
The Effect of Co Incorporation on the CO Oxidation Activity of LaFe1−xCoxO3 Perovskites

 ,
,  , ,
, ,
Abstract

1. Introduction
2. Results
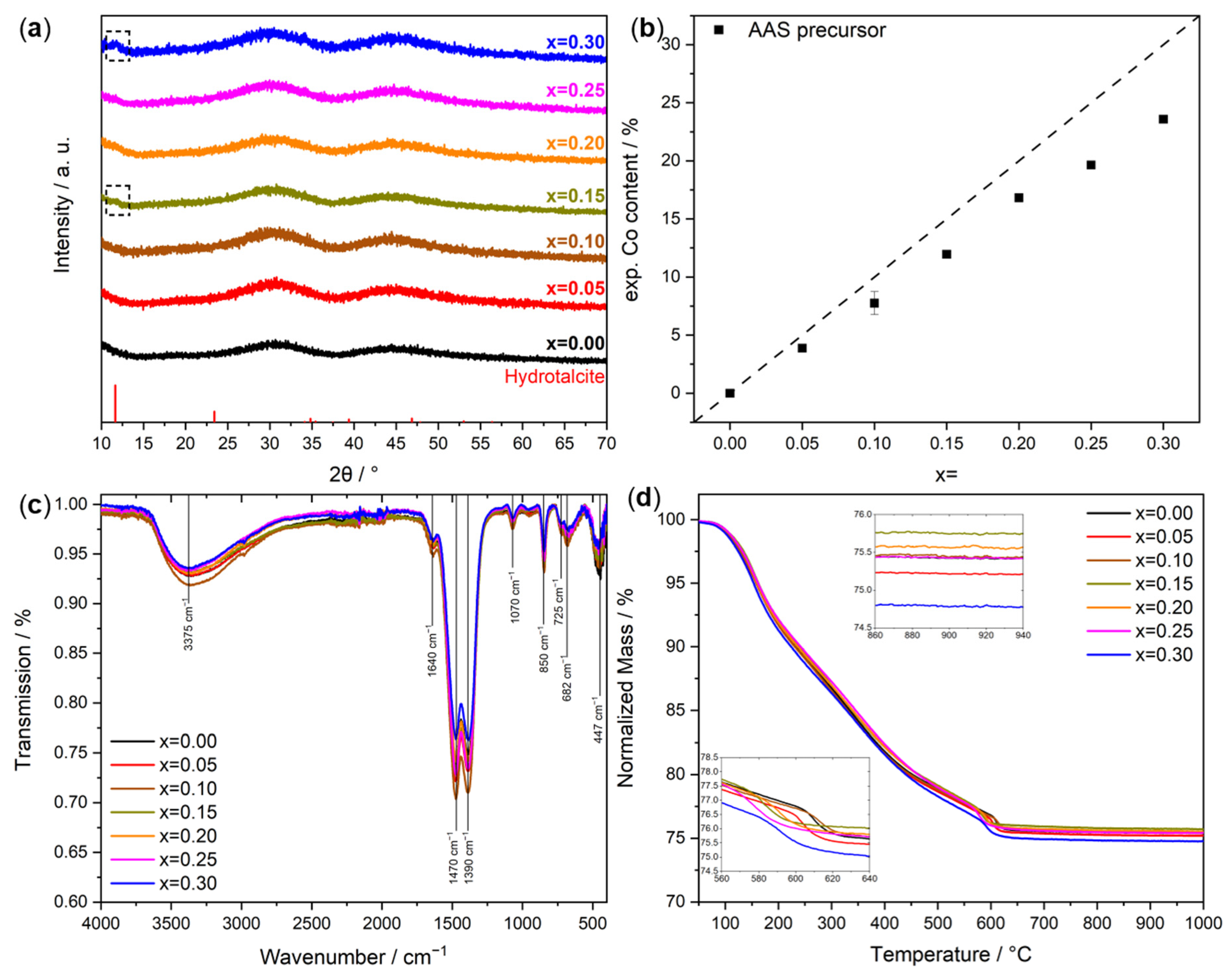
2.1. Co-Precipitation and Precursor Characterization
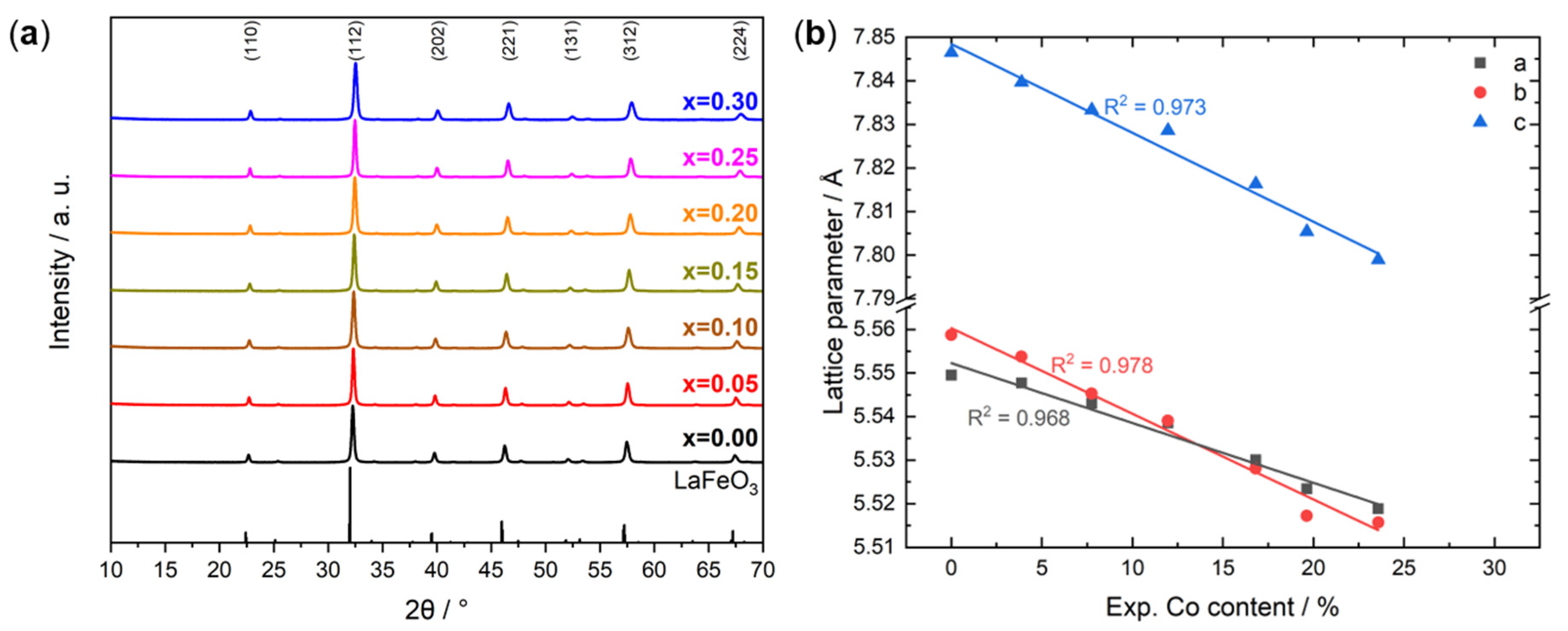
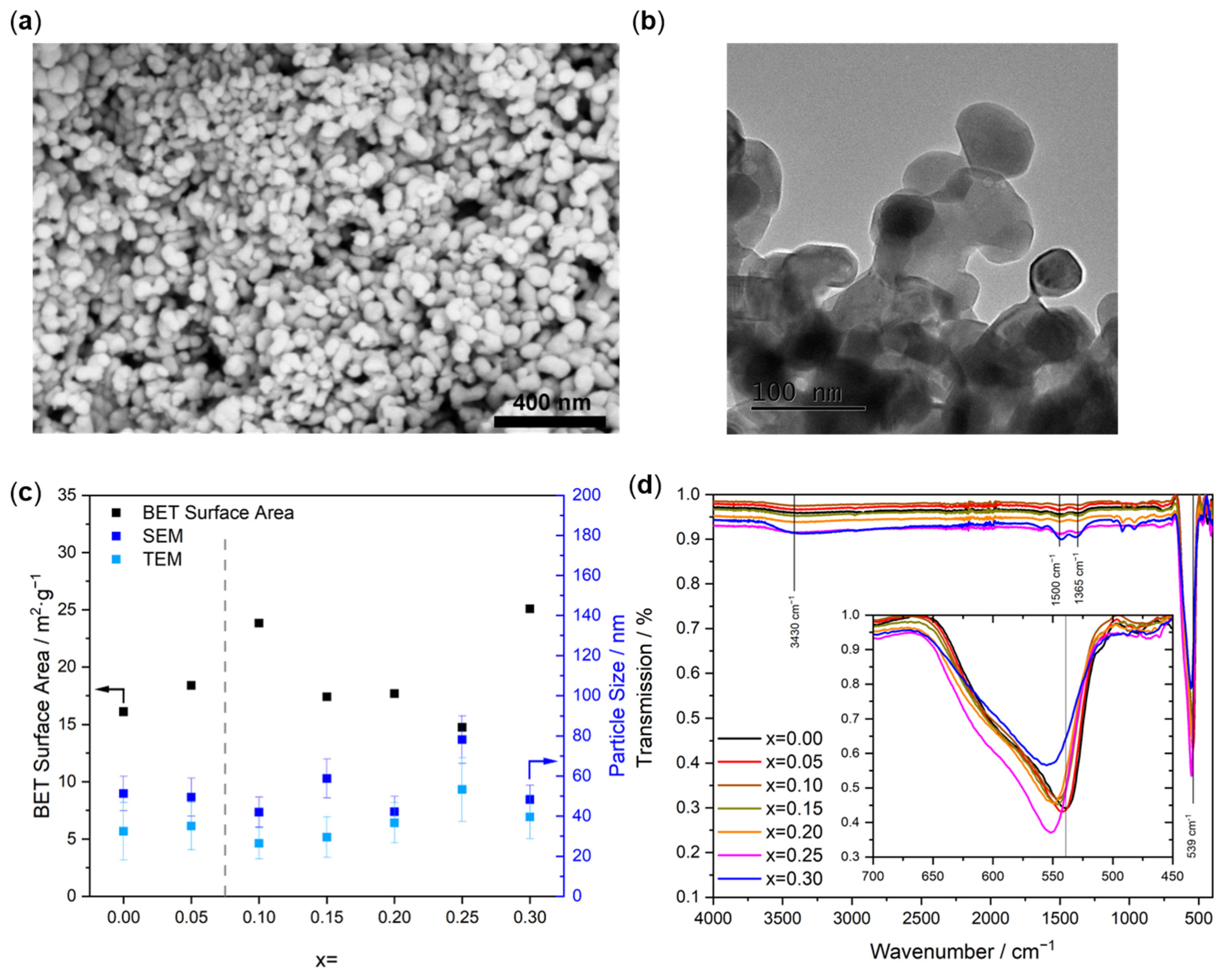
2.2. Catalyst Characterization
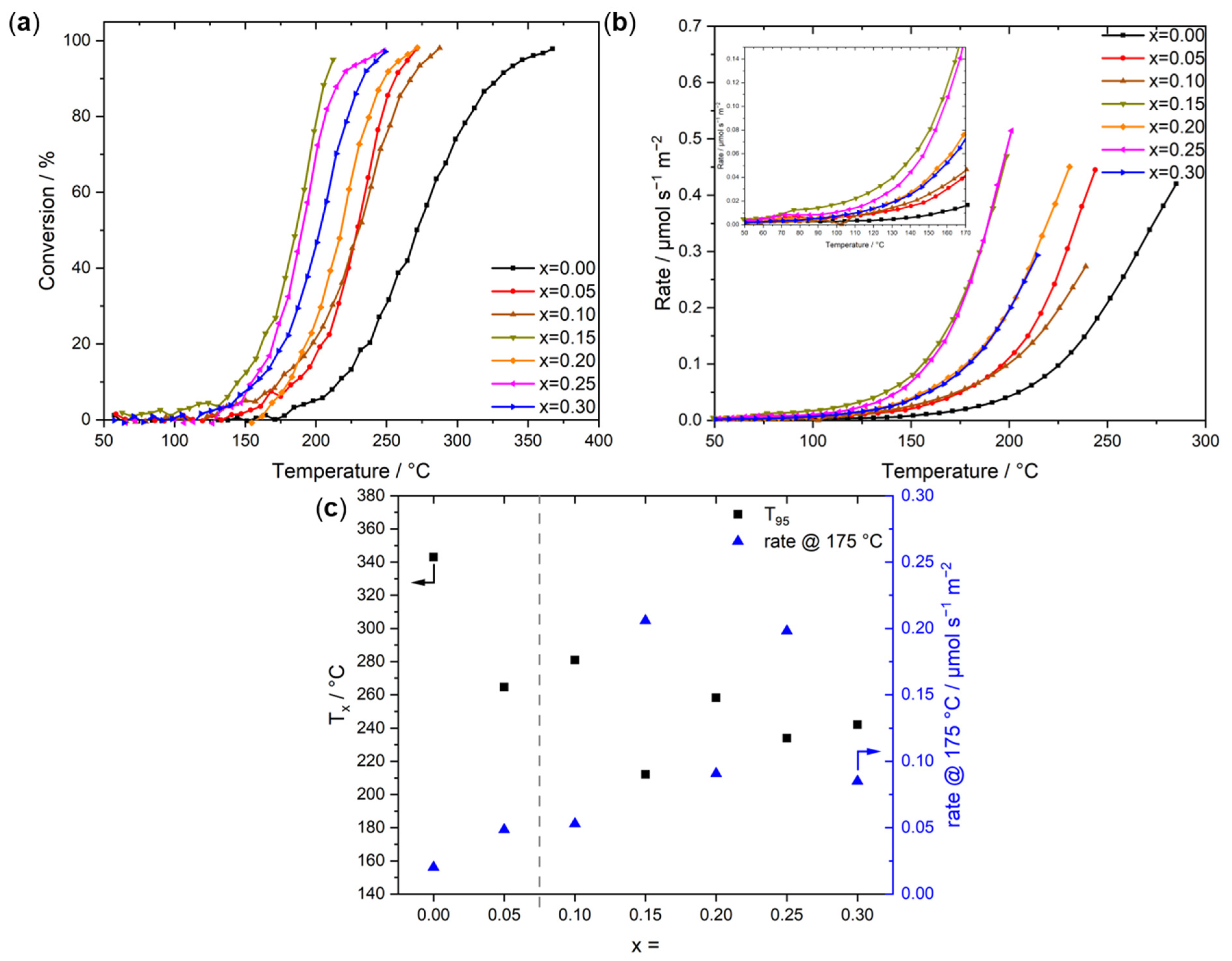
2.3. CO Oxidation Activity
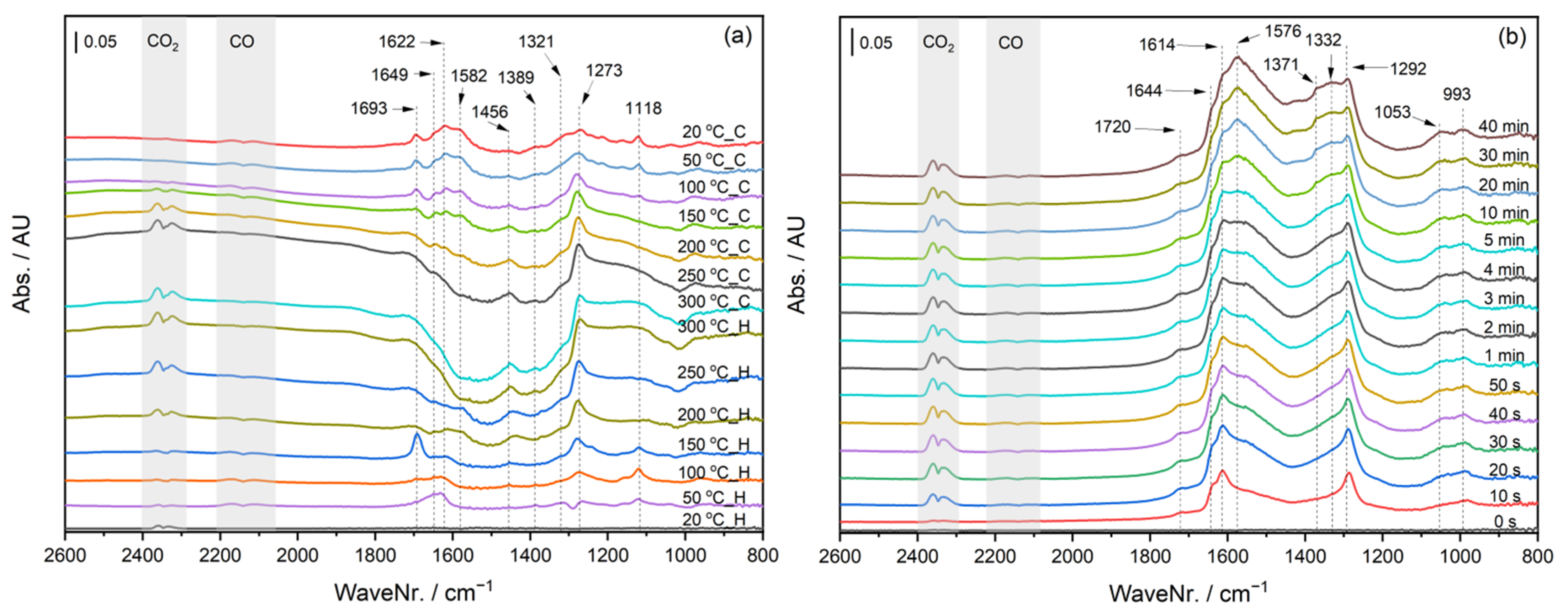
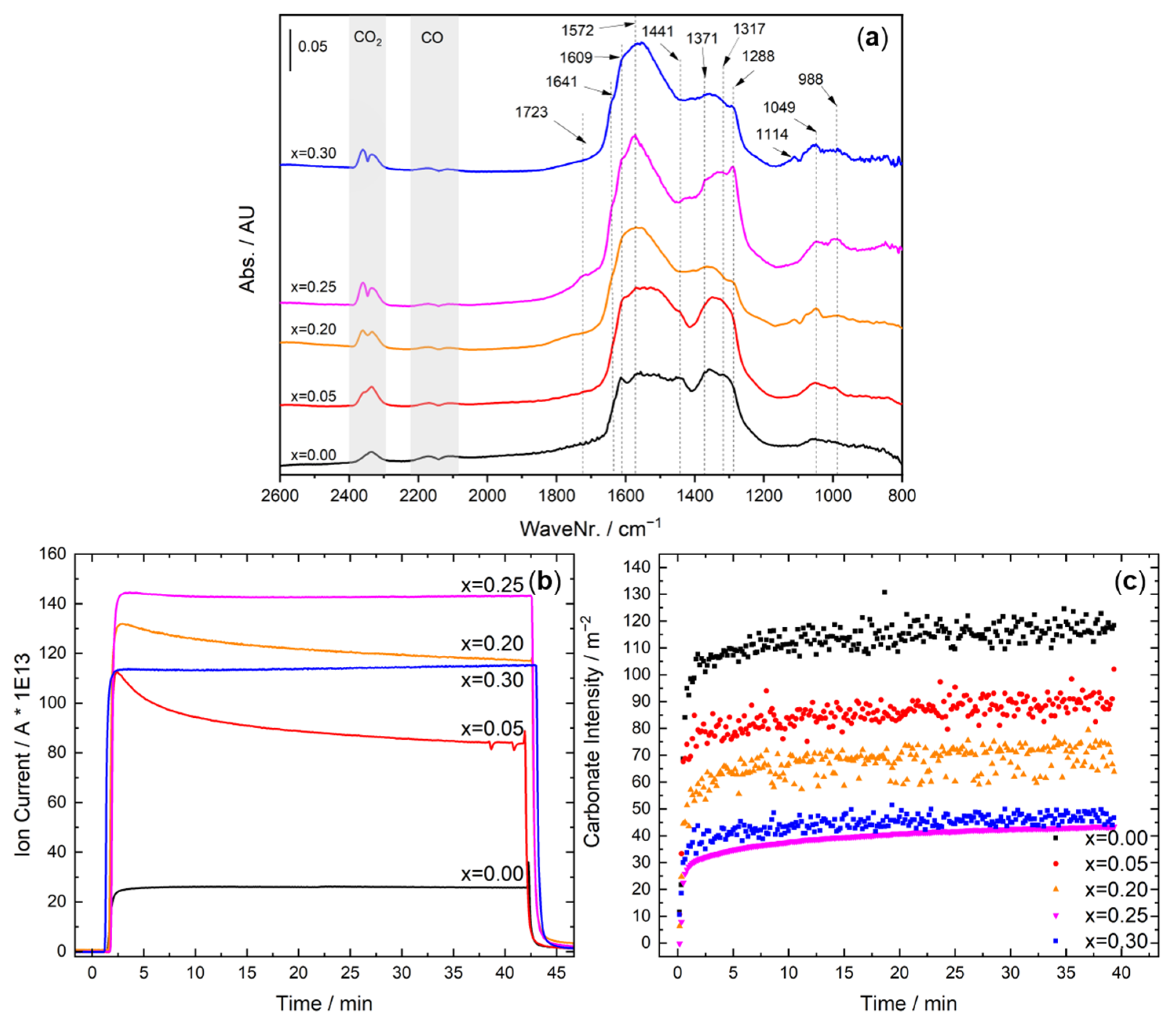
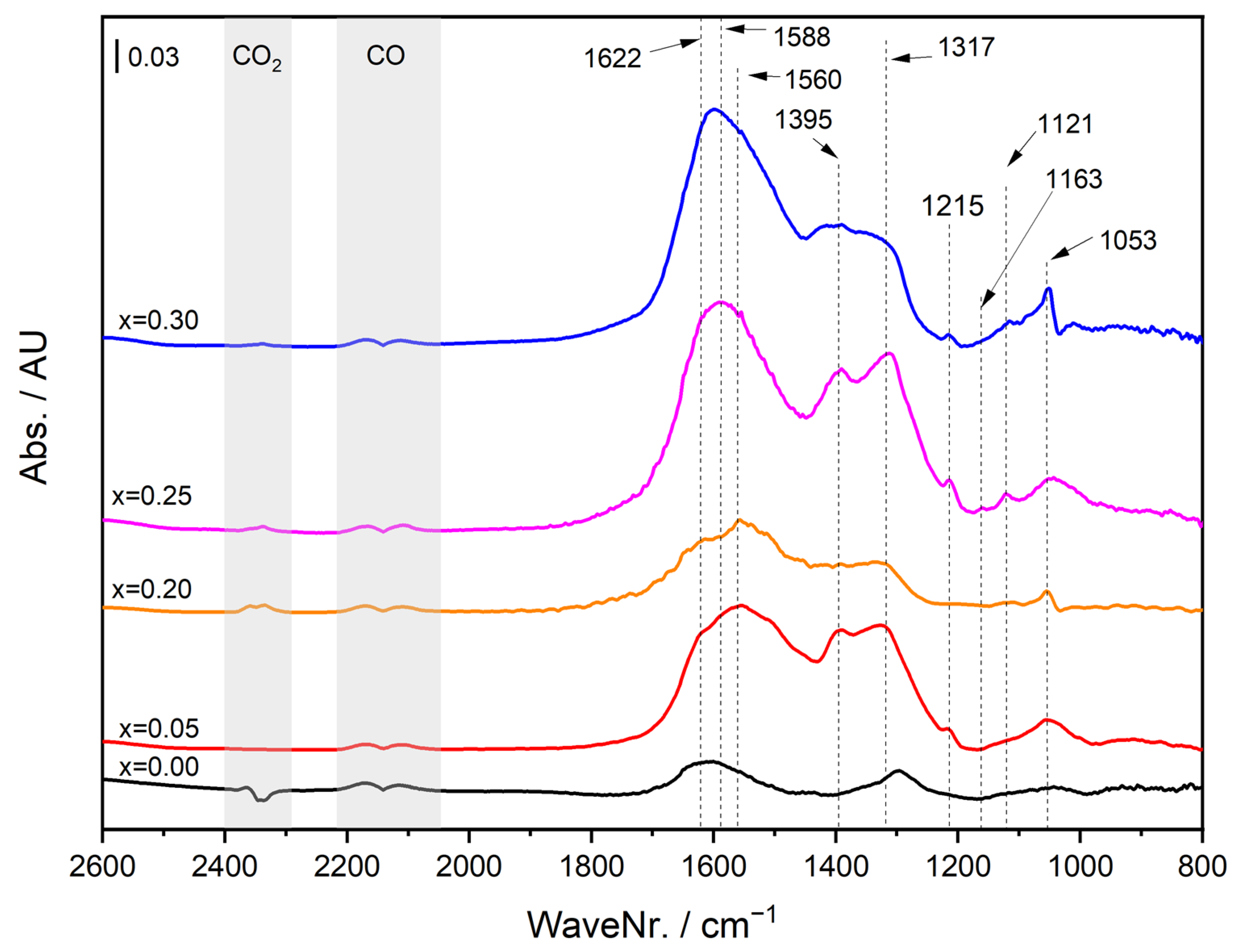
2.4. Operando DRIFTS during CO Oxidation
3. Materials and Methods
3.1. Raw Materials
3.2. Synthesis and Sample Preparation
3.3. Catalyst Characterization
3.4. Catalytic CO Oxidation at Ambient Pressure
3.5. Operando DRIFTS during CO Oxidation
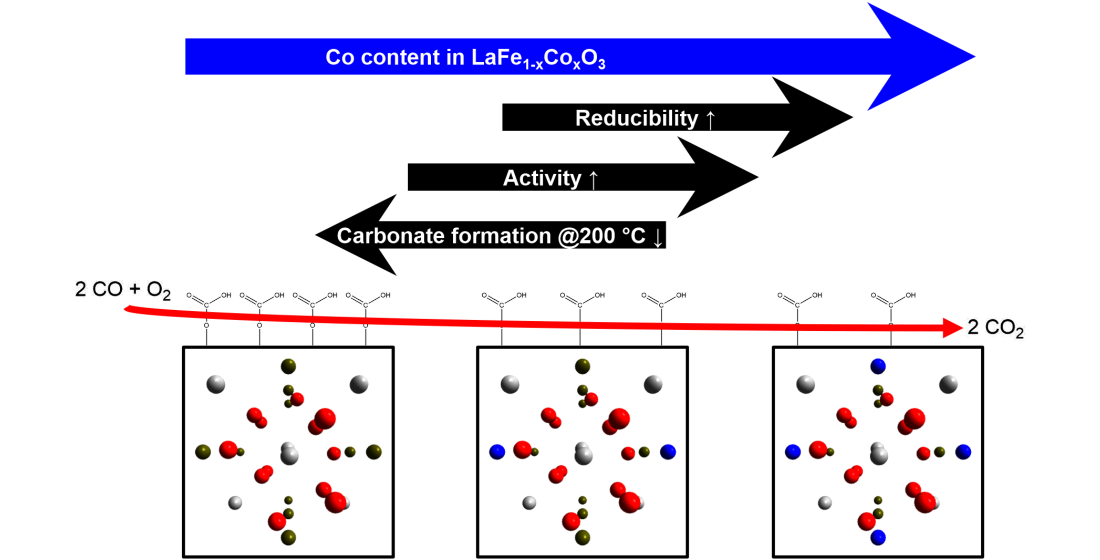
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Royer, S.; Duprez, D.; Can, F.; Courtois, X.; Batiot-Dupeyrat, C.; Laassiri, S.; Alamdari, H. Perovskites as substitutes of noble metals for heterogeneous catalysis: Dream or reality. Chem. Rev. 2014, 114, 10292–10368. [Google Scholar] [CrossRef] [PubMed]
- Kayaalp, B.; Lee, S.; Klauke, K.; Seo, J.; Nodari, L.; Kornowski, A.; Jung, W.; Mascotto, S. Template-free mesoporous La0.3Sr0.7Ti1−xFexO3±δ for CH4 and CO oxidation catalysis. Appl. Catal. B Environ. 2019, 245, 536–545. [Google Scholar] [CrossRef]
- Zhao, K.; He, F.; Huang, Z.; Wei, G.; Zheng, A.; Li, H.; Zhao, Z. Perovskite-type oxides LaFe1−CoO3 for chemical looping steam methane reforming to syngas and hydrogen co-production. Appl. Energy 2016, 168, 193–203. [Google Scholar] [CrossRef]
- Suntivich, J.; Gasteiger, H.A.; Yabuuchi, N.; Nakanishi, H.; Goodenough, J.B.; Shao-Horn, Y. Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal-air batteries. Nat. Chem. 2011, 3, 546–550. [Google Scholar] [CrossRef]
- Suntivich, J.; May, K.J.; Gasteiger, H.A.; Goodenough, J.B.; Shao-Horn, Y. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science 2011, 334, 1383–1385. [Google Scholar] [CrossRef]
