
Unconventional Gold-Catalyzed One-Pot/Multicomponent Synthesis of Propargylamines Starting from Benzyl Alcohols




Abstract
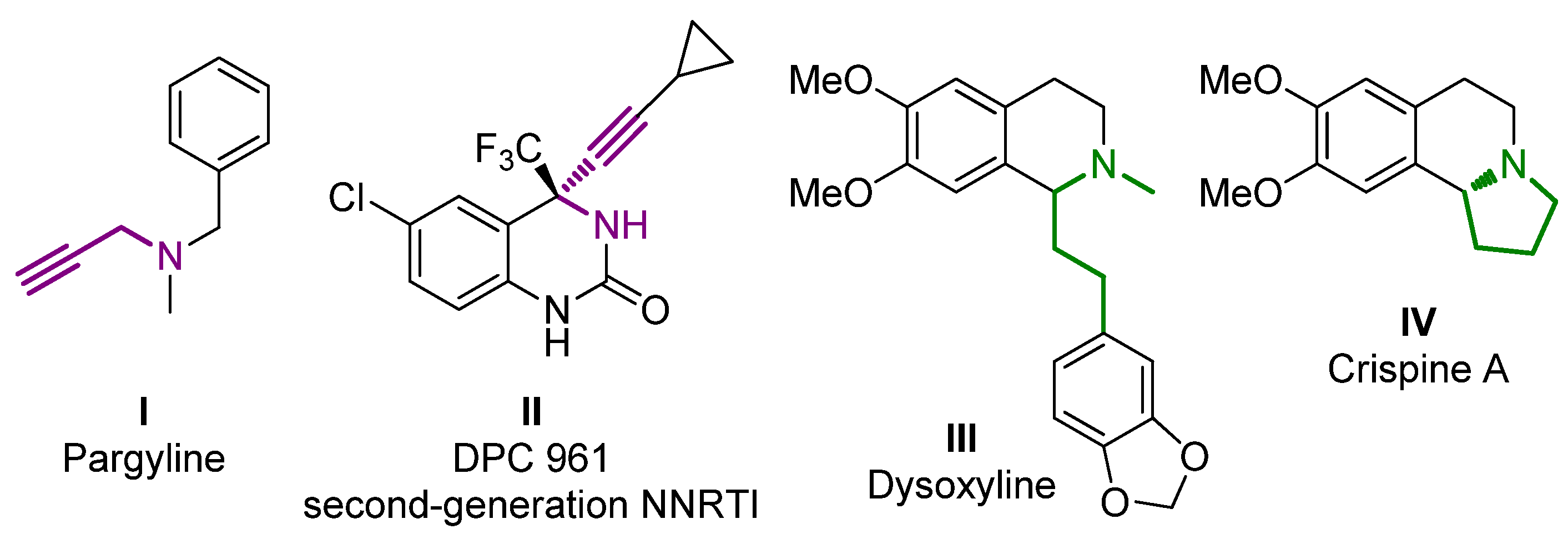
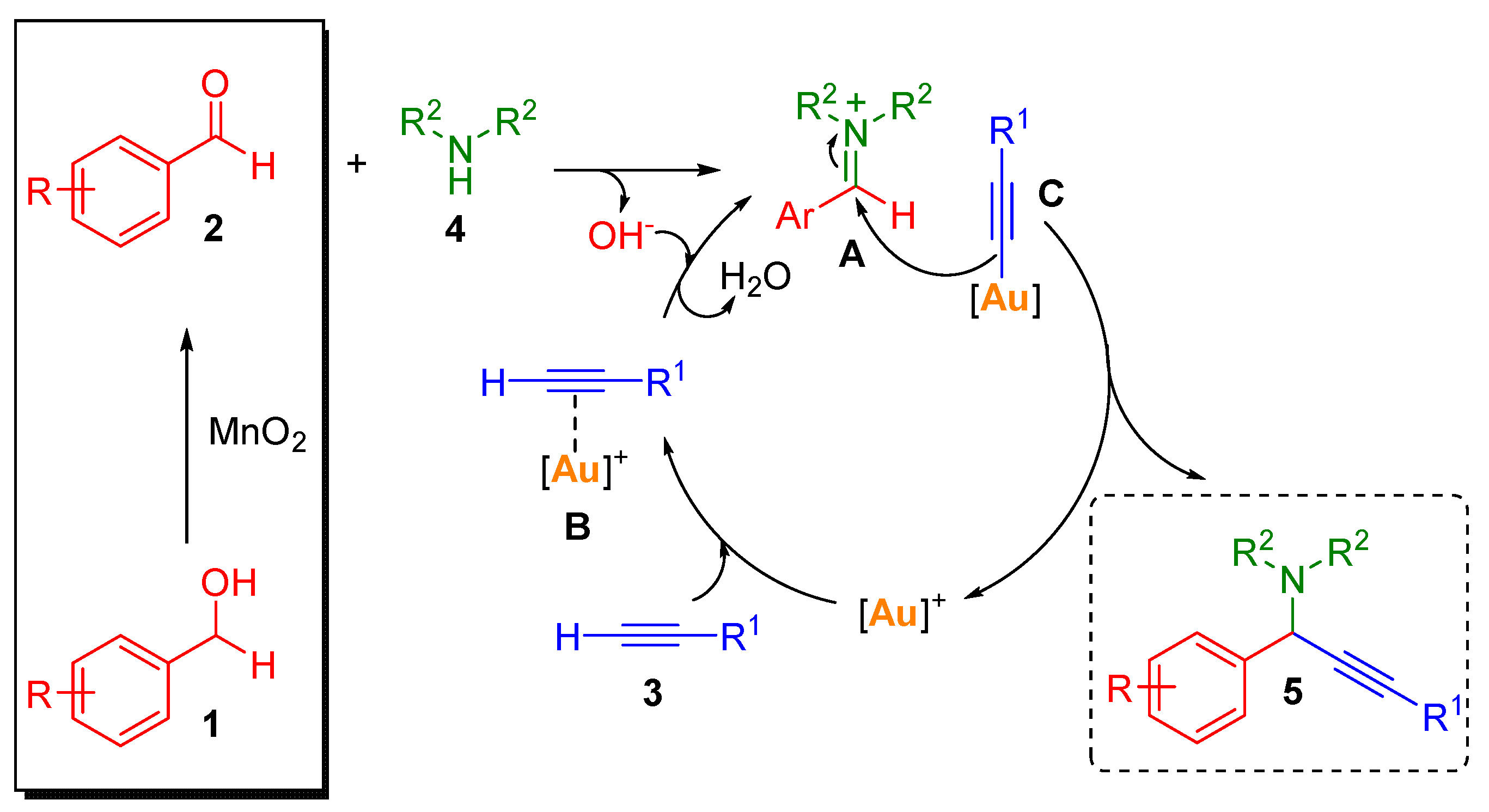
1. Introduction
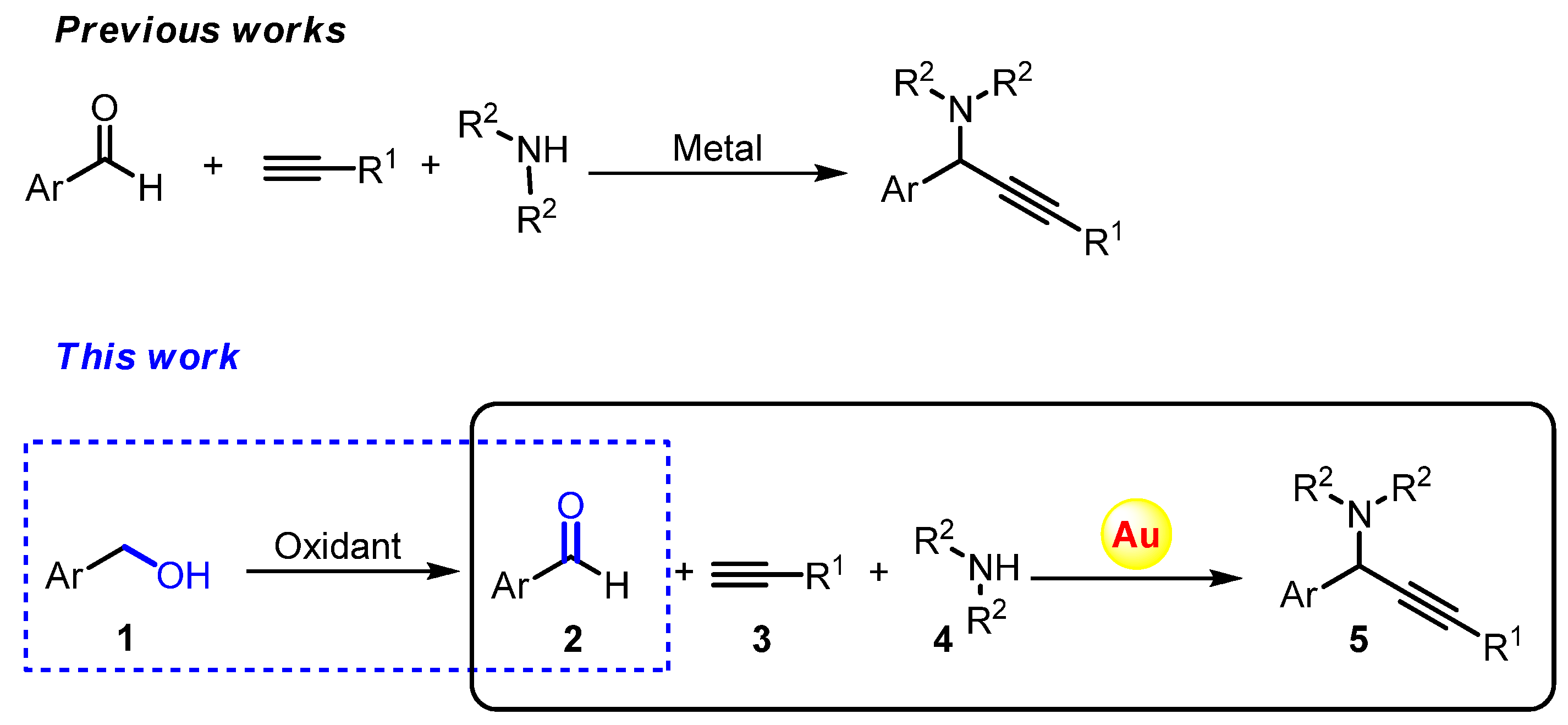
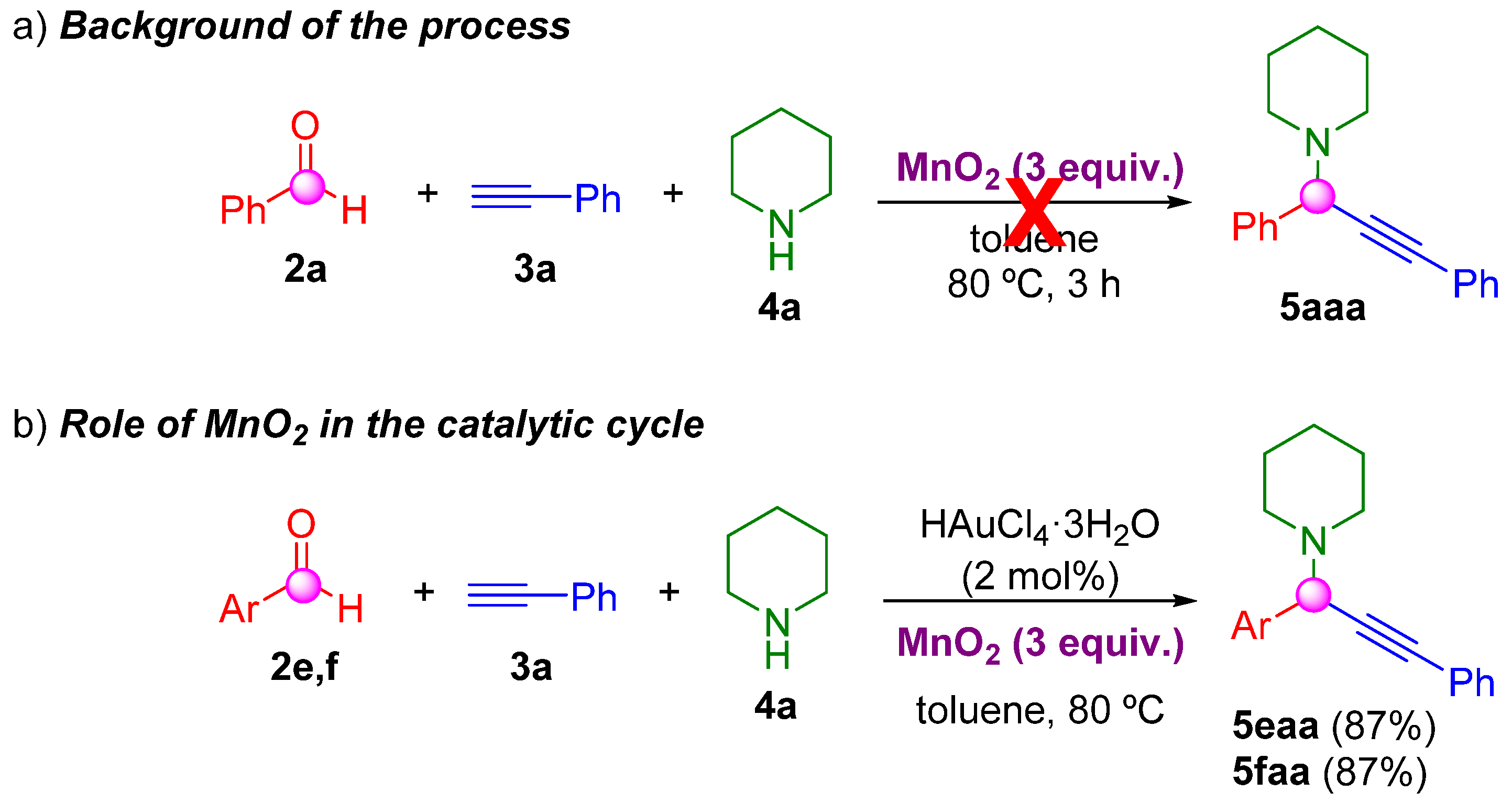
2. Results and Discussion
3. Materials and Methods
3.1. General Procedure for the Au-Catalyzed One-Pot/Multicomponent A3 Synthesis of Propargylamines 5
3.2. Characterization of Propargylamines 5
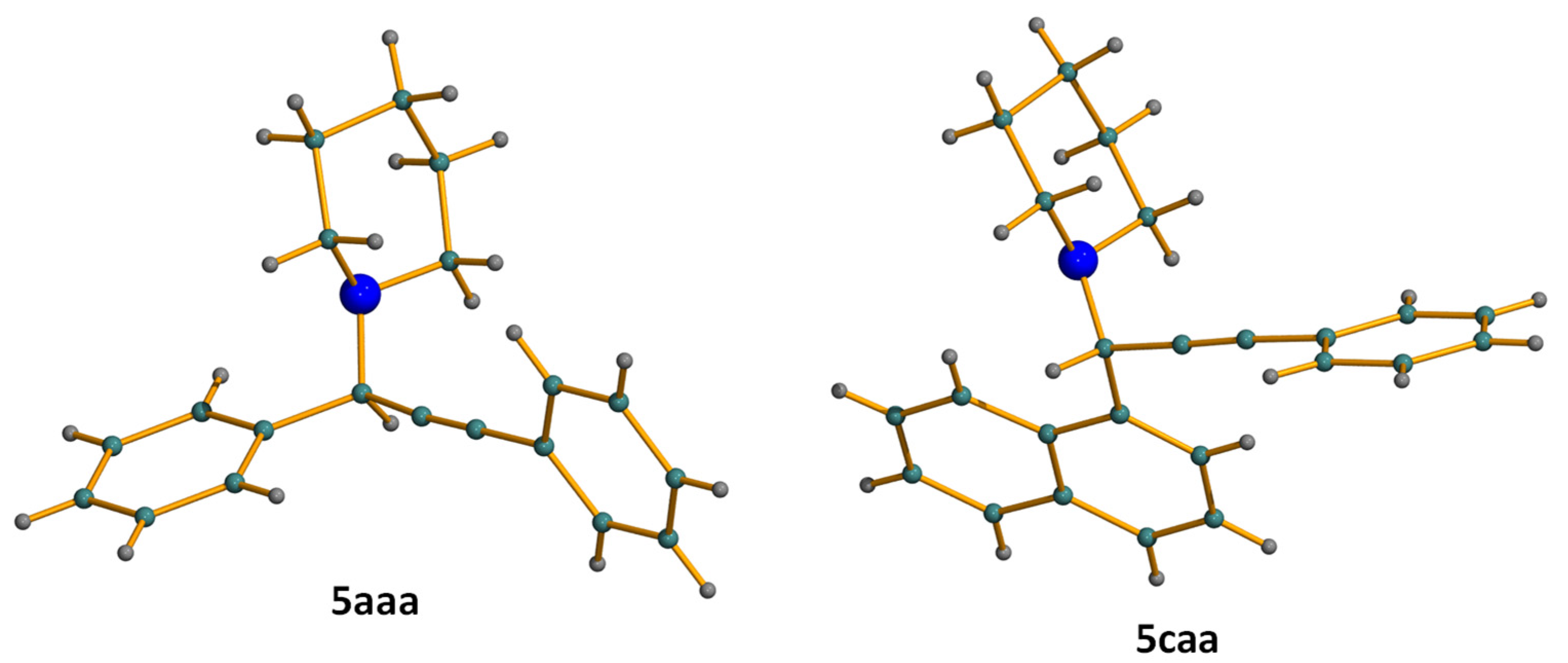
3.3. Crystal Structure Determinations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walji, A.M.; MacMillan, W.C. Strategies to Bypass the Taxol Problem. Enantioselective Cascade Catalysis, a New Approach for the Efficient Construction of Molecular Complexity. Synlett 2007, 1477–1489. [Google Scholar] [CrossRef]
- Zhao, W.; Chen, F.-E. One-pot Synthesis and its Practical Application in Pharmaceutical Industry. Curr. Org. Chem. 2012, 9, 873–897. [Google Scholar] [CrossRef]
- Hayashi, Y. Pot economy and one-pot synthesis. Chem. Sci. 2016, 7, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; Navas, J.; Sabater, M.J. Advances in One-Pot Synthesis through Borrowing Hydrogen Catalysis. Chem. Rev. 2018, 118, 1410–1459. [Google Scholar] [CrossRef]
- Zhu, J.; Bienaymé, H. (Eds.) Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Zhu, J.; Wang, Q.; Wang, M.-X. (Eds.) Multicomponent Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Herrera, R.P.; Marqués-López, E. (Eds.) Multicomponent Reactions: Concepts and Applications for Design and Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Weber, L. The application of multi-component reactions in drug discovery. Curr. Med. Chem. 2002, 9, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Hulme, C.; Gore, V. Multi-component reactions: Emerging chemistry in drug discovery from xylocain to crixivan. Curr. Med. Chem. 2003, 10, 51–80. [Google Scholar] [CrossRef]
- Touré, B.B.; Hal, D.G. Natural Product Synthesis Using Multicomponent Reaction Strategies. Chem. Rev. 2009, 109, 4439–4486. [Google Scholar] [CrossRef]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and Biology of Multicomponent Reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef]
- Tojo, G.; Fernandez, M.I. (Eds.) Oxidation of Alcohols to Aldehydes and Ketones: A Guide to Current Common Practice; Springer: New York, NY, USA, 2006. [Google Scholar]
