
Towards the Large-Scale Electrochemical Reduction of Carbon Dioxide


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Current Challenges in the Large-Scale Electrochemical CO2 Conversion Process
2.1. Problems at the Device Level
2.1.1. Low Current Density
2.1.2. Electrolyte pH Management
2.2. Problems at the Process Level
2.2.1. Product Separation
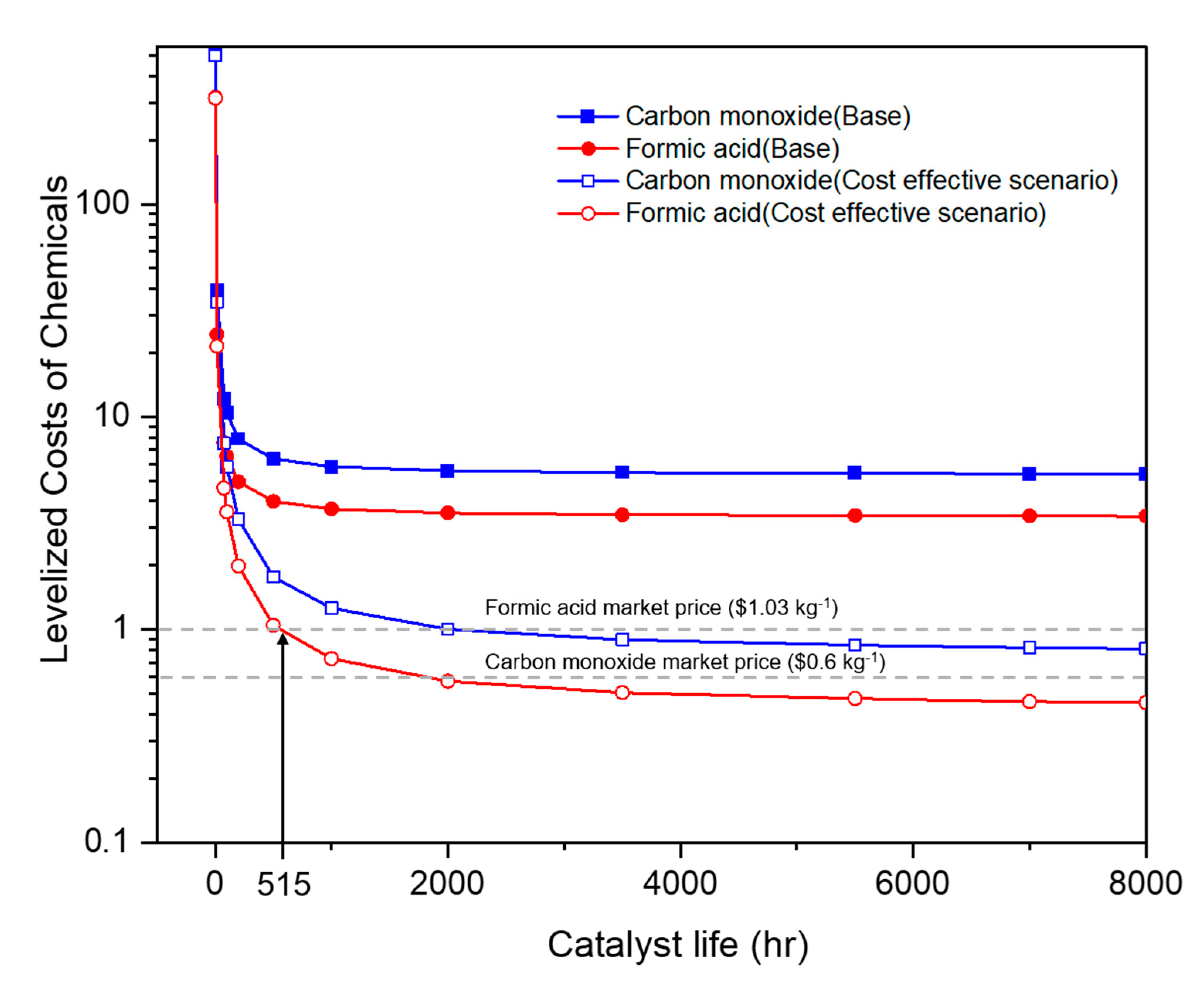
2.2.2. Electricity Costs
2.2.3. Integrated Process Design
3. Cutting Edge Devices for an Electrochemical CO2-Reduction Reactor
3.1. The Membrane
3.2. The Device
3.2.1. The Microfluidic Cell
3.2.2. The Zero-Gap Electrolyzer
3.2.3. The Multilayer Electrolyzer Stack
4. Conceptual Process Design and Techno-Economic Analysis for Mass Production
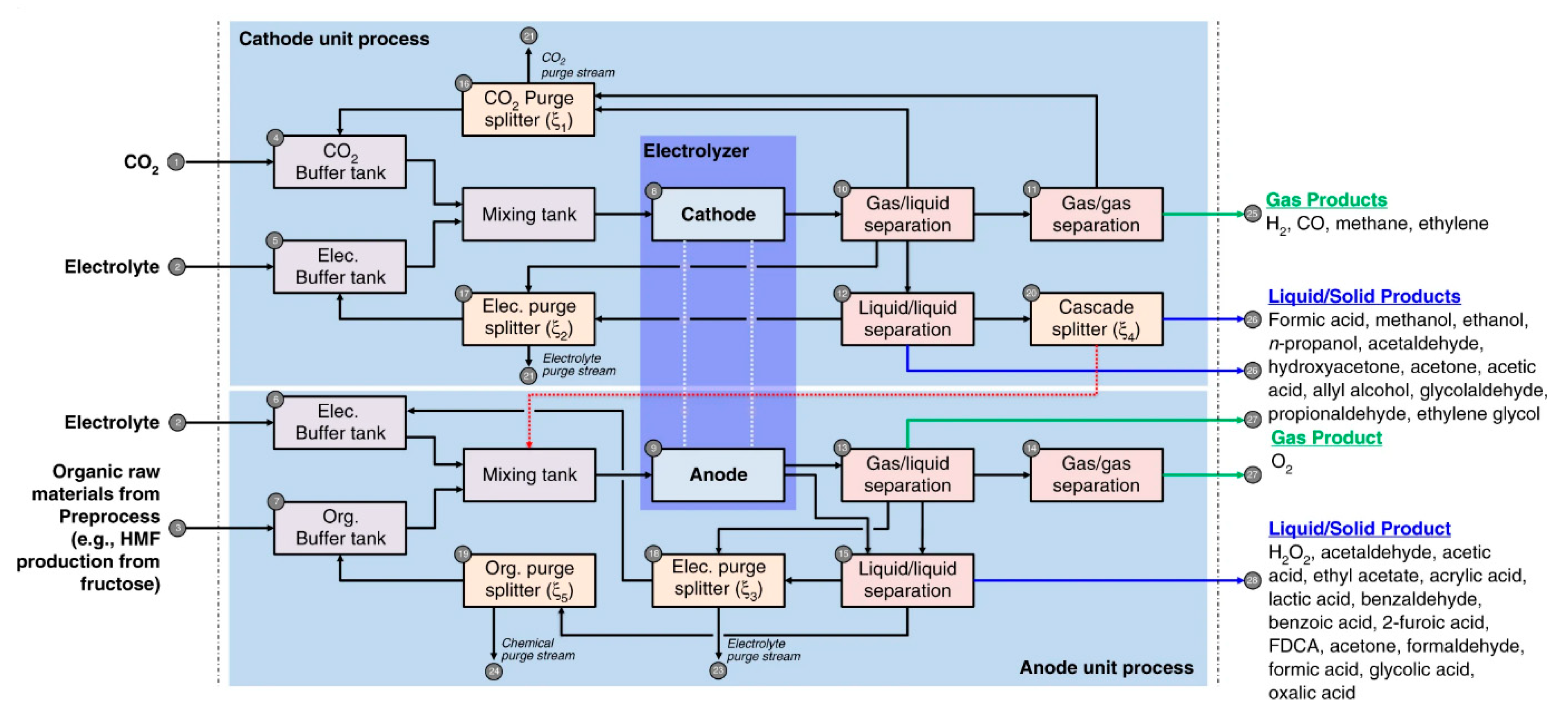
4.1. Product Separation
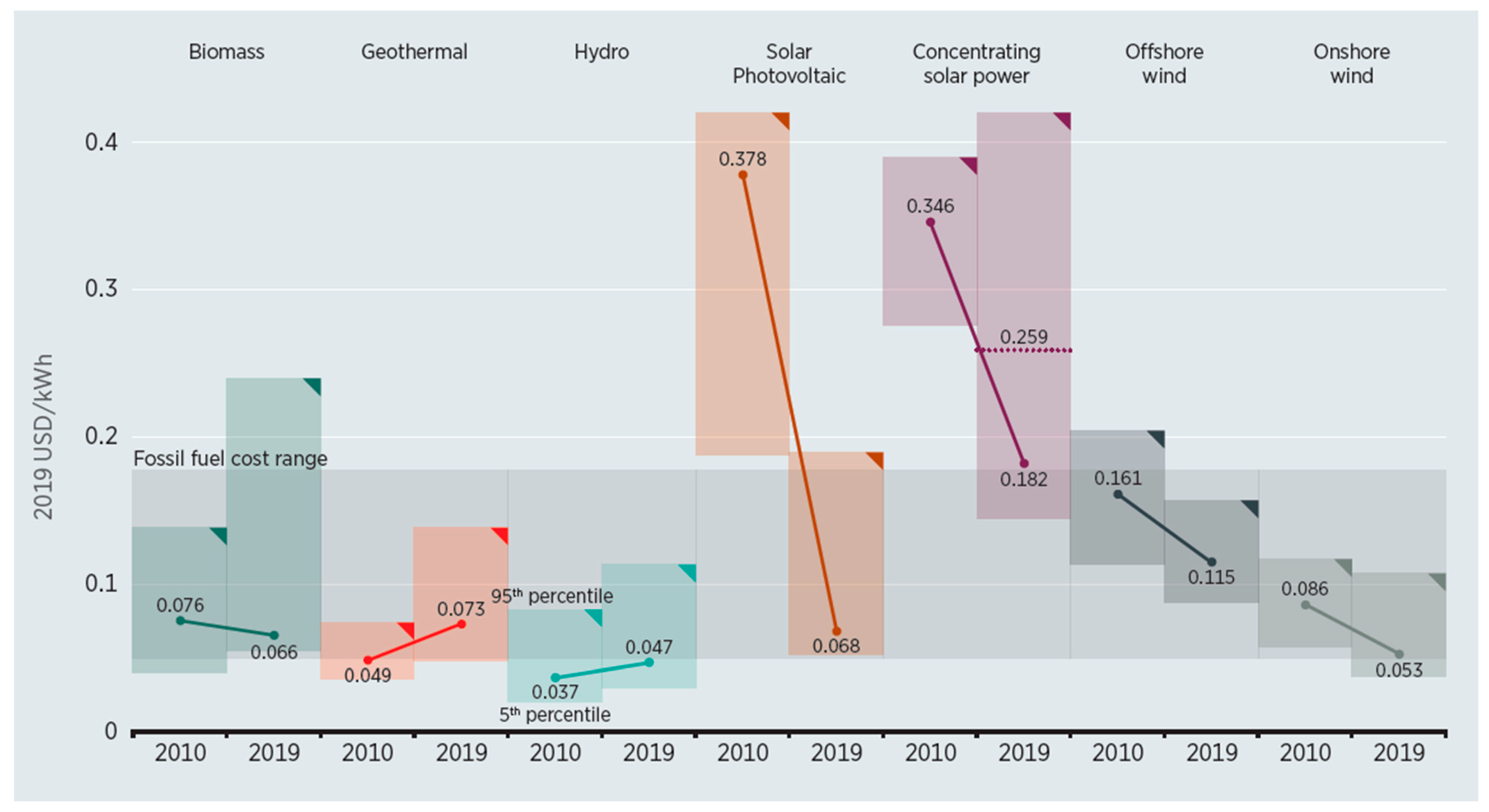
4.2. Renewable Energy for Electricity
4.3. Integrated Process Design
5. Stability Issues
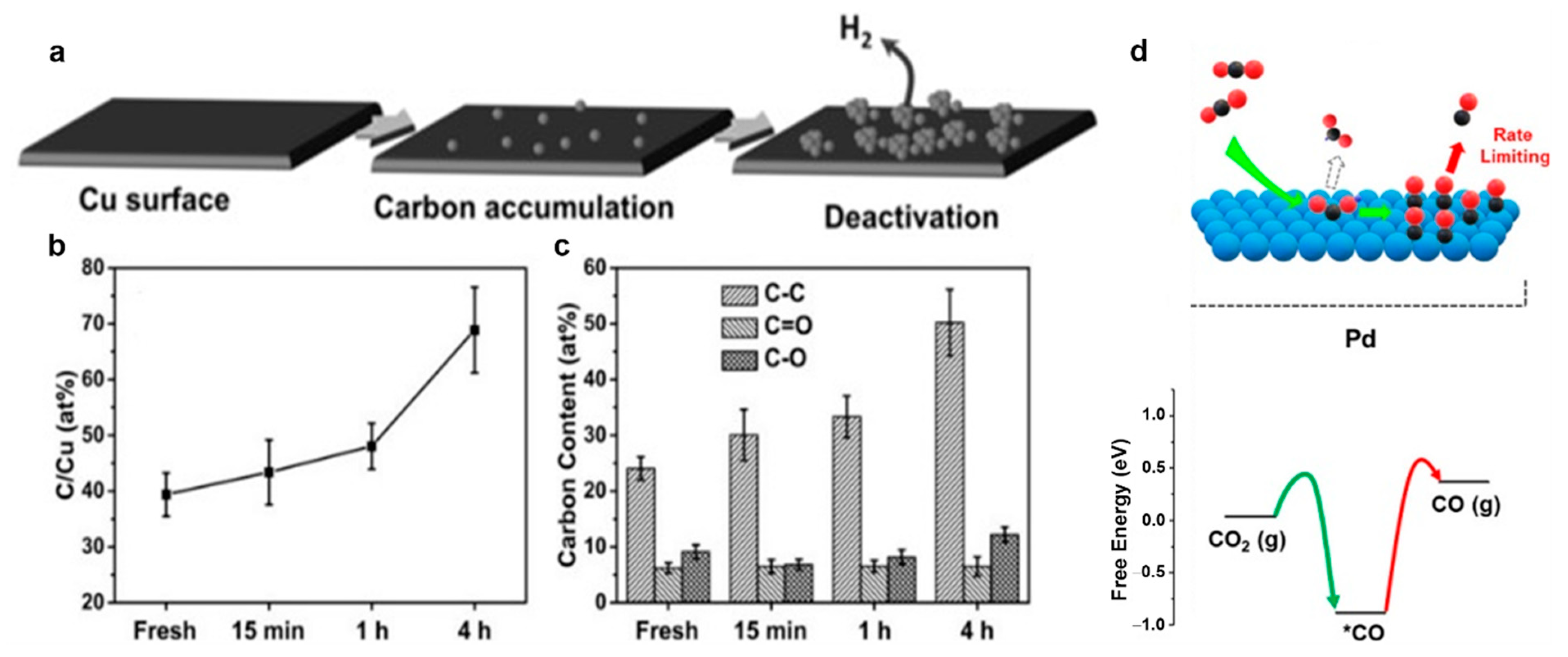
5.1. Catalyst Poisoning
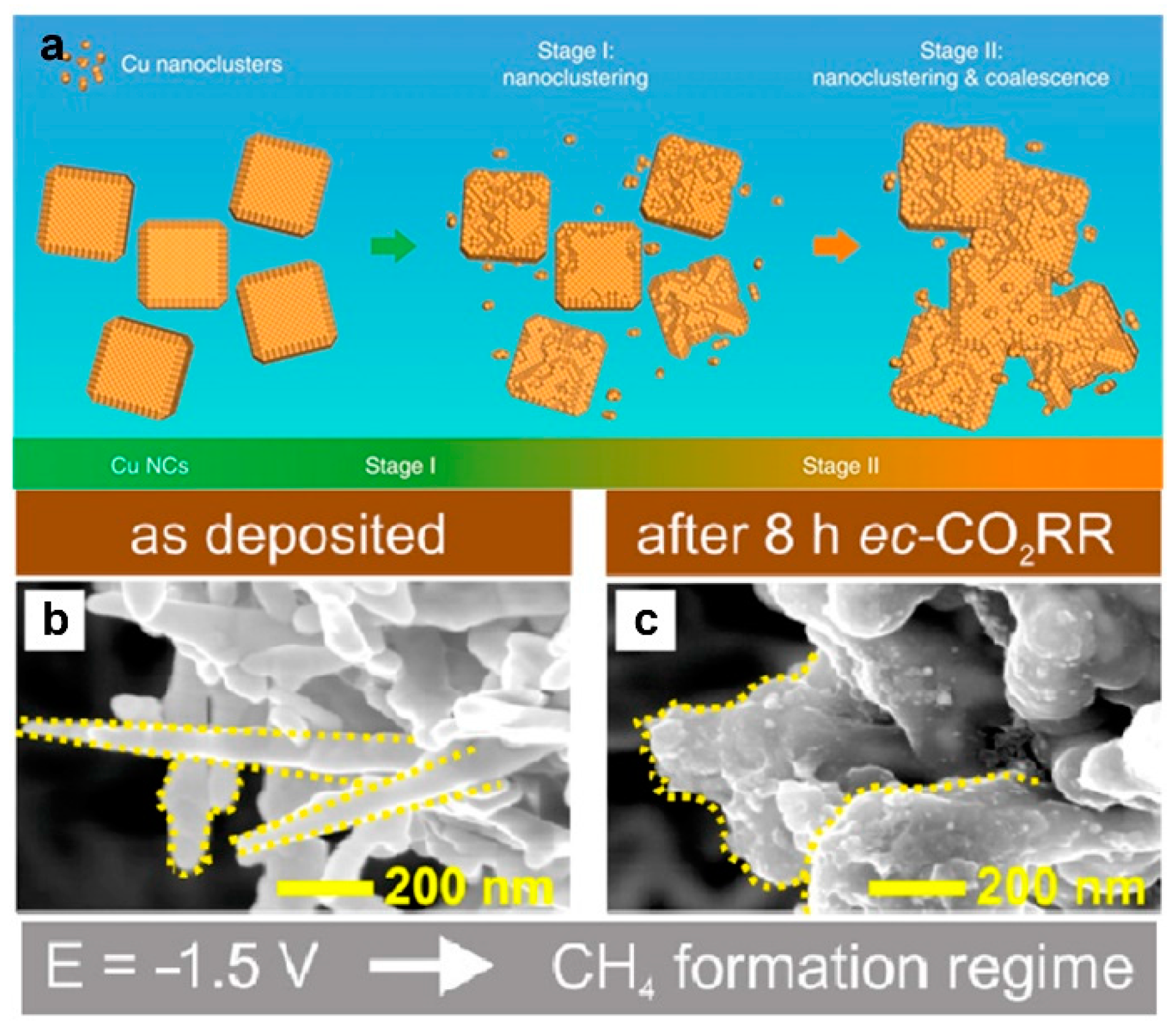
5.2. Catalyst Restructuring
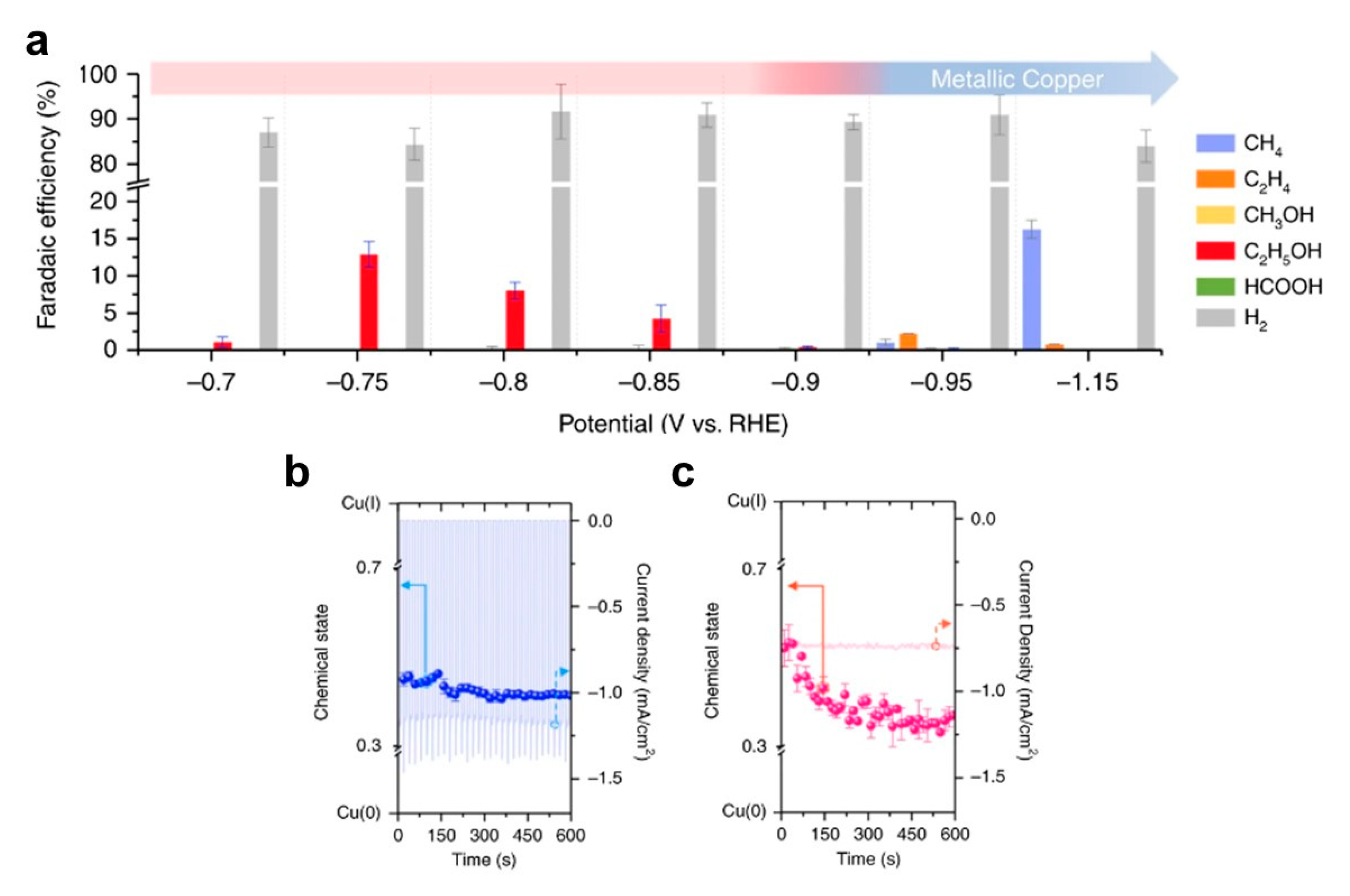
5.3. Changes in Catalyst Composition and Valency
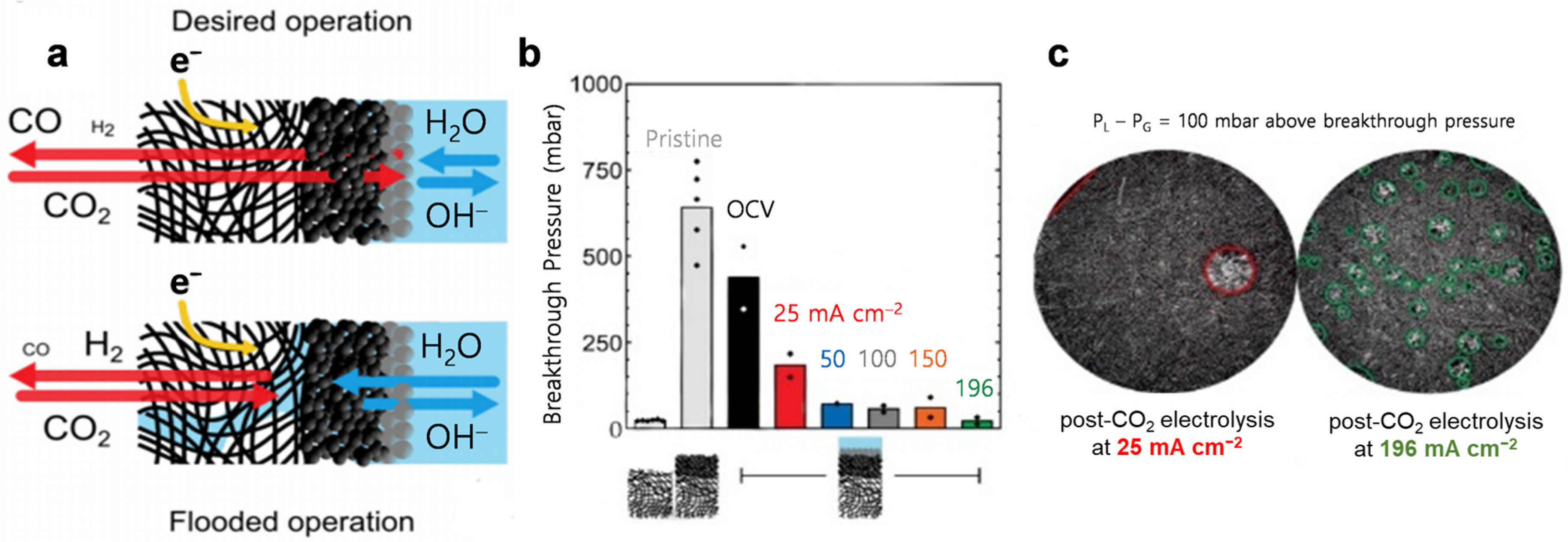
5.4. Electrolyte Flooding
6. Existing Strategies in Overcoming Performance Stability Issues
6.1. Modifying the Electronic Structure
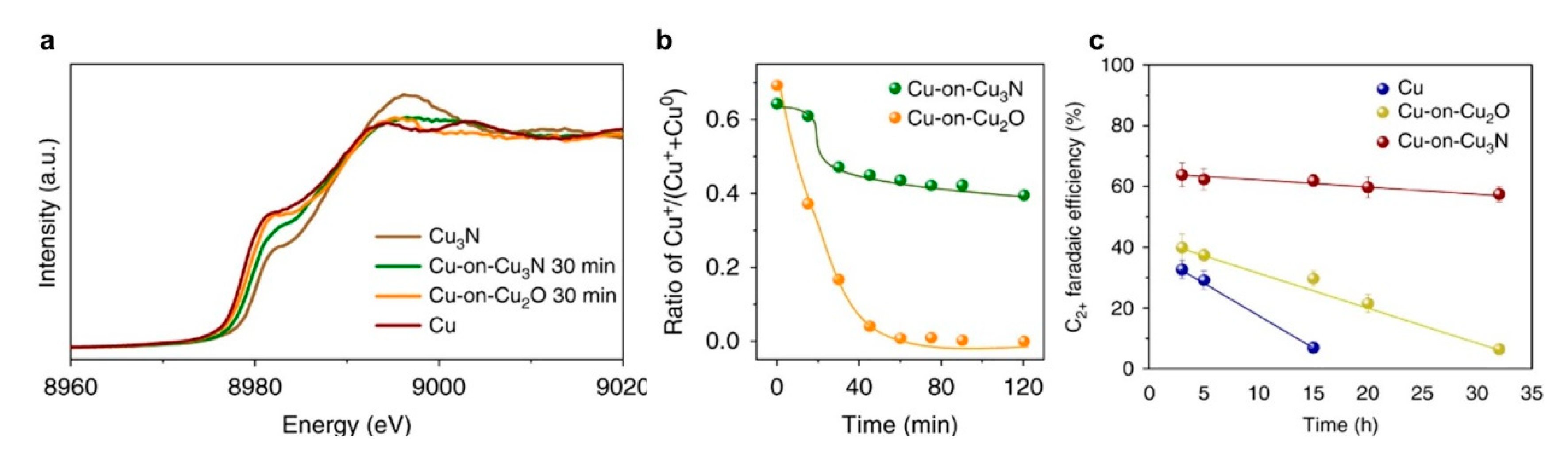
6.2. Utilization of Support Materials
6.3. Single Atom Fixation
6.4. Electrode, Electrolyte and Process Modifications
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gaulin, N.; Le Billon, P. Climate change and fossil fuel production cuts: Assessing global supply-side constraints and policy implications. Clim. Policy 2020, 20, 888–901. [Google Scholar] [CrossRef]
- Rahaman, M.; Dutta, A.; Zanetti, A.; Broekmann, P. Electrochemical Reduction of CO2 into Multicarbon Alcohols on Activated Cu Mesh Catalysts: An Identical Location (IL) Study. ACS Catal. 2017, 7, 7946–7956. [Google Scholar] [CrossRef]
- Lee, C.W.; Hong, J.S.; Yang, K.D.; Jin, K.; Lee, J.H.; Ahn, H.-Y.; Seo, H.; Sung, N.-E.; Nam, K.T. Selective Electrochemical Production of Formate from Carbon Dioxide with Bismuth-Based Catalysts in an Aqueous Electrolyte. ACS Catal. 2018, 8, 931–937. [Google Scholar] [CrossRef]
- Lee, C.W.; Shin, S.-J.; Jung, H.; Nguyen, D.L.T.; Lee, S.Y.; Lee, W.H.; Won, D.H.; Kim, M.G.; Oh, H.-S.; Jang, T.; et al. Metal–Oxide Interfaces for Selective Electrochemical C–C Coupling Reactions. ACS Energy Lett. 2019, 4, 2241–2248. [Google Scholar] [CrossRef]
- Zhao, K.; Nie, X.; Wang, H.; Chen, S.; Quan, X.; Yu, H.; Choi, W.; Zhang, G.; Kim, B.; Chen, J.G. Selective electroreduction of CO2 to acetone by single copper atoms anchored on N-doped porous carbon. Nat. Commun. 2020, 11, 2455. [Google Scholar] [CrossRef]
- Lin, S.-C.; Chang, C.-C.; Chiu, S.-Y.; Pai, H.-T.; Liao, T.-Y.; Hsu, C.-S.; Chiang, W.-H.; Tsai, M.-K.; Chen, H.M. Operando time-resolved X-ray absorption spectroscopy reveals the chemical nature enabling highly selective CO2 reduction. Nat. Commun. 2020, 11, 3525. [Google Scholar] [CrossRef]
- Na, J.; Seo, B.; Kim, J.; Lee, C.W.; Lee, H.; Hwang, Y.J.; Min, B.K.; Lee, D.K.; Oh, H.-S.; Lee, U. General technoeconomic analysis for electrochemical coproduction coupling carbon dioxide reduction with organic oxidation. Nat. Commun. 2019, 10, 5193. [Google Scholar] [CrossRef] [PubMed]
- Bushuyev, O.S.; De Luna, P.; Dinh, C.T.; Tao, L.; Saur, G.; van de Lagemaat, J.; Kelley, S.O.; Sargent, E.H. What Should We Make with CO2 and How Can We Make It? Joule 2018, 2, 825–832. [Google Scholar] [CrossRef]
- Jouny, M.; Luc, W.W.; Jiao, F. General Techno-Economic Analysis of CO2 Electrolysis Systems. Ind. Eng. Chem. Res. 2018, 57, 2165–2177. [Google Scholar] [CrossRef]
- Verma, S.; Kim, B.; Jhong, H.-R.; Ma, S.; Kenis, P.J.A. A Gross-Margin Model for Defining Technoeconomic Benchmarks in the Electroreduction of CO2. ChemSusChem 2016, 9, 1972–1979. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Lu, S.; Kenis, P.J.A. Co-electrolysis of CO2 and glycerol as a pathway to carbon chemicals with improved technoeconomics due to low electricity consumption. Nat. Energy 2019, 4, 466–474. [Google Scholar] [CrossRef]
