1. Introduction
Chiral compounds with one or several stereogenic centers consist of pairs of enantiomers. Many drugs on the market today have one or several stereogenic centers, β-blockers normally have one stereogenic center, and then consist of two enantiomers. The enantiomers may have the same effect on the patient, or the enantiomers may have different effects, or worse, one enantiomer may have several unwanted side effects. FDA considers the “wrong” enantiomer as an impurity and demands for pure enantiomers as the active pharmaceutical ingredient (API) in the marketed drugs, not racemates. The demand for enantiomerically pure drugs has increased year-by-year since the 1990s when FDA demanded manufacturers to evaluate the pharmacokinetics of a single enantiomer or mixture of enantiomers in a chiral drug. Quantitative assays for individual enantiomers should be developed for studies in in vivo samples early in drug development. It is postulated that the lower the effective dose of a drug, the greater the difference in the pharmacological effect of the optical isomers when drug receptor interactions are considered [
1]. The ratio of the more active enantiomer (eutomer) compared to the less active enantiomer (distomer) is known as the eudismic ratio. The higher the eudismic ratio, the higher the effectiveness of the drug [
2] and the “right” enantiomer should be provided.
High blood pressure (hypertension) and heart failure are big global health problems. In Norway, approximately 30% of all deaths are caused by cardiovascular diseases [
3]. Approximately 26 million people worldwide live with heart failure, and approximately seven million deaths are caused by hypertension annually [
4]. Heart failure cannot be cured but may be treated by medications in order to increase quality of life for the patients. Both heritage (genetic) and lifestyle may lead to increased risk of cardiovascular diseases. Risk factors for cardiovascular diseases, such as reduced activity, eating more sugar and salt, increased stress, obesity and overweight, may be reasons for the increasing health problems worldwide [
5].
A class of drugs that have been used in treatment of cardiovascular diseases for a long time, β-adrenergic receptor antagonists, are so-called β-blockers. The β
1-receptors in the human body are mainly located in the heart and regulate the contraction of the heart muscle. The β
1-blockers are antagonists that affect the β
1-receptors in the heart and are suitable for the treatment of hypertension and heart failure. When a β-blocker inhibits the binding of adrenaline and noradrenaline, the stress hormone level in the body will decrease, and thus the blood pressure and heart rate will decrease. β
2-receptors are mainly found in the lungs, and most β
2-agonists are mainly used to treat asthma by relaxing the smooth muscles in the lungs [
6].
Some β-blockers are non-selective and will inhibit both β
1- and β
2-receptors, while others are selective to either β
1- or β
2-receptors. Propranolol is an example of a so-called non-selective β-blocker. A problem for the non-selective β-blockers is that they may cause negative effects to the lungs, especially for asthma patients. β-receptors are found in all parts of the nervous system, and β-blockers will therefore give side effects even if the right drug is chosen. Selective β-blockers lead to fewer side effects and are safer in use. Choosing the right β-blocker for every type of treatment is therefore important [
6].
We have for some time worked with optimization of biocatalytic processes for several of these drugs. Lipase-catalyzed kinetic resolution of racemates has been successful for several of these [
7,
8]. In attempts to obtain enantiopure building blocks for the β-agonist (
R)-clenbuterol, asymmetrizations of ketones have been performed with ketoreductases giving high enantiomeric excess [
9].
In general, the
S-enantiomers of the β-blockers are known to be more active than the
R-enantiomers, and have the opposite stereo configuration for the β-agonists. Common for these compounds is a side chain on an aromatic group which consists of a secondary alcohol and an amine on the omega carbon. Traditionally, most of these drugs have been manufactured in racemic form, however, e.g., (
S)-atenolol, (
S)-propranolol and (
S)-metoprolol are also on the market as single enantiomers. When referring to a drug, one should refer to the API [
10].
Because enantiomers are mirror images of each other and not identical, each of them may act differently on the receptor. The pharmacological effect, clinical effect and toxicity of both enantiomers need to be investigated separately according to the U.S. Food and Drug Administration (FDA) [
4]. Today, chiral drugs are promoted increasingly with enantiopure API. For enantiopure drugs there is a requirement for enantiomeric excess (
ee) > 96%. Therefore, development of environmentally friendly synthetic methods for each of the enantiomers is important.
Practolol is a selective β
1-antagonist and was the first β
1-selective β-blocker used in the treatment of cardiovascular diseases at the beginning of the 1970s [
11]. Practolol showed effective treatment of heart failure and arrhythmic heart rate [
12]. The drug showed later some critical side effects, such as culomucocutaneous syndrome, in some patients, and was withdrawn from the market [
13].
Mulik et al. reported in 2016 a four-step synthesis of (
S)-practolol in 100%
ee with the use of
Pseudomonas cepacia sol-gel AK lipase as enantioselective catalyst. However, they report that the produced ester from the transesterification reaction is hydrolyzed and aminated to give the enantiopure building block with
S-configuration [
14]. According to previous reports, it is the slower reacting enantiomer, (
R)-
N-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide, which is aminated to give (
S)-practolol. They also claim that the configuration of the chlorohydrin is inverted in the amination step, which will not be the case since the amine is not attacking the stereocenter, but the primary carbon with the chloro atom. Ader and Schneider reported in 1992 enzymatic kinetic resolution of racemic
N-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide with
Pseudomonas sp. to give (
R)-
N-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide which was subsequently aminated to yield (
S)-practolol in 30% yield and >99%
ee [
15]. However, no optical rotation values of the building blocks or the drug have been reported. In order to develop greener and more sustainable processes for drugs with secondary alcohol side chains, we wanted to include the synthesis of practolol despite the regulations of the drug.
Pindolol, 1-(1
H-Indol-4-yloxy)-3-(isopropylamino)-2-propanol, was first released for clinical use in USA in 1982. It is a non-selective β-blocker, used in the treatment of high blood pressure, chest pain and irregular heartbeat. Pindolol has its substituents on the aromatic ring in the
ortho- and
meta-position, which is common for non-selective β-blockers. Selective β-blockers usually have a substituent in the
para-position on the aromatic ring [
4]. Pindolol is also a partial agonist and will therefore slow the resting heart rate less than other β-blockers like atenolol or metoprolol [
16]. Pindolol is usually sold under the brand names Visken (Sandoz) or Barbloc (Alpha) and is often used to treat high blood pressure during pregnancy because it does not affect the fetal heart function or blood flow. Although pindolol is a non-selective β-blocker, other uses for the drug have been reported. It has been tested in the treatment of fibromyalgia and related fatigue diseases, as well as in the treatment of depression in combination with selective serotonin reuptake inhibitors [
17,
18]. Pindolol is a rapidly absorbed drug and after oral ingestion it can be detected in the blood after 30 min. In patients with normal renal function, pindolol has a plasma half-life of three to four hours. The drug is also lipophilic and enters the central nervous system rapidly. Reported side effects include unwanted lowering of heart function or changes in the respiratory system. These side effects are related to its β-adrenergic blocking activity; other side effects have also been reported, such as dizziness, vivid dreams, feeling of weakness or fatigue, muscle cramps, as well as nausea [
16]. Precursors of β-blocker pindolol were synthesized by biocatalysis in 2017 by Lima et al. They performed hydrolysis of 2-acetoxy-1-(1
H-indol-4-yloxy)-3-chloropropane using lipase from
Pseudomonas fluorescens which yielded (2
S)-1-(1
H-indol-4-yloxy)-3-chloro-2-propanol in 96%
ee and (2
R)-2-acetoxy-1-(1
H-indol-4-yloxy)-3-chloropropane in 97%
ee, which was hydrolysed giving 97%
ee of the
R-chlorohydrin for the synthesis of (
S)-pindolol with retention of
ee [
17]. However, we have some doubts about the stereochemistry in this report which will be discussed.
