
A Stereoselective, Multicomponent Catalytic Carbonylative Approach to a New Class of α,β-Unsaturated γ-Lactam Derivatives

 ,
, 
 ,
,  , ,
, ,  ,
, 
Abstract
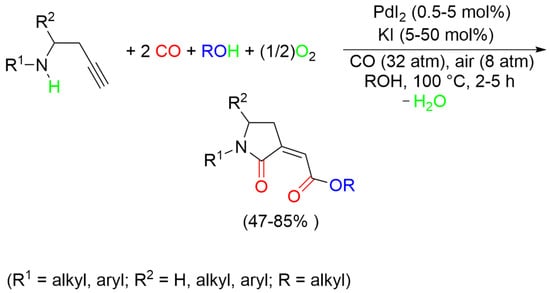
1. Introduction
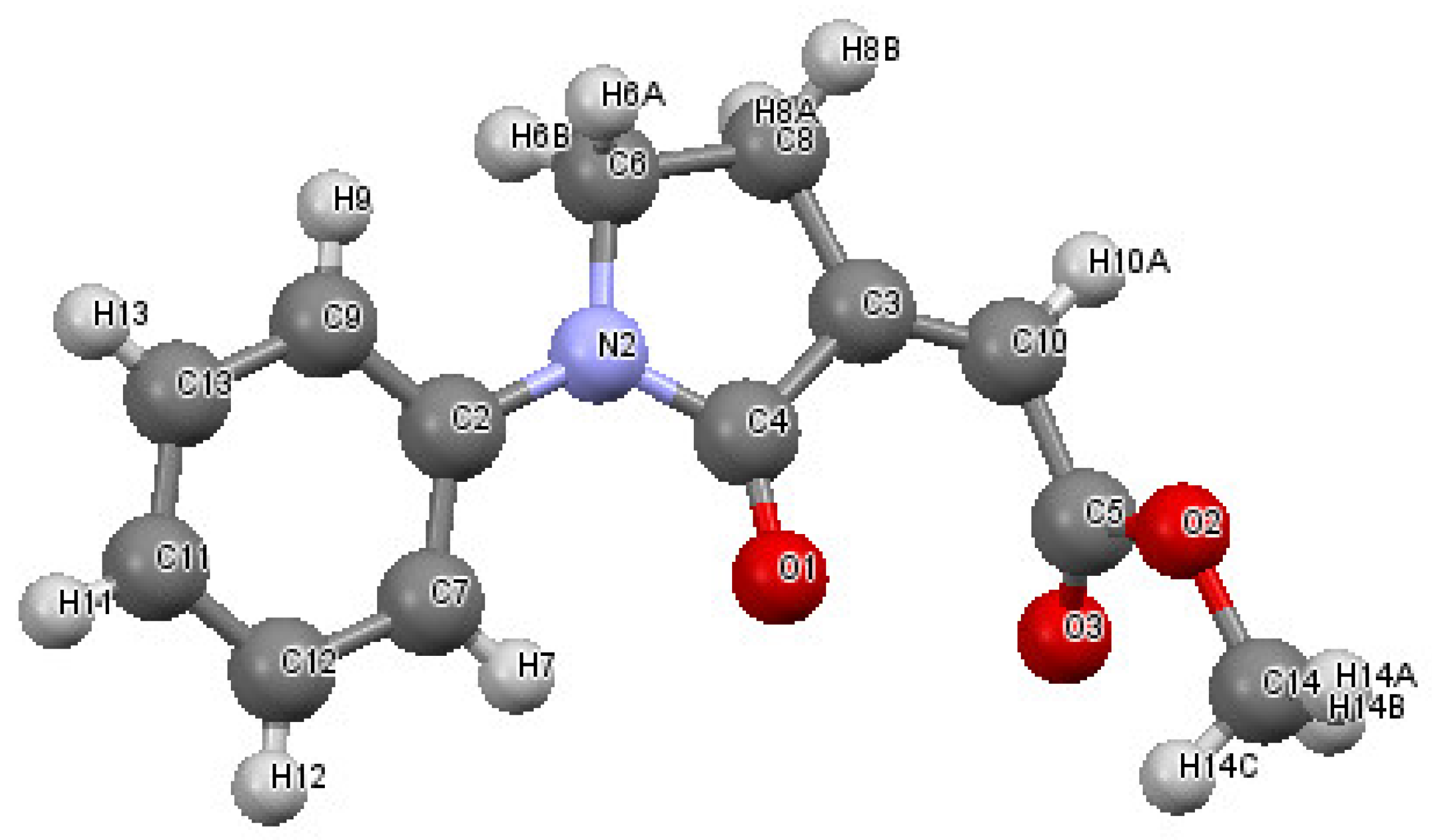
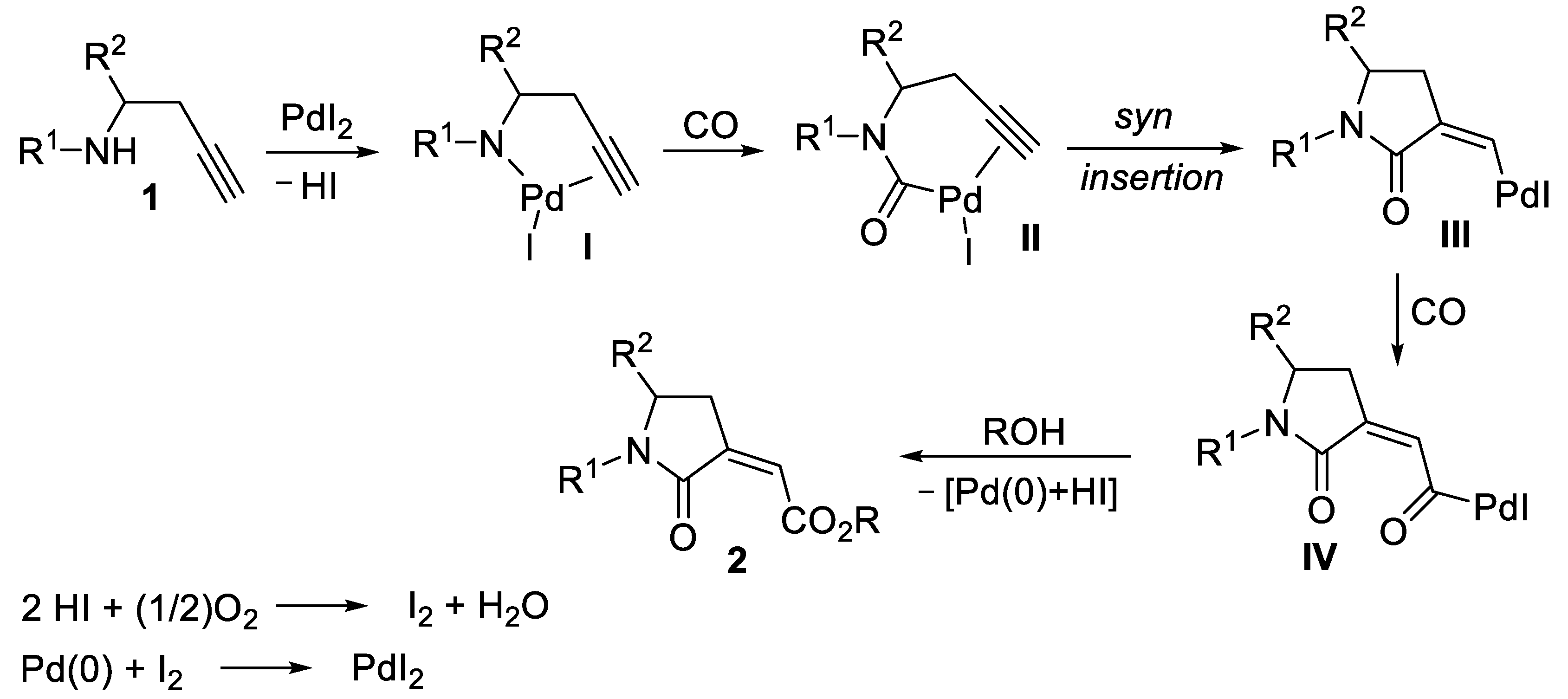
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Methods
3.2. Preparation of Substrates 1a–g and 1l–n
3.2.1. N-(But-3-yn-1-yl)aniline 1a
3.2.2. N-(But-3-yn-1-yl)-4-Chloroaniline 1b
3.2.3. N-(But-3-yn-1-yl)-4-Isopropylaniline 1c
3.2.4. N-(But-3-yn-1-yl)-4-(tert-Butyl)aniline 1d
3.2.5. 3-Bromo-N-(but-3-yn-1-yl)-5-Methylaniline 1e
3.2.6. N-(Pent-4-yn-2-yl)aniline 1f
3.2.7. N-(Hex-5-yn-3-yl)aniline 1g
3.2.8. N-Benzylbut-3-yn-1-Amine 1l
3.2.9. N-(1-Phenylethyl)but-3-yn-1-Amine 1m
3.2.10. N-tert-Butylbut-3-yn-1-Amine 1n
3.3. Preparation of Substrates 1h–k
3.3.1. N-(1-Phenylbut-3-yn-1-yl)aniline 1h
3.3.2. N-(1-(4-Bromophenyl)but-3-yn-1-yl)aniline 1i
3.3.3. N-(1-(4-Bromophenyl)but-3-yn-1-yl)aniline 1j
3.3.4. 4-Chloro-N-(1-(4-Methoxyphenyl)but-3-yn-1-yl)aniline 1k
3.4. General Procedure for the Palladium-Catalyzed Oxidative Carbonylation of N-Substituted 3-yn-1-Amines 1a–n in MeOH
3.4.1. (Z)-Methyl 2-(2-Oxo-1-Phenylpyrrolidin-3-ylidene)acetate 2a
3.4.2. (Z)-Methyl 2-(1-(4-Chlorophenyl)-2-Oxopyrrolidin-3-Ylidene)acetate 2b
3.4.3. (Z)-Methyl 2-(1-(4-Isopropylphenyl)-2-Oxopyrrolidin-3-Ylidene)acetate 2c
3.4.4. (Z)-Methyl 2-(1-(4-(tert-Butyl)phenyl)-2-Oxopyrrolidin-3-Ylidene)acetate 2d
3.4.5. (Z)-Methyl 2-(1-(3-Bromo-5-Methylphenyl)-2-Oxopyrrolidin-3-Ylidene)acetate 2e
3.4.6. (Z)-Methyl 2-(5-Methyl-2-Oxo-1-Phenylpyrrolidin-3-Ylidene)acetate 2f
3.4.7. (Z)-Methyl 2-(5-Ethyl-2-Oxo-1-Phenylpyrrolidin-3-Ylidene)acetate 2g
3.4.8. (Z)-Methyl 2-(2-Oxo-1,5-Diphenylpyrrolidin-3-Ylidene)acetate 2h
3.4.9. (Z)-Methyl 2-(5-(4-Bromophenyl)-2-Oxo-1-Phenylpyrrolidin-3-Ylidene)acetate 2i