- Alegre-Requena, J.V.; Marqués-López, E.; Herrera, R.P. Trifunctional Squaramide Catalyst for Efficient Enantioselective Henry Reaction Activation. Adv. Synth. Catal. 2016, 358, 1801–1809. [Google Scholar] [CrossRef]
- Alegre-Requena, J.V.; Marqués-López, E.; Herrera, R.P. Organocatalyzed Enantioselective Aldol and Henry Reactions Starting from Benzylic Alcohols. Adv. Synth. Catal. 2018, 360, 124–129. [Google Scholar] [CrossRef]
- Quintard, A.; Alexakis, A.; Mazet, C. Access to high levels of molecular complexity by one-pot iridium/enamine asymmetric catalysis. Angew. Chem. Int. Ed. 2011, 50, 2354–2358. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Sundén, H.; Sugiono, E. Unifying Metal- and Organocatalysis for Asymmetric Oxidative Iminium Activation: A Relay Catalytic System Enabling the Combined Allylic Oxidation of Alcohols and Prolinol Ether Catalyzed Iminium Reactions. Chem. Eur. J. 2012, 18, 3649–3653. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Sundén, H.; Hubener, L.; Sugiono, E. Asymmetric oxidative Lewis base catalysis—unifying iminium and enamine organocatalysis with oxidations. Chem. Commun. 2012, 48, 2201–2203. [Google Scholar] [CrossRef] [PubMed]
- Suh, C.W.; Kim, D.Y. Enantioselective One-Pot Synthesis of Ring-Fused Tetrahydroquinolines via Aerobic Oxidation and 1,5-Hydride Transfer/Cyclization Sequences. Org. Lett. 2014, 16, 5374–5377. [Google Scholar] [CrossRef] [PubMed]
- Rana, N.K.; Joshi, H.; Jha, R.K.; Singh, V.K. Enantioselective Tandem Oxidation/Michael–Aldol Approaches to Tetrasubstituted Cyclohexanes. Chem. Eur. J. 2017, 23, 2040–2043. [Google Scholar] [CrossRef]
- Wei, C.; Li, Z.; Li, C.-J. The Development of A3 Coupling (Aldehyde-Alkyne-Amine) and AA3 Coupling (Asymmetric Aldehyde-Alkyne-Amine). Synlett 2004, 1472–1483. [Google Scholar] [CrossRef]
- Yoo, W.-Y.; Zhao, L.; Li, C.-J. The A3-Coupling (Aldehyde–Alkyne–Amine) Reaction: A Versatile Method for the Preparation of Propargylamines. Aldrichim. Acta 2011, 44, 43–51. [Google Scholar]
- Peshkov, V.A.; Pereshivko, O.P.; Van der Eycken, E.V. A walk around the A3-coupling. Chem. Soc. Rev. 2012, 41, 3790–3807. [Google Scholar] [CrossRef]
- Lauder, K.; Toscani, A.; Scalacci, N.; Castagnolo, D. Synthesis and Reactivity of Propargylamines in Organic Chemistry. Chem. Rev. 2017, 117, 14091–14200. [Google Scholar] [CrossRef]
- Saha, T.K.; Das, R. Progress in Synthesis of Propargylamine and Its Derivatives by Nanoparticle Catalysis via A3 coupling: A Decade Update. ChemistrySelect 2018, 3, 147–169. [Google Scholar] [CrossRef]