- Sánchez, O.G.; Birdja, Y.Y.; Bulut, M.; Vaes, J.; Breugelmans, T.; Pant, D. Recent advances in industrial CO2 electroreduction. Curr. Opin. Green Sustain. Chem. 2019, 16, 47–56. [Google Scholar] [CrossRef]
- Kim, K.; Lee, W.H.; Na, J.; Hwang, Y.J.; Oh, H.-S.; Lee, U. Data-driven pilot optimization for electrochemical CO mass production. J. Mater. Chem. A 2020, 8, 16943–16950. [Google Scholar] [CrossRef]
- Liang, S.; Altaf, N.; Huang, L.; Gao, Y.; Wang, Q. Electrolytic cell design for electrochemical CO2 reduction. J. CO2 Util. 2020, 35, 90–105. [Google Scholar] [CrossRef]
- Higgins, D.C.; Hahn, C.; Xiang, C.; Jaramillo, T.F.; Weber, A.Z. Gas-Diffusion Electrodes for Carbon Dioxide Reduction: A New Paradigm. ACS Energy Lett. 2019, 4, 317–324. [Google Scholar] [CrossRef]
- Ren, W.; Zhao, C. Paths towards enhanced electrochemical CO2 reduction. Natl. Sci. Rev. 2020, 7, 7–9. [Google Scholar] [CrossRef]
- Garg, S.; Li, M.; Weber, A.Z.; Ge, L.; Li, L.; Rudolph, V.; Wang, G.; Rufford, T.E. Advances and challenges in electrochemical CO2 reduction processes: An engineering and design perspective looking beyond new catalyst materials. J. Mater. Chem. A 2020, 8, 1511–1544. [Google Scholar] [CrossRef]
- Lin, R.; Guo, J.; Li, X.; Patel, P.; Seifitokaldani, A. Electrochemical Reactors for CO2 Conversion. Catalysts 2020, 10, 473. [Google Scholar] [CrossRef]
- Ramdin, M.; Morrison, A.R.T.; de Groen, M.; van Haperen, R.; de Kler, R.; van den Broeke, L.J.P.; Trusler, J.P.M.; de Jong, W.; Vlugt, T.J.H. High Pressure Electrochemical Reduction of CO2 to Formic Acid/Formate: A Comparison between Bipolar Membranes and Cation Exchange Membranes. Ind. Eng. Chem. Res. 2019, 58, 1834–1847. [Google Scholar] [CrossRef]
- Yan, Z.; Zhu, L.; Li, Y.C.; Wycisk, R.J.; Pintauro, P.N.; Hickner, M.A.; Mallouk, T.E. The balance of electric field and interfacial catalysis in promoting water dissociation in bipolar membranes. Energy Environ. Sci. 2018, 11, 2235–2245. [Google Scholar] [CrossRef]
- Hernández-Pagán, E.A.; Vargas-Barbosa, N.M.; Wang, T.; Zhao, Y.; Smotkin, E.S.; Mallouk, T.E. Resistance and polarization losses in aqueous buffer–membrane electrolytes for water-splitting photoelectrochemical cells. Energy Environ. Sci. 2012, 5, 7582–7589. [Google Scholar] [CrossRef]
- Li, Y.C.; Zhou, D.; Yan, Z.; Gonçalves, R.H.; Salvatore, D.A.; Berlinguette, C.P.; Mallouk, T.E. Electrolysis of CO2 to Syngas in Bipolar Membrane-Based Electrochemical Cells. ACS Energy Lett. 2016, 1, 1149–1153. [Google Scholar] [CrossRef]
- Li, Y.C.; Yan, Z.; Hitt, J.; Wycisk, R.; Pintauro, P.N.; Mallouk, T.E. Bipolar Membranes Inhibit Product Crossover in CO2 Electrolysis Cells. Adv. Sustain. Syst. 2018, 2, 1700187. [Google Scholar] [CrossRef]
- Liu, Z.; Masel, R.I.; Chen, Q.; Kutz, R.; Yang, H.; Lewinski, K.; Kaplun, M.; Luopa, S.; Lutz, D.R. Electrochemical generation of syngas from water and carbon dioxide at industrially important rates. J. CO2 Util. 2016, 15, 50–56. [Google Scholar] [CrossRef]
- Kutz, R.B.; Chen, Q.; Yang, H.; Sajjad, S.D.; Liu, Z.; Masel, I.R. Sustainion Imidazolium-Functionalized Polymers for Carbon Dioxide Electrolysis. Energy Technol. 2017, 5, 929–936. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, H.; Kutz, R.; Masel, R.I. CO2 Electrolysis to CO and O2 at High Selectivity, Stability and Efficiency Using Sustainion Membranes. J. Electrochem. Soc. 2018, 165, J3371–J3377. [Google Scholar] [CrossRef]
- Weng, L.-C.; Bell, A.T.; Weber, A.Z. Towards membrane-electrode assembly systems for CO2 reduction: A modeling study. Energy Environ. Sci. 2019, 12, 1950–1968. [Google Scholar] [CrossRef]
- Spurgeon, J.M.; Kumar, B. A comparative technoeconomic analysis of pathways for commercial electrochemical CO2 reduction to liquid products. Energy Environ. Sci. 2018, 11, 1536–1551. [Google Scholar] [CrossRef]
- Fan, L.; Xia, C.; Zhu, P.; Lu, Y.; Wang, H. Electrochemical CO2 reduction to high-concentration pure formic acid solutions in an all-solid-state reactor. Nat. Commun. 2020, 11, 3633. [Google Scholar] [CrossRef]
