1. Introduction
Low-temperature fuel cells (FCs) with a proton-exchange membrane are popular energy-producing devices. Fuel cells, which use liquid fuels such as methanol, ethanol, or formic acid are attracting an increasing attention as alternative energy sources for portable electronic devices [
1] due to a relatively high current density at a very compact size [
2].
Several problems that impede widespread commercialization of alcohol fuel cells should be noted:
- -
Crossover of methanol or ethanol to the cathode region through the polymer membrane can lead to poisoning not only the cathode catalyst, but also the membrane itself [
3], which leads to a reduction in the life of alcohol FCs; and
- -
Poisoning of the catalyst surface by intermediate products of the oxidation reactions of alcohols, for example, HCHO and CO can decrease catalyst’s activity [
4,
5].
Another important type of organic fuel suitable for use in fuel cells is formic acid. It is less prone to crossover through Nafion
® membranes compared to alcohols. The lower flux is due to the anionic nature of the formic acid [
6,
7]. This allows the use of higher concentrations of formic acid to increase the current density in the fuel cell. Unfortunately, the problem of poisoning the catalyst surface with the intermediate products is still a pressing issue for both formic acid oxidation and alcohols. Note that the problem of catalyst poisoning is important both for the anode, on which the organic fuel is directly oxidized, and for the cathode, due to crossover. In these cases, the oxidation of organic molecules at the anode and cathode should proceed at different potentials.
One of the ways to solve the problem of poisoning platinum-containing catalysts and increase their activity is to use bi- and trimetallic nanoparticles based on Pt [
8,
9]. In the case of electrooxidation of methanol (
MOR) and ethanol (
EOR), PtRu alloys are well-known anodic catalysts with a lower sensitivity to CO poisoning compared to pure Pt [
10,
11]. However, in view of a relatively high cost of ruthenium, other alloys with transition d-metals, such as PtNi [
12], PtCu [
8,
13,
14], and PtCo [
14,
15], were also investigated. For example, it was shown in [
15] that the PtCo/C and PtCu/C bimetallic catalysts demonstrated higher activity in MOR compared to PtRu/C material. However, the authors noted that in the process of recording cyclic voltammograms, copper dissolved from the surface of the catalyst and copper ions passing into the solution could distort the experimental results [
15].
Pd/C is an effective catalyst for the electrooxidation reaction of formic acid (
FAOR) [
16,
17]. At the same time, the oxidation of formic acid was widely studied on some bimetallic alloys: PtAu [
18,
19], PtFe [
20], PtPb [
21], PtCo [
22], and others. The authors [
23] showed that the PtNi/C materials, that they had obtained, demonstrated significantly greater catalytic activity and increased stability in POMC compared to a commercial Pd/C catalyst.
The influence of the alloying component on the Pt electrocatalytic activity, can be explained by two effects. The first is a bifunctional catalysis mechanism, in which rapid oxidation is caused by an easier OH adsorption on inclusions of the doping component and easier oxidation of CO molecules chemisorbed on the neighboring Pt atoms [
24]. The second one is the electronic effect associated with the electronic interaction of Pt atoms and a doping metal, which leads to a decrease in the binding energy of adsorbed particles with the surface of metal nanoparticles [
25].
Another way to increase the activity and stability of Pt-containing electrocatalysts in the reactions of the alcohols electrooxidation is to use composite carbon and non-carbon supports. Thus, in [
26], the authors concluded that the contact of Pt and SnO
2 nanoparticles could reduce the oxidation potential of CO due to the bifunctional mechanism of the catalysis. The resulting catalysts showed four times greater activity in the ethanol electrooxidation reaction than the Pt/C material with a similar platinum content. The use of a Pt/C-Cu
3P composite catalyst also made it possible to considerably increase the platinum activity and the catalyst’s tolerance with regard to the intermediates, which were formed as a result of the oxidation process of the small organic fuel molecules [
27].
Thus, at present, the use of methanol, ethanol, and formic acid as a fuel in low-temperature fuel cells has already been implemented in practice, and the development of high-performance catalysts is an urgent task for researchers from all over the world. A very interesting question is whether it is possible to find an electrocatalyst that is the most effective one for the oxidation of many organic reducing agents. Ruixue Li et al., who studied composite Pt/C-Cu
3P catalysts of different composition, showed that the sample, which contained 50% of Cu
3P, presented a highly enhanced catalytic performance for electro-oxidation of methanol, ethanol, glycol and formic acid [
27]. Renan G. C. et al. [
28] measured the activity of three different catalysts: PtCu/C, PtSn/C, and Pt/C in the electrooxidation of methanol, ethanol, propanol, and butanol. It was shown that the PtCu/C catalyst, which showed high activity in MOR, was less active than the PtSn/C catalyst in EOR. At the same time, in the electrooxidation reaction of propanol and butanol, both bimetallic catalysts showed similar activity. The study of methanol and formic acid oxidation on PtCu/C and Pt/C catalysts carried out in [
29] showed that the bimetallic PtCu/C material exhibits better catalytic activity in these reactions than the analogous Pt/C. Kuznetsov V.V. et al. [
30] investigated the electrochemical behavior of nPd
0*(H
x−2nMoO
3)/GC (glassy carbon) electrodes. It was found that the nPd
0*(H
x−2nMoO
3)
cryst/GC catalyst has a greater catalytic effect in the methanol electrooxidation reaction than other molybdenum bronzes, but shows only a slight increase in activity for FAOR. It was shown that the Pt/C and PtRu/C catalysts obtained in [
31] exhibited higher activity and stability in the electrooxidation of formic acid than in the reactions of alcohols electrooxidation.
Unfortunately, it is difficult to compare the absolute values of catalysts activity given in different publications, since the conditions of such measurements can differ significantly. A more correct approach seems to be appropriate when the activity of bimetallic catalysts is studied under the same experimental conditions and compared to that of a certain standard, for which a well-known commercial Pt/C catalyst could be chosen. In our previous works we studied the influence of the nanostructured tin oxide content in Pt/(SnO
2/C) [
32,
33], the nature of the alloying component in PtM/C (where M = Ni, Cu) [
33,
34,
35,
36] and acid treatment conditions of PtCu/C catalysts [
34,
35] on the activity of platinum-containing catalysts in the reactions of methanol electrooxidation and oxygen electroreduction. These two types of catalysts are in good agreement with the two previously mentioned approaches aimed at increasing the platinum activity in electrooxidation reactions of simple organic substances: By alloying the metal itself or by using a composite metal oxide support.
The aim of this work is to compare electrooxidation features of such important organic substances as methanol, ethanol and formic acid on Pt/C, Pt/(SnO2/C) and PtCu/C catalysts, as well as to compare activity of these materials in the corresponding reactions. Let us note one more another important circumstance. PtRu/C catalysts are not usually used on the oxygen electrode of the fuel cells due to their low activity in the oxygen electroreduction reaction compared to Pt/C. Nevertheless, due to the crossover of organic molecules, their fast and efficient oxidation is also of importance for the cathode catalyst of alcohol fuel cells. In this regard, the study of effective “oxygen” catalysts activity in the reactions of organic compounds oxidation at high positive potentials, close to the potential of the oxygen electrode, is also of considerable interest.
