
Ordered Nanostructure Catalysts Efficient for NOx Storage/Reduction (NSR) Processes

Abstract
:1. Introduction
1.1. Overview of the Development on NSR Catalysts
1.2. Application of Nanotechnology for NSR Processes
2. Results and Discussion
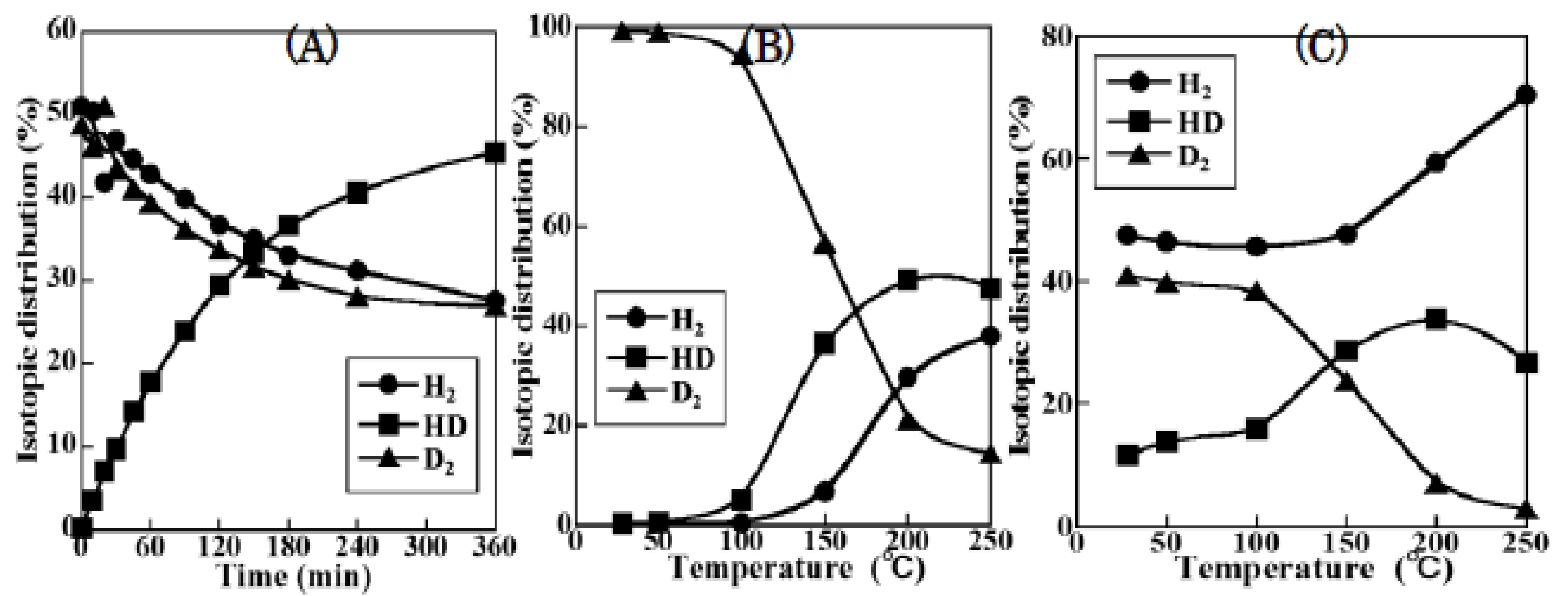
2.1. NSR Behavior over Various Nano Structured Catalysts
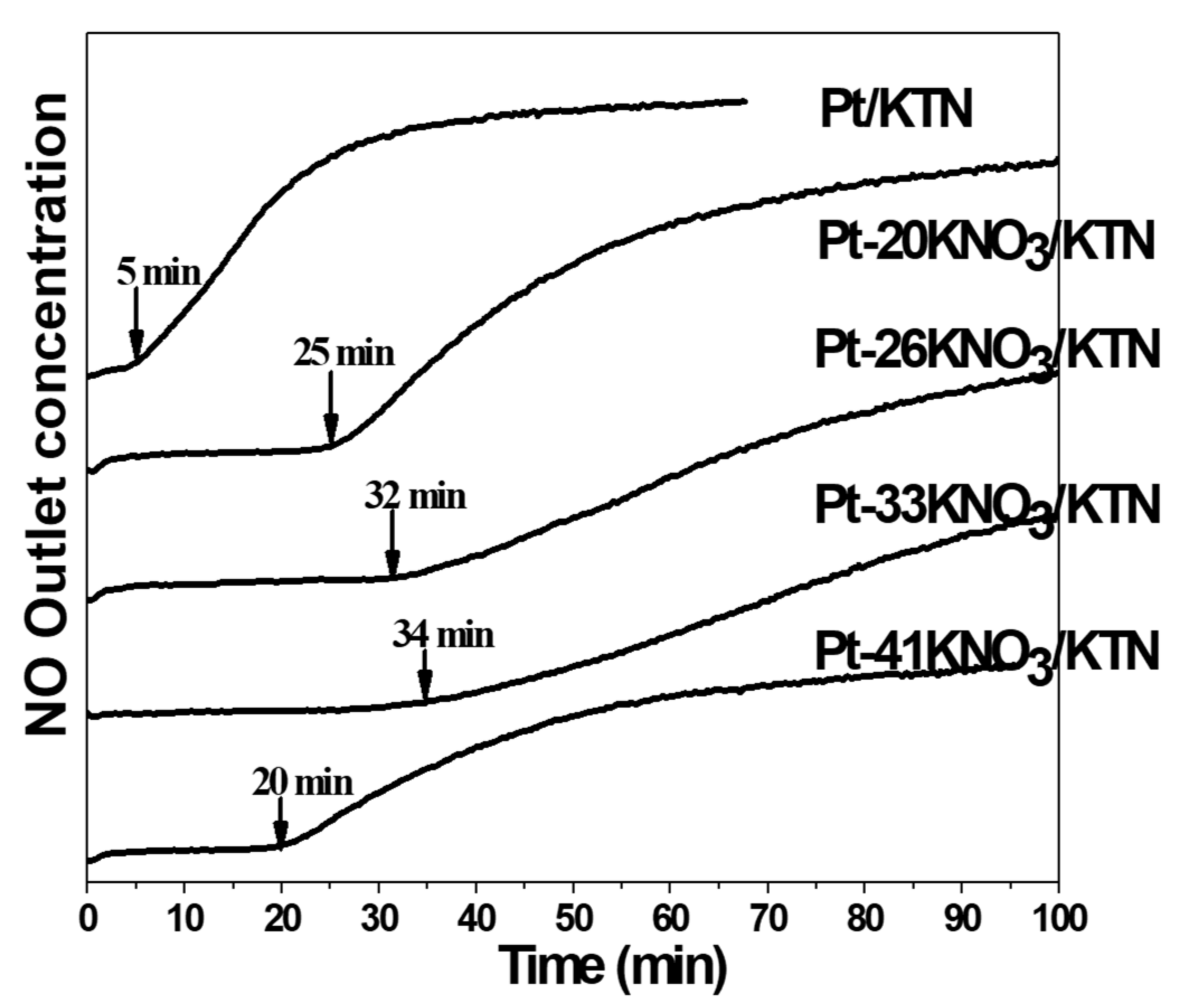
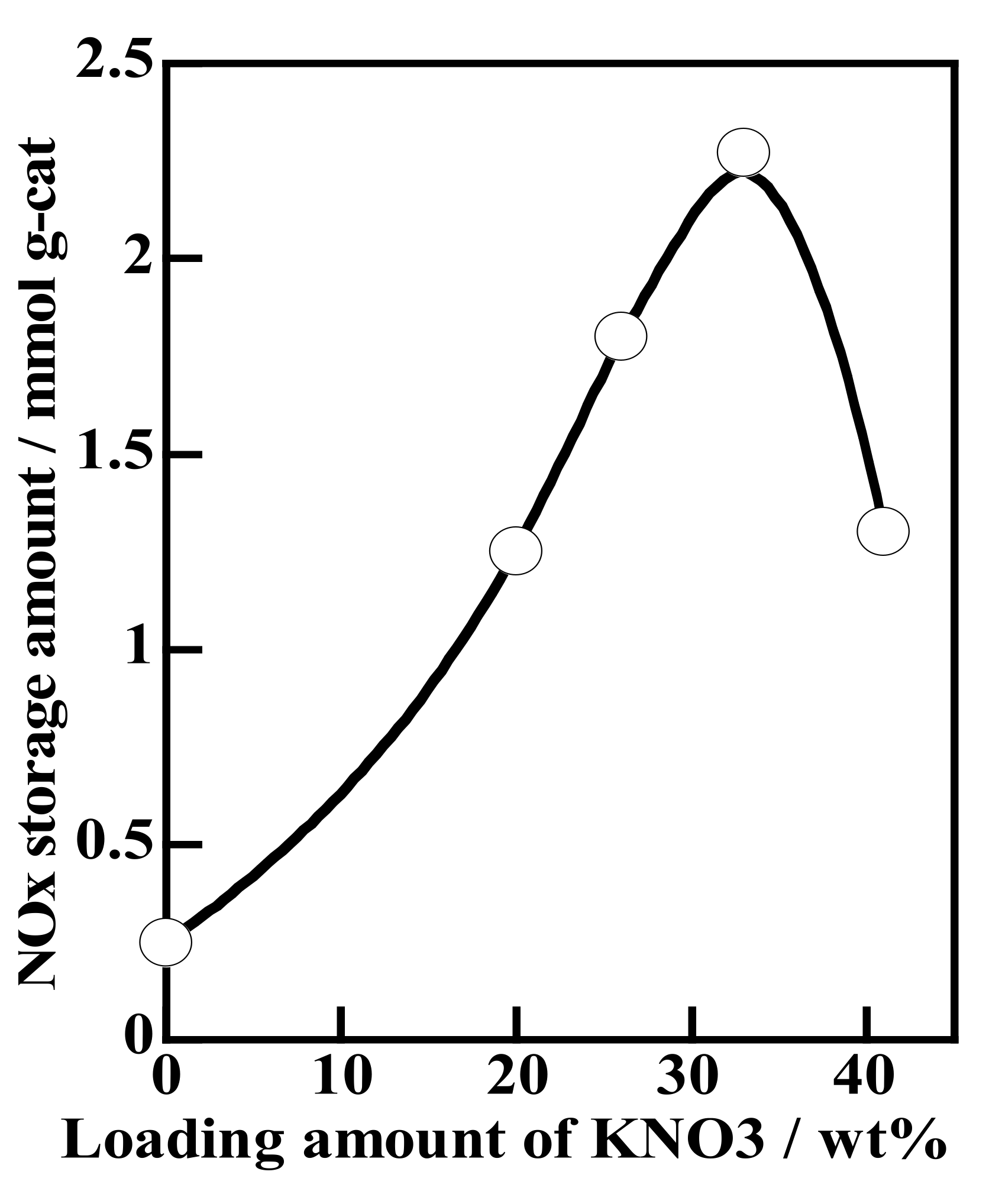
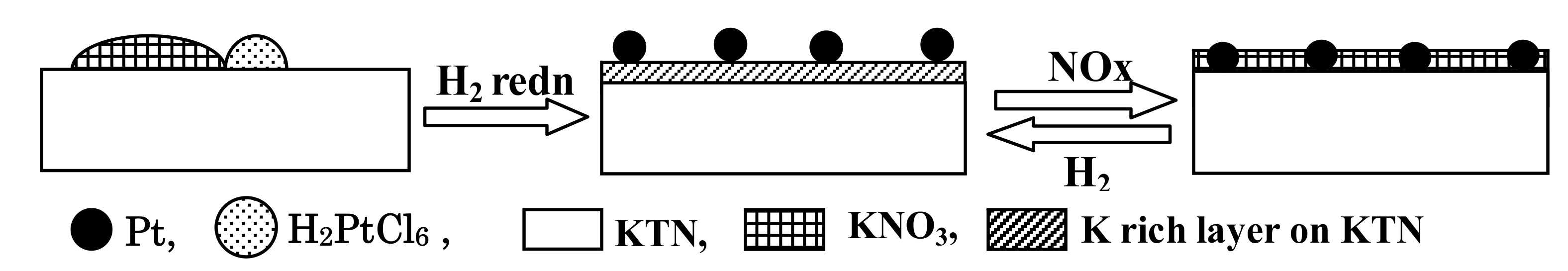
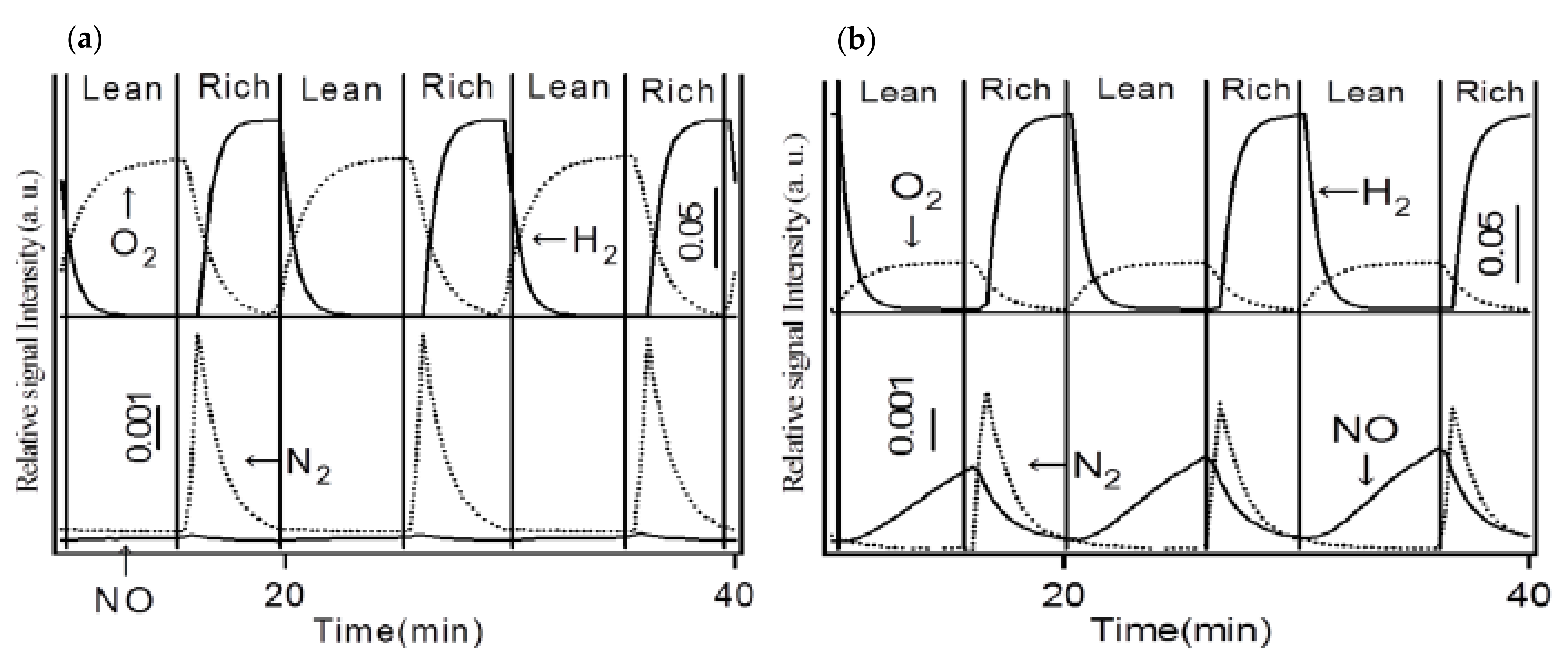
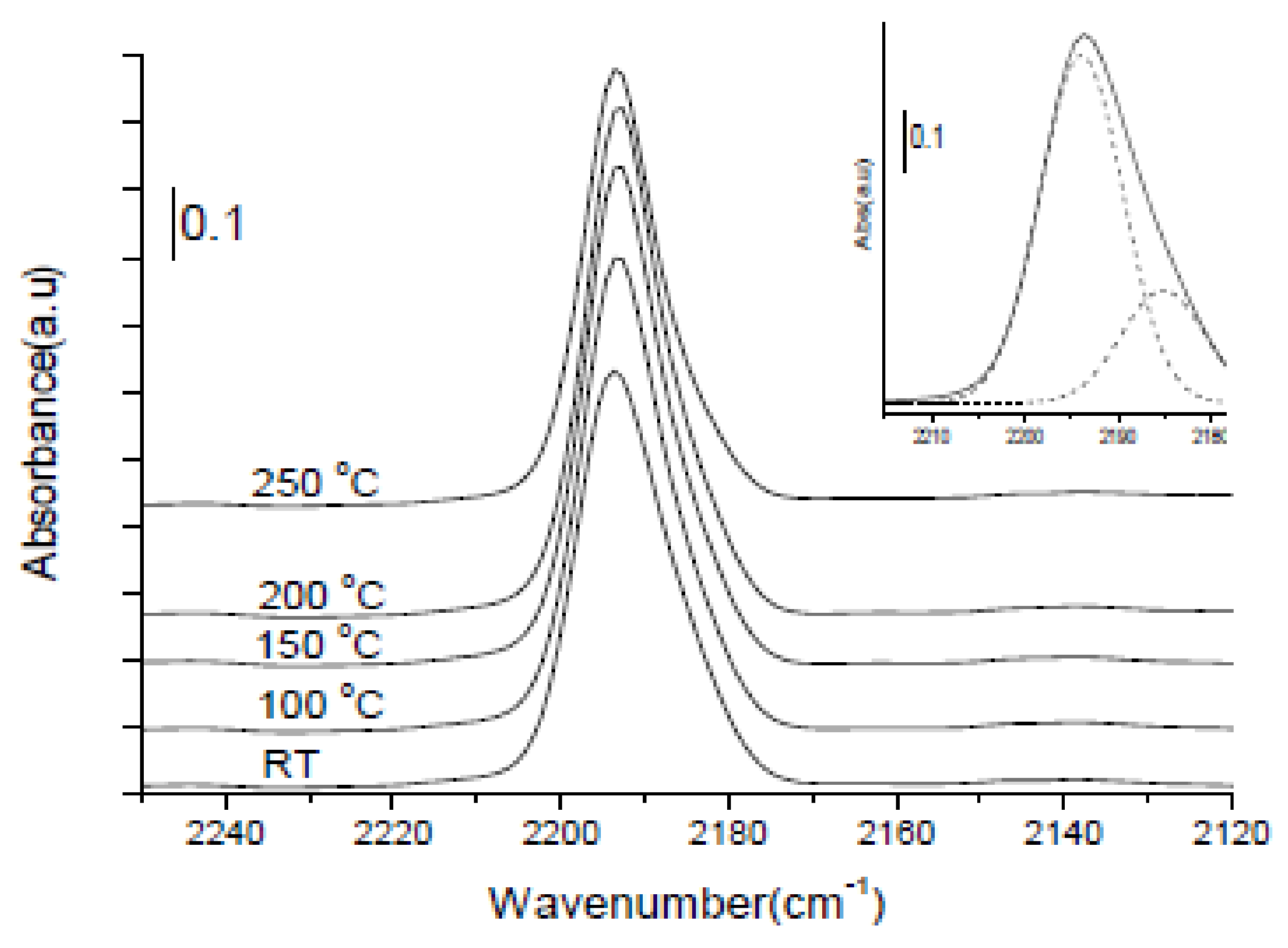
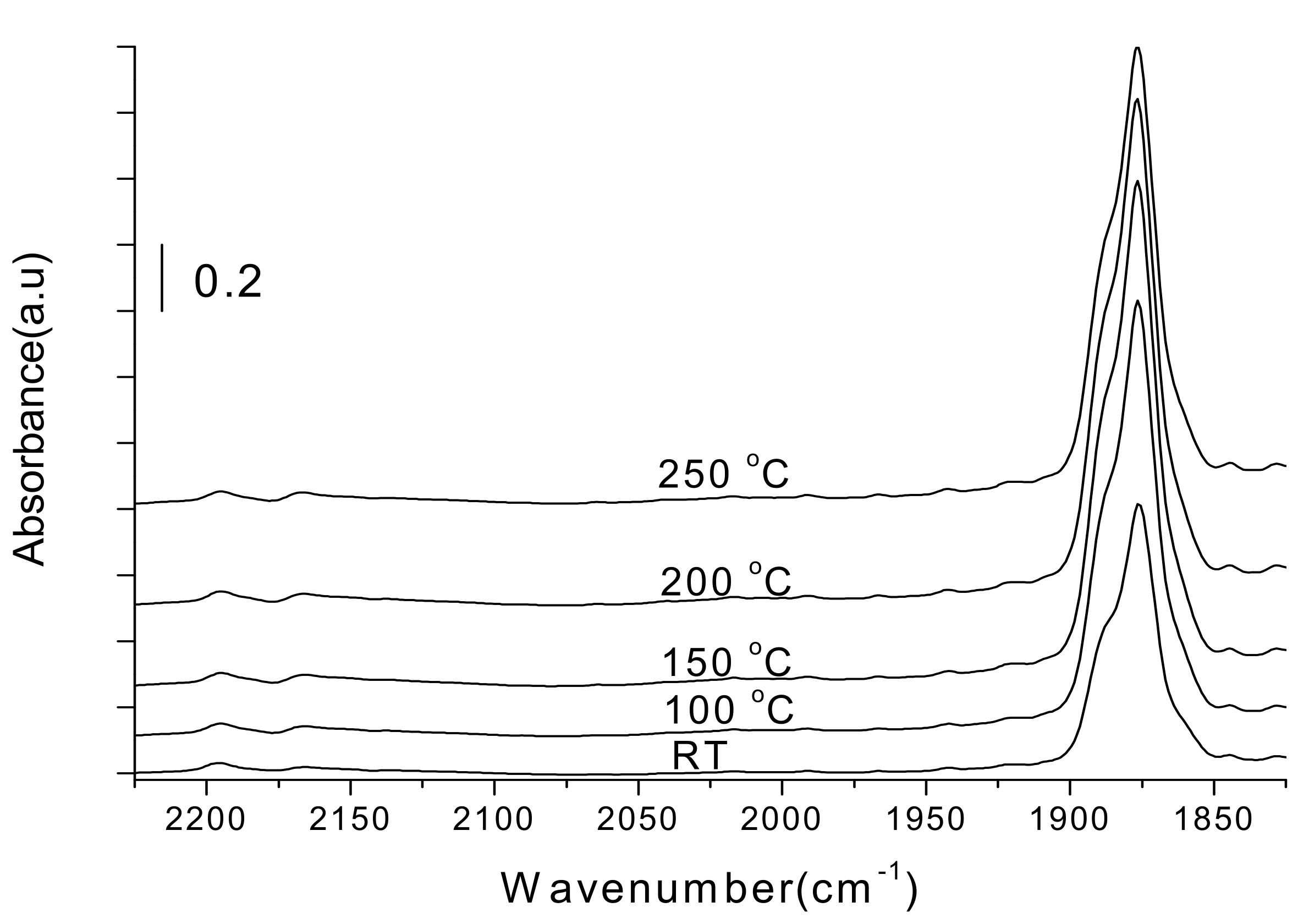
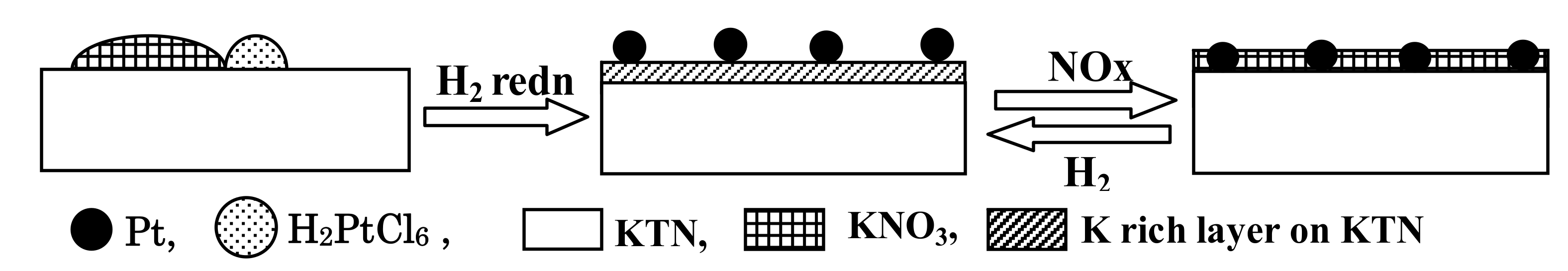
2.1.1. Potassium Cation Transfer Mechanism in NOx Storage and Reduction Processes over Pt/K-Titania Nanobelt Catalysts