- Rumayor, M.; Dominguez-Ramos, A.; Perez, P.; Irabien, A. A techno-economic evaluation approach to the electrochemical reduction of CO2 for formic acid manufacture. J. CO2 Util. 2019, 34, 490–499. [Google Scholar] [CrossRef]
- Salvatore, D.A.; Weekes, D.M.; He, J.; Dettelbach, K.E.; Li, Y.C.; Mallouk, T.E.; Berlinguette, C.P. Electrolysis of Gaseous CO2 to CO in a Flow Cell with a Bipolar Membrane. ACS Energy Lett. 2018, 3, 149–154. [Google Scholar] [CrossRef]
- McDonald, M.B.; Ardo, S.; Lewis, N.S.; Freund, M.S. Use of Bipolar Membranes for Maintaining Steady-State pH Gradients in Membrane-Supported, Solar-Driven Water Splitting. ChemSusChem 2014, 7, 3021–3027. [Google Scholar] [CrossRef]
- Weekes, D.M.; Salvatore, D.A.; Reyes, A.; Huang, A.; Berlinguette, C.P. Electrolytic CO2 Reduction in a Flow Cell. Acc. Chem. Res. 2018, 51, 910–918. [Google Scholar] [CrossRef]
- Yang, H.; Kaczur, J.J.; Sajjad, S.D.; Masel, R.I. Performance and long-term stability of CO2 conversion to formic acid using a three-compartment electrolyzer design. J. CO2 Util. 2020, 42, 101349. [Google Scholar] [CrossRef]
- Lee, J.; Lim, J.; Roh, C.-W.; Whang, H.S.; Lee, H. Electrochemical CO2 reduction using alkaline membrane electrode assembly on various metal electrodes. J. CO2 Util. 2019, 31, 244–250. [Google Scholar] [CrossRef]
- Xiang, H.; Rasul, S.; Hou, B.; Portoles, J.; Cumpson, P.; Yu, E.H. Copper–Indium Binary Catalyst on a Gas Diffusion Electrode for High-Performance CO2 Electrochemical Reduction with Record CO Production Efficiency. ACS Appl. Mater. Interfaces 2020, 12, 601–608. [Google Scholar] [CrossRef]
- Lu, X.; Leung, D.Y.C.; Wang, H.; Maroto-Valer, M.M.; Xuan, J. A pH-differential dual-electrolyte microfluidic electrochemical cells for CO2 utilization. Renew. Energy 2016, 95, 277–285. [Google Scholar] [CrossRef]
- Lv, J.-J.; Jouny, M.; Luc, W.; Zhu, W.; Zhu, J.-J.; Jiao, F. A Highly Porous Copper Electrocatalyst for Carbon Dioxide Reduction. Adv. Mater. 2018, 30, e1803111. [Google Scholar] [CrossRef]
- Lu, X.; Leung, D.Y.; Wang, H.; Xuan, J. A high performance dual electrolyte microfluidic reactor for the utilization of CO2. Appl. Energy 2017, 194, 549–559. [Google Scholar] [CrossRef]
- Lee, W.H.; Ko, Y.-J.; Choi, Y.; Lee, S.Y.; Choi, C.H.; Hwang, Y.J.; Min, B.K.; Strasser, P.; Oh, H.-S. Highly selective and scalable CO2 to CO—Electrolysis using coral-nanostructured Ag catalysts in zero-gap configuration. Nano Energy 2020, 76, 105030. [Google Scholar] [CrossRef]
- Delafontaine, L.; Asset, T.; Atanassov, P. Metal–Nitrogen–Carbon Electrocatalysts for CO2 Reduction towards Syngas Generation. ChemSusChem 2020, 13, 1688–1698. [Google Scholar] [CrossRef]
- Endrődi, B.; Kecsenovity, E.; Samu, A.A.; Darvas, F.; Jones, R.V.; Török, V.; Danyi, A.; Janáky, C. Multilayer Electrolyzer Stack Converts Carbon Dioxide to Gas Products at High Pressure with High Efficiency. ACS Energy Lett. 2019, 4, 1770–1777. [Google Scholar] [CrossRef]
- De Mot, B.; Ramdin, M.; Hereijgers, J.; Vlugt, T.J.H.; Breugelmans, T. Direct Water Injection in Catholyte-Free Zero-Gap Carbon Dioxide Electrolyzers. ChemElectroChem 2020, 7, 3839–3843. [Google Scholar] [CrossRef]
- Ju, H.; Kaur, G.; Kulkarni, A.P.; Giddey, S. Challenges and trends in developing technology for electrochemically reducing CO2 in solid polymer electrolyte membrane reactors. J. CO2 Util. 2019, 32, 178–186. [Google Scholar] [CrossRef]
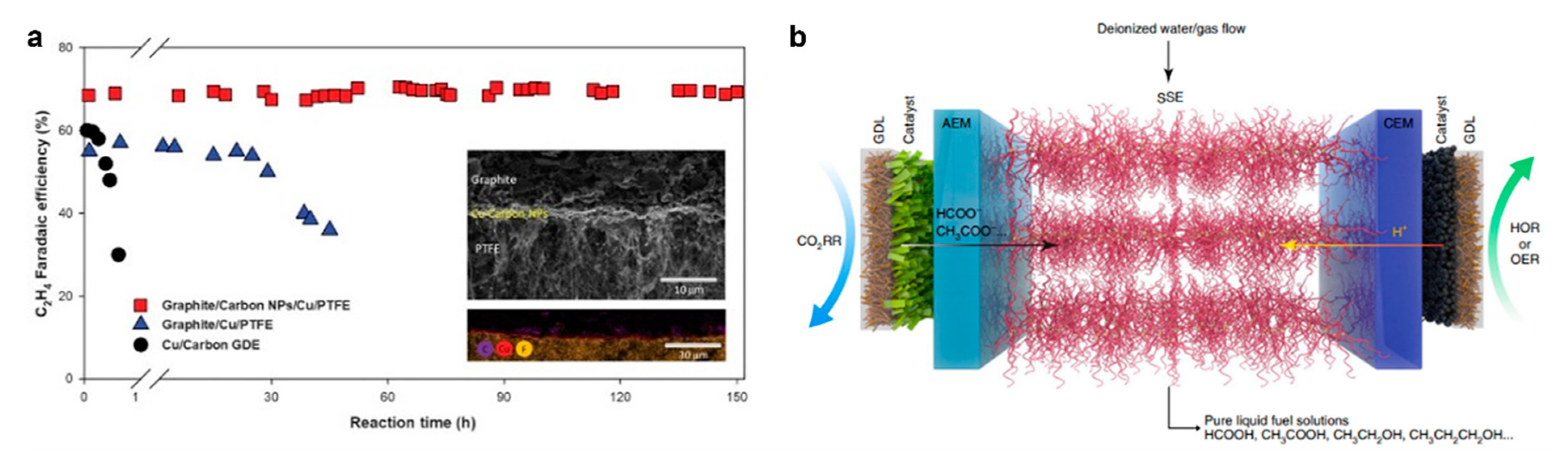
- Xia, C.; Zhu, P.; Jiang, Q.; Pan, Y.; Liang, W.; Stavitski, E.; Alshareef, H.N.; Wang, H. Continuous production of pure liquid fuel solutions via electrocatalytic CO2 reduction using solid-electrolyte devices. Nat. Energy 2019, 4, 776–785. [Google Scholar] [CrossRef]
- IRENA. Renewable Power Generation Costs in 2019; IRENA: Abu Dhabi, United Arab Emirates, 2020. [Google Scholar]
- Song, Y.; Zhang, X.; Xie, K.; Wang, G.; Bao, X. High-Temperature CO2 Electrolysis in Solid Oxide Electrolysis Cells: Developments, Challenges, and Prospects. Adv. Mater. 2019, 31, e1902033. [Google Scholar] [CrossRef]