- Rokade, B.V.; Barker, J.; Guiry, P.J. Development of and recent advances in asymmetric A3 coupling. Chem. Soc. Rev. 2019, 48, 4766–4790. [Google Scholar] [CrossRef]
- Jesin, I.; Nandi, G.C. Recent Advances in the A3 Coupling Reactions and their Applications. Eur. J. Org. Chem. 2019, 2704–2720. [Google Scholar] [CrossRef]
- Edmondson, D.E.; Mattevi, A.; Binda, C.; Li, M.; Hubálek, F. Structure and mechanism of monoamine oxidase. Curr. Med. Chem. 2004, 11, 1983–1993. [Google Scholar] [CrossRef] [PubMed]
- Orhan, I.E. Potential of Natural Products of Herbal Origin as Monoamine Oxidase Inhibitors. Curr. Pharm. Des. 2016, 22, 268–276. [Google Scholar] [CrossRef]
- King, R.W.; Klabe, R.M.; Reid, C.D.; Erickson-Viitanen, S.K. Potency of Nonnucleoside Reverse Transcriptase Inhibitors (NNRTIs) Used in Combination with Other Human Immunodeficiency Virus NNRTIs, NRTIs, or Protease Inhibitors. Antimicrob. Agents Chemother. 2002, 46, 1640–1646. [Google Scholar] [CrossRef][Green Version]
- Li, S.; Ma, J.-A. Core-structure-inspired asymmetric addition reactions: Enantioselective synthesis of dihydrobenzoxazinone- and dihydroquinazolinone-based anti-HIV agents. Chem. Soc. Rev. 2015, 44, 7439–7448. [Google Scholar] [CrossRef]
- Nugent, W.A. Exploring Chiral Space en route to DPC 963: A Personal Account. Adv. Synth. Catal. 2003, 345, 415–424. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Hutchings, G.J. Gold Catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K. Gold-Catalyzed Organic Reactions. Chem. Rev. 2007, 107, 3180–3211. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A.; Davies, P.W. Catalytic Carbophilic Activation: Catalysis by Platinum and Gold π Acids. Angew. Chem. Int. Ed. 2007, 119, 3478–3519. [Google Scholar] [CrossRef]
- Li, Z.; Brouwer, C.; He, C. Gold-Catalyzed Organic Transformations. Chem. Rev. 2008, 108, 3239–3265. [Google Scholar] [CrossRef]
- Arcadi, A. Alternative Synthetic Methods through New Developments in Catalysis by Gold. Chem. Rev. 2008, 108, 3266–3325. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Nuñez, E.; Echavarren, A.M. Gold-Catalyzed Cycloisomerizations of Enynes: A Mechanistic Perspective. Chem. Rev. 2008, 108, 3326–3350. [Google Scholar] [CrossRef]
- Gorin, D.J.; Sherry, B.D.; Toste, F.D. Ligand Effects in Homogeneous Au Catalysis. Chem. Rev. 2008, 108, 3351–3378. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Shi, X. Recent Advances in Asymmetric Gold Catalysis. ChemCatChem. 2010, 2, 609–619. [Google Scholar] [CrossRef]