2.1.2. Activity and Selectivity-Controlling Factors for NOx Storage-Reduction Process by Pt-Alkali Metal Nanocomposites Supported on TiO2 with Different Crystal Structures
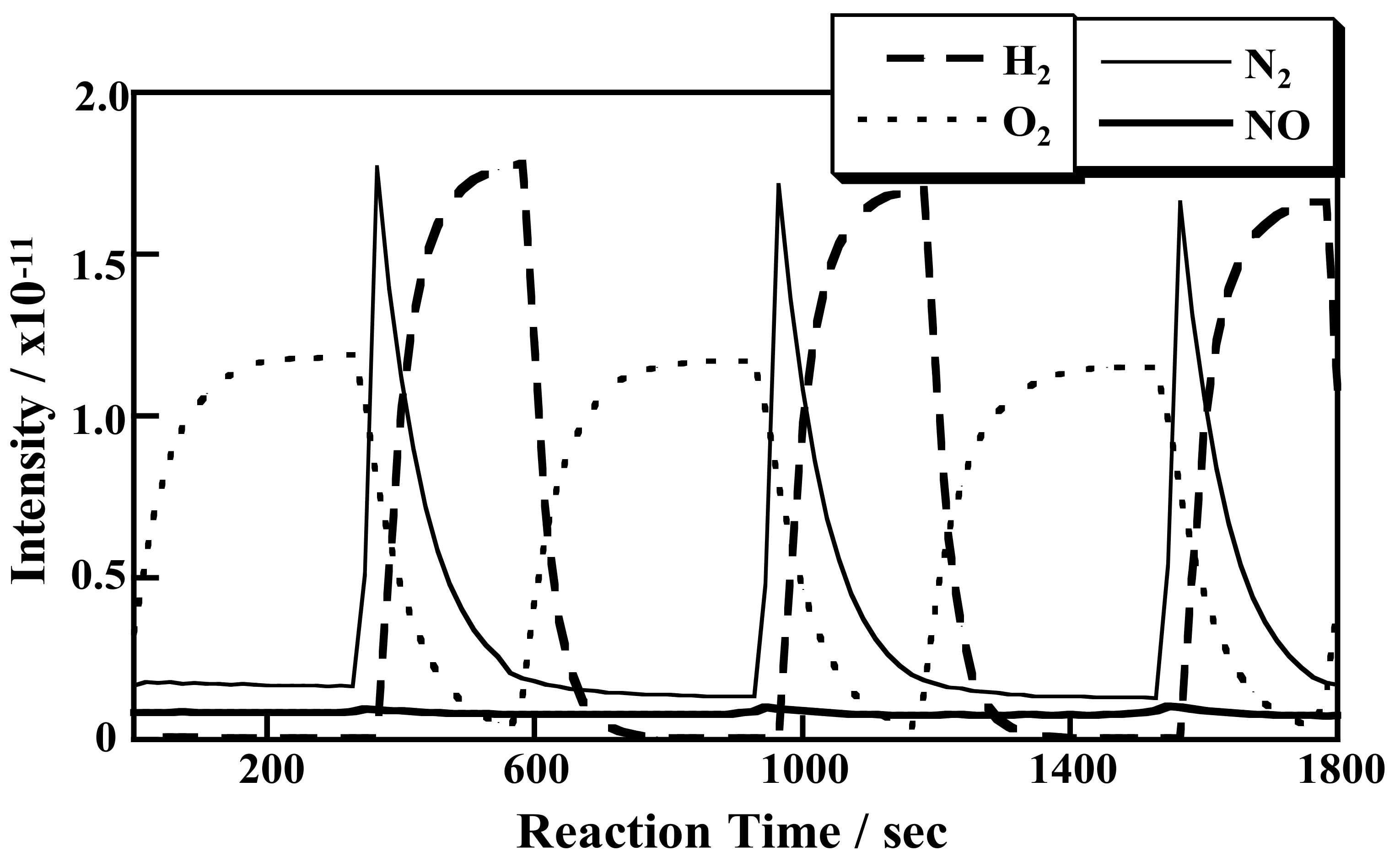
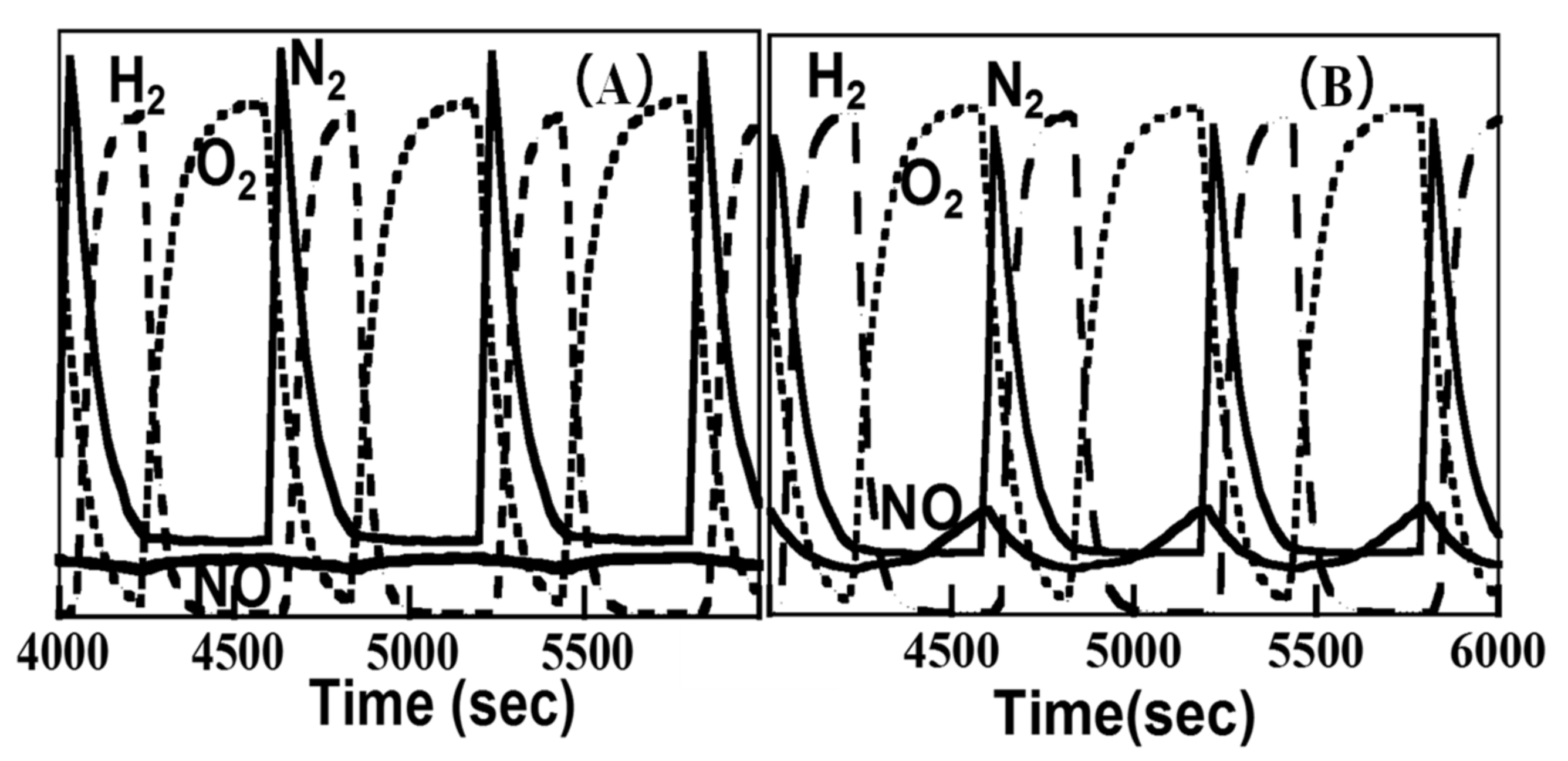
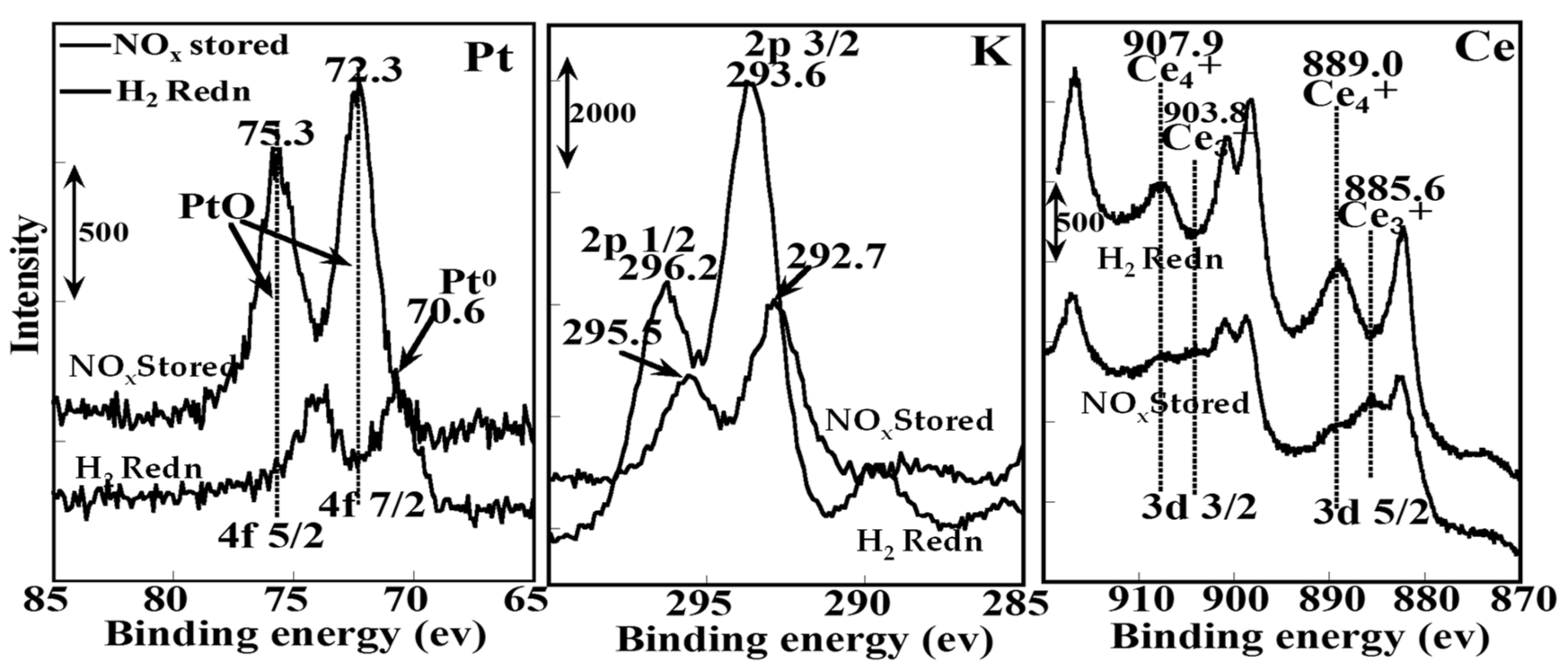
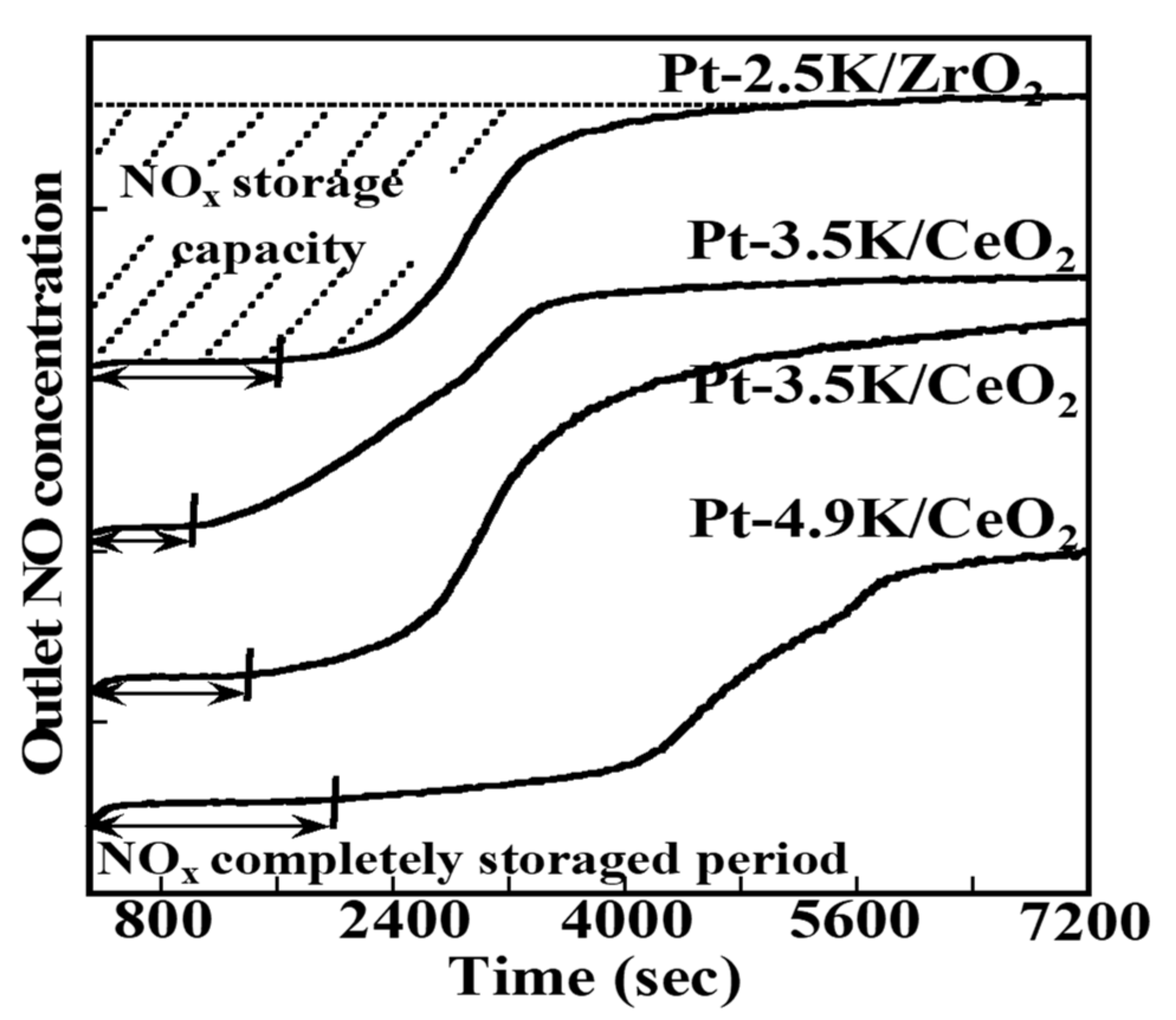
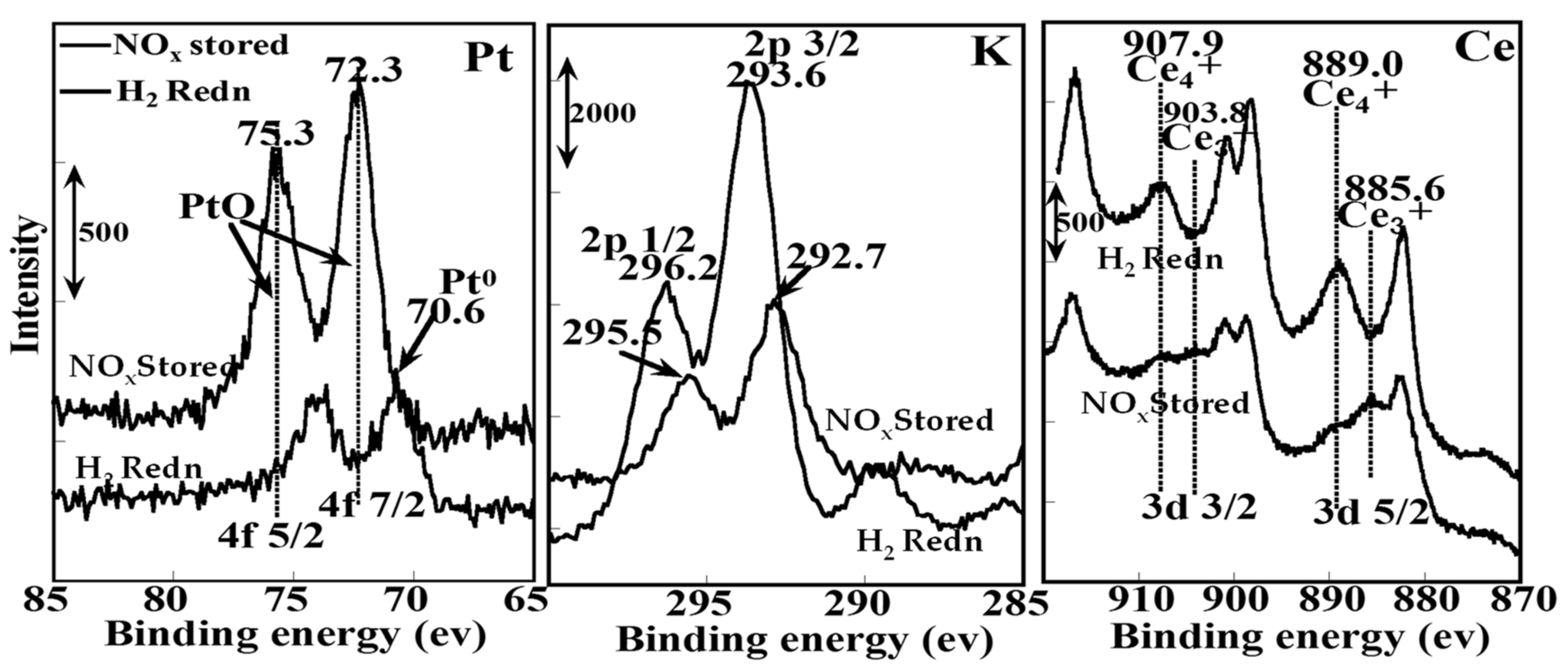
2.1.3. Mechanistic Study of NOx Storage and Reduction Behavior by In Situ XPS and FTIR over Pt/K/CeO2 and Pt/K/ZrO2 NSR Catalysts
2.1.4. Comparison of NOx Storage and Reduction Behaviors among Ceria-Supported Pt-KNO3, Cu-KNO3, and Co-KNO3 NSR Catalysts
2.1.5. Preparation of Nanoporous Nickel(II) Phosphate VSB-5 and Their Adsorption and Catalytic Behavior in NO-CO Reactions
2.1.6. Unusual Storage of NO and Its Efficient Removal by Various Reducing Agents inside One-Dimensional Micropores of Nickel (II) Phosphate VSB-5 Catalysts