- Mohammadi, A.; Mehrpooya, M. A comprehensive review on coupling different types of electrolyzer to renewable energy sources. Energy 2018, 158, 632–655. [Google Scholar] [CrossRef]
- Cheng, W.-H.; Richter, M.H.; Sullivan, I.; Larson, D.M.; Xiang, C.; Brunschwig, B.S.; Atwater, H.A. CO2 Reduction to CO with 19% Efficiency in a Solar-Driven Gas Diffusion Electrode Flow Cell under Outdoor Solar Illumination. ACS Energy Lett. 2020, 5, 470–476. [Google Scholar] [CrossRef]
- Chatterjee, S.; Griego, C.; Hart, J.L.; Li, Y.; Taheri, M.L.; Keith, J.A.; Snyder, J.D. Free Standing Nanoporous Palladium Alloys as CO Poisoning Tolerant Electrocatalysts for the Electrochemical Reduction of CO2 to Formate. ACS Catal. 2019, 9, 5290–5301. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, R.; Zhu, X.; Han, N.; Song, B.; Liu, T.; Hu, G.; Li, Y.; Lu, J.; Li, Y. Mesoporous PdAg Nanospheres for Stable Electrochemical CO2 Reduction to Formate. Adv. Mater. 2020, 32, e2000992. [Google Scholar] [CrossRef]
- Pavesi, D.; Ali, F.S.M.; Anastasiadou, D.; Kallio, T.; Figueiredo, M.C.; Gruter, G.-J.M.; Koper, M.T.M.; Schouten, K.J.P. CO2 electroreduction on bimetallic Pd–In nanoparticles. Catal. Sci. Technol. 2020, 10, 4264–4270. [Google Scholar] [CrossRef]
- Weng, Z.; Zhang, X.; Wu, Y.; Huo, S.; Jiang, J.; Liu, W.; He, G.; Liang, Y.; Wang, H. Self-Cleaning Catalyst Electrodes for Stabilized CO2 Reduction to Hydrocarbons. Angew. Chem. Int. Ed. 2017, 56, 13135–13139. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, L.; Libretto, N.J.; Li, X.; Li, C.; Wang, Y.; He, C.; Lee, J.; Gregg, J.; Zong, H.; et al. Ensemble Effect in Bimetallic Electrocatalysts for CO2 Reduction. J. Am. Chem. Soc. 2019, 141, 16635–16642. [Google Scholar] [CrossRef]
- Dutta, A.; Morstein, C.E.; Rahaman, M.; López, A.C.; Broekmann, P. Beyond Copper in CO2 Electrolysis: Effective Hydrocarbon Production on Silver-Nanofoam Catalysts. ACS Catal. 2018, 8, 8357–8368. [Google Scholar] [CrossRef]
- Akhade, S.A.; Luo, W.; Nie, X.; Bernstein, N.J.; Asthagiri, A.; Janik, M.J. Poisoning effect of adsorbed CO during CO2 electroreduction on late transition metals. Phys. Chem. Chem. Phys. 2014, 16, 20429–20435. [Google Scholar] [CrossRef]
- Koper, M.T.; Van Santen, R.R. Electric field effects on CO and NO adsorption at the Pt(111) surface. J. Electroanal. Chem. 1999, 476, 64–70. [Google Scholar] [CrossRef]
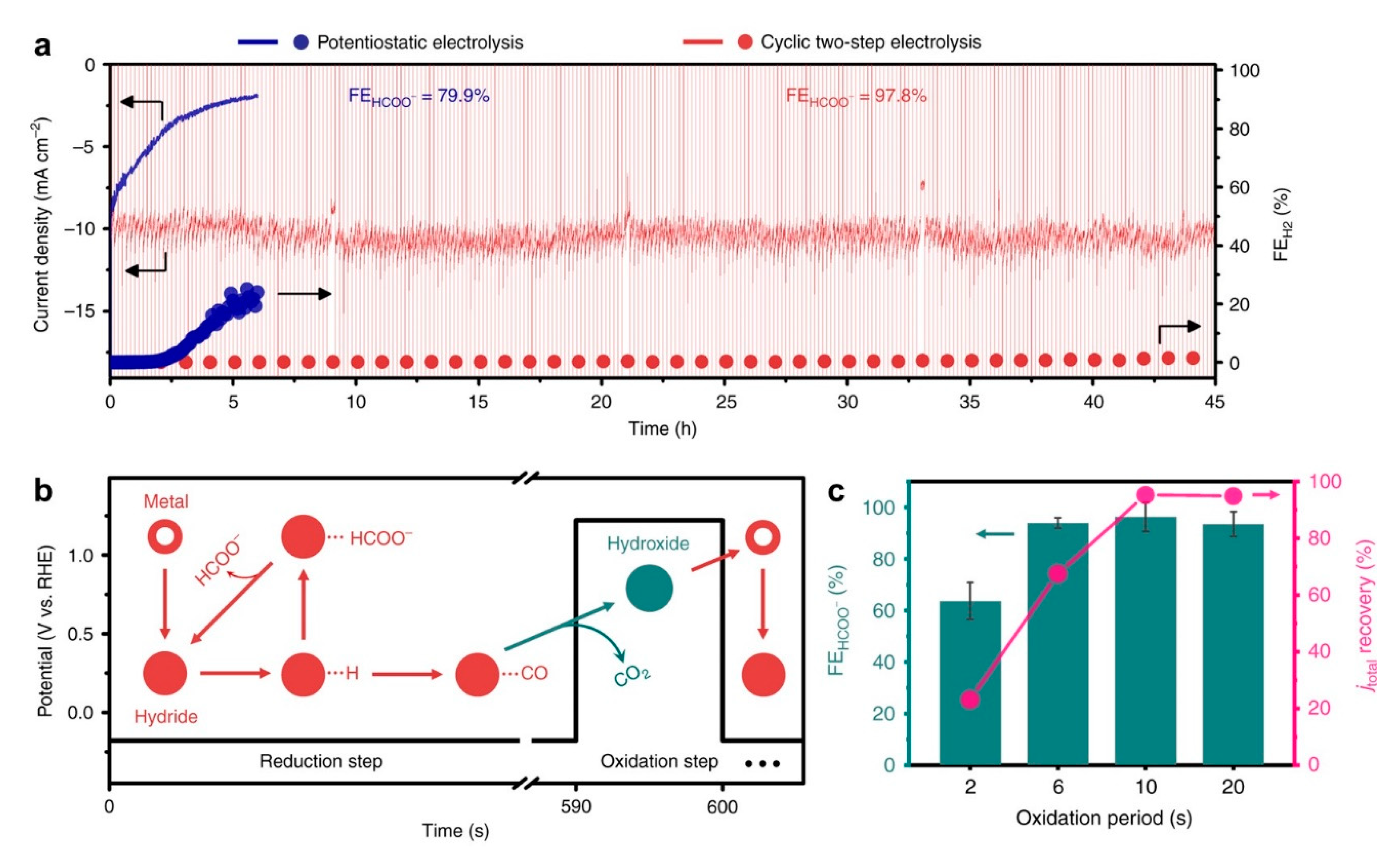
- Lee, C.W.; Cho, N.H.; Nam, K.T.; Hwang, Y.J.; Min, B.K. Cyclic two-step electrolysis for stable electrochemical conversion of carbon dioxide to formate. Nat. Commun. 2019, 10, 3919. [Google Scholar] [CrossRef]
- Jiang, S.; Klingan, K.; Pasquini, C.; Dau, H. New aspects of operando Raman spectroscopy applied to electrochemical CO2 reduction on Cu foams. J. Chem. Phys. 2019, 150, 041718. [Google Scholar] [CrossRef]
- Hori, Y.; Konishi, H.; Futamura, T.; Murata, A.; Koga, O.; Sakurai, H.; Oguma, K. “Deactivation of copper electrode” in electrochemical reduction of CO2. Electrochim. Acta 2005, 50, 5354–5369. [Google Scholar] [CrossRef]
- Lucas, F.W.S.; Lima, F.H.B. Electrodeposited Tin-Antimony Alloys as Novel Electrocatalysts for Selective and Stable Carbon Dioxide Reduction to Formate. ChemElectroChem 2020, 7, 3733–3742. [Google Scholar] [CrossRef]
- Lum, Y.; Kwon, Y.; Lobaccaro, P.; Chen, L.; Clark, E.L.; Bell, A.T.; Ager, J.W. Trace Levels of Copper in Carbon Materials Show Significant Electrochemical CO2 Reduction Activity. ACS Catal. 2016, 6, 202–209. [Google Scholar] [CrossRef]
- Bai, X.; Chen, W.; Zhao, C.; Li, S.; Song, Y.; Ge, R.; Wei, W.; Sun, Y. Exclusive Formation of Formic Acid from CO2 Electroreduction by a Tunable Pd-Sn Alloy. Angew. Chem. 2017, 129, 12387–12391. [Google Scholar] [CrossRef]
- Choi, W.; Won, D.H.; Hwang, Y.J. Catalyst design strategies for stable electrochemical CO2 reduction reaction. J. Mater. Chem. A 2020, 8, 15341–15357. [Google Scholar] [CrossRef]