Carteolol is another β-adrenergic antagonist (β-blocker) manufactured mostly with racemic API and administered as eye-drops for reduction of aqueous production in the eye (glaucoma) [
19]. In these patients, an elevated intraocular pressure (IOP) leads to damage to the optic nerve, reducing the visual field gradually until the patient is completely blind. It is the second most common cause of irreversible blindness after age-related macular degeneration in western Europe. In 2010, 2.1 million people worldwide went irreversibly blind because of glaucoma [
20]. In 2019, the majority of Norwegian glaucoma patients (68%) were treated with β-blockers betaxolol or timolol, either as single drugs or in combination with other drugs such as prostaglandin analogues or carboanhydrase inhibitors [
21].
With the aim of sustainable production of enantiopure β-blockers, we have performed several synthetic strategies with lower amounts of reactants and shorter reaction times than previously reported. The general mechanism of base catalyzed deprotonation of phenolic protons with subsequent nucleophilic attachment of epichlorohydrin has been studied with different bases and different concentrations of epichlorohydrin. Lipase B from
Candida antarctica has been shown to catalyze reactions of similar compounds with high
ee of both product and remaining starting material (hydrolysis and transesterification reactions) [
22,
23].
2. Results
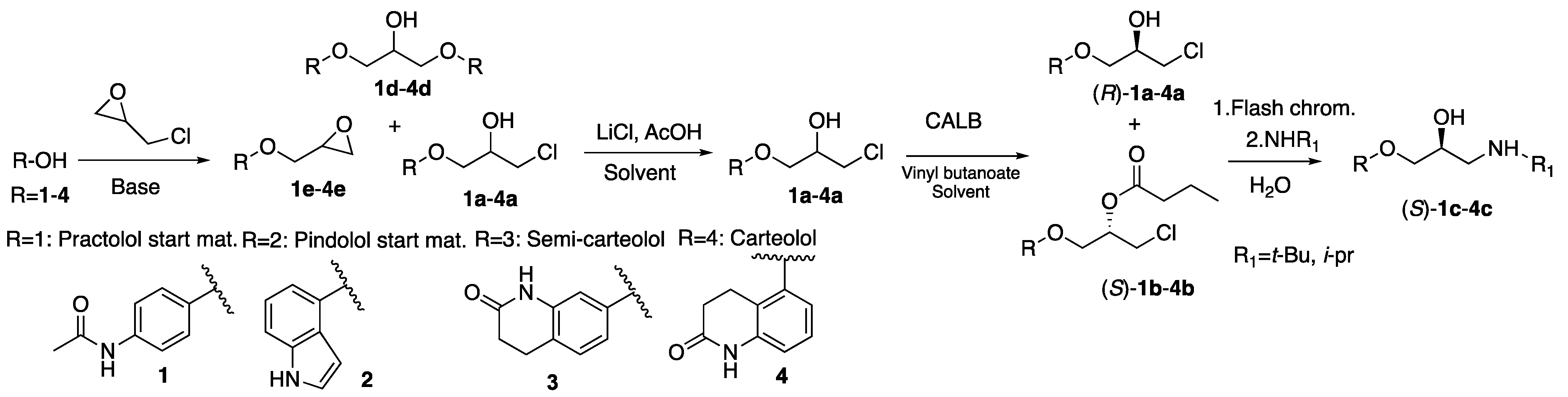
Chlorohydrin building blocks (
R)-
1a-
4a for the synthesis of enantiomers of the β-blockers practolol ((
S)-
1c), pindolol and derivatives of carteolol ((
S)-
3c-4c) have been synthesized in 92–97%
ee by chemo-enzymatic methods (
Scheme 1). The highest yields of the racemic chlorohydrins were obtained with 0.3–1 equivalents of base in the deprotonation step of the starting materials
1–
4, 2 equivalents of epichlorohydrin, 12–26 h reaction time and 30 °C reaction temperature. The intermediate epoxides
1e–
4e were protonated with acetic acid and then opened with lithium chloride. Recently, we reduced the amount of acetic acid from 10 to 5 equivalents giving the same yields of the chlorohydrins. Kinetic resolutions of the racemic halohydrins were performed in different solvents with lipase B from
Candida antarctica and vinyl butanoate as the acyl donor. Amination of the
R-chlorohydrins (
R)-
1a and (
R)-
3a-
4a gave the
S-β-blockers with preserved or increased
ee. Due to the low
ee (92%), the amination step of (
R)-
2a was not performed. Previously, we have published the synthesis of the building block for (
S)-atenolol in > 98%
ee by a similar protocol [
7].
Analysis of the reaction mixtures from the syntheses of
1a–
4a on LC-MS showed that the most abundant by-products in these reactions were the dimers
1d–
4d (
Scheme 1) of the deprotonated starting materials
1–
4. In order to ensure full conversion of the starting materials and to avoid the formation of the dimer by-products in the syntheses, concentrations of base and 2-(chloromethyl)oxirane (epichlorohydrin), reaction time and temperature have been varied. When high concentration of base was used, the intramolecular cyclization of the anions of
1a–
4a was observed to boost by increased reaction time; otherwise, the acid and lithium chloride were added immediately after full conversion of the starting materials. Investigations of reaction conditions in synthesis of the racemic practolol precursor
1a are shown in
Table 1. When 0.5–1.0 equivalents of sodium hydroxide dissolved in water was used in the reaction of
1 with epichlorohydrin with a reaction temperature of 80 °C for 24 h, only the dimer
N,N-(((2-hydroxypropane-1,3-diyl)bis(oxy))-bis(4,1-phenylene))diacetamide (
1d) was obtained in addition to a small fraction of the epoxide
1e (
Table 1). Dimer
1d was characterized by NMR-, MS- and IR-analyses. The chemical shifts for
1d were assigned using
1H-NMR-,
13C-NMR-, COSY-, HSQC- and HMBC-spectra. The analyses were performed on a 600 MHz NMR instrument from Bruker, Germany, with deuterated dimethyl sulfoxide as solvent.
The strategy for avoiding the formation of the dimers 1d–4d and also to generate a high ratio of halohydrin to epoxide was to use only 0.3 equivalents of base. The chlorohydrins 1a–4a were synthesized in 59–77% yield.
The starting material 5-(3-chloro-2-hydroxypropoxy)-3,4-dihydroquinolin-2(1H)-one (4) for the synthesis of carteolol is quite expensive, so we wanted to investigate similar reactions of 7-(3-chloro-2-hydroxypropoxy)-3,4-dihydroquinolin-2(1H)-one (3) as a model substrate.
Table 2 entry 3 shows that the highest relative rate of the formation of halohydrin
1a over the formation of the epoxide
1e is obtained with one eq of base.
