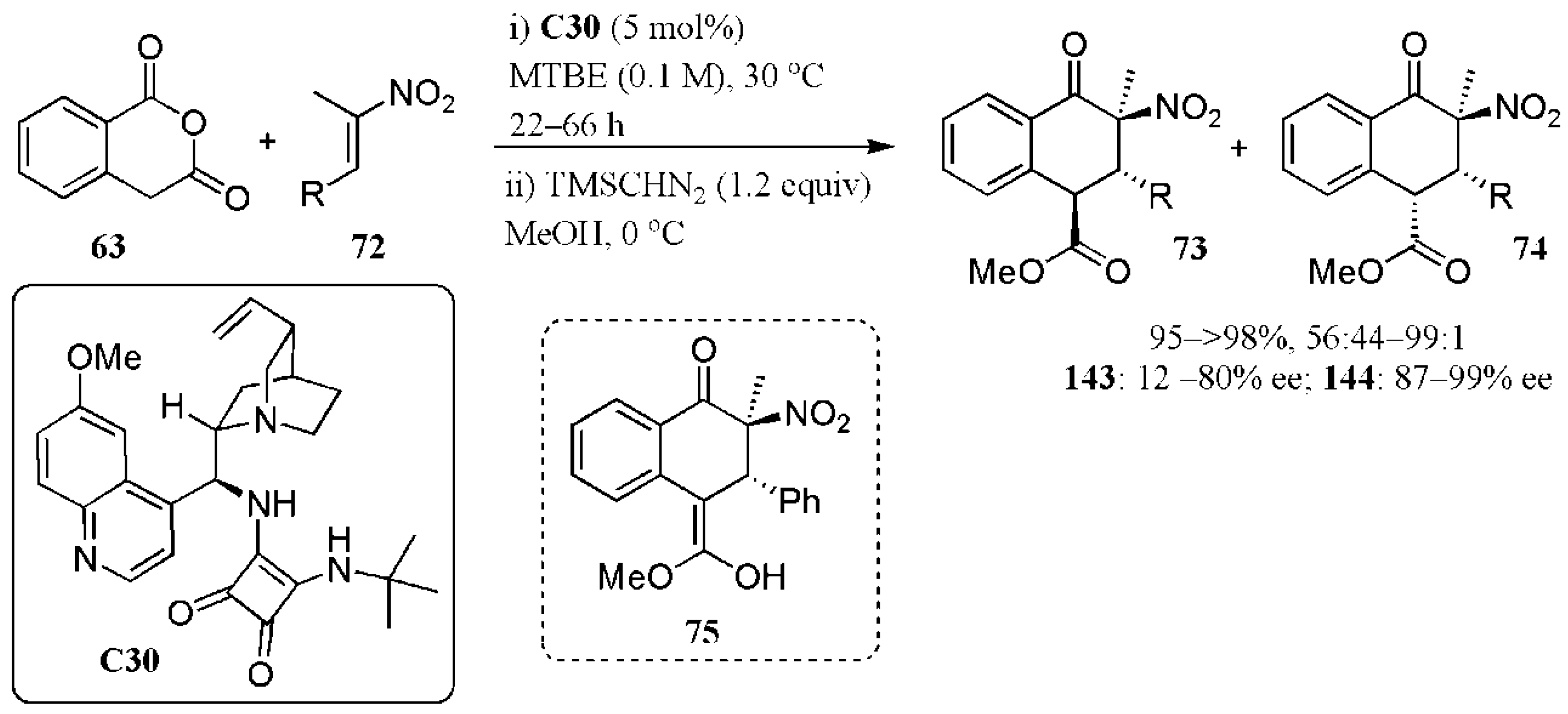{kind=link}
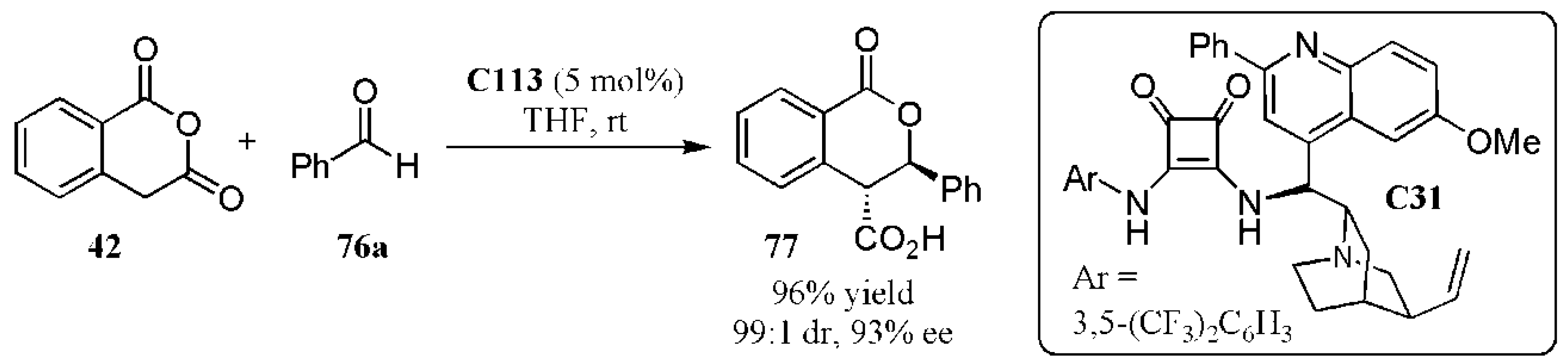{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phillips, A.M.F.; Prechtl, M.H.G.; Pombeiro, A.J.L. Non-Covalent Interactions in Enantioselective Organocatalysis: Theoretical and Mechanistic Studies of Reactions Mediated by Dual H-Bond Donors, Bifunctional Squaramides, Thioureas and Related Catalysts. Catalysts 2021, 11, 569. https://doi.org/10.3390/catal11050569
Phillips AMF, Prechtl MHG, Pombeiro AJL. Non-Covalent Interactions in Enantioselective Organocatalysis: Theoretical and Mechanistic Studies of Reactions Mediated by Dual H-Bond Donors, Bifunctional Squaramides, Thioureas and Related Catalysts. Catalysts. 2021; 11(5):569. https://doi.org/10.3390/catal11050569
Chicago/Turabian StylePhillips, Ana Maria Faisca, Martin H. G. Prechtl, and Armando J. L. Pombeiro. 2021. "Non-Covalent Interactions in Enantioselective Organocatalysis: Theoretical and Mechanistic Studies of Reactions Mediated by Dual H-Bond Donors, Bifunctional Squaramides, Thioureas and Related Catalysts" Catalysts 11, no. 5: 569. https://doi.org/10.3390/catal11050569
APA StylePhillips, A. M. F., Prechtl, M. H. G., & Pombeiro, A. J. L. (2021). Non-Covalent Interactions in Enantioselective Organocatalysis: Theoretical and Mechanistic Studies of Reactions Mediated by Dual H-Bond Donors, Bifunctional Squaramides, Thioureas and Related Catalysts. Catalysts, 11(5), 569. https://doi.org/10.3390/catal11050569