3.4.10. (Z)-Methyl 2-(5-(4-Methoxyphenyl)-2-Oxo-1-Phenylpyrrolidin-3-Ylidene)acetate 2j
3.4.11. (Z)-Methyl 2-(1-(4-Chlorophenyl)-5-(4-Methoxyphenyl)-2-Oxopyrrolidin-3-Ylidene)acetate 2k
3.4.12. (Z)-Methyl 2-(1-Benzyl-2-Oxopyrrolidin-3-Ylidene)acetate 2l
3.4.13. (Z)-Methyl 2-(2-Oxo-1-(1-Phenylethyl)pyrrolidin-3-Ylidene)acetate 2m
3.4.14. (Z)-Methyl 2-(1-(tert-Butyl)-2-Oxopyrrolidin-3-Ylidene)acetate 2n
3.5. General Procedure for the Palladium-Catalyzed Oxidative Carbonylation of N-Substituted 3-yn-1-Amines 1a and 1f in Different Alcoholic Solvents
3.5.1. (Z)-Ethyl 2-(2-Oxo-1-Phenylpyrrolidin-3-Ylidene)acetate 2a′
3.5.2. (Z)-Isopropyl 2-(2-Oxo-1-Phenylpyrrolidin-3-Ylidene)acetate 2a″
3.5.3. (Z)-tert-Butyl 2-(2-Oxo-1-Phenylpyrrolidin-3-Ylidene)acetate 2a‴
3.5.4. (Z)-Ethyl 2-(1-Benzyl-2-Oxopyrrolidin-3-Ylidene)acetate 2l′
3.5.5. (Z)-Isopropyl 2-(1-Benzyl-2-Oxopyrrolidin-3-Ylidene)acetate 2l″
3.6. Palladium-Catalyzed Oxidative Carbonylation of N-(but-3-yn-1-yl)-4-(tert-Butyl)aniline 1d to (Z)-Methyl 2 (1-(4-(tert-Butyl)phenyl)-2-Oxopyrrolidin-3-Ylidene)acetate 2d in Larger Scale
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Das, D.; Bhanage, B. Double carbonylation reactions: Overview and recent advances. Adv. Synth. Catal. 2020, 362, 3022–3058. [Google Scholar] [CrossRef]
- Peng, J.-B. Recent advances in carbonylative difunctionalization of Alkenes. Adv. Synth. Catal. 2020, 362, 3059–3080. [Google Scholar] [CrossRef]
- Zhang, S.; Neumann, H.; Beller, M. Synthesis of α,β-unsaturated carbonyl compounds by carbonylation reactions. Chem. Soc. Rev. 2020, 49, 3187–3210. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Xu, J.; Wu, X.-F. No making without breaking: Nitrogen-centered carbonylation reactions. ACS Catal. 2020, 10, 6510–6531. [Google Scholar] [CrossRef]
- Peng, J.B.; Geng, H.-Q.; Wu, X.-F. The chemistry of CO: Carbonylation. Chem 2019, 5, 526–552. [Google Scholar] [CrossRef]