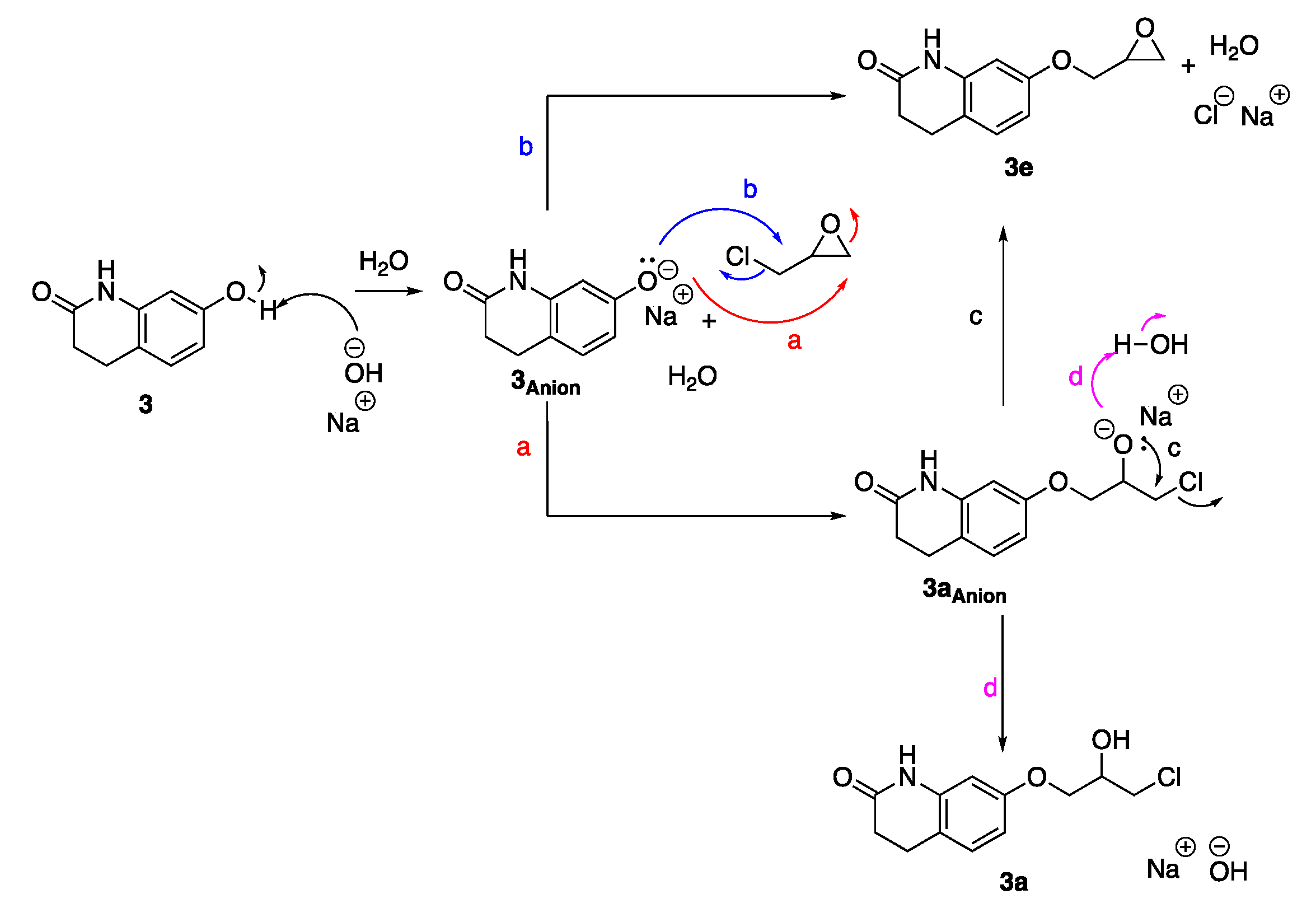
We saw the same trend in the synthesis of
2a/
2e. However, in the synthesis of
3a/
3e and
4a/4e, 0.3 eq of base gave the highest yield. When catalytic amounts of base are used, the anions of the halohydrins formed will likely deprotonate a water molecule which regenerates the base for new deprotonations of the starting materials. A plausible mechanism for regeneration of the base in these reactions is shown for the reaction of 7-(3-chloro-2-hydroxypropoxy)-3,4-dihydroquinolin-2(1
H)-one (
3,
Scheme 2). After deprotonation of phenol
3 by the base,
3Anion can react with the least substituted epoxide-carbon in epichlorohydrin (path a) forming
3aAnion. 7-(Oxiran-2-ylmethoxy)-3,4-dihydroquinolin-2(1H)-one,
3e, is formed by a Williamson ether synthesis reaction between
3 and epichlorohydrin, as shown in path b. An internal cyclization of
3aAnion also forms epoxide
3e (path c), while protonation of
3aAnion yields chlorohydrin
3a and regenerates the base allowing for the use of catalytic amounts of base (<1 equivalent).
Addition of lithium chloride and acetic acid before any work-up of all the reactions forming 1a–4a gave higher yields than when the reactions were stopped after the nucleophilic attack of epichlorohydrin in the first step.
As the hydroxide ion may also attack epichlorohydrin directly both on carbon 1 in the oxirane and on the α-carbon, the by-products 2-(chloromethyl)oxirane (
6), 3-chloropropane-1,2-diol (
7) and propane-1,2,3-triol (
8) may also be formed, see
Scheme 3. All impurities were removed by flash chromatography.
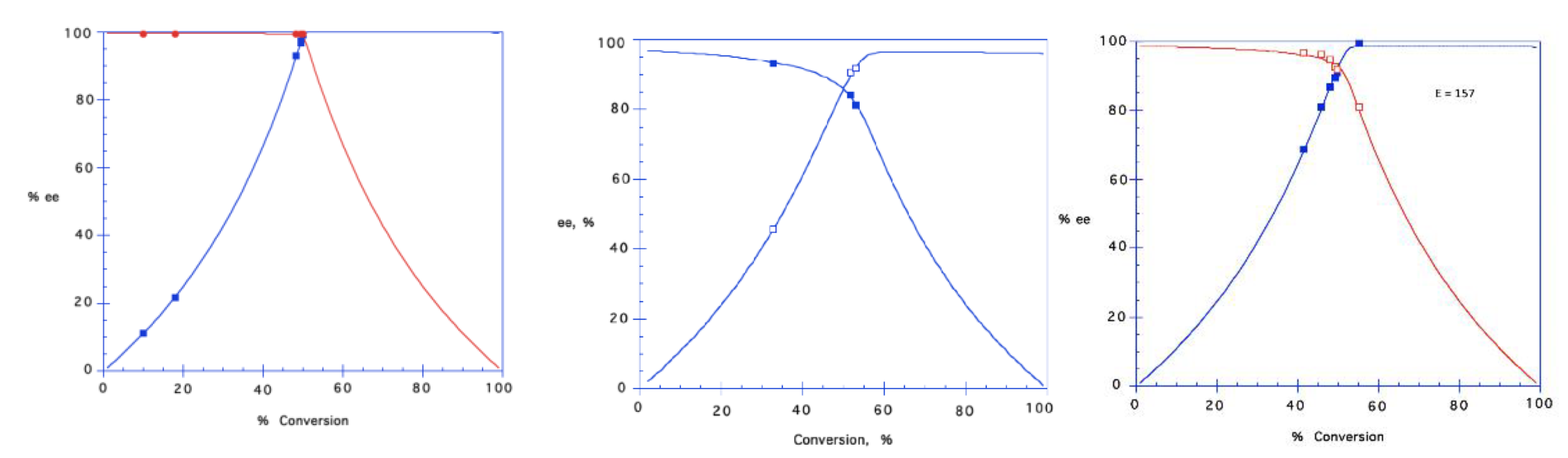
Kinetic resolutions of
1a–
4a have been performed in different solvents catalyzed by CALB with moderate to high
E-values (calculated by
E&K Calculator, 2.1b0 PPC) [
24], giving moderate to high
ee-values of the chiral building blocks. (
Figure 1). Optical rotation values of (
R)-
1a, (
S)-
1a, (
S)-
1b, (
R)-
3a, (
S)-
3b, (
S)-
3c, (
R)-
4a and (
S)-
4b have not been reported previously, and the determination of optical rotation and predictions of absolute configuration of these pure enantiomers should be of interest to both academia and industry. Compounds with >96%
ee are regarded as “enantiopure” by pharmaceutical means. Due to the relatively low
ee of (
R)-
2a in our hands (92%
ee) we did not proceed with the synthesis of the pindolol enantiomer; however, we would have expected to retain the
ee from the amination of (
R)-
2a giving (
S)-pindolol of 92%
ee.