- Won, D.H.; Shin, H.; Chung, M.W.; Jung, H.; Chae, K.H.; Oh, H.-S.; Hwang, Y.J.; Min, B.K. Achieving tolerant CO2 electro-reduction catalyst in real water matrix. Appl. Catal. B Environ. 2019, 258, 117961. [Google Scholar] [CrossRef]
- Yoo, C.J.; Dong, W.J.; Park, J.Y.; Lim, J.W.; Kim, S.; Choi, K.S.; Ngome, F.O.O.; Choi, S.-Y.; Lee, J.-L. Compositional and Geometrical Effects of Bimetallic Cu–Sn Catalysts on Selective Electrochemical CO2 Reduction to CO. ACS Appl. Energy Mater. 2020, 3, 4466–4473. [Google Scholar] [CrossRef]
- Alfath, M.; Lee, C.W. Recent Advances in the Catalyst Design and Mass Transport Control for the Electrochemical Reduction of Carbon Dioxide to Formate. Catalysts 2020, 10, 859. [Google Scholar] [CrossRef]
- Ouyang, R.; Liu, J.-X.; Li, W.-X. Atomistic Theory of Ostwald Ripening and Disintegration of Supported Metal Particles under Reaction Conditions. J. Am. Chem. Soc. 2013, 135, 1760–1771. [Google Scholar] [CrossRef]
- Zhang, A.; Liang, Y.; Li, H.; Zhang, B.; Liu, Z.; Chang, Q.; Zhang, H.; Zhu, C.-F.; Geng, Z.; Zhu, W.; et al. In-Situ Surface Reconstruction of InN Nanosheets for Efficient CO2 Electroreduction into Formate. Nano Lett. 2020, 20, 8229–8235. [Google Scholar] [CrossRef]
- Zhong, H.; Qiu, Y.; Li, X.; Pan, L.; Zhang, H. Ordered cone-structured tin directly grown on carbon paper as efficient electrocatalyst for CO2 electrochemical reduction to formate. J. Energy Chem. 2021, 55, 236–243. [Google Scholar] [CrossRef]
- Pan, F.; Yang, Y. Designing CO2 reduction electrode materials by morphology and interface engineering. Energy Environ. Sci. 2020, 13, 2275–2309. [Google Scholar] [CrossRef]
- Dutta, A.; Montiel, I.Z.; Erni, R.; Kiran, K.; Rahaman, M.; Drnec, J.; Broekmann, P. Activation of bimetallic AgCu foam electrocatalysts for ethanol formation from CO2 by selective Cu oxidation/reduction. Nano Energy 2020, 68, 104331. [Google Scholar] [CrossRef]
- Yang, H.; Huang, Y.; Deng, J.; Wu, Y.; Han, N.; Zha, C.; Li, L.; Li, Y. Selective electrocatalytic CO2 reduction enabled by SnO2 nanoclusters. J. Energy Chem. 2019, 37, 93–96. [Google Scholar] [CrossRef]
- Huang, J.; Hörmann, N.; Oveisi, E.; Loiudice, A.; De Gregorio, G.L.; Andreussi, O.; Marzari, N.; Buonsanti, R. Potential-induced nanoclustering of metallic catalysts during electrochemical CO2 reduction. Nat. Commun. 2018, 9, 3117. [Google Scholar] [CrossRef]
- Duan, Y.-X.; Meng, F.-L.; Liu, K.-H.; Yi, S.-S.; Li, S.-J.; Yan, J.-M.; Jiang, Q. Amorphizing of Cu Nanoparticles toward Highly Efficient and Robust Electrocatalyst for CO2 Reduction to Liquid Fuels with High Faradaic Efficiencies. Adv. Mater. 2018, 30, e1706194. [Google Scholar] [CrossRef] [PubMed]
- Matsubu, J.C.; Yang, V.N.; Christopher, P. Isolated Metal Active Site Concentration and Stability Control Catalytic CO2 Reduction Selectivity. J. Am. Chem. Soc. 2015, 137, 3076–3084. [Google Scholar] [CrossRef] [PubMed]
- Spoeri, C.; Kwan, J.T.H.; Bonakdarpour, A.; Wilkinson, D.P.; Strasser, P. The Stability Challenges of Oxygen Evolving Catalysts: Towards a Common Fundamental Understanding and Mitigation of Catalyst Degradation. Angew. Chem. Int. Ed. 2017, 56, 5994–6021. [Google Scholar] [CrossRef] [PubMed]
- De Luna, P.; Quintero-Bermudez, R.; Dinh, C.-T.; Ross, M.B.; Bushuyev, O.S.; Todorović, P.; Regier, T.; Kelley, S.O.; Yang, P.; Sargent, E.H. Catalyst electro-redeposition controls morphology and oxidation state for selective carbon dioxide reduction. Nat. Catal. 2018, 1, 103–110. [Google Scholar] [CrossRef]
- Andrews, E.M.; Flake, J.; Fang, Y. CO2 Electrocatalytic Reduction at Gold and Copper Electrodes: Role of Particle Size and Surface Chemistry. ECS Trans. 2015, 66, 67–70. [Google Scholar] [CrossRef]
- Kauffman, D.R.; Thakkar, J.; Siva, R.; Matranga, C.; Ohodnicki, P.R.; Zeng, C.; Jin, R. Efficient Electrochemical CO2 Conversion Powered by Renewable Energy. ACS Appl. Mater. Interfaces 2015, 7, 15626–15632. [Google Scholar] [CrossRef]
- Mistry, H.; Reske, R.; Zeng, Z.; Zhao, Z.-J.; Greeley, J.; Strasser, P.; Cuenya, B.R. Exceptional Size-Dependent Activity Enhancement in the Electroreduction of CO2 over Au Nanoparticles. J. Am. Chem. Soc. 2014, 136, 16473–16476. [Google Scholar] [CrossRef]