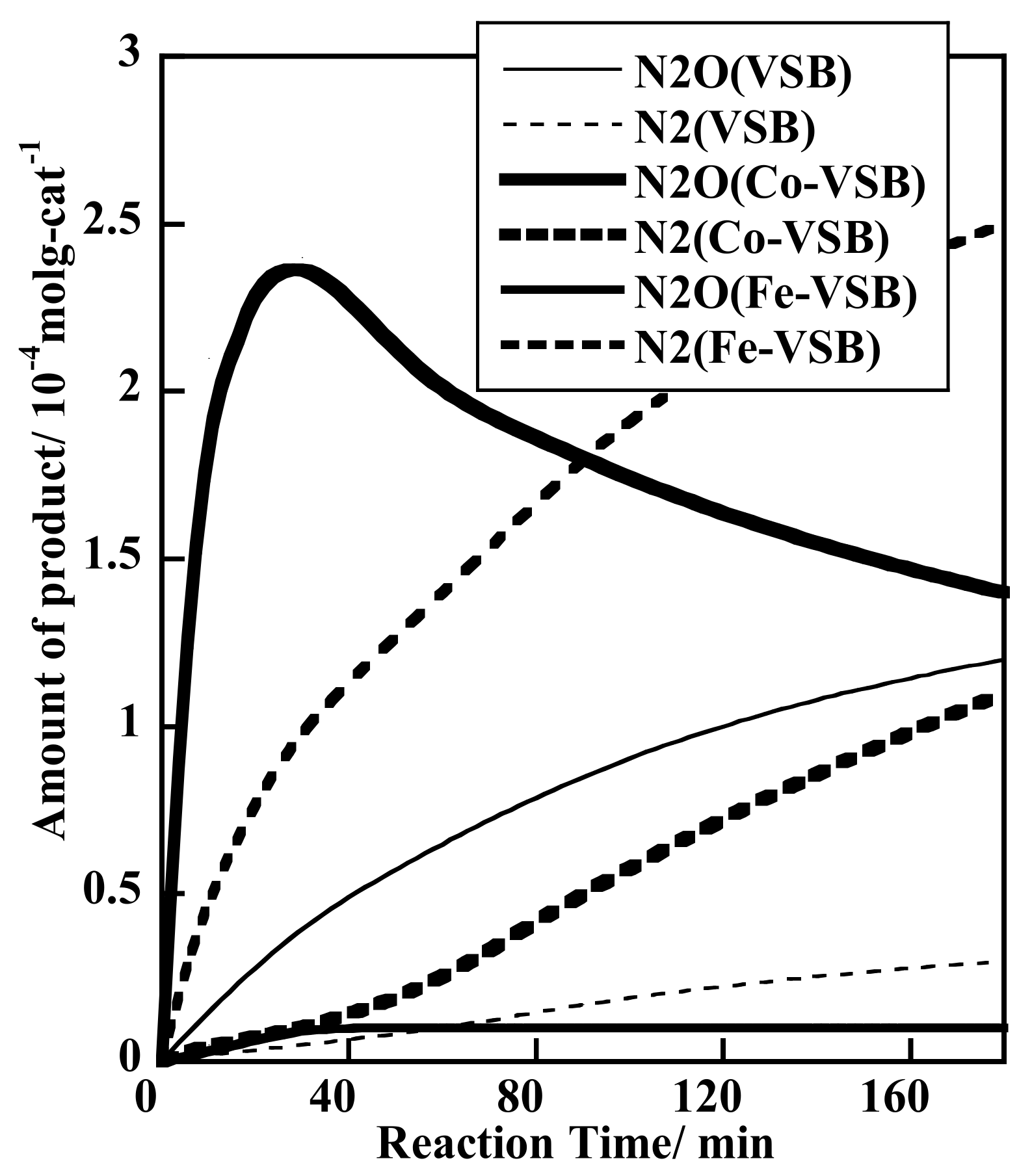
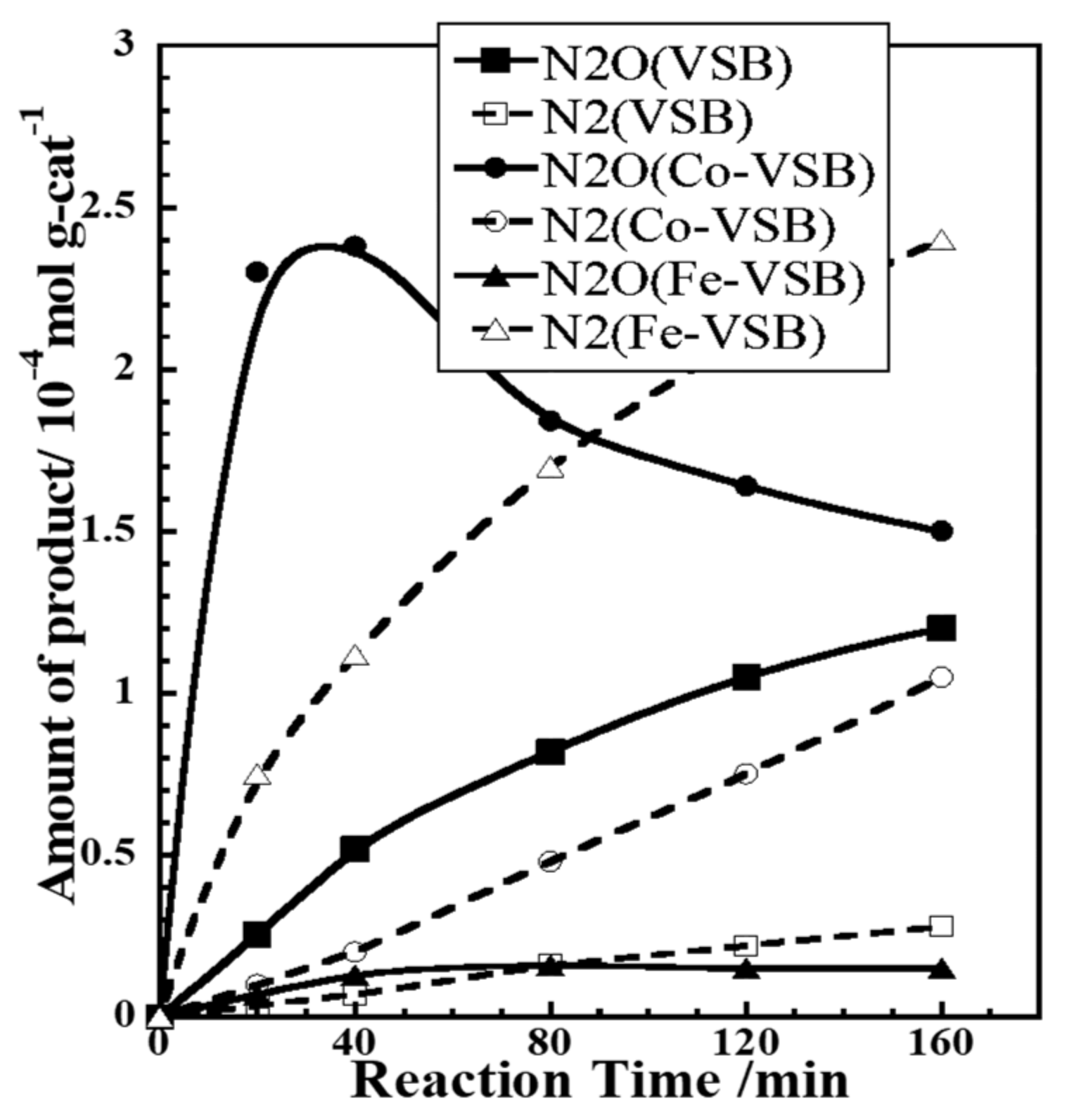
2.1.7. Effect of Co(II), Fe(II), and V(II) Substitution upon Unusual NOx Storage and Efficient Removal by Various Reducing Agents inside 1-D Channels of Microporous Nickel (II) Phosphate Catalysts
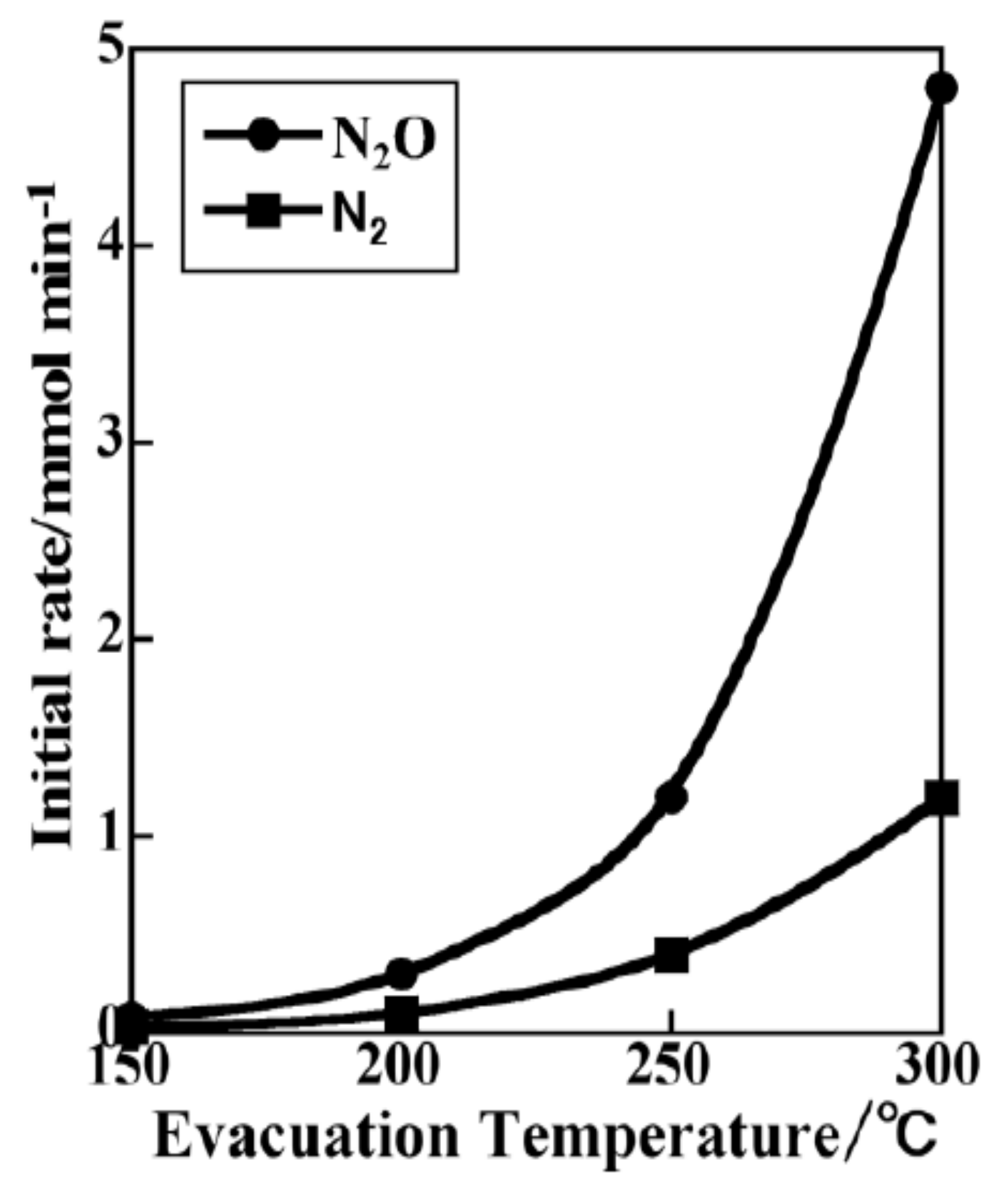
2.1.8. Catalytic Reduction of NO by H2 over Co-Substituted Nanoporous Ni Phosphates, VSB-5
3. Mechanistic Investigation and Design of NSR Catalysts
3.1. Transition-Metal Redox Sites
3.2. NOx Trapping Sites
- (a)
- NO oxidation step; as reviewed in the previous section, BaO presents some negative effects in NO oxidation as a result of its blockage of the Pt surface and its stabilization effect on Pt oxides (inactive for NO oxidation).