In the amination reactions of (
R)-
1a and (
R)-
3a, the
ee was retained and (
S)-practolol ((
S)-
1c) and (
S)-semi-carteolol ((
S)-
3c) were produced in 96 and 97%
ee, respectively. (
R)-
4a was obtained in 96%
ee, and (
S)-
4c was obtained in 96%
ee from amination of (
R)-
4a (
Table 3). Reaction times of the kinetic resolutions varied by the amounts of lipase added; however, 12 h should be a proper reaction time. Danilewicz and Kemp (1973) reported that the optical rotation of (
R)-practolol was +4.3° at 25 °C [
25]. We conclude that the negative rotation of (
S)-practolol determined by us (–3.9°) accounts for the S-configuration.
We noticed that other research groups have reported data for synthesis of enantiopure practolol and pindolol [
14,
17]. Reaction time for the enzymatic hydrolyses of the acetate of 1-(1
H-indol-4-yloxy)-3-chloro-2-propanol reported by Lima et al. varied from 12 to 25 h and the authors reported an
E-value of 30 in the hydrolytic kinetic resolution of the racemic acetylester of
2a using Novozym
® 435 [
17]. By using lipase from
Pseudomonas fluorescens in acetylation of racemic
2a, an
E-value of 11 was obtained, with an
ees of the chlorohydrin
2a of 72%
ees and 69%
eep for 51% conversion, 24 h reaction time at 40 ℃. However, there is a misunderstanding of stereochemistry in Lima´s report. The authors report that hydrolysis of the racemic acetate and transesterification reaction of the chlorohydrin
2a give enantiomers with opposite stereochemistry. The product and the remaining alcohol will have the same configuration in hydrolysis of the acetyl ester and transesterification of the chlorohydrin
2a. Hydrolysis of the ester enantiomer from the hydrolysis of racemic ester should not be the
R-acetate of
2a, but the
S-ester. We claim that they have produced (
S)-
2a in 97%
ee instead of (
R)-
2a in their hydrolysis of the acetic ester of 1-((1
H-indol-4-yl)oxy)-3-chloropropan-2-ol. The authors must have used the
S-halohydrin in the synthesis and would then have achieved (
R)-pindolol. The optical rotation value is not reported. In our project we used CALB from SyncoZymes as catalyst and obtained an
E-value of 66 in the esterification of
2a. At 53% conversion (24 h),
ees and
eep values were 92% and 81%, respectively. The optimization of this synthesis is underway. We encourage researchers to report both optical rotation values and determination of absolute configuration of enantiomers.
3. Materials and Methods
All chemicals used in this project are commercially available, of analytical grade and were purchased from Sigma-Aldrich Norway (Oslo, Norway) or vwr Norway (Oslo, Norway). HPLC grade of solvents were used for the HPLC-analyses. Dry MeCN was acquired from a solvent purifier, MBraun MD-SPS800 (München, Germany), and stored in a flask containing molecular sieves (4Å).
3.1. Enzymes
Candida antarctica Lipase B (CALB) (activity ≥ 10,000 PLU/g, lot#20170315) immobilized at high hydrophobic macroporous resin, produced in fermentation with genetically modified Pichia pastoris. The enzyme is a gift from SyncoZymes Co, Ltd. (Shanghai, China).
3.2. Chromatographic Analyses
All analyses were performed on an Agilent HPLC 1100 (Santa Clara, CA, USA). Manual injector (Rheodyne 77245i/Agilent 10 μL loop) and a variable wavelength detector (VWD) set to 254 nm were used.
3.2.1. Achiral HPLC
Separation was performed on a XDB C18-column (250 × 4.6 mm ID, 5 μm particle size, 80 Å, Phenomenex, Oslo, Norway).
Dimer 1d: eluent gradient: H2O:acetonitrile (90:10)-H2O:acetonitrile (75:15) over 20 min: tR 15.71 min.
3.2.2. Chiral HPLC
Separation of enantiomers was performed on a Chiralcel OD-H column (250 × 4.6 mm ID, 5 μm particle size, Daicel, Chiral Technologies Europe, Gonthier d’Andernach, Illkirch, France). Baseline separation was obtained; if not, R
s is given. Chlorohydrin
1a:
tR (
S)-
1a = 31.1 min,
tR (
R)-
1a = 38.8 min, butanoate ester
1b:
tR (
S)-
1b = 18.9 min and (
R)-
1b 23.7 min, eluent: hexane:
i-PrOH, 83:17, 1 mL/min flow, R
s = 12.05 and
tR (
S)-
1b = 24.8 min and (
R)-
1b 31.6 min with eluent: hexane:
i-PrOH, 85:15, 1 mL/min flow, R
s = 8.91. Practolol
1c,
tR (
R)-
1c = 13.6 min,
tR (
S)-
1c = 17.2 min, eluent: hexane:
i-PrOH, 83:17, 1 mL/min flow, R
s = 6.17. Chlorohydrin
2a:
tR (
S)-
2a = 50.7 min,
tR (
R)
-2a = 22.5 min, butanoate ester
2b: tR (
S)-
2b = 14.0 min,
tR (
R)-
2b = 14.7 min, eluent: hexane:
i-PrOH, 80:20, 1 mL/min flow. Chlorohydrin
3a: tR (
S)-
3a = 20.10 min,
tR (
R)-
3a = 25.83 min, eluent: hexane:
i-PrOH:DEA, 60:40:0.4, 0.4 mL/min flow, butanoate ester
3b:
tR (
S)-
3b = 56.30 min,
tR (
R)-
3b = 63.47 min, eluent: hexane:
i-PrOH:DEA, 90:10:0.4, 0.4 mL/min flow. Chlorohydrin
4a: tR (
S)-
4a = 63.43 min,
tR (
R)-
4a = 68.68 min, eluent: hexane:EtOH:TFA, 90:10:0, 0.4 mL/min flow, R
s = 1.70. Butanoate ester
4b:
tR (
S)-
4b = 110.33 min,
tR (
R)-
4b = 118.44 min, R
s = 1.65, eluent: hexane:
i-PrOH:DEA, 95:5:0.4, 0.4 mL/min flow. Selected chromatograms can be found from
Supplementary Materials.
3.3. TLC-Analyses and Column Chromatography
TLC-analyses were performed on Merck silica 60 F254 (Sigma-Aldrich Norway, (Oslo, Norway) and detection with UV at λ = 254 nm. Flash chromatography was performed using silica gel from Sigma-Aldrich Norway, (Oslo, Norway) (pore size 60Å, 230–400 mesh particle size, 40–63 μm particle size).
A New Brunswick G24 Environmental Incubator Shaker (New Brunswick co. Inc., Edison, NJ, USA) was used for enzymatic reactions.
3.4. Analyses
NMR-analyses were recorded on a Bruker 400 MHz Avance III HD instrument equipped with a 5 mm SmartProbe Z-gradient probe operating at 400 MHz for
1H and 100 MHz for
13C, respectively, or on a Bruker 600 MHz Avance III HD instrument equipped with a 5 mm cryogenic CP-TCI Z-gradient probe operating at 600 MHz for
1H and 150 MHz for
13C (Bruker, Rheinstetten, Germany). Chemical shifts are in ppm relative to TMS and coupling constants are in hertz (Hz).