- Li, X.; Wang, S.; Li, L.; Sun, Y.; Xie, Y. Progress and Perspective for In Situ Studies of CO2 Reduction. J. Am. Chem. Soc. 2020, 142, 9567–9581. [Google Scholar] [CrossRef] [PubMed]
- Fiordaliso, E.M.; Sharafutdinov, I.; Carvalho, H.W.P.; Grunwaldt, J.-D.; Hansen, T.W.; Chorkendorff, I.; Wagner, J.B.; Damsgaard, C.D. Intermetallic GaPd2 Nanoparticles on SiO2 for Low-Pressure CO2 Hydrogenation to Methanol: Catalytic Performance and In Situ Characterization. ACS Catal. 2015, 5, 5827–5836. [Google Scholar] [CrossRef]
- Luc, W.; Collins, C.; Wang, S.; Xin, H.; He, K.; Kang, Y.; Jiao, F. Ag-Sn bimetallic catalyst with a core-shell structure for CO2 reduction. J. Am. Chem. Soc. 2017, 139, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Varela, A.S.; Bonifacio, C.S.; Zegkinoglou, I.; Sinev, I.; Choi, Y.W.; Kisslinger, K.; Stach, E.A.; Yang, J.C.; Strasser, P.; et al. Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat. Commun. 2016, 7, 12123. [Google Scholar] [CrossRef]
- Gao, D.; Zegkinoglou, I.; Divins, N.J.; Scholten, F.; Sinev, I.; Grosse, P.; Cuenya, B.R. Plasma-Activated Copper Nanocube Catalysts for Efficient Carbon Dioxide Electroreduction to Hydrocarbons and Alcohols. ACS Nano 2017, 11, 4825–4831. [Google Scholar] [CrossRef] [PubMed]
- Armbrüster, M.; Schlögl, R.; Grin, Y. Intermetallic compounds in heterogeneous catalysis—A quickly developing field. Sci. Technol. Adv. Mater. 2014, 15, 034803. [Google Scholar] [CrossRef] [PubMed]
- Varela, A.S.; Schlaup, C.; Jovanov, Z.P.; Malacrida, P.; Horch, S.; Stephens, I.E.L.; Chorkendorff, I. CO2 Electroreduction on Well-Defined Bimetallic Surfaces: Cu Overlayers on Pt(111) and Pt(211). J. Phys. Chem. C 2013, 117, 20500–20508. [Google Scholar] [CrossRef]
- Jovanov, Z.P.; Hansen, H.A.; Varela, A.S.; Malacrida, P.; Peterson, A.A.; Nørskov, J.K.; Stephens, I.E.; Chorkendorff, I. Opportunities and challenges in the electrocatalysis of CO2 and CO reduction using bifunctional surfaces: A theoretical and experimental study of Au–Cd alloys. J. Catal. 2016, 343, 215–231. [Google Scholar] [CrossRef]
- Clark, E.L.; Hahn, C.; Jaramillo, T.F.; Bell, A.T. Electrochemical CO2 Reduction over Compressively Strained CuAg Surface Alloys with Enhanced Multi-Carbon Oxygenate Selectivity. J. Am. Chem. Soc. 2017, 139, 15848–15857. [Google Scholar] [CrossRef]
- Peterson, A.A.; Nørskov, J.K. Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. [Google Scholar] [CrossRef]
- Leonard, M.E.; Orella, M.J.; Aiello, N.; Román-Leshkov, Y.; Forner-Cuenca, A.; Brushett, F.R. Editors’ Choice—Flooded by Success: On the Role of Electrode Wettability in CO2 Electrolyzers that Generate Liquid Products. J. Electrochem. Soc. 2020, 167, 124521. [Google Scholar] [CrossRef]
- Leonard, M.E.; Clarke, L.E.; Forner-Cuenca, A.; Brown, S.M.; Brushett, F.R. Investigating Electrode Flooding in a Flowing Electrolyte, Gas-Fed Carbon Dioxide Electrolyzer. ChemSusChem 2020, 13, 400–411. [Google Scholar] [CrossRef]
- Dinh, C.-T.; Burdyny, T.; Kibria, G.; Seifitokaldani, A.; Gabardo, C.M.; De Arquer, F.P.G.; Kiani, A.; Edwards, J.P.; De Luna, P.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef]
- Burchardt, T. An evaluation of electrocatalytic activity and stability for air electrodes. J. Power Sources 2004, 135, 192–197. [Google Scholar] [CrossRef]
- Bidault, F.; Brett, D.; Middleton, P.; Brandon, N. Review of gas diffusion cathodes for alkaline fuel cells. J. Power Sources 2009, 187, 39–48. [Google Scholar] [CrossRef]
- Jeanty, P.; Scherer, C.; Magori, E.; Wiesner-Fleischer, K.; Hinrichsen, O.; Fleischer, M. Upscaling and continuous operation of electrochemical CO2 to CO conversion in aqueous solutions on silver gas diffusion electrodes. J. CO2 Util. 2018, 24, 454–462. [Google Scholar] [CrossRef]
- Albo, J.; Perfecto-Irigaray, M.; Beobide, G.; Irabien, A. Cu/Bi metal-organic framework-based systems for an enhanced electrochemical transformation of CO2 to alcohols. J. CO2 Util. 2019, 33, 157–165. [Google Scholar] [CrossRef]