- Garayalde, D.; Nevado, C. Synthetic applications of gold-catalyzed ring expansions. Beilstein J. Org. Chem. 2011, 7, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.; Hashmi, A.S.K. Gold catalysis in total synthesis—An update. Chem. Soc. Rev. 2012, 41, 2448–2462. [Google Scholar] [CrossRef] [PubMed]
- Visbal, R.; Graus, S.; Herrera, R.P.; Gimeno, M.C. Gold Catalyzed Multicomponent Reactions beyond A3 Coupling. Molecules 2018, 23, 2255. [Google Scholar] [CrossRef]
- Herrera, R.P.; Gimeno, M.C. Main Avenues in Gold Coordination Chemistry. Chem. Rev. 2021. [Google Scholar] [CrossRef]
- Wei, C.; Li, C.-J. A Highly Efficient Three-Component Coupling of Aldehyde, Alkyne, and Amines via C−H Activation Catalyzed by Gold in Water. J. Am. Chem. Soc. 2003, 125, 9584–9585. [Google Scholar] [CrossRef] [PubMed]
- Lo, V.K.-Y.; Liu, Y.; Wong, M.-K.; Che, C.-M. Gold(III) Salen Complex-Catalyzed Synthesis of Propargylamines via a Three-Component Coupling Reaction. Org. Lett. 2006, 8, 1529–1532. [Google Scholar] [CrossRef]
- Elie, B.T.; Levine, C.; Ubarretxena-Belandia, I.; Varela-Ramírez, A.; Aguilera, R.J.; Ovalle, R.; Contel, M. Water Soluble Phosphane-Gold(I) Complexes. Applications as Recyclable Catalysts in a Three-component Coupling Reaction and as Antimicrobial and Anticancer Agents. Eur. J. Inorg. Chem. 2009, 3421–3430. [Google Scholar] [CrossRef]
- Oña-Burgos, P.; Fernández, I.; Roces, L.; Fernández, L.T.; García-Granada, S.; Ortiz, F.L. An unprecedented phosphinamidic gold(III) metallacycle: Synthesis via tin(IV) precursors, structure, and multicomponent catalysis. Organometallics 2009, 28, 1739–1747. [Google Scholar] [CrossRef]
- Ko, H.-M.; Kung, K.K.-Y.; Cui, J.-F.; Wong, M.-K. Bis-cyclometallated gold(III) complexes as efficient catalysts for synthesis of propargylamines and alkylated indoles. Chem. Commun. 2013, 49, 8869–8871. [Google Scholar] [CrossRef] [PubMed]
- Abbiati, G.; Rossi, E. Silver and gold-catalyzed multicomponent reactions. Beilstein J. Org. Chem. 2014, 10, 481–513. [Google Scholar] [CrossRef]
- Kung, K.K.-Y.; Lo, V.K.-Y.; Ko, H.-M.; Li, G.-L.; Chan, P.-Y.; Leung, K.-C.; Zhou, Z.; Wang, M.-Z.; Che, C.-M.; Wong, M.-K. Cyclometallated Gold(III) Complexes as Effective Catalysts for Synthesis of Propargylic Amines, Chiral Allenes and Isoxazoles. Adv. Synth. Catal. 2013, 355, 2055–2070. [Google Scholar] [CrossRef]
- von Wachenfeldt, H.; Polukeev, A.V.; Loganathan, N.; Paulsen, F.; Röse, P.; Garreau, M.; Wendt, O.F.; Strand, D. Cyclometallated gold(III) aryl-pyridine complexes as efficient catalysts for three-component synthesis of substituted oxazoles. Dalton Trans. 2015, 44, 5347–5353. [Google Scholar] [CrossRef]