1H and
13C NMR spectra can be found from
Supplementary Materials. Infrared spectroscopy was performed at a Nexus FT-IR instrument (Madison, WI, USA). Exact masses were analyzed with a Synapt G2-S Q-TOF mass spectrometer from Waters
TM (Waters Norway, Oslo, Norway). Ionization of samples was done with an ASAP probe (APCI), and calculation of exact masses and spectra processing were performed with Waters
TM Software (Masslynxs V4.1 SCN871). IR and MS spectra details can be found from
Supplementary Materials.
3.5. Optical Rotations
Optical rotations were measured on an Anton Paar (MCP 5100) polarimeter with a 2.5-centimeter-long cell (Dipl. Ing. Houm AS, Oslo, Norway). The analyses were performed at 20–23 °C and the samples were dissolved in different solvents, c = 1.0g/100 mL, if not stated otherwise. Wavelength of the light was 589 nm (D).
3.6. Absolute Configurations
Absolute configuration of (
S)-
1c was determined by comparing the optical rotation with previously reported data. Optical rotation values of (
R)-
1a, (
S)-
1a, (
S)-
1b, (
R)-
3a, (
S)-
3b, (
S)-
3c, (
R)-
4a and (
S)-
4b have not been reported previously and the absolute configurations were determined by the enantioselectivity of CALB which we have reported previously [
22,
23].
3.7. Synthesis Protocols
N-(4-Hydroxyphenyl)acetamide (paracetamol, 151.163 g/mol, 1.998 g, 13.22 mmol) was dissolved in NaOH solution (0.3 eq), and 2-(chloromethyl)oxirane (epichlorohydrin) (26.44 mmol, 2 equiv.) was added dropwise with stirring. The reaction mixture was stirred at room temperature until TLC (CH2Cl2: MeOH, 10:1) showed full conversion of paracetamol. The reaction mixture was filtrated and washed with MeCN or distilled H2O. The filtrate was extracted with EtOAc and the organic layer was dried over MgSO4, filtrated and concentrated under reduced pressure. The product mixture was analyzed by HPLC with EclipseXDB C18-column and gradient H2O:MeCN, 90:10-H2O:MeCN, 75:25 over 20 min, 0.5 mL/min flow, showing both the chlorohydrin N-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide, 1a, and the epoxide N-(4-(oxiran-2-ylmethoxy)phenyl)acetamide, 1e. The product mixture was reacted further without purification. The amount of reagents in step 2 are calculated from the assumption that the starting material contains only N-(4-(oxiran-2-ylmethoxy)phenyl)acetamide, 1e, even if the starting material also contained N-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide, 1a. The mixture of 1a/1e (0.681 g, 3.29 mmol) was dissolved in MeCN (10 mL), and LiCl (0.912 g, 21.5 mmol) and AcOH (3.0 mL, 53.1 mmol) were added. The reaction mixture was stirred at room temperature for 26 h and TLC (CH2Cl2:MeOH, 10:1) showed only the chlorohydrin 1a, Rf = 0.50. The reaction was stopped by adding Na2CO3 until reaching neutral pH. The precipitated salt was filtrated off. The reaction mixture was then extracted with EtOAc and washed with satd. NaCl solution. The organic layer was dried over MgSO4, filtrated, and concentrated under reduced pressure. A yellow-brown viscous liquid was collected, which was purified by flash chromatography (CH2Cl2:MeOH, 10:1, v/v). After purification, the product 1a was collected as a white solid (0.543 g, 2.24 mmol, 68% yield, >99% purity). (HPLC, tR = 13.4 min). TLC (CH2Cl2:MeOH, 10:1) Rf = 0.43 for N-(4-(3-chloro-2-hydroxypropoxy)phenyl) acetamide. 1H NMR (400 MHz, DMSOd6): δ 9.78 (s, 1H, NH), 7.47 (d, 2H, AR-H, J = 10.2 Hz), 6.88 (d, 2H, Ar-H, J = 9.6 Hz), 5.52 (d, 1H, OH, J = 5.4 Hz), 4.01 (sext, 1H, CH, J = 5.3 Hz), 3.92 (d, 2H, CH2, J = 5.6 Hz), 3.74 (dd, 1H, CH2, J1 = 5.4 Hz, J2 = 11.8 Hz), 3.66 (dd, 1H, CH2, J1 = 5.4 Hz, J2 = 10.8 Hz), 2.00 (s, 3H, CH3). 13C NMR (100 MHz, DMSOd6): δ 168.1, 154.6, 133.2, 120.9, 114.9, 69.6, 69.1, 47.2, 24.3. HRMS (TOF-ASAP+): [M+H]+ = 244.0739 m/z (calc. mass: 244.0740, C11H15NO3Cl).
1-((1
H-indol-4-yl)oxy)-3-chloropropan-2-ol (
2a) [
17]
1H-Indol-4-ol (0.51 g, 3.80 mmol) was dissolved in 1,4-dioxane (3 mL) and NaOH (0.16 g, 3.93 mmol, 1 eq) was dissolved in water (5 mL) and epichlorohydrin (2.98 mL, 38 mmol) was added. The mixture was stirred at rt for 5 h until TLC showed full conversion of starting material (CH2Cl2, Rf = 0.21). The product was extracted using CH2Cl2 (50 mL) and washed with EtOAc (3 × 30 mL) and water (3 × 30 mL). The CH2Cl2-phase was dried over anhydrous MgSO4 and evaporated under reduced pressure, yielding 0.48 g of a mixture of 1-((1H-indol-4-yl)oxy)-3-chloropropan-2-ol (2a) and 4-(oxiran-2-ylmethoxy)-1H-indole (2e) as a brown oil.
A mixture of 2a/2e (0.48 g, 2.56 mmol) was dissolved in THF (8 mL). AcOH (1.46 mL, 25.6 mmol) and LiCl (0.22 g, 5.12 mmol) were added. The mixture was stirred at rt for 72 h. NaCO3 was added until neutral pH was obtained. The product was extracted with CH2Cl2 (50 mL) and washed with satd. NaCl solution (3 × 30 mL). The CH2Cl2 phase was then dried over anhydrous MgSO4 and evaporated. After purification by flash chromatography with CH2Cl2 as eluent, the product was obtained as a slightly yellow oil of 1-((1H-indol-4-yl)oxy)-3-chloropropan-2-ol (2a) (0.5701 g, 2.52 mmol, 98.5% yield). Spectroscopic data for 2a: 1H NMR (400 MHz, DMSOd6) δ 11.07 (s, 1H, NH), 7.22 (t, 1H, CH, J = 2.6 Hz), 7.02 (m, 2H, Ar-H), 6.47 (t, 1H, CH, J = 2.3 Hz), 6.49 (dd, 1H, Ar-H, J1 = 1.2 Hz, J2 = 7.1 Hz), 5.5 (s, 1H, OH), 4.09 (m, 3H, CH and CH2), 3.84 (dd, 1H, CH, J1 = 5.2 Hz, J2 = 11.3 Hz), 3.75 (dd, 1H, CH, J1 = 4.3 Hz, J2 = 11.3 Hz). 13C NMR (100 MHz, DMSOd6): δ 152.4, 137.9, 124.1, 122.5, 118.9, 105.6, 100.4, 98.7, 69.3, 69.2, 47.5. HRMS (TOF ASAP+): [M+H]+ = 226.0632 m/z.