- Bienen, F.; Kopljar, D.; Löwe, A.; Geiger, S.; Wagner, N.; Klemm, E.; Friedrich, K.A. Revealing Mechanistic Processes in Gas-Diffusion Electrodes During CO2 Reduction via Impedance Spectroscopy. ACS Sustain. Chem. Eng. 2020, 8, 13759–13768. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, S.-X.; Gandionco, K.A.; Bond, A.M.; Zhang, J. Electrocatalytic carbon dioxide reduction: From fundamental principles to catalyst design. Mater. Today Adv. 2020, 7, 100074. [Google Scholar] [CrossRef]
- Karamad, M.; Tripkovic, V.; Rossmeisl, J. Intermetallic Alloys as CO Electroreduction Catalysts—Role of Isolated Active Sites. ACS Catal. 2014, 4, 2268–2273. [Google Scholar] [CrossRef]
- Gu, J.; Hsu, C.-S.; Bai, L.; Chen, H.M.; Hu, X. Atomically dispersed Fe3+ sites catalyze efficient CO2 electroreduction to CO. Science 2019, 364, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Bellini, M.; Folliero, M.G.; Evangelisti, C.; He, Q.; Hu, Y.; Pagliaro, M.V.; Oberhauser, W.; Marchionni, A.; Filippi, J.; Miller, H.A.; et al. A Gold–Palladium Nanoparticle Alloy Catalyst for CO Production from CO2 Electroreduction. Energy Technol. 2019, 7, 1800859. [Google Scholar] [CrossRef]
- Ma, X.; Li, Z.; Achenie, L.E.K.; Xin, H. Machine-Learning-Augmented Chemisorption Model for CO2 Electroreduction Catalyst Screening. J. Phys. Chem. Lett. 2015, 6, 3528–3533. [Google Scholar] [CrossRef]
- Zheng, X.; Ji, Y.; Tang, J.; Wang, J.; Liu, B.; Steinrück, H.-G.; Lim, K.; Li, Y.; Toney, M.F.; Chan, K.; et al. Theory-guided Sn/Cu alloying for efficient CO2 electroreduction at low overpotentials. Nat. Catal. 2019, 2, 55–61. [Google Scholar] [CrossRef]
- Bernal, M.; Bagger, A.; Scholten, F.; Sinev, I.; Bergmann, A.; Ahmadi, M.; Rossmeisl, J.; Cuenya, B.R. CO2 electroreduction on copper-cobalt nanoparticles: Size and composition effect. Nano Energy 2018, 53, 27–36. [Google Scholar] [CrossRef]
- Zhu, W.; Tackett, B.M.; Chen, J.G.; Jiao, F. Bimetallic Electrocatalysts for CO2 Reduction. Top. Curr. Chem. 2018, 376, 105–125. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jung, H.; Kim, N.-K.; Oh, H.-S.; Min, B.K.; Hwang, Y.J. Mixed Copper States in Anodized Cu Electrocatalyst for Stable and Selective Ethylene Production from CO2 Reduction. J. Am. Chem. Soc. 2018, 140, 8681–8689. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Thoi, V.S. Non-Innocent Role of Porous Carbon Towards Enhancing C2-3 Products in Electroreduction of Carbon Dioxide. ACS Appl. Mater. Interfaces 2020, 12, 45929–45935. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, X.; Zhou, X.; Li, Q.; Wang, H.; Yi, J.; Liu, Y.; Zhang, J. Simply and effectively electrodepositing Bi-MWCNT-COOH composite on Cu electrode for efficient electrocatalytic CO2 reduction to produce HCOOH. J. CO2 Util. 2020, 37, 106–112. [Google Scholar] [CrossRef]
- Liu, T.; Ali, S.; Lian, Z.; Li, B.; Su, D.S. CO2 electoreduction reaction on heteroatom-doped carbon cathode materials. J. Mater. Chem. A 2017, 5, 21596–21603. [Google Scholar] [CrossRef]
- Vasileff, A.; Zheng, Y.; Qiao, S.Z. Carbon Solving Carbon’s Problems: Recent Progress of Nanostructured Carbon-Based Catalysts for the Electrochemical Reduction of CO2. Adv. Energy Mater. 2017, 7, 1700759. [Google Scholar] [CrossRef]
- Klinkova, A.; De Luna, P.; Dinh, C.-T.; Voznyy, O.; Larin, E.M.; Kumacheva, E.; Sargent, E.H. Rational Design of Efficient Palladium Catalysts for Electroreduction of Carbon Dioxide to Formate. ACS Catal. 2016, 6, 8115–8120. [Google Scholar] [CrossRef]
- Malhotra, M.; Sharma, R.; Sanduja, M.; Kumar, R.; Jain, J.; Deep, A. Synthesis, characterization and evaluation of mannich bases as potent antifungal and hydrogen peroxide scavenging agents. Acta Pol. Pharm. Drug Res. 2012, 69, 355–361. [Google Scholar]
- Hegenberger, E.; Wu, N.L.; Phillips, J. Evidence of strong interaction between iron particles and an activated carbon support. J. Phys. Chem. 1987, 91, 5067–5071. [Google Scholar] [CrossRef]
- Sharma, S.; Pollet, B.G. Support materials for PEMFC and DMFC electrocatalysts—A review. J. Power Sources 2012, 208, 96–119. [Google Scholar] [CrossRef]