- Reimert, R.; Marschner, F.; Renner, H.-J.; Boll, W.; Supp, E.; Brejc, M.; Liebner, W.; Schaub, G. Gas production, 2. In Ullmann’s Encyclopedia of Industrial Chemistry; Baltes, H., Göpel, W., Hesse, J., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp. 423–479. [Google Scholar]
- Mancuso, R.; Della Ca’, N.; Veltri, L.; Ziccarelli, I.; Gabriele, B. PdI2-based catalysis for carbonylation reactions: A personal account. Catalysts 2019, 9, 610. [Google Scholar] [CrossRef]
- Mancuso, R.; Strangis, R.; Ziccarelli, I.; Della Ca’, N.; Gabriele, B. Palladium catalysis with sulfurated substrates under aerobic conditions: A direct oxidative carbonylation approach to thiophene-3-carboxylic esters. J. Catal. 2021, 393, 335–343. [Google Scholar] [CrossRef]
- Lei, H.; Xin, S.; Qiu, Y.; Zhang, X. Enantioselective total synthesis of (−)-kainic acid and (+)-acromelic acid C via Rh(I)-catalyzed asymmetric enyne cycloisomerization. Chem. Commun. 2018, 54, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Madec, D.; Poli, G.; Thuong, M.; Sottocornola, S.; Prestat, G.; Broggini, G. New access to kainic acid via intramolecular palladium-catalyzed allylic alkylation. Synlett 2007, 2007, 1521–1524. [Google Scholar] [CrossRef]
- Baker, R.; MacLeod, A.M.; Saunders, J.; Merchant, K. Thiadiazoles Useful in the Treatment of Senile Dementia. U.S. Patent 5405853, 11 April 1995. [Google Scholar]
- Danishefsky, S.; Berman, E.; Clizbe, L.A.; Hirama, M. A simple synthesis of L-.gamma.-carboxyglutamate and derivatives thereof. J. Am. Chem. Soc. 1979, 101, 4385–4386. [Google Scholar] [CrossRef]
- De Marco, R.; Mazzotti, G.; Greco, A.; Gentilucci, L. Heterocyclic scaffolds in the design of peptidomimetic integrin ligands: Synthetic strategies, structural aspects, and biological activity. Curr. Top. Med. Chem. 2015, 16, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Lindvall, M.; Manning, J.R.; McEnroe, G.; Novartis, A.G. Novel Dihydroisoxazole Compounds and Their Use for the Treatment of Hepatitis B. PCT Patent Application WO2019/97479 A1, 23 May 2019. [Google Scholar]