2. Results and Discussion
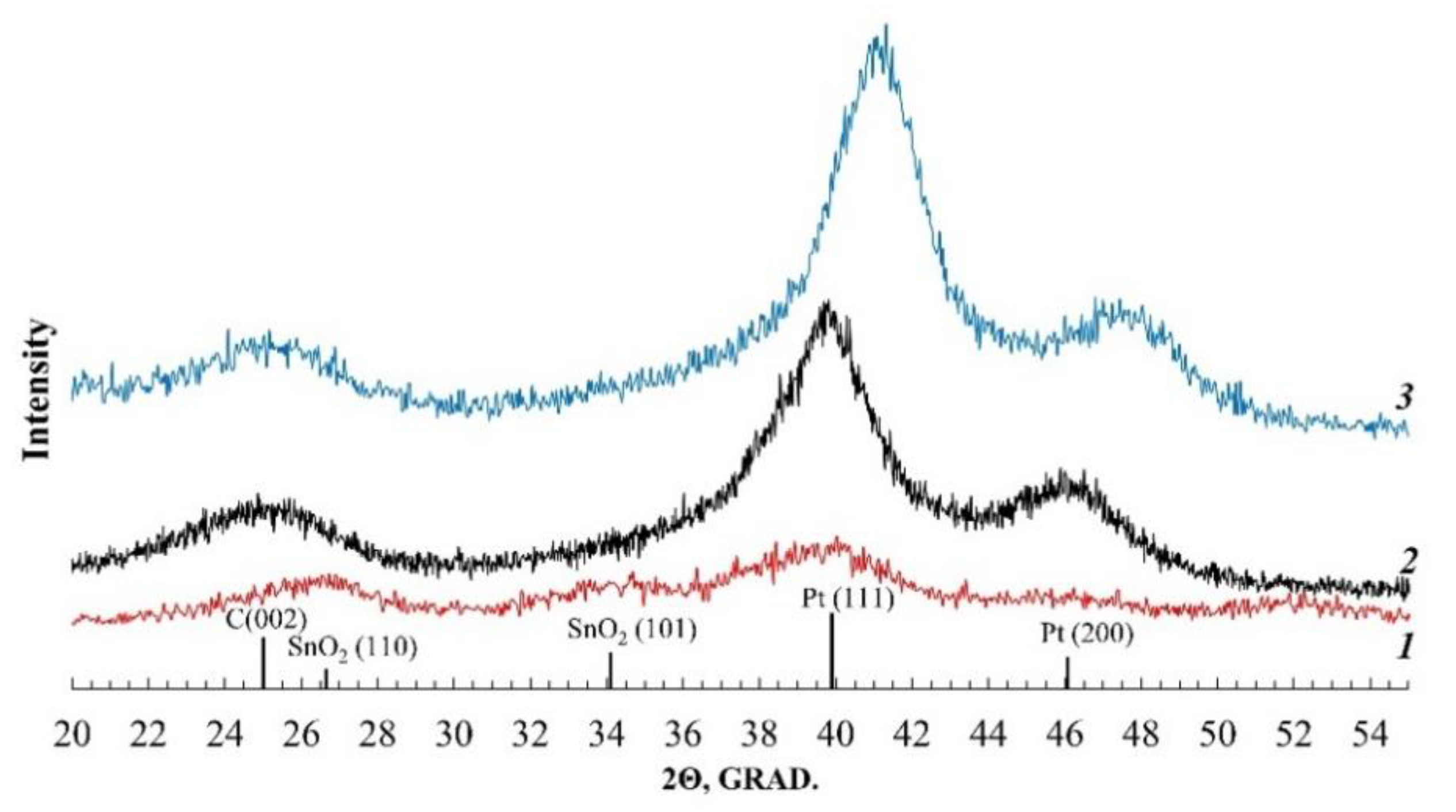
According to the results of the analysis performed, all the studied materials contain about 20% wt. of platinum (
Table 1). On the X-ray diffraction patterns of the samples (
Figure 1), the reflections corresponding to the phases of carbon and platinum (face-centered cubic lattice, space group Fm3m) are clearly visible. For the Pt/(SnO
2/C) material, the broadened reflection corresponding to the SnO
2 phase is also observed. Note that for the PtCu/C catalyst, the (111) and (200) reflections of the platinum are shifted to the region of large 2 theta angles, compared to the Pt/C material. Such a shift indicates a decrease in the crystal lattice parameter of the metal (
Table 1) and is associated with the formation of a PtCu solid solution.
The average crystallite size of the metal phase was determined by the width of the reflections at half their height (FWHM) (
Figure 1) using the Scherrer formula. The average crystallite size of platinum is from 1.9 to 2.4 nm, while the calculated size of the bimetallic PtCu crystallites is slightly higher—2.9 nm (
Table 1).
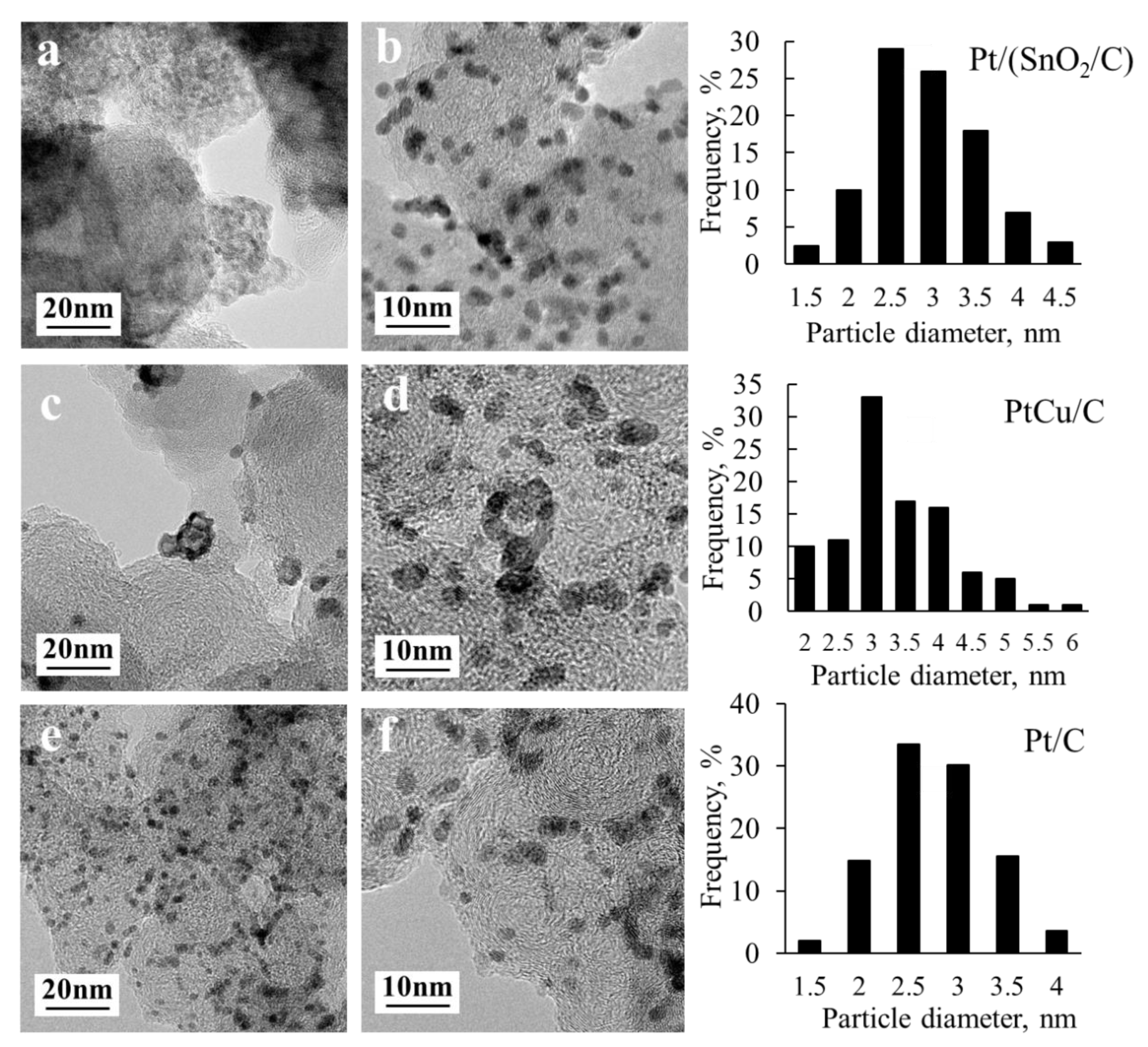
The presence of metal nanoparticles on the surface of finely dispersed supports was also confirmed by transmission electron microscopy (
Figure 2). TEM microphotographs show individual metal nanoparticles uniformly distributed on the surface of the particles of the carbon support. Note that SnO
2 nanoparticles are more difficult to detect in the micrographs of a Pt/(SnO
2/C) catalyst than the platinum nanoparticles, due to a significant difference in contrast.
Nevertheless, the presence of SnO
2 nanoparticles in such catalysts was proved by the results of a high resolution TEM [
32]. Size determination of metal nanoparticles in TEM photographs made it possible to construct histograms of the size distribution (
Figure 2) and determine their average size for each catalyst (
Table 1). A comparison of the average crystallite size according to XRD and the average nanoparticle size according to TEM (
Table 1) confirms a good agreement between the results obtained by these methods: The largest particle size is characteristic of the PtCu/C material. At the same time, the values of the average sized nanoparticles, determined by TEM, are slightly larger than the average crystallite sizes for all catalysts studied (
Table 1). Such a situation is characteristic of similar materials [
37,
38]. In both cases, the particle size increases in the row Pt/(SnO
2/C) < Pt/C < PtCu/C.