- Peña, M.A.; Fierro, J.L. Chemical structures and performance of perovskite oxides. Chem. Rev. 2001, 101, 1981–2017. [Google Scholar] [CrossRef] [PubMed]
- Szabo, V.; Bassir, M.; van Neste, A.; Kaliaguine, S. Perovskite-type oxides synthesized by reactive grinding: Part II: Catalytic properties of LaCo(1−x)FexO3 in VOC oxidation. Appl. Catal. B Environ. 2002, 37, 175–180. [Google Scholar] [CrossRef]
- Sumathi, R.; Johnson, K.; Viswanathan, B.; Varadarajan, T.K. Selective oxidation and dehydrogenation of benzyl alcohol on ABB′O3 (A=Ba, B=Pb, Ce, Ti and B′=Bi, Cu, Sb)-type perovskite oxides-temperature programmed reduction studies. Appl. Catal. A Gen. 1998, 172, 15–22. [Google Scholar] [CrossRef]
- Waffel, D.; Alkan, B.; Fu, Q.; Chen, Y.-T.; Schmidt, S.; Schulz, C.; Wiggers, H.; Muhler, M.; Peng, B. Towards Mechanistic Understanding of Liquid-Phase Cinnamyl Alcohol Oxidation with tert-Butyl Hydroperoxide over Noble-Metal-Free LaCo1-x FexO3 Perovskites. ChemPlusChem 2019, 84, 1155–1163. [Google Scholar] [CrossRef]
- Voorhoeve, R.J.; Remeika, J.P.; Johnson, D.W. Rare-Earth manganites: Catalysts with low ammonia yield in the reduction of nitrogen oxides. Science 1973, 180, 62–64. [Google Scholar] [CrossRef]
- Voorhoeve, R.J.H.; Remeika, J.P.; Trimble, L.E. Defect chemistry and catalysis in oxidation and reduction over perovskite-type oxides. Ann. N. Y. Acad. Sci. 1976, 272, 3–21. [Google Scholar] [CrossRef]
- Baythoun, M.S.G.; Sale, F.R. Production of strontium-substituted lanthanum manganite perovskite powder by the amorphous citrate process. J. Mater. Sci. 1982, 17, 2757–2769. [Google Scholar] [CrossRef]
- Taguchi, H.; Yamada, S.; Nagao, M.; Ichikawa, Y.; Tabata, K. Surface characterization of LaCoO3 synthesized using citric acid. Mater. Res. Bull. 2002, 37, 69–76. [Google Scholar] [CrossRef]
- Gallagher, P.K.; Johnson, D.W.; Remeika, J.P.; Schrey, F.; Trimble, L.E.; Vogel, E.M.; Voorhoeve, R. The activity of La0.7Sr0.3MnO3 without Pt and La0.7Pb0.3MnO3 with varying Pt contents for the catalytic oxidation of CO. Mater. Res. Bull. 1975, 10, 529–538. [Google Scholar] [CrossRef]