- Ciccone, A.; Motto, C.; Abraha, I.; Cozzolino, F.; Santilli, I. Glycoprotein IIb-IIIa inhibitors for acute ischaemic stroke. Cochrane Database Syst. Rev. 2014, CD005208. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, T.I.; Kumar, A.; Kumar, K.S.; Dikshit, D.K. Platelets and atherothrombosis: Causes, targets and treatments for thrombosis. Curr. Med. Chem. 2013, 20, 2779–2797. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.A.; Widen, J.C.; Harki, D.A.; Brummond, K.M. Covalent modifiers: A chemical perspective on the reactivity of α,β-unsaturated carbonyls with thiols via hetero-Michael addition reactions. J. Med. Chem. 2017, 60, 839–885. [Google Scholar] [CrossRef]
- Yuan, B.; Jiang, Y.; Qi, Z.; Guan, X.; Wang, T.; Yan, R. External oxidant-free oxidative tandem cyclization: NaI-catalyzed thiolation for the synthesis of 3-thiosubstituted pyrroles. Adv. Synth. Catal. 2019, 361, 5112–5117. [Google Scholar] [CrossRef]
- Azuma, M.; Yoshikawa, T.; Kogure, N.; Kitajima, M.; Takayama, H. Biogenetically inspired total syntheses of Lycopodium alkaloids, (+)-flabellidine and (−)-lycodine. J. Am. Chem. Soc. 2014, 136, 11618–11621. [Google Scholar] [CrossRef]
- Breuning, M.A.; Harms, K.; Koert, U. The imidato-alkenyllithium route for the synthesis of the isoquinocycline-pyrrolopyrrole substructure. Org. Lett. 2011, 13, 1402–1405. [Google Scholar] [CrossRef] [PubMed]
- Courtois, G.; Mesnard, D.; Dugue, B.; Miginiac, L. Aminomethylation secondaire ou primaire d’organoaluminiques α-insatures a l’aide de gem-aminoethers N-trimethylsilyles: Synthese d’amines secondaires ou primaires, β-ethyleniques, β-acetyleniques ou α-alleniques. Bull. Soc. Chim. Fr. 1987, 124, 93–98. [Google Scholar]