In the process of electrochemical standardization of Pt/C and PtM/C catalysts in the HClO
4 solutions, the platinum surface is cleaned and developed, which leads to an increase in the currents on the CVs, which is more pronounced in the first cycles [
36]. It is known that, on CVs of PtCu/C catalysts, peaks of anodic dissolution from the intrinsic phase and from a solid solution of copper in platinum can be observed in the potential ranges of 0.25–0.45 and 0.7–0.8 V, respectively [
34,
36,
39]. In this work no similar anodic maxima were observed for the studied PtCu/C catalyst (
Figure S1), which is due to the previous dissolution of “weakly bound” copper at the stage of preliminary treatment of the PtCu/C material in acid.
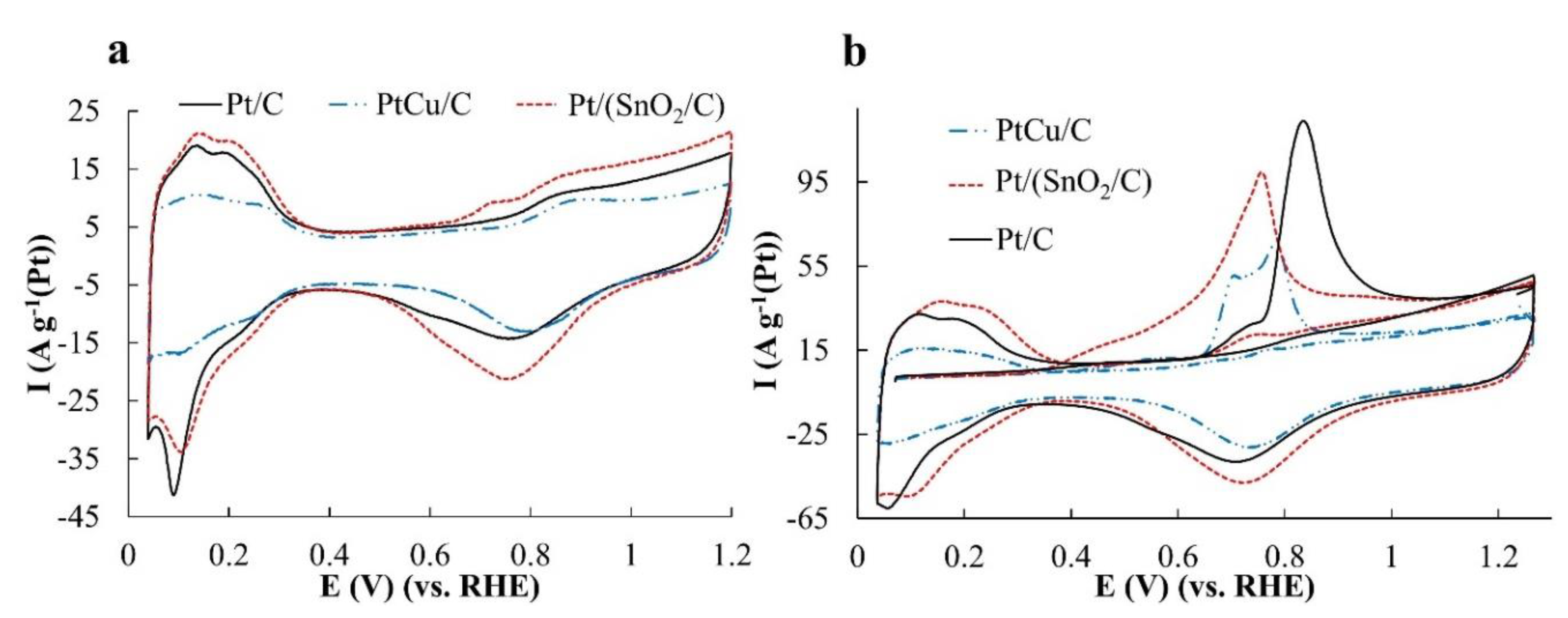
The cyclic voltammograms obtained on the standardized samples have the form characteristic of platinum-containing catalysts and include the regions of hydrogen adsorption/desorption, the double electric layer charging, as well as the region of platinum surface oxidation and its reduction during the reverse course of the potential (
Figure 3a). The ESA values calculated for the hydrogen regions of CVs turned out to be the highest for Pt/(SnO
2/C) and Pt/C, which is consistent with the small average size of metal nanoparticles in these materials (
Table 2).
The correct determination of the ESA values is confirmed by the results of a similar calculation made for the electrochemical desorption of CO (
Figure 3b). Note, that CO oxidation on the Pt/(SnO
2/C) catalyst begins much earlier than that on the PtCu/C and Pt/C, and the anodic maxima on the voltammograms associated with CO oxidation for Pt/(SnO
2/C) and PtCu/C are shifted into the region of lower potentials as compared to Pt/C (
Figure 3b). This proves that CO oxidation on the catalysts, which we have synthesized, goes in a much easier way in comparison with Pt/C. It would be only natural to associate the early onset of oxidation on Pt/(SnO
2/C) with the bifunctional mechanism of the catalysis, in which the oxidation of CO molecules occurs with the participation of OH
ads groups adsorbed on SnO
2 nanoparticles on the sites of a three-boundary contact [
26].
Before comparing the catalysts activity in the reactions of methanol, ethanol, and formic acid electrooxidation, one should analyze the criteria for such an assessment, as well as discuss the effect of the electrode potential on the features of the platinum surface state and the nature of reactions that take place on it. The easiest way to do it, is with the MOR taken as an example, and then note the specific features of EOR and FAOR. Cyclic voltammetry is often used to study electrooxidation kinetics of the methanol and other organic compounds [
2,
7,
9,
10,
11].