- Angel, S.; Neises, J.; Dreyer, M.; Friedel Ortega, K.; Behrens, M.; Wang, Y.; Arandiyan, H.; Schulz, C.; Wiggers, H. Spray-flame synthesis of La(Fe, Co)O3 nano-perovskites from metal nitrates. AIChE J. 2019, 10, 441. [Google Scholar] [CrossRef]
- Alkan, B.; Cychy, S.; Varhade, S.; Muhler, M.; Schulz, C.; Schuhmann, W.; Wiggers, H.; Andronescu, C. Spray-Flame-Synthesized LaCo1−xFexO3 Perovskite Nanoparticles as Electrocatalysts for Water and Ethanol Oxidation. ChemElectroChem 2019, 6, 4266–4274. [Google Scholar] [CrossRef]
- Junwu, Z.; Xiaojie, S.; Yanping, W.; Xin, W.; Xujie, Y.; Lude, L. Solution-Phase Synthesis and Characterization of Perovskite LaCoO3 Nanocrystals via A Co-Precipitation Route. J. Rare Earths 2007, 25, 601–604. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Uphade, B.S.; Belhekar, A.A. Oxidative Conversion of Methane to Syngas over LaNiO3Perovskite with or without Simultaneous Steam and CO2Reforming Reactions: Influence of Partial Substitution of La and Ni. J. Catal. 1996, 163, 312–318. [Google Scholar] [CrossRef]
- Sim, Y.; Yoo, J.; Ha, J.-M.; Jung, J.C. Oxidative coupling of methane over LaAlO3 perovskite catalysts prepared by a co-precipitation method: Effect of co-precipitation pH value. J. Energy Chem. 2019, 35, 1–8. [Google Scholar] [CrossRef]
- Kühl, S.; Düdder, H.; Girgsdies, F.; Kähler, K.; Muhler, M.; Behrens, M. Perovskites as Precursors for Ni/La2O3 Catalysts in the Dry Reforming of Methane: Synthesis by Constant pH Co-Precipitation, Reduction Mechanism and Effect of Ru-Doping. Z. Anorg. Allg. Chem. 2017, 643, 1088–1095. [Google Scholar] [CrossRef]
- Bedel, L.; Roger, A.; Estournes, C.; Kiennemann, A. Co0 from partial reduction of La(Co,Fe)O3 perovskites for Fischer–Tropsch synthesis. Catal. Today 2003, 85, 207–218. [Google Scholar] [CrossRef]
- Derakhshi, Z.; Tamizifar, M.; Arzani, K.; Baghshahi, S. Synthesis and Characterization of LaCoxFe1−xO3 (0≤x≤1) Nano-Crystal Powders by Pechini Type Sol–Gel Method. Synth. React. Inorg. M. 2016, 46, 25–30. [Google Scholar] [CrossRef]