- (b)
- NOx storage step; because of the complexity of this step, it is difficult to identify and differentiate mechanisms that occurred during the NOx trapping process. Both nitrites and nitrates are detected on the NSR catalyst after NOx trapping. Therefore, it is well accepted that there must be a nitrite route and a nitrate route for the NOx trapping on BaO. Within the nitrite route, it is proposed that NO is oxidized on Pt sites and directly trapped by nearby BaO sites to form Ba nitrites. The Ba nitrites are finally oxidized to Ba nitrates. Within the nitrate route, it is proposed that NO is oxidized to NO2 on Pt sites. NO2 spills over to the BaO site to form Ba nitrate with the evolution of NO. When Ba loading is high or low, there are more or fewer Ba sites that are close to the Pt sites, which leads to the predominance of the nitrite route or nitrate route. At high temperatures, no matter whether NO or NO2 is used as the NOx precursor, similar NOx trapping capacity is observed on the Pt/BaO/Al2 O3 catalyst. This is because NO can be easily oxidized to NO2 at high temperatures. However, at low temperatures, the NOx-trapping capacity of BaO is low when using NO as a NOx precursor due to limited NO oxidation into NO2. With NOx trapping BaO, the H2O and CO2 in the exhaust could react to produce barium hydroxide and carbonate. The thermodynamic stability of BaCO3 is higher than that of Ba (OH)2. In contrast to BaO, in order to trap NOx, Ba(OH)2 and BaCO3 must decompose first and then react with NOx. Therefore, the presence of H2O and CO2 in the exhaust gas phase will affect the rate of NOx trapping. In addition, CO2 decreases more NOx-trapping capacity than H2O.
- (c)
- NOx release step; the release of NOx is very important for efficient regeneration of NOx-trapping sites and the following NOx reduction process. NOx release is induced by the decomposition of Ba nitrites and Ba nitrates upon high temperature and rich excursion (reductant introduction). The stability of Ba nitrites and Ba nitrates decreases with increasing temperature and decreasing O2 partial pressure, causing their decomposition so as to release NOx. Apart from the reductant gases and O2, the presence of other gases, such as H2O and CO2, also affects NOx release during the rich-burn cycle. The presence of H2O reduces NOx release, while CO2 increases NOx release.