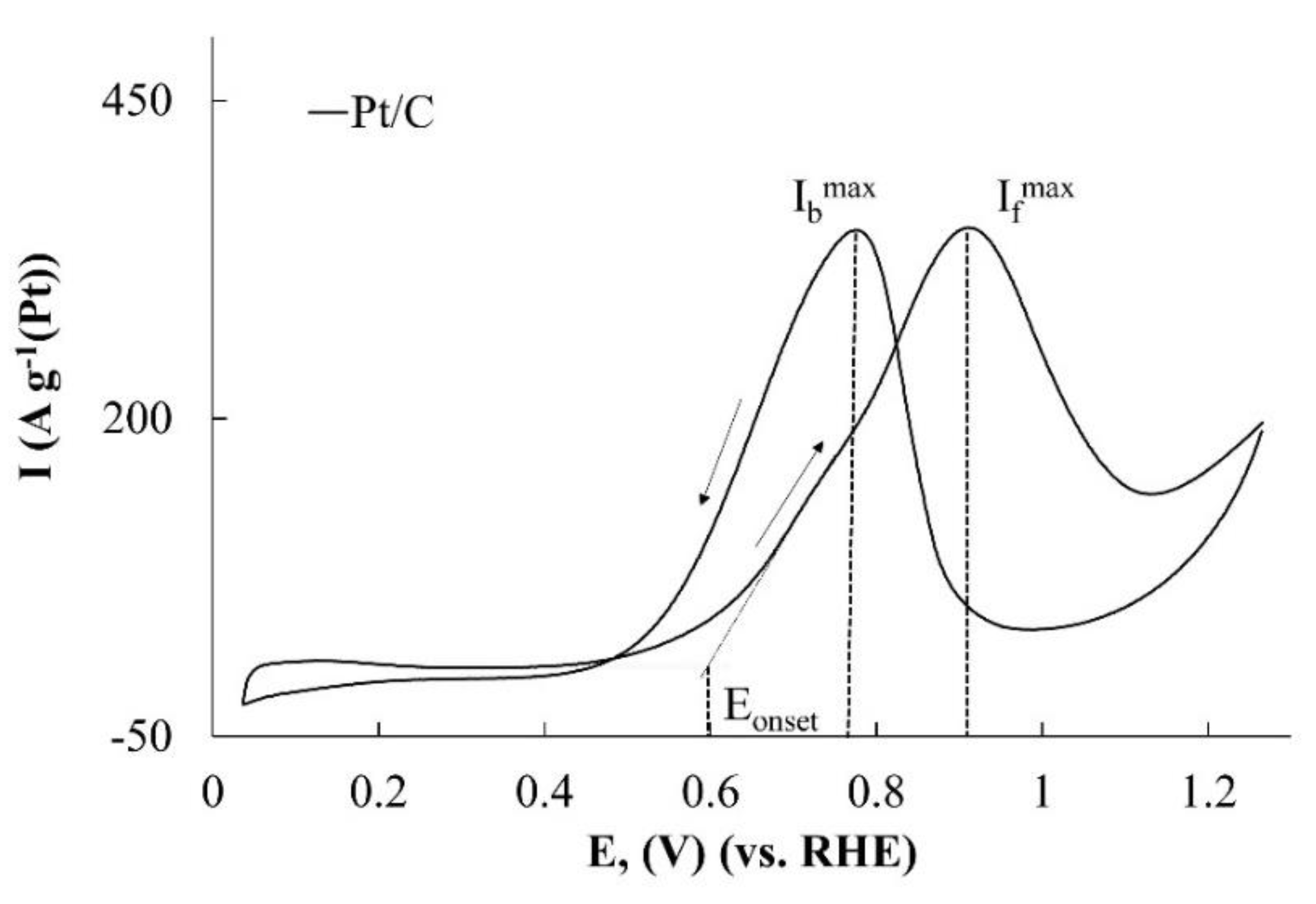
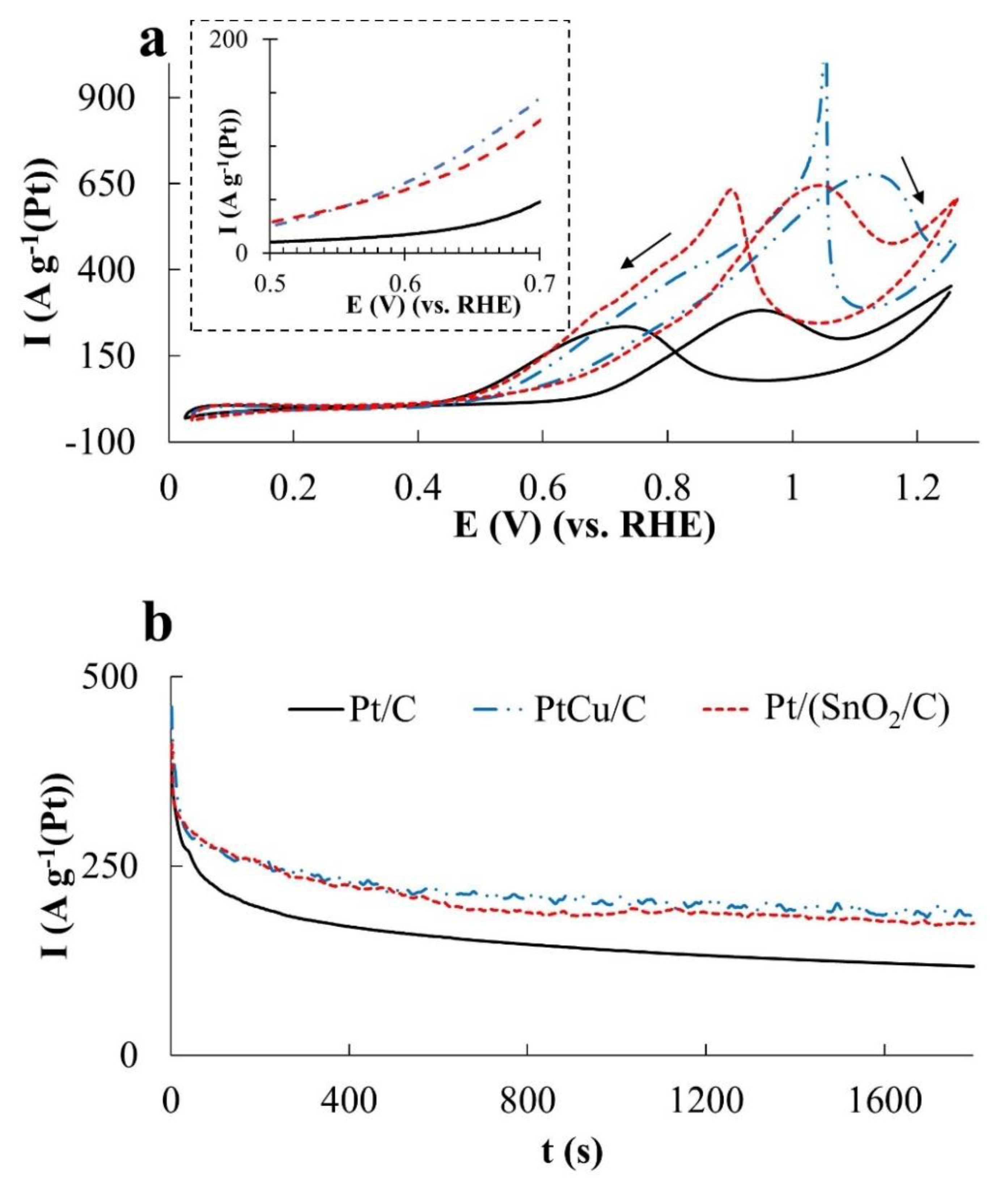
A typical CV of methanol oxidation in an acidic solution is shown in
Figure 4. The first anodic peak on the forward curve at a potential of about 0.90 V (
Figure 4) is considered to be the main peak of methanol oxidation by the overwhelming majority of researchers. The higher its intensity is, the higher the catalyst activity is. At the same time, it should be noted that, along with the methanol electrooxidation, other processes can also occur on the surface of platinum-containing electrodes that have an effect on the state of this surface and its catalytic activity with respect to MOR. We have used the analysis results of these processes on the Pt/C electrode, carried out by Dong Young Chung et al. [
40].
In the potential region 0.40~0.50 V CO, formed during the electrooxidation of methanol, is adsorbed on the active sites of the platinum catalyst. A multistage process resulting in the CO formation can be described by a simplified equation:
As a result of chemisorption passivation of the platinum surface, the methanol oxidation rate is very low, in the potential range 0.60~0.75 V OH groups are adsorbed on the platinum surface, which are formed from water by the reaction:
In this case, the limiting step of the MOR is the reaction:
When the potential is shifted towards higher values, the reaction rate increases, and among other things, due to the increase in the degree of the platinum surface being filled with OH groups. In the potential range of 0.80~0.85 V the oxidation of the intermediate CO by OH in accordance with reaction III is accelerated to such an extent that the electrooxidation of methanol on the Pt surface becomes a limiting stage for it. However, with the further increase in the electrode potential (range 0.90~0.975 V), strong adsorption of OH groups and an increase in the degree of filling the platinum surface with these oxygen-containing particles reduce the access to this surface for methanol molecules adsorption.
This causes a decrease in the methanol conversion rate and formation of a maximum on the CV (
Figure 4). The subsequent increase in the potential leads to an even greater coverage of platinum with oxygen-containing particles and to Pt-OH conversion into Pt-O
x. Meanwhile, the methanol oxidation rate on the oxidized metal surface decreases. At potentials of 1.10~1.20 V, the methanol oxidation takes place already on Pt-O
x, the mechanism of this reaction unfortunately is not entirely clear.
A change in the composition of the platinum-containing catalyst, apparently, can affect both the length of the corresponding potential ranges and the rate of certain processes that determine the kinetics and the CV form transformation.
While selecting parameters that characterize the catalysts activity in the MOR, the onset potential of methanol oxidation is of the greatest interest (
Figure 4). The smaller it is, the more promising the use of this catalyst in DMFC is. The current magnitude at the CV maximum on the forward curve corresponds to the moment, when the decrease in the rate of methanol oxidation, caused by the closure of the platinum surface by OH group begins to prevail over the increase in the rate of its electrooxidation on the free platinum surface due to an increase in the electrode potential. It is natural to expect that the higher the specific rate of methanol electrooxidation on the platinum is, the higher the
Ifmax value is. Some researchers use the ratio of
Ifmax/Ibmax values to assess the catalysts tolerance in the MOR. However, the results [
40,
41] indicate that the value of
Ibmax is mostly caused not by the rate of methanol oxidation on bare platinum but by the degree of the platinum surface coverage with oxygen-containing particles. This makes the use of the
Ifmax/Ibmax criterion rather doubtful in assessing the catalysts tolerance. Note that, despite the change in the character of the stage that limits the methanol electrooxidation with the increasing potential, the total amount of the converted reagent is proportional to the electric charge passed through the electrode. Therefore, we suggest using the amount of electricity, having passed through the anode in the range of potentials, which correspond to the maximum on the forward path CV, as a criterion to evaluate the catalysts activity. It is important that this criterion (
QCH3OH) takes into account not only the maximum current value, but also the shape (extent) of the maximum.