- Wu, Y.; Cordier, C.; Berrier, E.; Nuns, N.; Dujardin, C.; Granger, P. Surface reconstructions of LaCo1−xFexO3 at high temperature during N2O decomposition in realistic exhaust gas composition: Impact on the catalytic properties. Appl. Catal. B Environ. 2013, 140–141, 151–163. [Google Scholar] [CrossRef]
- Yang, Y.; Shi, K. Preparation of LaFeXCo1−XO3 Perovskite by Coprecipitation. Open, J. Nat. Sci. 2016, 4, 378–383. [Google Scholar] [CrossRef]
- Kaiwen, Z.; Xuehang, W.; Wenwei, W.; Jun, X.; Siqi, T.; Sen, L. Nanocrystalline LaFeO3 preparation and thermal process of precursor. Adv. Powder Technol. 2013, 24, 359–363. [Google Scholar] [CrossRef]
- Kaliaguine, S.; Szabo, V.; van Neste, A.; Gallot, J.E.; Bassir, M.; Muzychuk, R. Perovskite-Type Oxides Synthesized by Reactive Grinding. Appl. Catal. A Gen. 2001, 11, 39–56. [Google Scholar] [CrossRef]
- Xu, J.; Liu, J.; Zhao, Z.; Zheng, J.; Zhang, G.; Duan, A.; Jiang, G. Three-dimensionally ordered macroporous LaCoxFe1−xO3 perovskite-type complex oxide catalysts for diesel soot combustion. Catal. Today 2010, 153, 136–142. [Google Scholar] [CrossRef]
- Staykov, A.; Téllez, H.; Akbay, T.; Druce, J.; Ishihara, T.; Kilner, J. Oxygen Activation and Dissociation on Transition Metal Free Perovskite Surfaces. Chem. Mater. 2015, 27, 8273–8281. [Google Scholar] [CrossRef]
- Seo, M.H.; Park, H.W.; Lee, D.U.; Park, M.G.; Chen, Z. Design of Highly Active Perovskite Oxides for Oxygen Evolution Reaction by Combining Experimental and ab Initio Studies. ACS Catal. 2015, 5, 4337–4344. [Google Scholar] [CrossRef]
- Yang, J.; Hu, S.; Fang, Y.; Hoang, S.; Li, L.; Yang, W.; Liang, Z.; Wu, J.; Hu, J.; Xiao, W.; et al. Oxygen Vacancy Promoted O2 Activation over Perovskite Oxide for Low-Temperature CO Oxidation. ACS Catal. 2019, 9, 9751–9763. [Google Scholar] [CrossRef]
- Freund, H.-J.; Meijer, G.; Scheffler, M.; Schlögl, R.; Wolf, M. CO oxidation as a prototypical reaction for heterogeneous processes. Angew. Chem. Int. Ed. 2011, 50, 10064–10094. [Google Scholar] [CrossRef]