- Bittencourt, C.; Hecq, M.; Felten, A.; Pireaux, J.J.; Ghijsen, J.; Felicissimo, M.P.; Rudolf, P.; Drube, W.; Ke, X.; Van Tendeloo, G. Platinum-carbon nanotube interaction. Chem. Phys. Lett. 2008, 462, 260–264. [Google Scholar] [CrossRef]
- Yuan, J.; Yang, M.-P.; Zhi, W.-Y.; Wang, H.; Wang, H.; Lu, J.-X. Efficient electrochemical reduction of CO2 to ethanol on Cu nanoparticles decorated on N-doped graphene oxide catalysts. J. CO2 Util. 2019, 33, 452–460. [Google Scholar] [CrossRef]
- Liang, Z.-Q.; Zhuang, T.-T.; Seifitokaldani, A.; Li, J.; Huang, C.-W.; Tan, C.-S.; Li, Y.; De Luna, P.; Dinh, C.T.; Hu, Y.; et al. Copper-on-nitride enhances the stable electrosynthesis of multi-carbon products from CO2. Nat. Commun. 2018, 9, 3828. [Google Scholar] [CrossRef] [PubMed]
- Lewera, A.; Timperman, L.; Roguska, A.; Alonso-Vante, N. Metal–Support Interactions between Nanosized Pt and Metal Oxides (WO3 and TiO2) Studied Using X-ray Photoelectron Spectroscopy. J. Phys. Chem. C 2011, 115, 20153–20159. [Google Scholar] [CrossRef]
- Wen, G.; Ren, B.; Park, M.G.; Yang, J.; Dou, H.; Zhang, Z.; Deng, Y.; Bai, Z.; Yang, L.; Gostick, J.; et al. Ternary Sn-Ti-O Electrocatalyst Boosts the Stability and Energy Efficiency of CO2 Reduction. Angew. Chem. Int. Ed. 2020, 59, 12860–12867. [Google Scholar] [CrossRef]
- Pan, Y.; Lin, R.; Chen, Y.; Liu, S.; Zhu, W.; Cao, X.; Chen, W.; Wu, K.; Cheong, W.-C.; Wang, Y.; et al. Design of Single-Atom Co–N5 Catalytic Site: A Robust Electrocatalyst for CO2 Reduction with Nearly 100% CO Selectivity and Remarkable Stability. J. Am. Chem. Soc. 2018, 140, 4218–4221. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.N.; Oh, S.; Kaminsky, C.J.; Chu, S.B.; Zhang, G.; Miller, J.T.; Surendranath, Y. Strong Electronic Coupling of Molecular Sites to Graphitic Electrodes via Pyrazine Conjugation. J. Am. Chem. Soc. 2018, 140, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
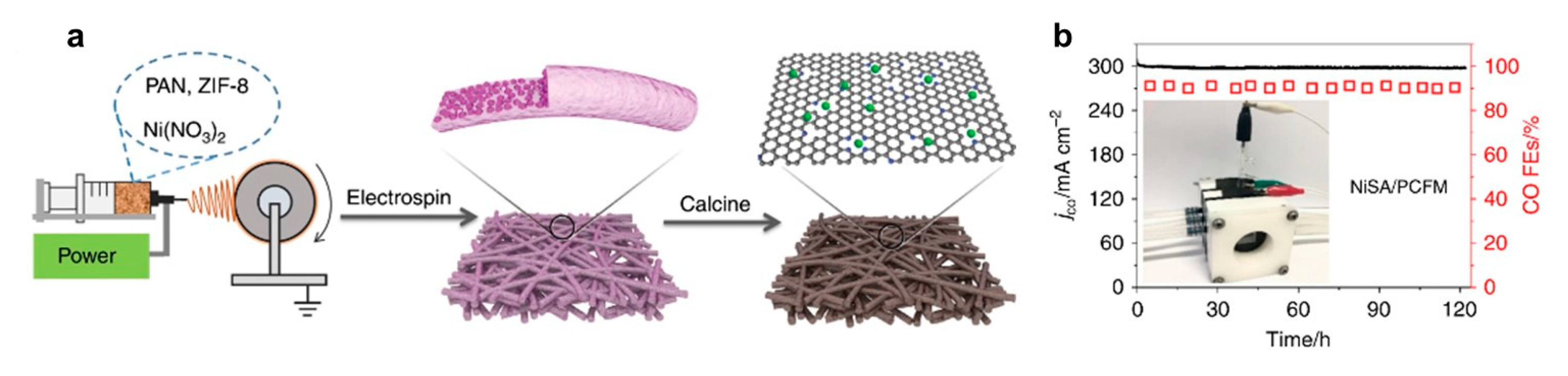
- Yang, H.; Lin, Q.; Zhang, C.; Yu, X.; Cheng, Z.; Li, G.; Hu, Q.; Ren, X.; Zhang, Q.; Liu, J.; et al. Carbon dioxide electroreduction on single-atom nickel decorated carbon membranes with industry compatible current densities. Nat. Commun. 2020, 11, 593. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Li, J.-X.; Ding, L.-L.; Liu, L.; Wang, S.-M.; Han, Z.-B. Palladium Nanoparticles Encapsulated in the MIL-101-Catalyzed One-Pot Reaction of Alcohol Oxidation and Aldimine Condensation. Inorg. Chem. 2018, 57, 13586–13593. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Tamura, S.; Yamanaka, S.; Niitsuma, Y.; Sone, Y.; Umeda, M. Minimization of Pt-electrocatalyst deactivation in CO2 reduction using a polymer electrolyte cell. React. Chem. Eng. 2020, 5, 1064–1070. [Google Scholar] [CrossRef]
- Lu, X.; Yu, T.; Wang, H.; Qian, L.; Lei, P. Electrochemical Fabrication and Reactivation of Nanoporous Gold with Abundant Surface Steps for CO2 Reduction. ACS Catal. 2020, 10, 8860–8869. [Google Scholar] [CrossRef]
- Hou, Y.; Bolat, S.; Bornet, A.; Romanyuk, Y.E.; Guo, H.; Moreno-García, P.; Montiel, I.Z.; Lai, Z.; Müller, U.; Grozovski, V.; et al. Photonic Curing: Activation and Stabilization of Metal Membrane Catalysts (MMCs) for the Electrochemical Reduction of CO2. ACS Catal. 2019, 9, 9518–9529. [Google Scholar] [CrossRef]

















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Wijaya, D.T.; Na, J.; Lee, C.W. Towards the Large-Scale Electrochemical Reduction of Carbon Dioxide. Catalysts 2021, 11, 253. https://doi.org/10.3390/catal11020253
Park S, Wijaya DT, Na J, Lee CW. Towards the Large-Scale Electrochemical Reduction of Carbon Dioxide. Catalysts. 2021; 11(2):253. https://doi.org/10.3390/catal11020253
Chicago/Turabian StylePark, Subin, Devina Thasia Wijaya, Jonggeol Na, and Chan Woo Lee. 2021. "Towards the Large-Scale Electrochemical Reduction of Carbon Dioxide" Catalysts 11, no. 2: 253. https://doi.org/10.3390/catal11020253
APA StylePark, S., Wijaya, D. T., Na, J., & Lee, C. W. (2021). Towards the Large-Scale Electrochemical Reduction of Carbon Dioxide. Catalysts, 11(2), 253. https://doi.org/10.3390/catal11020253