- Hui, T.-W.; Cui, J.-F.; Wong, M.-K. Modular synthesis of propargylamine modified cyclodextrins by a gold(III)-catalyzed three-component coupling reaction. RSC Adv. 2017, 7, 14477–14480. [Google Scholar] [CrossRef]
- Grirrane, A.; Álvarez, E.; García, H.; Corma, A. Double A3-Coupling of Primary Amines Catalysed by Gold Complexes. Chem. Eur. J. 2018, 24, 16356–16367. [Google Scholar] [CrossRef] [PubMed]
- Montanel-Pérez, S.; Herrera, R.P.; Laguna, A.; Villacampa, M.D.; Gimeno, M.C. The fluxional amine gold(iii) complex as an excellent catalyst and precursor of biologically active acyclic carbenes. Dalton Trans. 2015, 44, 9052–9062. [Google Scholar] [CrossRef]
- Aliaga-Lavrijsen, M.; Herrera, R.P.; Villacampa, M.D.; Gimeno, M.C. Efficient Gold(I) Acyclic Diaminocarbenes for the Synthesis of Propargylamines and Indolizines. ACS Omega 2018, 3, 9805–9813. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; Navas, J.; Sabater, M.J. Coupling of Two Multistep Catalytic Cycles for the One-Pot Synthesis of Propargylamines from Alcohols and Primary Amines on a Nanoparticulated Gold Catalyst. Chem. Eur. J. 2012, 18, 14150–14156. [Google Scholar] [CrossRef]
- Lili, L.; Xin, Z.; Jinsen, G.; Chunming, X. Engineering metal-organic frameworks immobilize gold catalysts for highly efficient one-pot synthesis of propargylamines. Green Chem. 2012, 14, 1710–1720. [Google Scholar] [CrossRef]
- Karimi, B.; Gholinejad, M.; Khorasani, M. Highly efficient three-component coupling reaction catalyzed by gold nanoparticles supported on periodic mesoporous organosilica with ionic liquid framework. Chem. Commun. 2012, 48, 8961–8963. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Bejar, M.; Peters, K.; Hallett-Tapley, G.L.; Grenier, M.; Scaiano, J.C. Rapid one-pot propargylamine synthesis by plasmon mediated catalysis with gold nanoparticles on ZnO under ambient conditions. Chem. Commun. 2013, 49, 1732–1734. [Google Scholar] [CrossRef]
- Anand, N.; Ramudu, P.; Reddy, K.H.P.; Rao, K.S.R.; Jagadeesh, B.; Babu, V.S.P.; Burri, D.R. Gold nanoparticles immobilized on lipoic acid functionalized SBA-15: Synthesis, characterization and catalytic applications. Appl. Catal. A Gen. 2013, 454, 119–126. [Google Scholar] [CrossRef]
- Borah, B.J.; Borah, S.J.; Saikia, K.; Dutta, D.K. Efficient one-pot synthesis of propargylamines catalysed by gold nanocrystals stabilized on montmorillonite. Catal. Sci. Technol. 2014, 4, 4001–4009. [Google Scholar] [CrossRef]