The electrooxidation of ethanol and the formic acid proceeds according to the mechanisms that are different from the methanol oxidation [
24,
42,
43]. However, in these cases the following three features are of importance too: (1) Formation of adsorption active intermediates, their electrooxidation being performed in the presence of OH groups; (2) strong adsorption of OH groups with increasing potential; (3) oxidation of the platinum surface (formation of PtO
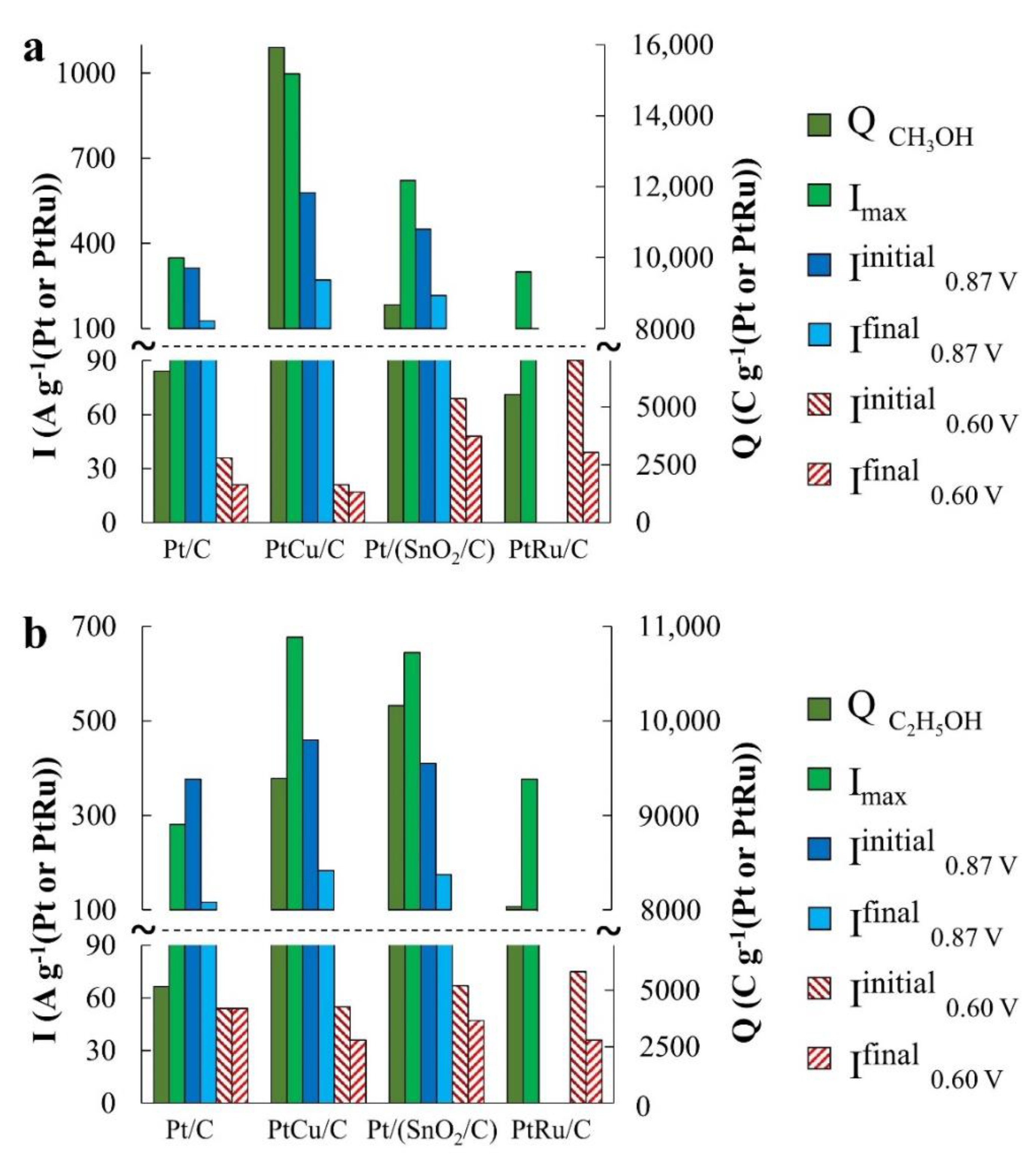
x), which negatively affects electrooxidation rate of the corresponding organic molecules. Therefore, the catalysts activity in the EOR and FAOR was estimated according to the same parameters as for the MOR, including the amount of electricity spent on the oxidation of the corresponding organic compound during the forward potential sweep (during CV registration) (
Figure 5a,
Figure 6a and
Figure 7a,
Table 2). In addition, the current–time dependences (chronoamperograms) measured at potentials 0.87 V and 0.60 V were compared for all the catalysts used (
Figure 5b,c,
Figure 6b,c and
Figure 7b,c and
Table 2). When carrying out chronoamperometric measurements, it was assumed that the behavior of catalysts at a potential of 0.87 V can provide information useful for their use on the cathode of a corresponding fuel cell under the conditions of an organic reducing agent crossover. Studying the behavior of catalysts at a potential of 0.60 V can provide information useful for using a catalyst at the anode. In the latter case, the chronoamperograms were compared with those not only for Pt/C, but also for the commercial PtRu/C catalyst containing 40 wt% Pt, 20 wt% Ru.
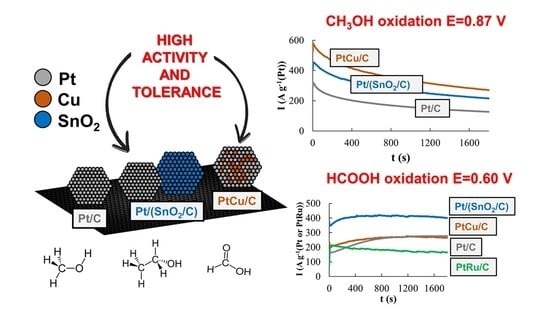
In according with the voltammetry data both modified catalysts demonstrated much higher activity compared to Pt/C in the electrooxidation reactions of all organic compounds. In this case, the differences in CVs are clearly manifested at potentials above 0.70 V. Nevertheless, it makes sense to discuss in more detail the relationship between the activities of each material in regard to the oxidation of each specific reagent, as well as the specifics of each reagent oxidation on different catalysts.