- Lukashuk, L.; Yigit, N.; Rameshan, R.; Kolar, E.; Teschner, D.; Hävecker, M.; Knop-Gericke, A.; Schlögl, R.; Föttinger, K.; Rupprechter, G. Operando Insights into CO Oxidation on Cobalt Oxide Catalysts by NAP-XPS, FTIR, and XRD. ACS Catal. 2018, 8, 8630–8641. [Google Scholar] [CrossRef]
- Föttinger, K.; Schlögl, R.; Rupprechter, G. The mechanism of carbonate formation on Pd-Al2O3 catalysts. Chem. Commun. 2008, 3, 320–322. [Google Scholar] [CrossRef]
- Yi, Y.; Zhang, P.; Qin, Z.; Yu, C.; Li, W.; Qin, Q.; Li, B.; Fan, M.; Liang, X.; Dong, L. Low temperature CO oxidation catalysed by flower-like Ni–Co–O: How physicochemical properties influence catalytic performance. RSC Adv. 2018, 8, 7110–7122. [Google Scholar] [CrossRef]
- Kuhn, J.; Ozkan, U. Surface properties of Sr- and Co-doped LaFeO3. J. Catal. 2008, 253, 200–211. [Google Scholar] [CrossRef]
- Allmann, R.; Jepsen, H.P. Die Struktur des Hydrotalkits. Neues Jahrb. Miner. Mon. 1969, 1969, 544–551. [Google Scholar]
- Xie, J.; Wu, Q.; Zhang, D.; Ding, Y. Biomolecular-Induced Synthesis of Self-Assembled Hierarchical La(OH)CO3 One-Dimensional Nanostructures and Its Morphology-Held Conversion toward La2O3 and La(OH)3. Cryst. Growth Des. 2009, 9, 3889–3897. [Google Scholar] [CrossRef]
- del Arco, M.; Trujillano, R.; Rives, V. Cobalt-iron hydroxycarbonates and their evolution to mixed oxides with spinel structure. J. Mater. Chem. 1998, 8, 761–767. [Google Scholar] [CrossRef]
- Uzunova, E.; Klissurski, D.; Mitov, I.; Stefanov, P. Cobalt-iron hydroxide carbonate as a precursor for the synthesis of high-dispersity spinel mixed oxides. Chem. Mater. 1993, 5, 576–582. [Google Scholar] [CrossRef]
- Wang, C.-B.; Lin, H.-K.; Tang, C.-W. Thermal Characterization and Microstructure Change of Cobalt Oxides. Catal. Lett. 2004, 94, 69–74. [Google Scholar] [CrossRef]
- Sangaletti, L.; Depero, L.E.; Allieri, B.; Nunziante, P.; Traversa, E. An X-ray study of the trimetallic LaxSm1−xFeO3 orthoferrites. J. Eur. Ceram. Soc. 2001, 21, 719–726. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta. Cryst. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Gaikwad, V.M.; Acharya, S.A. Novel perovskite–spinel composite approach to enhance the magnetization of LaFeO3. RSC Adv. 2015, 5, 14366–14373. [Google Scholar] [CrossRef]