- Tong, S.; Piemontesi, C.; Wang, Q.; Wang, M.-X.; Zhu, J. Silver-catalyzed three-component 1,1-aminoacylation of homopropargylamines: α-additions for both terminal alkynes and isocyanides. Angew. Chem. Int. Ed. 2017, 56, 7958–7962. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | PdI2/KI/1a Molar Ratio | Concn of 1a b | T (°C) | t (h) | PCO (atm) | Pair (atm) | Yield (%) c |
|---|---|---|---|---|---|---|---|
| 1 | 1/10/20 | 0.04 | 100 | 2 | 32 | 8 | 74 |
| 2 | 1/5/20 | 0.04 | 100 | 2 | 32 | 8 | 65 |
| 3 | 1/2/20 | 0.04 | 100 | 2 | 32 | 8 | 55 |
| 4 | 1/10/20 | 0.04 | 100 | 2 | 48 | 12 | 66 |
| 5 | 1/10/20 | 0.04 | 100 | 2 | 16 | 4 | 67 |
| 6 | 1/10/20 | 0.02 | 100 | 2 | 32 | 8 | 59 |
| 7 | 1/10/20 | 0.10 | 100 | 2 | 32 | 8 | 85 |
| 8 | 1/10/20 | 0.04 | 80 | 2 | 32 | 8 | 55 |
| 9 | 1/10/20 | 0.04 | 100 | 1 | 32 | 8 | 42 d |
| 10 | 1/10/20 | 0.04 | 100 | 8 | 32 | 8 | 33 |

| Entry | 1 | ROH | PdI2/KI/1 Molar Ratio | Substrate Concentration c | Time (h) | 2 | Yield of 2 (%) d |
|---|---|---|---|---|---|---|---|
| 1 |  | MeOH | 1/10/20 | 0.1 | 2 |  | 85 |
| 2 | 1a | MeOH | 1/10/100 | 0.1 | 2 | 2a | 72 |
| 3 | 1a | MeOH | 1/10/200 | 0.1 | 2 | 2a | 64 |
| 4 | 1a | EtOH | 1/10/20 | 0.1 | 2 |  | 82 |
| 5 | 1a | iPrOH | 1/10/20 | 0.1 | 2 |  | 72 |
| 6 | 1a | tBuOH | 1/10/20 | 0.1 | 2 |  | 33 |
| 7 | 1a | tBuOH | 1/10/20 | 0.04 | 2 | 2a″ | 54 |
| 8 |  | MeOH | 1/10/20 | 0.1 | 2 |  | 77 |
| 9 | 1b | MeOH | 1/10/100 | 0.1 | 2 | 2b | 72 |
| 10 |  | MeOH | 1/10/20 | 0.1 | 2 |  | 74 |
| 11 | 1c | MeOH | 1/10/100 | 0.1 | 2 | 2c | 72 |
| 12 |  | MeOH | 1/10/20 | 0.1 | 2 |  | 70 |
| 13 |  | MeOH | 1/10/20 | 0.1 | 2 |  | 78 |
| 14 | 1e | MeOH | 1/10/100 | 0.1 | 2 | 2e | 75 |
| 15 |  | MeOH | 1/10/20 | 0.1 | 2 |  | 77 |
| 16 |  | MeOH | 1/10/20 | 0.1 | 2 |  | 78 |
| 17 |  | MeOH | 1/10/20 | 0.1 | 2 |  | 82 |