- Moghaddam, F.M.; Ayati, S.E.; Hosseini, S.H.; Pourjavadi, A. Gold immobilized onto poly(ionic liquid) functionalized magnetic nanoparticles: A robust magnetically recoverable catalyst for the synthesis of propargylamine in water. RSC Adv. 2015, 5, 34502–34510. [Google Scholar] [CrossRef]
- Feiz, A.; Bazgir, A. Gold nanoparticles supported on mercaptoethanol directly bonded to MCM-41: An efficient catalyst for the synthesis of propargylamines. Catal. Commun. 2016, 73, 88–92. [Google Scholar] [CrossRef]
- Loni, M.; Yazdani, H.; Bazgir, A. Gold Nanoparticles-Decorated Dithiocarbamate Nanocomposite: An Efficient Heterogeneous Catalyst for the Green A3-Coupling Synthesis of Propargylamines. Catal. Lett. 2018, 148, 3467–3476. [Google Scholar] [CrossRef]
- Soengas, R.; Navarro, Y.; Iglesias, M.J.; López-Ortiz, F. Immobilized Gold Nanoparticles Prepared from Gold(III)-Containing Ionic Liquids on Silica: Application to the Sustainable Synthesis of Propargylamines. Molecules 2018, 23, 2975. [Google Scholar] [CrossRef] [PubMed]
- Aghahosseini, H.; Rezaei, S.J.T.R.; Tadayyon, M.; Ramazani, A.; Amani, V.; Ahmadi, R.; Abdolahnjadian, D. Highly Efficient Aqueous Synthesis of Propargylamines through C–H Activation Catalyzed by Magnetic Organosilica-Supported. Gold Nanoparticles as an Artificial Metalloenzyme. Eur. J. Inorg. Chem. 2018, 2589–2598. [Google Scholar] [CrossRef]
- Bensaad, M.; Berrichi, A.; Bachir, R.; Bedrane, S. Nano and Sub‑nano Gold–Cobalt Particles as Effective Catalysts in the Synthesis of Propargylamines via AHA Coupling. Catal. Lett. 2021, 151, 1068–1079. [Google Scholar] [CrossRef]
- Sagadevan, A.; Pampana, V.K.K.; Hwang, K.C. Copper Photoredox Catalyzed A3’ Coupling of Arylamines, Terminal Alkynes, and Alcohols through a Hydrogen Atom Transfer Process. Angew. Chem. Int. Ed. 2019, 58, 3838–3842. [Google Scholar] [CrossRef]
- Hosseinzadeh, S.Z.; Babazadeh, M.; Shahverdizadeh, G.H.; Es’haghi, M.; Hossinzadeh-Khanmiri, R. Silica Encapsulated-Gold Nanoparticles as a Nano-reactor for Aerobic Oxidation of Benzyl alcohols and Tandem Oxidative A3 coupling Reactions in Water. Catal. Lett. 2020, 150, 2784–2791. [Google Scholar] [CrossRef]
- Movahed, S.K.; Lehi, N.F.; Dabiri, M. Gold nanoparticle supported on ionic liquidmodified graphene oxide as an efficient and recyclable catalyst for one-pot oxidative A3- coupling reaction of benzyl alcohols. RSC Adv. 2014, 4, 42155–42158. [Google Scholar] [CrossRef]