3.3. Surface Structure for Storage
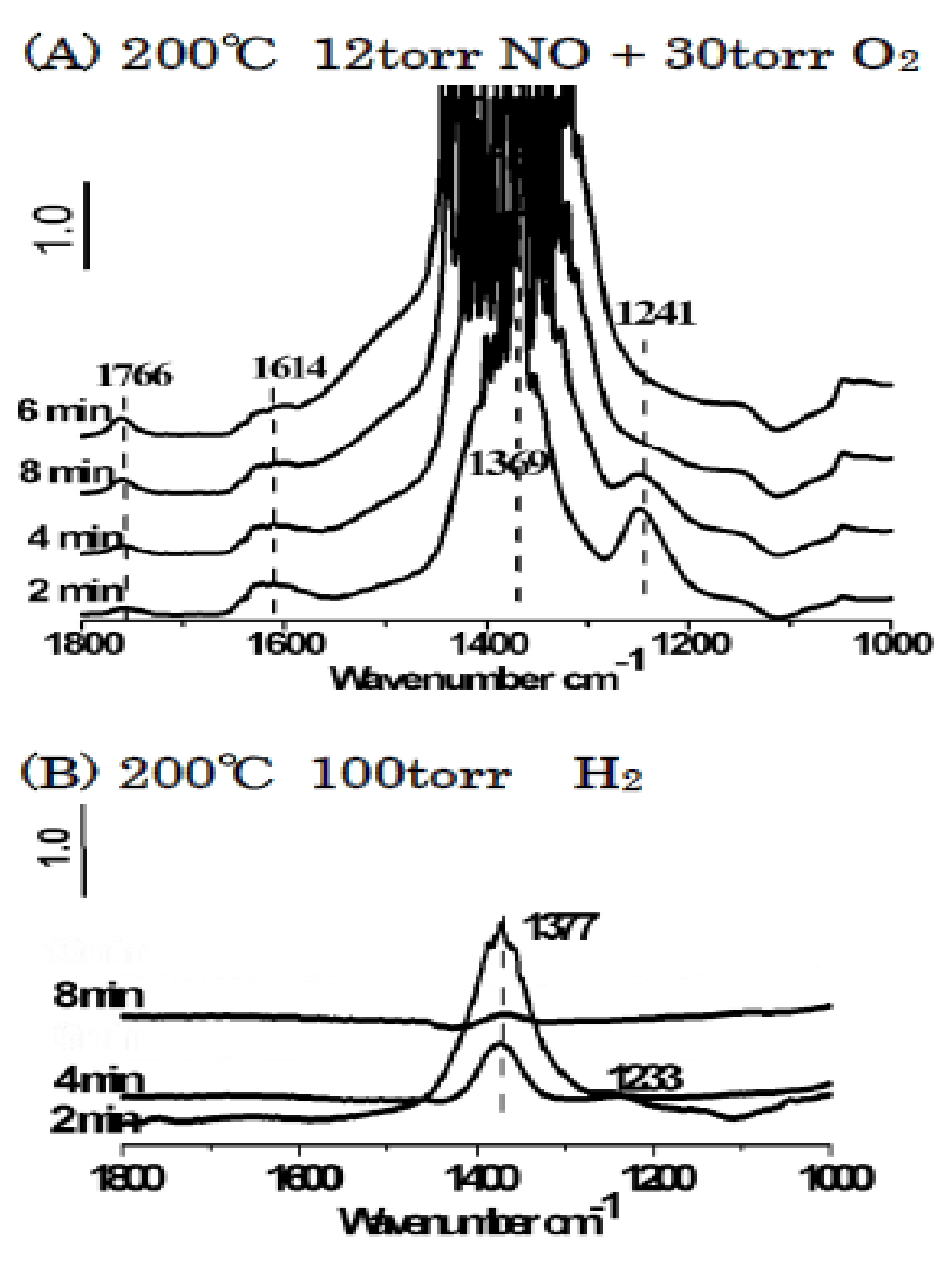
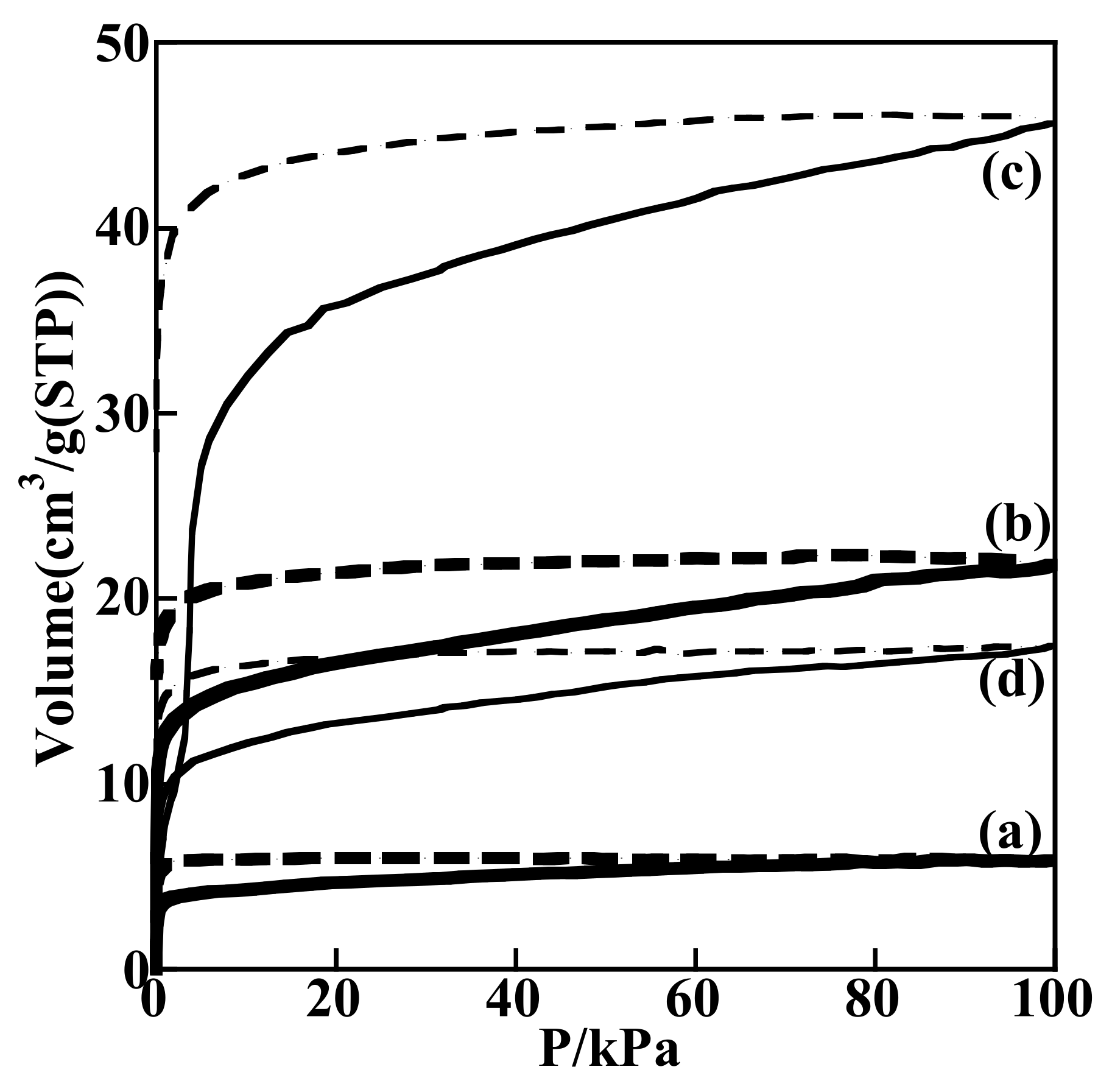