1-((1H-Indol-4-yl)oxy)-3-chloropropan-2-ol (2a) (0.08 g, 0.37 mmol) and butyric anhydride (0.075 mL, 0.46 mmol) were added to pyridine (0.05 mL, 0.62 mmol). The mixture was stirred for 24 h at rt. Extraction was performed with hexane and CH2Cl2 and washed with satd. NaCl solution. The organic phase was dried over anhydrous MgSO4 and evaporated, yielding 0.08 g of a mixture of 2a (84.6%, HPLC) and 2b (11.0%, HPLC). Separation by flash chromatography using CH2Cl2 as eluent yielded 1% of 2b (0.80 mg, 0.003 mmol). Spectroscopic data for 2b: 1H NMR (400 MHz, CDCl3): δ 8.22 (s, 1H, NH), 7.03-7.16 (m, 3H, Ar-H and CH), 6.64 (m, 1H, CH), 6.56 (dd, 1H, Ar-H), 4.19-4.37 (m, 3H, CH and CH2), 3.75-3.90 (m, 2H, CH2), 2.37 (td, 2H, CH2, J1 = 11.7 Hz, J2 = 7.33, J3 = 1.34 Hz), 1.69 (sext, 2H, CH2, J = 7.34 Hz), 0.97 (t, 3H, CH3, J = 7.64 Hz). 13C NMR (100 MHz, CDCl3): δ 151.9, 137.4, 122.9, 122.8, 118.7, 105.2, 101.0, 99.7, 70.1, 68.6, 46.2, 36.4, 18.4, 13.6.
To a solution of 3 (1.6541 g, 10.1 mmol) in MeOH (30 mL), NaOH-solution (0.17 M, 30 mL, 0.5 eq) was added. Epichlorohydrin (1.565 mL, 20.0 mmol) was added dropwise to the reaction mixture which was then stirred at rt for 24 h. TLC (CHCl3:CH2Cl2:EtOH, 10:9:1) showed full conversion of 3a with two products: Rf (3e) = 0.31, Rf (3a) = 0.41. Insoluble by-products were filtered off. The filtrate was extracted with CH2Cl2 (3 × 20 mL). The organic phase was washed with satd. NaCl solution (2 × 10 mL), dried over anhydrous MgSO4, and filtered before the solvent was removed under reduced pressure. This resulted in white crystals and a yellow, highly viscous oil in a mixture. The mixture was recrystallized from EtOH to yield 3a as white crystals (1.1074 g, 5.05 mmol, 50% yield). 1H NMR (400 MHz, CD3OD): δ 7.08 (m, 1H, Ar-H), 6.59 (m, 1H, Ar-H), 6.49 (m, 1H, Ar-H), 3.84–4.27 (m, 2 H, CH2), 2.88 (m, 2H, CH2), 2.74–2.88 (m, 2H, CH2), 2.55 (m, 2H, CH2). 13C NMR (100 MHz, CD3OD): δ 172.7, 158.2, 138.4, 128.3, 116.4, 108.5, 102.0, 68.9, 49.8, 43.5, 30.5, 24.0.
LiCl (1.0440g, 24.6 mmol) and AcOH (2.810 mL, 49.1 mmol) were added to a solution of 3a/3e (1.0766 g, 4.91 mmol) in MeCN (10 mL). The reaction was stirred at rt and TLC showed full conversion of the starting material after 24 h. The reaction mixture was extracted with CH2Cl2 (3 × 20 mL), dried over anhydrous MgSO4 and the solvent was removed under reduced pressure. 3a was obtained as white crystals (0.9647 g, 3.77 mmol, 77% yield). 1H NMR (400 MHz, CD3OD): δ 7.09 (m, 1H, Ar-H), 6.60 (m, 1H, Ar-H), 6.51 (m, 1H, Ar-H), 4.13 (m, 1H, CH), 4.06 (m, 2H, CH2), 3.69–3.77 (m, 2H, CH2), 2.87–2.90 (m, 2H, CH2), 2.53–2.57 (m, 2H, CH2). 13C NMR (400 MHz, CD3OD) δ: 172.7, 158.2, 138.4, 128.3, 116.4, 108.5, 102.0, 69.6, 68.9, 45.4, 30.5, 24.0. HRMS (TOF-ASAP+): [M+H]+ = 256.0793 m/z (calc. mass: 256.0740, C11H15NO3Cl).
5-(3-Chloro-2-hydroxypropoxy)-3,4-dihydroquinolin-2(1
H)-one (
4a) [
26]
Epichlorohydrin (0.134 mL, 1.7 mmol) was added to a solution of 4 (0.1665g, 1.0 mmol) in H2O (0.585 mL) and DMSO (0.375 mL). Aqueous solution of NaOH (9.5 M, 0.090 mL, 0.5 eq) was added to the reaction mixture. The solution was stirred at rt for 24 h where 4a and 4e slowly precipitated from the solution. TLC (hexane:i-PrOH, 4:1) showed full conversion after 24 h and the product mixture was filtered off, yielding 4a and 4e (0.1505 g) as white crystals in a 1:4 ratio, determined by 1H NMR. TLC (CHCl3:acetone, 4:1), Rf (4a) = 0.32, Rf (4e) = 0.46. The mixture of 4e/4a (1.0979 g, 5.01 mmol) was then dissolved in MeCN (5 mL). LiCl (1.0741 g, 25 mmol) and AcOH (2.865 mL, 50 mmol) were added, and the reaction mixture was stirred for 24 h until TLC showed full conversion of 4e. Na2CO3-solution (pH 12) was added until the reaction mixture reached pH 7. The product was filtered off and 4a was obtained as white crystals (0.7807 g, 3.1 mmol, 61% yield). 1H NMR (400 MHz, CD3OD) δ 7.01 (m, 1H, Ar-H), 6.56 (m, 1H, Ar-H), 6.41 (m, 1H, Ar-H), 4.05 (m, 1H, CH), 3.98 (m, 2H, CH2), 3.60–3.69 (m, 2H, CH2), 2.86 (t, 2H, CH2, J = 7.6 Hz), 2.43 (t, 2H, CH2, J = 7.6 Hz). 13C NMR (100 MHz, CD3OD) δ 172.5, 155.8, 138.6, 127.6, 111.9, 108.5, 106.3, 69.6, 69.0, 45.4, 29.6, 18.1. HRMS (TOF-ASAP+): [M+H]+ = 256.0743 m/z (calc. mass: 256.0740, C11H15NO3Cl).
Racemic N-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide, 1a, (0.220 g, 0.905 mmol) was dissolved in isopropylamine (0.519 mL, 6.33 mmol, 7.0 eq) and distilled H2O (0.150 mL). The reaction mixture was stirred at rt for 48 h, and was concentrated under reduced pressure. The crude product was recrystallized from MeCN, and N-(4-(2-hydroxy-3-(isopropylamino)propoxy)phenyl)acetamide, 1c, was collected as a white solid (0.115 g, 0.432 mmol, 48% yield). 1H-NMR (400 MHz, DMSOd6). δ 9.74 (s, 1H, NH), 7.44 (d, 2H, Ar-H, J = 10.8 Hz), 6.84 (d, 2H, Ar-H, J = 10.8 Hz), 4.91 (d, 1H, OH, J = 5.3 Hz), 3.89–3.86 (m, 1H, CH), 3.80 (t, 2H, CH2, J = 6.7 Hz), 2.73–2.62 (m, 2H, CH2), 2.53–2.51 (m, 1H, CH), 1.99 (s, 3H, CH3), 1.46 (s, 1H, NH), 0.95 (dd, 6H, 2 × CH3, J1 = 2.2 Hz, J2 = 6.2 Hz). 13C-NMR (150 MHz, DMSOd6): δ 167.7 (C = O), 154.4 (Ar-C), 132.4 (Ar-C), 120.4 (2 × Ar-C-H), 114.3 (2 × Ar-C-H), 70.9 (CH), 68.4 (CH2), 50.0 (CH), 48.1 (CH2), 23.7 (CH3), 22.9 (2 × CH3). IR (cm−1, diluted): 3343 (m), 2969 (m), 1127 (m), 950 (s), 816 (m). HRMS (TOF-ASAP+): [M+H]+ = 267.1712 m/z (calc. mass: 267.1709, C14H23N2O3).