- Fatiadi, A.J. Active Manganese Dioxide Oxidation in Organic Chemistry-Part I. Synthesis 1976, 65–104. [Google Scholar] [CrossRef]
- Leadbeater, N.E.; Torenius, H.M.; Tye, H. Microwave-assisted Mannich-type three-component reactions. Mol. Divers. 2003, 7, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, T.; Ogawa, A. Copper(0)-Induced Deselenative Insertion of N,N-Disubstituted Selenoamides into Acetylenic C−H Bond Leading to Propargylamines. Org. Lett. 2009, 11, 2045–2048. [Google Scholar] [CrossRef]
- Chng, L.L.; Yang, J.; Wei, Y.; Ying, J.Y. Semiconductor-Gold Nanocomposite Catalysts for the Efficient Three-Component Coupling of Aldehyde, Amine and Alkyne in Water. Adv. Synth. Catal. 2009, 351, 2887–2896. [Google Scholar]
- Layek, K.; Chakravarti, R.; Kantam, M.L.; Maheswaran, H.; Vinu, A. Nanocrystalline magnesium oxide stabilized gold nanoparticles: An advanced nanotechnology based recyclable heterogeneous catalyst platform for the one-pot synthesis of propargylamines. Green Chem. 2011, 13, 2878–2887. [Google Scholar] [CrossRef]
- Zhang, X.; Corma, A. Supported gold(III) catalysts for highly efficient three-component coupling reactions. Angew. Chem. Int. Ed. 2008, 47, 4358–4361. [Google Scholar] [CrossRef] [PubMed]
- Ramu, E.; Varala, R.; Sreelatha, N.; Adapa, S.R. Zn(OAc)2·2H2O: A versatile catalyst for the one-pot synthesis of propargylamines. Tetrahedron Lett. 2007, 48, 7184–7190. [Google Scholar] [CrossRef]
- Feng, H.; Ermolat’ev, D.S.; Song, G.; Van der Eycken, E.V. Microwave-Assisted Decarboxylative Three-Component Coupling of a 2-Oxoacetic Acid, an Amine, and an Alkyne. J. Org. Chem. 2011, 76, 7608–7613. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Hirasawa, M.; Konakahara, T. InBr3–Et3N promoted alkynylation of aldehydes and N,O-acetals under mild conditions: Facile and simple preparation of propargylic alcohols and amines. Tetrahedron Lett. 2003, 44, 4171–4174. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst (mol%) | MnO2 (equiv.) | Yield (%) (b) |
|---|---|---|---|
| 1 | HAuCl4·3H2O (5) | 5 | >99 |
| 2 | CuI (5) | 5 | 61 |
| 3 | ZnI2 (5) | 5 | 46 |
| 4 | CuCl (5) | 5 | 34 |
| 5 | CF3COOAg (5) | 5 | 93 |
| 6 | HAuCl4·3H2O (4) | 5 | >99 |
| 7 | HAuCl4·3H2O (3) | 5 | >99 |
| 8 | HAuCl4·3H2O (2) | 5 | >99 |
| 9 | HAuCl4·3H2O (1) | 5 | >99 |
| 10 | HAuCl4·3H2O (3) | 4 | >99 |
| 11 | HAuCl4·3H2O (2) | 3 | >99 |
| 12 | HAuCl4·3H2O (2) | 2 | 75 |
| 13 | HAuCl4·3H2O (2) | 1 | 44 |