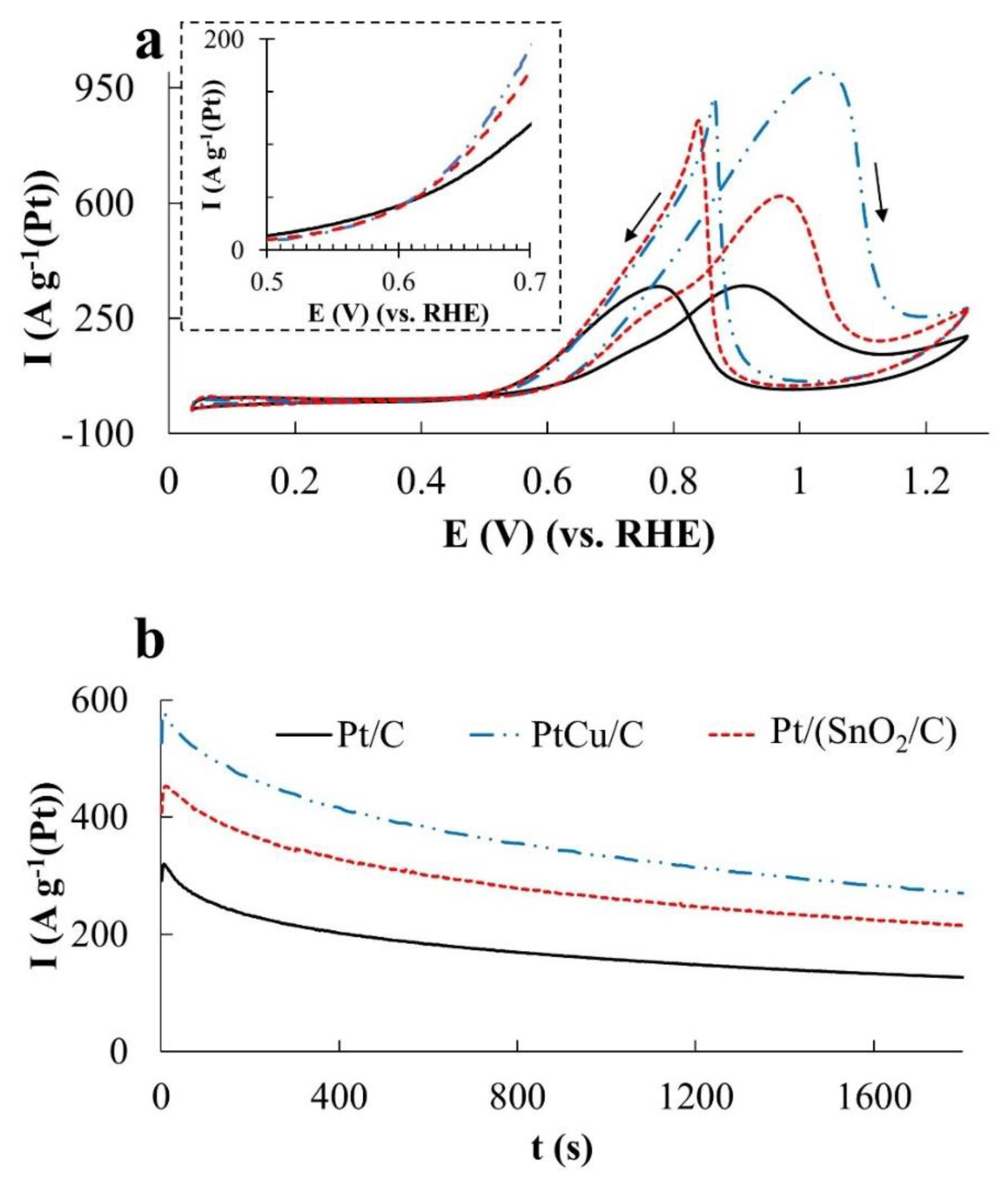
In the MOR, the differences in the catalysts’ activity manifest themselves most significantly, which follow both from a comparison of
Imax and
QCH3OH values (
Figure 5a and
Figure 6a), and a comparison of the electric current values in chronoamperograms (
Figure 5b) measured at a potential of 0.87 V. The catalysts are arranged in a row: Pt/C < Pt/(SnO
2/C) < PtCu/C according to the increase in mass activity. Despite the low ESA, the mass activity of PtCu/C, estimated from the
Imax and
QCH3OH values, is almost three times higher than that for Pt/C and almost twice as high for Pt/(SnO
2/C) (
Figure 5a and
Figure 6a).
In this case, the potentials for the onset of methanol oxidation during the forward sweep of the potential on all catalysts are close (
Table 2), and in the reverse sweep, they slightly decrease in the order PtCu/C > Pt/(SnO
2/C) > Pt/C (
Figure 5a). Peaks of methanol oxidation on the branches of the reverse sweep of the CV potential are smoothed in the case of Pt/C and become more abrupt (sharp) for Pt/(SnO
2/C) and PtCu/C catalysts (
Figure 5a). Similar features of the CV form in the reverse course of the potential sweep are observed during oxidation of both ethanol (
Figure 7a) and formic acid (
Figure 8a).
The ratio of the catalyst activities changes significantly if we compare the chronoamperograms of methanol oxidation at a potential of 0.60 V (
Figure 5c). Under these conditions, the activity of PtCu/C is minimal, Pt/C is slightly higher, and the activity of the Pt/(SnO
2/C) catalyst is much higher. In the latter case, the current strength weakly decreases with time, because of which the activity of the Pt/(SnO
2/C) electrode in MOR is slightly higher than that of PtRu/C, whose activity in MOR decreases faster (
Figure 5c).
Based on voltammograms, ethanol oxidation begins on Pt/(SnO
2/C) and PtCu/C catalysts at lower potentials (~0.50 V) than on Pt/C (~0.67 V) (the inset in
Figure 7a,
Table 2). In terms of mass activity in EOR in according with CV data, the catalysts are in the series PtCu/C ≈ Pt/(SnO
2/C) > Pt/C (
Figure 6b). The differences in mass activity between PtCu/C and Pt/(SnO
2/C) are not so great here, as in the case of MOR (
Figure 5a and
Figure 6a), and the decay current curves at a potential of 0.87 V for these catalysts practically coincide (
Figure 6b). At the same time, it is precisely during the oxidation of ethanol that the potentials for the onset of ethanol oxidation on the CVs return branches differ most significantly, substantially decreasing in the order PtCu/C > Pt/(SnO
2/C) > Pt/C (