3.8. Synthesis of Enantiomers
Racemic N-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide, 1a, (0.543 g, 2.24 mmol) was dissolved in dry MeCN (60 mL). Vinyl butanoate (2.56 g, 22.4 mmol) and molecular sieves (4Å) were added. The reaction was started by adding CALB (1.22 g) and stirred in an incubator shaker (30 ℃, 200 rpm). Samples (150 μL) were collected regularly, concentrated, and dissolved in i-PrOH before HPLC analyses. After 26 h, the reaction was stopped by filtering off the enzyme and removing the solvent under reduced pressure. The crude product was purified twice by flash chromatography (CH2Cl2:EtOAc, 1:1). (R)-1a was collected as a white solid (0.208 g, 0.852 mmol, 38% yield, ee = 97%). = −1.0 (c 1.0, i-PrOH). (S)-1-(4-Acetamidophenoxy)-3-chloropropan-2-yl butanoate, (S)-1b, was collected as a brown viscous liquid (0.33 g, 1.05 mmol, 47%yield, ee = 84%). = +16.3 (c 1.1, MeCN). (S)-N-(4-(3-Chloro-2-hydroxypropoxy)phenyl)acetamide, (S)-1a, was obtained by hydrolysis of (S)-1b with CALB: The crude product was purified by flash chromatography (CH2Cl2:EtOAc, 1:1) and (S)-1a was collected as a light brown solid (0.0475 g, 0.195 mmol, 26% yield, ee = 81%) = +11.99 (c 1.0, i-PrOH). The E-value was calculated by E&K calculator 2.1b0 PPC, E = 55. The NMR spectra for (S)-1a were in correspondence with the spectra for 1a.
Racemic 1-chloro-3-(1H-indol-4-yloxy)-propan-2-ol (2a) (18.5 mg, 0.08 mmol) was dissolved in dry CH2Cl2 (3 mL) and molecular sieves (4Å) were added. Vinyl butanoate (75 µL, 0.59 mmol) and immobilized CALB (36.8 mg) were added, and the reaction was stirred in the incubator shaker for 24 h (30 °C, 200 rpm) to reach 50% conversion. Samples (150 μL) were collected regularly for chiral HPLC analysis. (R)-2a was obtained in 92% ee and E = 66. NMR spectra were in accordance with the spectra for 2a.
Racemic 7-(3-chloro-2-hydroxypropoxy)-3,4-dihydroquinolin-2(1H)-one (3a) (0.7492 g, 2.93 mmol) was dissolved in dry MeCN (60 ml) and molecular sieves (4Å) were added. Vinyl butanoate (1.672 mg, 1.860 mL, 14.7 mmol) was added to the solution and the reaction was started by adding CALB (1.3724 g) and placing the container in an incubator shaker (30 °C, 200 rpm). The reaction was stopped after 24 h by filtering off CALB and the molecular sieves. The solvent was removed under reduced pressure yielding the mixture of ester (S)-3a and chlorohydrin (R)-3a as a brown oil, which were separated by flash chromatography (EtOAc:hexane:MeOH, 7:12:1). Chlorohydrin (R)-3a was isolated as white crystals (0.2573 g, 34% yield, ee = 96%), (c 1.0, MeOH). Ester (S)-3b was obtained as a yellow oil (0.4436 g, 51% yield, 91% purity, 86% ee). (c 1.0, MeOH). HPLC, eluent: hexane: i-PrOH:DEA, 90:10:0.4, tR (S)-3b = 56.30 min, tR (R)-3b = 63.47 min. E = 157. 1H NMR (R)-3a (600 MHz, CD3OD): δ 7.06 (m, 1H, Ar-H), 6.56 (m, 1H, Ar-H), 6.48 (m, 1H, Ar-H), 5.33 (p, 1H, CH, J = 5.0 Hz), 4.15 (d, 2H, CH2, J = 4.9 Hz), 3.83–3.86 (m, 2H, CH2), 2.84–2.87 (m, 2H, CH2), 2.52–2.54 (m, 2H, CH2), 2.35 (td, 2H, CH2, J = 7.4 Hz), 1.65 (sext, 2H, CH2, J1 = 7.4 Hz, J2 = 15.0 Hz), 0.96 (td, 3H, CH3, J1 = 7.4 Hz, J2 = 15.0 Hz). 13C NMR (150 MHz, CD3OD); δ 175.8, 172.3, 157.6, 138.2, 128.1, 116.3, 108.2, 101.8, 70.8, 66.1, 48.2, 35.2, 30.1, 23.7, 16.1, 12.3. 1H NMR (S)-3b (600 MHz, CD3OD). δ 7.06 (m, 1H, Ar-H), 6.56 (m, 1H, Ar-H) 6.48 (m, 1H, Ar-H), 5.33 (p, 1H, CH), 4.15 (d, 2H, CH2, J1 = 4.14 Hz), 3.83–3.86 (m, 2H, CH2-Cl), 2.85 (t, 2H, CH2, J = 7.10 Hz), 2.53 (t, 2H, CH2, J = 7.60 Hz), 2.35 (t, 2H, CH2, J = 7.10 Hz), 1.65 (sext, 2H, CH2, J = 7.10 Hz), 0.96 (t, 3H, CH3, J = 8.0 Hz).
Racemic 5-(3-chloro-2-hydroxypropoxy)-3,4-dihydroquinolin-2(1H)-one (4a) (0.1724 g, 0.67 mmol) was dissolved in dry MeCN (20 mL) and molecular sieves (4Å) were added. Vinyl butanoate (0.428 mL, 3.75 mmol) was added to the solution and the reaction was started by adding CALB (0.6745 g) and placing the container in an incubator shaker (37℃, 200 rpm). The reaction was stopped after 74 h by filtering off CALB and the molecular sieves. The solvent was removed under reduced pressure, yielding a mixture of ester (S)-4b and chlorohydrin (R)-4a as a brown oil, which were separated by flash column chromatography (EtOAc:hexane, 7:3). Chlorohydrin (R)-4a was isolated as white crystals (0.0652 g, 0.2549 mmol, 38% yield, ee = 96%), (c 1.0, DMSO). HPLC eluent: hexane:EtOH:TFA (90:10:0.1), 0.4 mL/min. tR(S)-4a = 64.10 min, tR(R)-4a = 69.48 min, Rs = 1.70. Ester (S)-4b was obtained as a colorless oil (0.0947 g, 0,029 mmol, 43% yield, 79% purity, 77% ee). (c 1.0, DMSO). HPLC eluent: hexane:iPrOH:DEA, 95:5:0.4, 0.4 mL/min. tR(S)-4b = 110.32 min, tR(R)-4b = 118.43 min, Rs = 1.65, E = 31. 1H NMR (R)-4a (400 MHz, DMSOd6). δ 10.02 (s, 1H, NH), 7.07 (t, 1H, Ar-H, J = 8.1 Hz), 6.59–6.61 (d, 1H, Ar-H, J = 8.2 Hz), 6.48–6.50 (d, 1H, Ar-H, J = 7.9 Hz), 5.57 (s, 1H, OH), 4.03 (m, 1H, CH), 3.96–3.97 (m, 2H, CH2), 3.75–3.79 (dd, 1H, CH2, J1 = 4.6 Hz, J2 = 11.1 Hz), 3.66–3.70 (dd, 1H, CH2, J1 = 5.5 Hz, J2 = 11.1 Hz), 2.82 (t, 2H, CH2, J = 7.7 Hz), 2.40 (t, 2H, CH2, J = 7.7 Hz). 13C NMR (100 MHz, DMSOd6): 170.5, 155.9, 139.8, 128.1, 111.7, 108.8, 106.2, 69.6, 69.1, 47.3, 30.3, 18.7. (0.0947 g, 0.29 mmol, 43% yield, ee = 77%). (c 1.0, DMSO) HPLC, eluent: hexane: i-PrOH:DEA, 95:5:0.4, 0.4 mL/min tR(S)-4b = 110.32 min, tR(R)-4b = 118.43 min, Rs = 1.65. 1H NMR (S)-4b (400 MHz, DMSOd6). δ 10.04 (s, 1H), 7.08 (t, 1H, Ar-H, J = 8.12 Hz), 6.60–6.62 (d, 1H, Ar-H, J = 8.01 Hz), 6.49–6.51 (d, 1H, Ar-H, J = 7.84 Hz), 5.35 (m, 1H, CH), 4.17–4.21 (dd, 1H, CH2, J1 = 4.14 Hz, J2 = 10.62 Hz), 4.11–4.15 (dd, 1H, CH2, J1 = 6.16 Hz, J2 = 10.60 Hz), 3.94–3.98 (dd, 1H, CH2, J1 = 4.16 Hz, J2 = 11.84 Hz), 3.86–3.91(dd, 1H, CH2, J1 = 6.50 Hz, J2 = 11.82 Hz), 2.77 (t, 2H, CH2, J = 7.67 Hz), 2.39 (m, 2H, CH2), 2.35–2.31 (m, 2H, CH2), 1.61–1.51 (sext, 2H, CH2, J = 7.32 Hz), 0.89 (t, 3H, CH3, J = 7.39 Hz). 13C NMR (100 MHz, DMSOd6): 172.6, 170.5, 155.5, 139.9, 128.2, 111.8, 109.1, 106.3, 71.1, 67.4, 43.8, 35.8, 30.2, 18.6, 18.4, 13.8.