- Rietveld, H.M. Line profiles of neutron powder-diffraction peaks for structure refinement. Acta. Cryst. 1967, 22, 151–152. [Google Scholar] [CrossRef]
- Cui, H.; Zayat, M.; Levy, D. Epoxide assisted sol–gel synthesis of perovskite-type LaMxFe1−xO3 (M=Ni,Co) nanoparticles. J. Non-Cryst. Solids 2006, 352, 3035–3040. [Google Scholar] [CrossRef]
- Faye, J.; Baylet, A.; Trentesaux, M.; Royer, S.; Dumeignil, F.; Duprez, D.; Valange, S.; Tatibouët, J.-M. Influence of lanthanum stoichiometry in La1−xFeO3−δ perovskites on their structure and catalytic performance in CH4 total oxidation. Appl. Catal. B Environ. 2012, 126, 134–143. [Google Scholar] [CrossRef]
- Mihai, O.; Chen, D.; Holmen, A. Chemical looping methane partial oxidation: The effect of the crystal size and O content of LaFeO3. J. Catal. 2012, 293, 175–185. [Google Scholar] [CrossRef]
- Chang, H.; Bjørgum, E.; Mihai, O.; Yang, J.; Lein, H.L.; Grande, T.; Raaen, S.; Zhu, Y.-A.; Holmen, A.; Chen, D. Effects of Oxygen Mobility in La–Fe-Based Perovskites on the Catalytic Activity and Selectivity of Methane Oxidation. ACS Catal. 2020, 10, 3707–3719. [Google Scholar] [CrossRef]
- Khan, S.; Oldman, R.J.; Corà, F.; Catlow, C.R.A.; French, S.A.; Axon, S.A. A computational modelling study of oxygen vacancies at LaCoO3 perovskite surfaces. Phys. Chem. Chem. Phys. 2006, 8, 5207–5222. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, P.; Qiao, Z.-A.; Zhang, J.; Zhu, H.; Chen, J.; Chen, Y.; Dai, S. Ionic liquid-mediated synthesis of meso-scale porous lanthanum-transition-metal perovskites with high CO oxidation performance. Chem. Commun. (Camb.) 2015, 51, 5910–5913. [Google Scholar] [CrossRef]
- Ciambelli, P.; Cimino, S.; de Rossi, S.; Lisi, L.; Minelli, G.; Porta, P.; Russo, G. AFeO3 (A=La, Nd, Sm) and LaFe1−xMgxO3 perovskites as methane combustion and CO oxidation catalysts: Structural, redox and catalytic properties. Appl. Catal. B Environ. 2001, 29, 239–250. [Google Scholar] [CrossRef]