Figure 7a). Assuming that the oxidation of ethanol during the reverse course of the potential sweep begins after cleaning the platinum surface from oxygen-containing particles we can come to the conclusion that the surface of PtCu nanoparticles is cleaned most quickly (it is least passivated).
Comparison of the activity of catalysts at a potential of 0.60 V shows that, as in the methanol oxidation, the PtCu/C catalyst is the least active, while Pt/(SnO
2/C) and Pt/C exhibit close oxidation rates (
Figure 7c). In this case, the activity of the PtRu/C electrode initially exceeds the activity of the other studied catalysts. However, due to the most rapid drop in the current on the PtRu/C electrode, after 1800 s the activities of PtRu/C and PtCu/C in EOR practically do not differ (
Figure 7c).
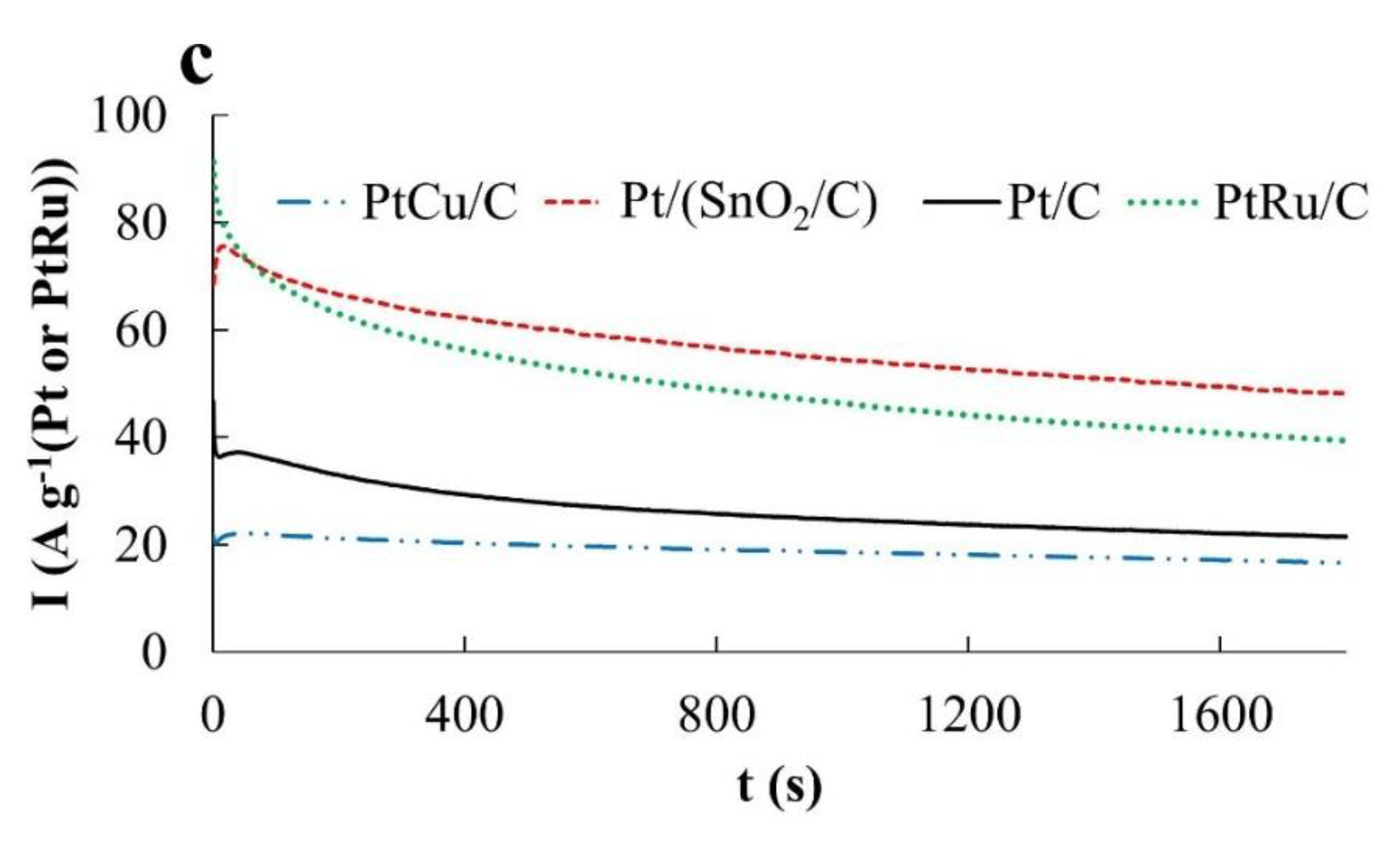
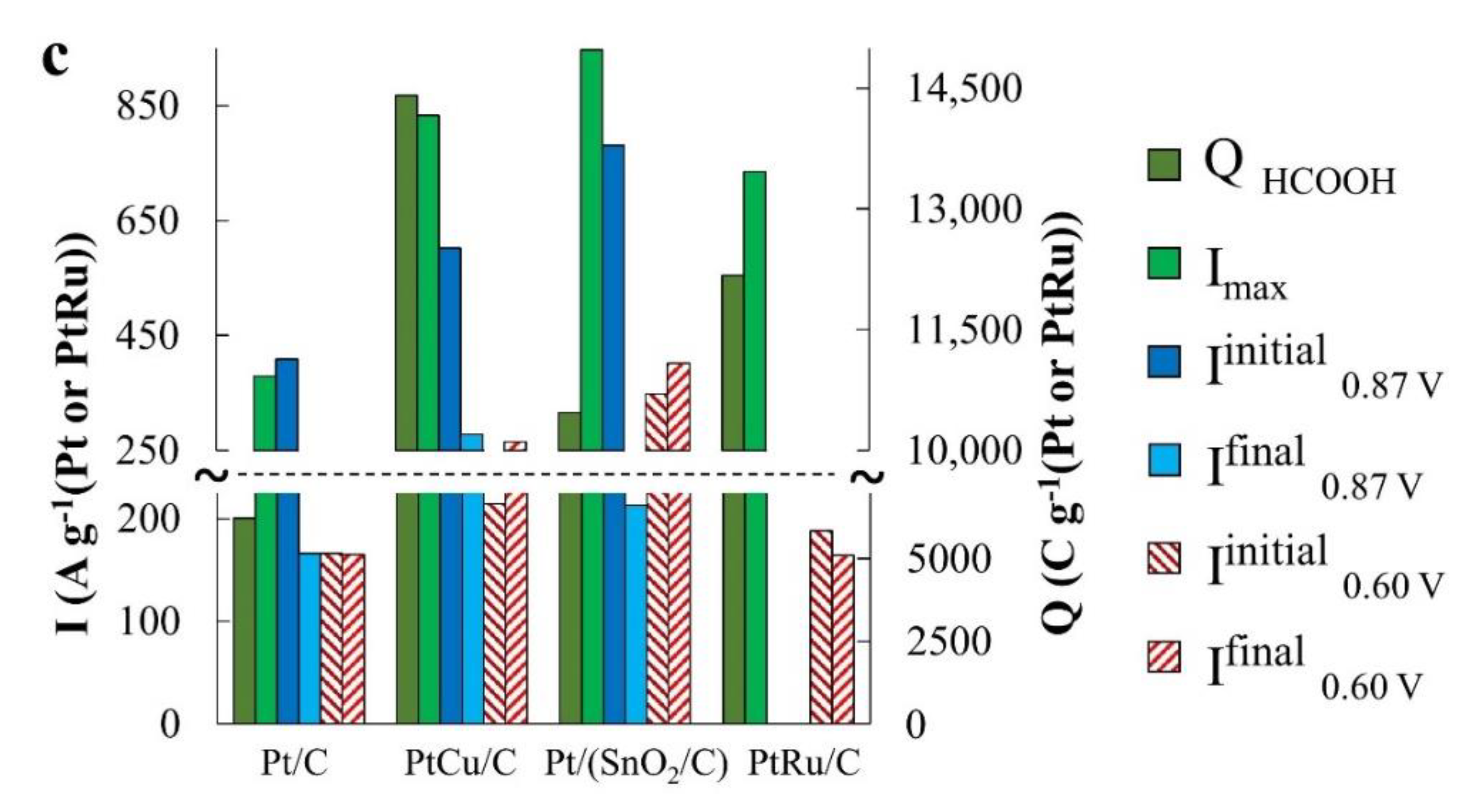
During the oxidation of formic acid, the mass activity of the catalysts, estimated from the
Imax and
QHCOOH values, increases in the order Pt/C < Pt/(SnO
2/C) ≈ PtCu/C (
Figure 8a and
Figure 6c). The electric current on the anode chronoamperograms at E = 0.87 V falls most slowly on the PtCu/C catalyst (
Figure 8b and
Figure 6c), which indicates its highest tolerance to oxidation products at this potential, most likely to CO. (Current fluctuations observed in chronoamperograms of formic acid oxidation (
Figure 8b,c) are due to intense gas evolution). We note that in the case of FAOR, the anode peaks on the CVs reverse branches are much higher than on the forward curves, and the potentials of the onset of oxidation decrease in a series corresponding to a decrease in mass activity: Pt/(SnO
2/C) > PtCu/C ≥ Pt/C (
Figure 8a). It is interesting that the peak of the reverse stroke and the corresponding amount of electricity for the Pt/(SnO
2/C) catalyst are much higher than for PtCu/C (
Figure 8a). A distinctive feature of the FAOR process is also the presence of two maxima on the branches of the forward course of the CV sweep at potentials of ~0.66 and ~1.1 V (inset in
Figure 8a). It is known that the oxidation of formic acid on the Pt surface can proceed by two mechanisms [
43]: I—dehydrogenation with the formation of CO
2 and II—dehydration with the formation of a catalytic poison—CO. Obviously, the dehydrogenation pathway is the preferable pathway for the oxidation of formic acid. The oxidation peak in the forward sweep of the CV potential at ~0.66 V corresponds to the dehydrogenation pathway [
43].
The greater its intensity, the greater is the activity of the catalyst in the implementation of the preferred reaction mechanism. In the voltammograms of PtCu/C and Pt/(SnO2/C), the intensity of this peak is higher than that of Pt/C, however, the second peak associated with formic acid dehydration is also significantly more intense on these catalysts.