3.9. Enantiopure Drug Derivatives
(
R)-
N-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide, (
R)-
1a, (0.175 g, 0.719 mmol) was dissolved in
i-PrNH
2 (0.470 mL, 5.73 mmol, 8.0 equiv) and distilled H
2O (0.075 mL). The reaction mixture was stirred at rt for 96 h, and the solvent was removed under reduced pressure. The crude product was recrystallized from MeCN and (
S)-
N-(4-(2-hydroxy-3-(isopropylamino)propoxy)phenyl)acetamide ((
S)-
1c) was collected as a white solid (0.0313 g, 0.117 mmol, 16% yield,
ee = 96%). Optical rotation of (
S)-
1c:
= −3.998° (
c 1.0, EtOH). mp: 124.7–124.9 ℃ (lit. 130–131 ℃) [
25].
1H NMR (600 MHz, DMSO
d6): δ 9.75 (s, 1H, NH), 7.45–7.44 (d, Ar-H,
J = 8.4 Hz), 6.85–6.84 (d, Ar-H,
J = 8.4 Hz), 4.95 (s, 1H, OH), 3.90–3.87 (m, 1H, CH), 3.81–3.80 (d, 2H, CH
2,
J = 6.8 Hz), 2.72–2.65 (m, 2H, CH
2), 2.55–2.52 (m, 1H, CH), 2.00 (s, 3H, CH
3), 0.97 (dd, 6H, CH
3,
J1 = 2.0 Hz,
J2 = 6.3 Hz).
13C NMR (150 MHz, DMSO
d6): δ 167.7, 154.4, 132.5, 120.4, 114.4, 70.9, 68.3, 49.9, 48.2, 23.7, 22.7. IR (cm
−1, diluted): 3343 (m), 2969 (m), 1127 (m), 950 (s), 816 (m). HRMS (TOF-ASAP
+): [M+H]
+ = 267.1711
m/z (calc. mass [M+H]
+ = 267.1709, C
14H
23N
2O
3).
Chlorohydrin (R)-3a (0.2573, 1.0063 mmol) was dissolved in t-BuNH2 (15.4 mL, 147 mmol, 147 equiv) and H2O (4.6 mL) and stirred at rt for 8 h. t-BuNH2 and H2O were removed under reduced pressure, yielding the crude product as a mixture of a yellow oil and white crystals. The crude product was purified by flash chromatography (EtOAc:hexane:MeOH:TEA, 80:7:10:3), yielding (S)-3c as a yellow oil (0.2419 g, 0.9460 mmol, 82% yield, 89% purity (NMR), ee = 97%). (c = 1.0, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.08 (m, 1H, Ar-H), 6.59 (m, 1H, Ar-H), 6.50 (m, 1H, Ar-H), 4.02 (m, 1H, CH), 3.95 (m, 2H, CH2), 2.88 (t, 2H, CH2, J = 7.6) 2.73–2.81 (m, 2H, CH2), 2.54 (t, 2H, CH2, J = 7.6 Hz), 1.18 (s, 9H, C(CH3)3). 13C NMR (150 MHz, CD3OD) δ 172.6, 158.3, 138.3, 128.1, 116.1, 108.4, 101.9, 70.8, 68.7, 50.4, 44.8, 30.7, 27.1, 24.2.
Chlorohydrin (R)-4a (0.0652 g, 0.2549 mmol) was dissolved in t-BuNH2 (3.75 mL, 37.75 mmol) and H2O (1.0 mL) and stirred at rt for 10 h. t-BuNH2 and H2O were removed under reduced pressure, yielding the crude product which was purified by flash chromatography (EtOAc:hexane:MeOH:TEA, 80:7:10:3), yielding (S)-4c as a yellow oil (0.0521 g, 0.1784 mmol, 70% yield, ee = 96%). 1H NMR (600 MHz, DMSOd6) δ 10.02 (s, 1H, NH), 7.08 (m, 1H, Ar-H), 6.59 (m, 1H, Ar-H), 6.50 (m, 1H, Ar-H), 5.54 (s, 1H, NH), 5.37 (s, 1H, OH) 4.03 (m, 1H, CH), 3.95–4.05 (m, 2H, CH2), 3.39 (t, 2H, CH2, J = 7.6 Hz), 2.73–2.81 (m, 2H, CH2), 2.54 (t, 2H, CH2, J = 7.6 Hz), 1.21 (s, 9H, C(CH3)3). 13C NMR (150 MHz, DMSOd6) δ 172.6, 158.3, 138.3, 128.1, 116.1, 108.4, 101.9, 70.8, 68.7, 58.2, 44.8, 30.7, 27.1 (3C), 20.4.



 ,
, 





{kind=link}
{kind=link}
{kind=link}
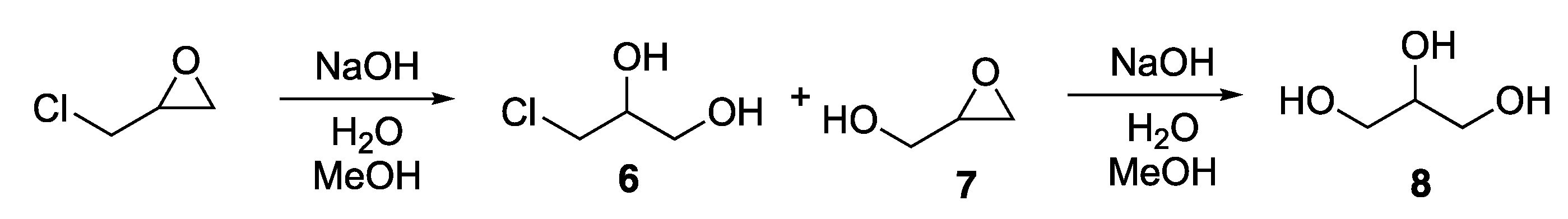{kind=link}