- Zhang, X.; Li, H.; Li, Y.; Shen, W. Structural Properties and Catalytic Activity of Sr-Substituted LaFeO3 Perovskite. Chin. J. Catal. 2012, 33, 1109–1114. [Google Scholar] [CrossRef]
- Li, P.; Hu, X.; Zhang, L.; Dai, H.; Zhang, L. Sol-gel nanocasting synthesis of patterned hierarchical LaFeO3 fibers with enhanced catalytic CO oxidation activity. Nanoscale 2011, 3, 974–976. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Rao, R.R.; Katayama, Y.; Lee, D.; Wang, X.R.; Crumlin, E.; Venkatesan, T.; Lee, H.N.; Shao-Horn, Y. CO2 Reactivity on Cobalt-Based Perovskites. J. Phys. Chem. C 2018, 122, 20391–20401. [Google Scholar] [CrossRef]
- Coenen, K.; Gallucci, F.; Mezari, B.; Hensen, E.; van Sint Annaland, M. An in-situ IR study on the adsorption of CO2 and H2O on hydrotalcites. J. CO2 Util. 2018, 24, 228–239. [Google Scholar] [CrossRef]
- Hadjiivanov, K.I.; Vayssilov, G.N. Characterization of oxide surfaces and zeolites by carbon monoxide as an IR probe molecule; Elsevier: Amsterdam, The Netherlands, 2002; pp. 307–511. ISBN 9780120078479. [Google Scholar]
- Signorile, M.; Zamirri, L.; Tsuchiyama, A.; Ugliengo, P.; Bonino, F.; Martra, G. On the Surface Acid–Base Properties of Amorphous and Crystalline Mg2SiO4 as Probed by Adsorbed CO, CO2, and CD3CN. ACS Earth Space Chem. 2020, 4, 345–354. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Mu, J.; Fan, S.; Chen, X.; Wang, L.; Yin, Z.; Tadé, M.; Liu, S. Oxygen Vacancy-rich Porous Co3O4 Nanosheets toward Boosted NO Reduction by CO and CO Oxidation: Insights into the Structure-Activity Relationship and Performance Enhancement Mechanism. ACS Appl. Mater. Interfaces 2019, 11, 41988–41999. [Google Scholar] [CrossRef]
- Drenchev, N.L.; Chakarova, K.K.; Lagunov, O.V.; Mihaylov, M.Y.; Ivanova, E.Z.; Strauss, I.; Hadjiivanov, K.I. In situ FTIR Spectroscopy as a Tool for Investigation of Gas/Solid Interaction: Water-Enhanced CO2 Adsorption in UiO-66 Metal-Organic Framework. J. Vis. Exp. 2020, 156. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| x = | a [Å] | b [Å] | c [Å] | Cell Volume [Å3] | Crystallite Size (LVol-IB) [nm] | SABET [m2 g−1] | Pore Volume [cm3 g−1] | Particle Size (TEM) [nm] | Particle Size (SEM) [nm] |