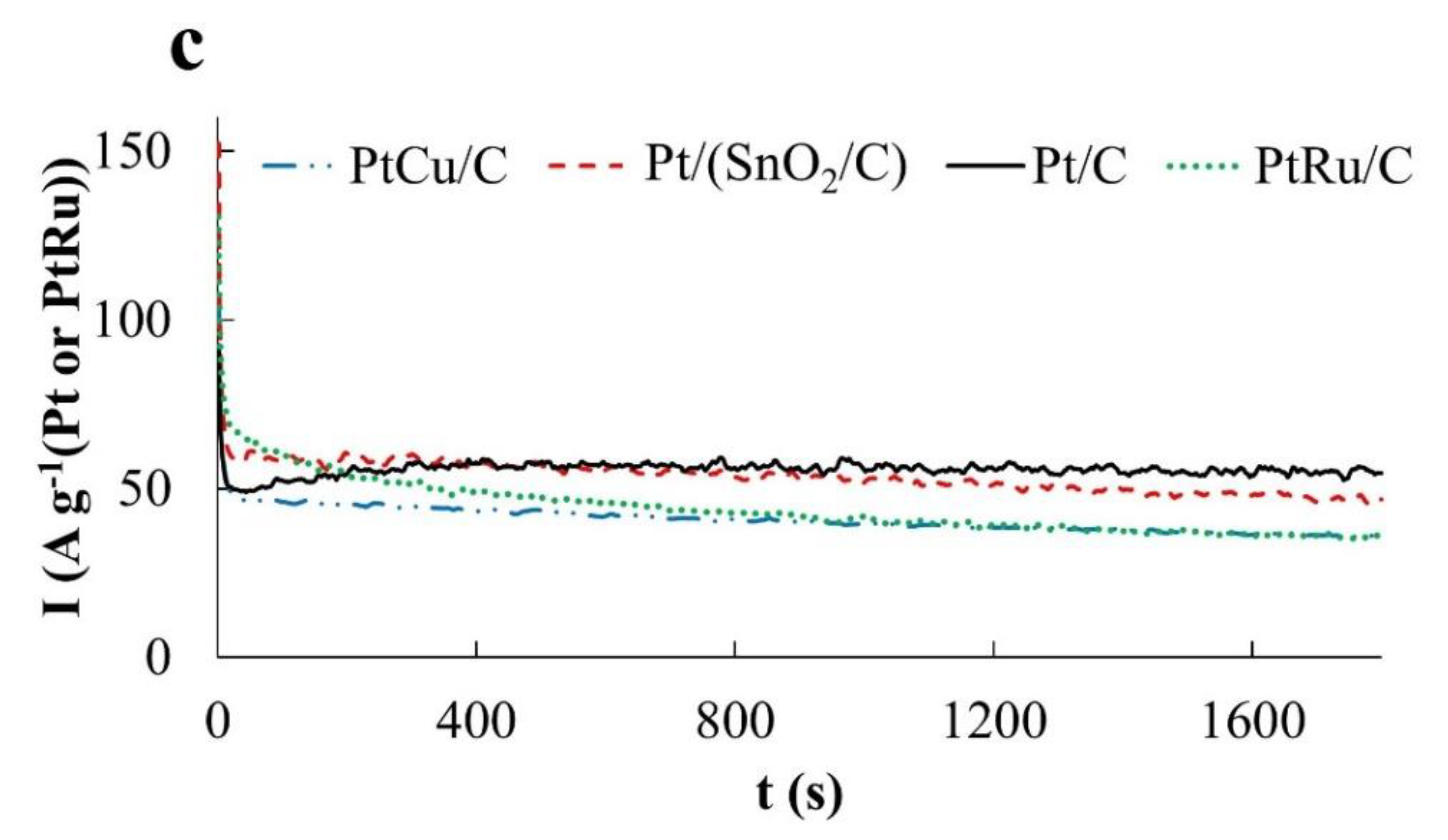
Chronoamperograms of FAOR at a potential of 0.60 V differ from those for MOR and EOR by a significantly higher reaction rate (high currents) (
Figure 8c). This may be due to the predominant oxidation of HCOOH to CO
2, that is, the absence of significant amounts of CO passivating the electrode. Note that already 300 s after the start of measurements, the FAOR rate on the PtCu/C and Pt/C catalysts exceeds that on the PtRu/C electrode. The highest activity in FAOR is demonstrated by the Pt/(SnO
2/C) catalyst: Its mass activity is about 1.6 times higher than the mass activity of PtCu/C, Pt/C, and PtRu/C catalysts (
Figure 8c). Interestingly, the oxidation current of formic acid on the Pt/(SnO
2/C) catalyst at a potential of 0.60 V is higher than at a more positive potential of 0.87 V.
The study showed that PtCu/C catalyst exhibits higher activity in the reactions of electrooxidation of organic substances in comparison with Pt/C at high potentials, while the Pt/(SnO
2/C) catalyst is the most active among the studied samples and is tolerant to the resulting intermediates under conditions where electrooxidation occurs at a potential of 0.60 V. The reasons for the increased activity of Pt/(SnO
2/C) and PtCu/C catalysts compared to Pt/C in MOR were previously discussed in [
32,
33,
44] and [
33,
34,
35], respectively. The presence of Pt and SnO
2 nanoparticles attached to the surface of a carbon support can facilitate the implementation of a bifunctional catalysis mechanism by facilitating the adsorption of hydroxyl groups on the surface of tin dioxide nanoparticles and their participation in the conversion of CO molecules (an intermediate product of organic substances oxidation) on the surface of neighboring platinum nanoparticles. High activity of bimetallic catalysts in MOR is associated with promoting effect of metal on platinum activity, due to the electronic interaction d-orbitals, and the decrease in the interatomic distance in the alloy structure, compared with the structure of pure platinum [
45]. The same reasons can result in increased activity of PtCu/C and Pt/(SnO
2/C) catalysts in EOR and FOR. While considering the previously noted specificity of CVs anodic branches during the reverse course of the potential sweep, one can also speak about the influence of the alloying component and SnO
2 nanoparticles both on the rate of electrooxidation reactions, and on the oxidation state of the platinum surface. In its turn, this affects the catalysts activity in the reactions of organic compounds electrooxidation.
4. Conclusions
The study of the methanol, ethanol and formic acid electrooxidation on Pt/C, Pt/(SnO2/C) and PtCu/C electrocatalysts containing about 20% wt. of platinum has shown that under certain conditions both modified catalysts significantly exceed Pt/C in terms of mass activity in all reactions. At the same time, the values and ratio of the catalyst activities substantially depend on the electrode potential.
The integral activity of catalysts in MOR, EOR, and FAOR can be estimated from the results of voltammetric measurements by comparing the maximum currents and the amount of electricity consumed for the oxidation of the organic reagent during the forward sweep of the potential over a wide range of values. The bimetallic catalyst demonstrates the highest activity in MOR, while the mass activities of both modified catalysts in EOR and FAOR are close to each other. The specific activity of the bimetallic catalyst differs even more from those for Pt/(SnO2/C) and, especially, Pt/C, since the PtCu/C ESA is almost 2 times lower than that of the other two samples. Under potentiostatic conditions at E = 0.87 V, PtCu/C and Pt/(SnO2/C) catalysts also exhibit higher activity as compared to Pt/C.
At a potential of 0.60 V, the oxidation of methanol and ethanol proceeds most slowly on the PtCu/C catalyst. The Pt/(SnO2/C) catalyst demonstrates in this case the specific activity and tolerance, which are practically not inferior or even superior to those of the commercial PtRu/C catalyst. No significant differences in the activities of Pt/C and Pt/(SnO2/C) in EOR at a potential of 0.60 V were found. At the same time, the mass activity of the Pt/(SnO2/C) catalyst in FAOR at 0.60 V is approximately 1.6 times higher than that of the PtCu/C, Pt/C catalysts and 2.4 times higher than mass activity of the PtRu/C.
Thus, the platinum–copper catalyst demonstrates the highest activity in the oxidation of organic substances at high potentials, while the Pt/(SnO2/C) catalyst exceeds Pt/C in activity and tolerance to intermediate oxidation products in a wider range of potentials. At the same time, at E = 0.60 V, it slightly exceeds the PtRu/C catalyst in MOR & EOR and significantly surpasses it and other catalysts in FAOR.
The influence of the reagent nature subjected to electrooxidation is manifested both in the difference of the absolute values of the rates of the corresponding electrooxidation reactions, decreasing in according with CVs results in the order CH3OH > HCOOH > C2H5OH, and in the different ratio of these rates on different catalysts.
The reasons for the positive effect of tin nanoparticles, which came into contact with platinum, and doping of platinum with copper on its activity in the studied electrooxidation reactions are apparently different. We believe that for Pt/(SnO2/C) catalyst this is a bifunctional catalysis mechanism; for PtCu/C it is the promoting effect of copper atoms on platinum activity due to the electronic interaction of d-orbitals and crystal lattice contraction. Apparently, platinum alloying with copper also affects the features of the chemisorption of oxygen-containing intermediates on the surface of the metal nanoparticles. In this regard, catalysts in which bimetallic PtCu nanoparticles are deposited on a SnO2/C composite support may be of essential interest. In conclusion, we would like to note that the advantages of the PtCu/C catalysts for the oxidation of simple organic substances in comparison with Pt/C are more pronounced at high positive potentials, thus it may be efficiently used on the oxygen electrode of the fuel cells with organic fuels. A Pt/(SnO2/C) catalyst can compete with PtRu/C at the anode of such fuel cell.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}