| Entry | Ar (1) | R1 (3) | Amine (4) | Time (h) | Yield (%) (b) |
|---|---|---|---|---|---|
| 1 | Ph, 1a | Ph, 3a | Piperidine, 4a | 3 | 97 |
| 2 | 4-MeC6H4, 1b | Ph, 3a | Piperidine, 4a | 6 | 87 |
| 3 (c) | 1-naphthyl, 1c | Ph, 3a | Piperidine, 4a | 4 | 90 |
| 4 (d) | 3-NO2C6H4, 1d | Ph, 3a | Piperidine, 4a | 3 | 90 |
| 5 | 4-BrC6H4, 1e | Ph, 3a | Piperidine, 4a | 6 | 94 |
| 6 | 4-ClC6H4, 1f | Ph, 3a | Piperidine, 4a | 3 | 93 |
| 7 | 3-ClC6H4, 1g | Ph, 3a | Piperidine, 4a | 3 | 98 |
| 8 | 4-FC6H4, 1h | Ph, 3a | Piperidine, 4a | 3 | 94 |
| 9 | Ph, 1a | Ph, 3a | Pyrrolidine, 4b | 4 | 98 |
| 10 | Ph, 1a | Ph, 3a | Morpholine, 4c | 3 | 96 |
| 11 | 4-CNC6H4, 1i | Ph, 3a | Morpholine, 4c | 6 | 85 |
| 12 | Ph, 1a | Ph, 3a | Bu2NH, 4d | 18 | 95 |
| 13 | Ph, 1a | Ph, 3a | Et2NH, 4e | 18 | 96 |
| 14 (c) | Ph, 1a | 4-MeC6H4, 3b | Piperidine, 4a | 5 | 98 |
| 15 (c,e) | Ph, 1a | Me3Si, 3c | Piperidine, 4a | 18 | 98 |
| 16 (f) | Ph, 1a | Ph, 3a | Piperidine, 4a | 6 | 95 |

| Entry | Ar | Amine (4) | Time (h) | Yield (%) (c) |
|---|---|---|---|---|
| 1 | 3-NO2C6H4, 1d | Piperidine, 4a | 3 | 94 |
| 2 | 3-NO2C6H4, 2d | Piperidine, 4a | 3 | 81 |
| 3 | 4-BrC6H4, 1e | Piperidine, 4a | 6 | 96 |
| 4 | 4-BrC6H4, 2e | Piperidine, 4a | 6 | 85 |
| 5 | 4-ClC6H4, 1f | Piperidine, 4a | 3 | 96 |
| 6 | 4-ClC6H4, 2f | Piperidine, 4a | 3 | 88 |
| 7 | 3-ClC6H4, 1g | Piperidine, 4a | 3 | >99 |
| 8 | 3-ClC6H4, 2g | Piperidine, 4a | 3 | 86 |
| 9 | 4-CNC6H4, 1i | Morpholine, 4c | 6 | 90 |
| 10 | 4-CNC6H4, 2i | Morpholine, 4c | 6 | 45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zárate-Roldán, S.; Gimeno, M.C.; Herrera, R.P. Unconventional Gold-Catalyzed One-Pot/Multicomponent Synthesis of Propargylamines Starting from Benzyl Alcohols. Catalysts 2021, 11, 513. https://doi.org/10.3390/catal11040513
Zárate-Roldán S, Gimeno MC, Herrera RP. Unconventional Gold-Catalyzed One-Pot/Multicomponent Synthesis of Propargylamines Starting from Benzyl Alcohols. Catalysts. 2021; 11(4):513. https://doi.org/10.3390/catal11040513
Chicago/Turabian StyleZárate-Roldán, Stephany, María Concepción Gimeno, and Raquel P. Herrera. 2021. "Unconventional Gold-Catalyzed One-Pot/Multicomponent Synthesis of Propargylamines Starting from Benzyl Alcohols" Catalysts 11, no. 4: 513. https://doi.org/10.3390/catal11040513
APA StyleZárate-Roldán, S., Gimeno, M. C., & Herrera, R. P. (2021). Unconventional Gold-Catalyzed One-Pot/Multicomponent Synthesis of Propargylamines Starting from Benzyl Alcohols. Catalysts, 11(4), 513. https://doi.org/10.3390/catal11040513