|---|---|---|---|---|---|---|---|---|---|
| 0.00 | 5.5494 (7) | 5.5587 (2) | 7.8464 (6) | 242.0473 | 46.6 | 16 | 0.13 | 32.4 ± 14.5 | 51.2 ± 8.6 |
| 0.05 | 5.5477 (0) | 5.5537 (6) | 7.8396 (2) | 241.5441 | 45.7 | 18 | 0.12 | 35.1 ± 11.9 | 49.4 ± 9.5 |
| 0.10 | 5.5429 (5) | 5.5452 (7) | 7.8332 (7) | 240.7734 | 33.2 | 24 | 0.16 | 26.4 ± 7.8 | 41.9 ± 7.5 |
| 0.15 | 5.5385 (1) | 5.5390 (2) | 7.8286 (1) | 240.1664 | 34.4 | 17 | 0.16 | 29.5 ± 10.2 | 58.7 ± 9.7 |
| 0.20 | 5.5300 (8) | 5.5280 (7) | 7.8163 (2) | 238.9508 | 32.3 | 18 | 0.15 | 36.6 ± 10.0 | 42.2 ± 7.6 |
| 0.25 | 5.5234 (3) | 5.5172 (5) | 7.8054 (2) | 237.8637 | 45.4 | 15 | 0.04 | 53.3 ± 15.9 | 78.2 ± 11.9 |
| 0.30 | 5.5188 (9) | 5.5157 (3) | 7.7989 (8) | 237.4070 | 33.8 | 25 | 0.26 | 39.6 ± 10.8 | 48.2 ± 7.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dreyer, M.; Krebs, M.; Najafishirtari, S.; Rabe, A.; Friedel Ortega, K.; Behrens, M. The Effect of Co Incorporation on the CO Oxidation Activity of LaFe1−xCoxO3 Perovskites. Catalysts 2021, 11, 550. https://doi.org/10.3390/catal11050550
Dreyer M, Krebs M, Najafishirtari S, Rabe A, Friedel Ortega K, Behrens M. The Effect of Co Incorporation on the CO Oxidation Activity of LaFe1−xCoxO3 Perovskites. Catalysts. 2021; 11(5):550. https://doi.org/10.3390/catal11050550
Chicago/Turabian StyleDreyer, Maik, Moritz Krebs, Sharif Najafishirtari, Anna Rabe, Klaus Friedel Ortega, and Malte Behrens. 2021. "The Effect of Co Incorporation on the CO Oxidation Activity of LaFe1−xCoxO3 Perovskites" Catalysts 11, no. 5: 550. https://doi.org/10.3390/catal11050550
APA StyleDreyer, M., Krebs, M., Najafishirtari, S., Rabe, A., Friedel Ortega, K., & Behrens, M. (2021). The Effect of Co Incorporation on the CO Oxidation Activity of LaFe1−xCoxO3 Perovskites. Catalysts, 11(5), 550. https://doi.org/10.3390/catal11050550