| 18 |  | MeOH | 1/10/20 | 0.1 | 2 |  | 70 |
| 19 |  | MeOH | 1/10/20 | 0.1 | 2 |  | 71 |
| 20 |  | MeOH | 1/10/20 | 0.1 | 2 |  | 70 |
| 21 |  | MeOH | 1/10/20 | 0.1 | 2 |  | 50 |
| 22 | 1l | EtOH | 1/10/20 | 0.1 | 5 |  | 47 |
| 23 | 1l | iPrOH | 1/10/20 | 0.1 | 5 |  | 48 |
| 24 | 1l | MeOH | 1/10/20 | 0.04 | 2 | 2l | 65 |
| 25 |  | MeOH | 1/10/20 | 0.1 | 2 |  | 52 |
| 26 | 1m | MeOH | 1/10/20 | 0.04 | 2 | 2m | 75 |
| 27 |  | MeOH | 1/10/20 | 0.04 | 5 |  | 47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancuso, R.; Ziccarelli, I.; Brindisi, M.; Altomare, C.D.; Frattaruolo, L.; Falcicchio, A.; Della Ca’, N.; Cappello, A.R.; Gabriele, B. A Stereoselective, Multicomponent Catalytic Carbonylative Approach to a New Class of α,β-Unsaturated γ-Lactam Derivatives. Catalysts 2021, 11, 227. https://doi.org/10.3390/catal11020227
Mancuso R, Ziccarelli I, Brindisi M, Altomare CD, Frattaruolo L, Falcicchio A, Della Ca’ N, Cappello AR, Gabriele B. A Stereoselective, Multicomponent Catalytic Carbonylative Approach to a New Class of α,β-Unsaturated γ-Lactam Derivatives. Catalysts. 2021; 11(2):227. https://doi.org/10.3390/catal11020227
Chicago/Turabian StyleMancuso, Raffaella, Ida Ziccarelli, Matteo Brindisi, Cosimo D. Altomare, Luca Frattaruolo, Aurelia Falcicchio, Nicola Della Ca’, Anna Rita Cappello, and Bartolo Gabriele. 2021. "A Stereoselective, Multicomponent Catalytic Carbonylative Approach to a New Class of α,β-Unsaturated γ-Lactam Derivatives" Catalysts 11, no. 2: 227. https://doi.org/10.3390/catal11020227
APA StyleMancuso, R., Ziccarelli, I., Brindisi, M., Altomare, C. D., Frattaruolo, L., Falcicchio, A., Della Ca’, N., Cappello, A. R., & Gabriele, B. (2021). A Stereoselective, Multicomponent Catalytic Carbonylative Approach to a New Class of α,β-Unsaturated γ-Lactam Derivatives. Catalysts, 11(2), 227. https://doi.org/10.3390/catal11020227