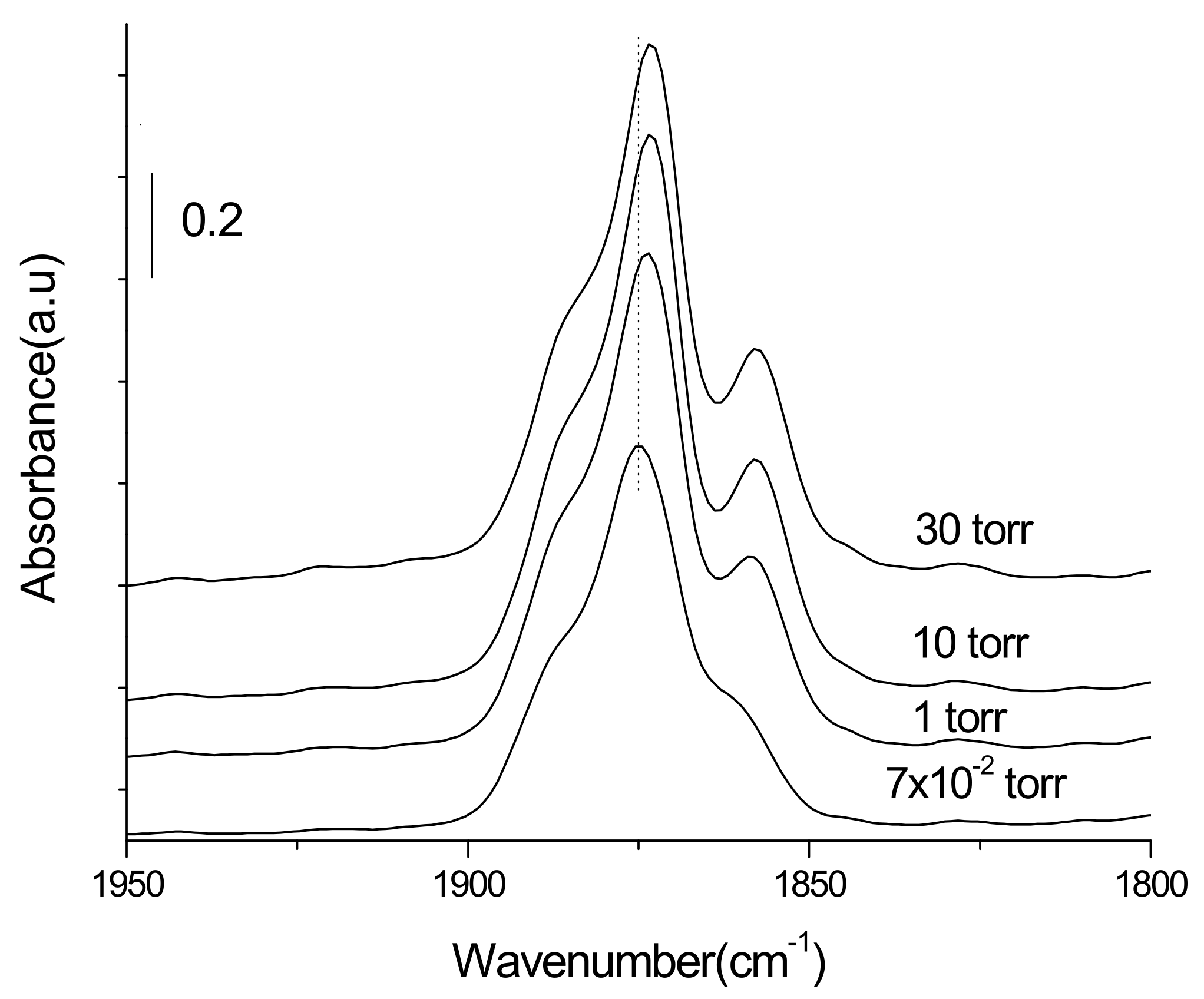
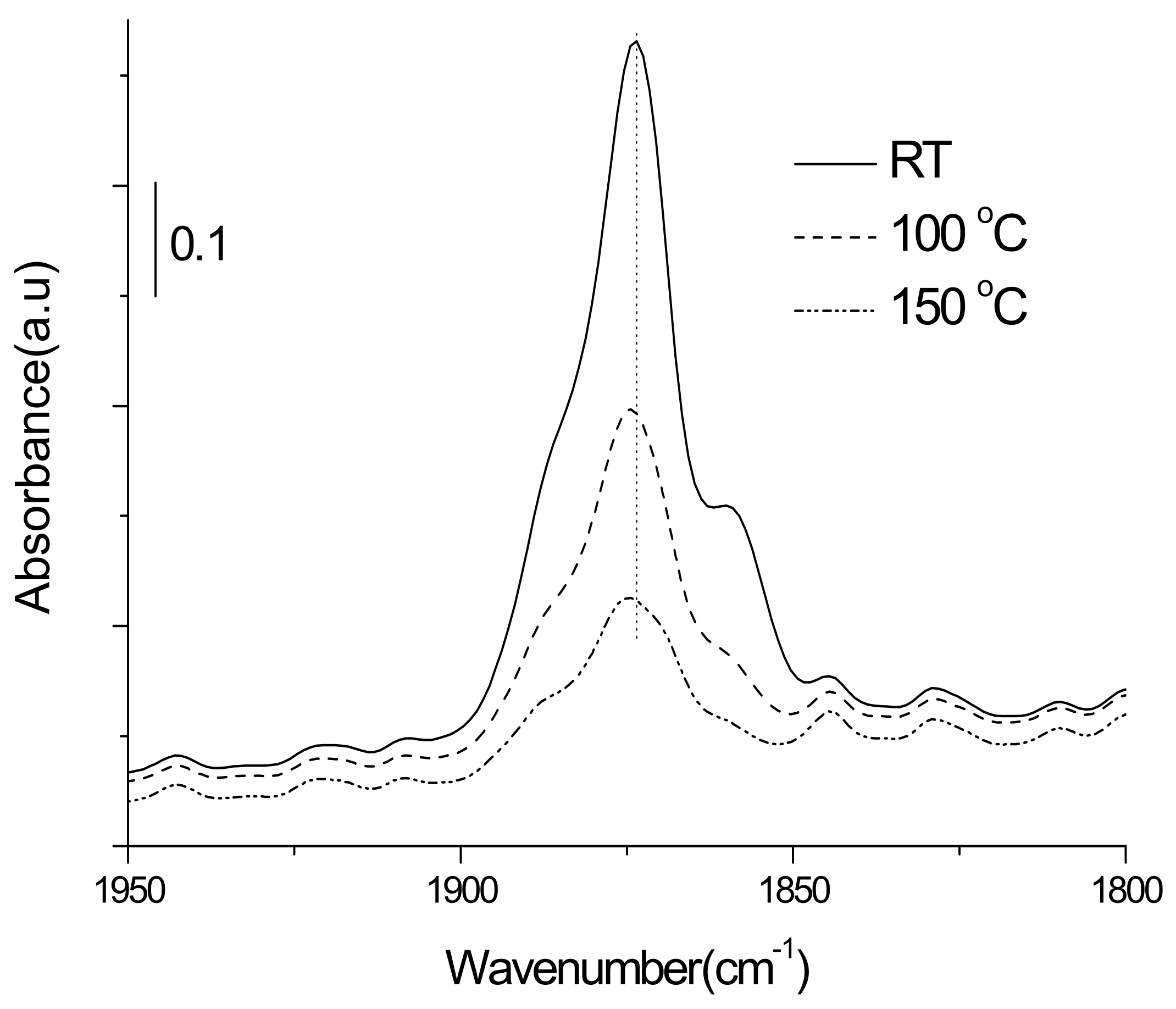
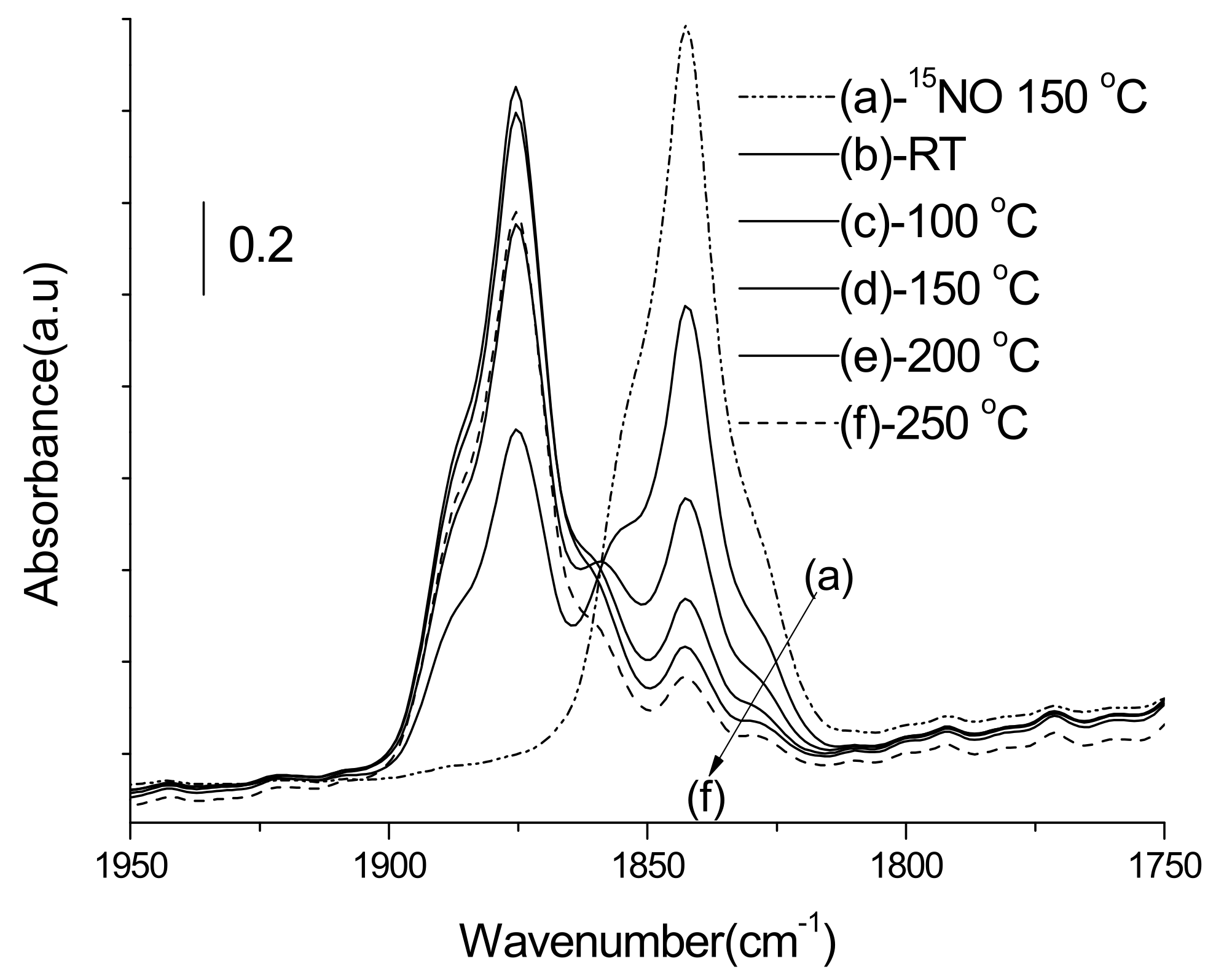
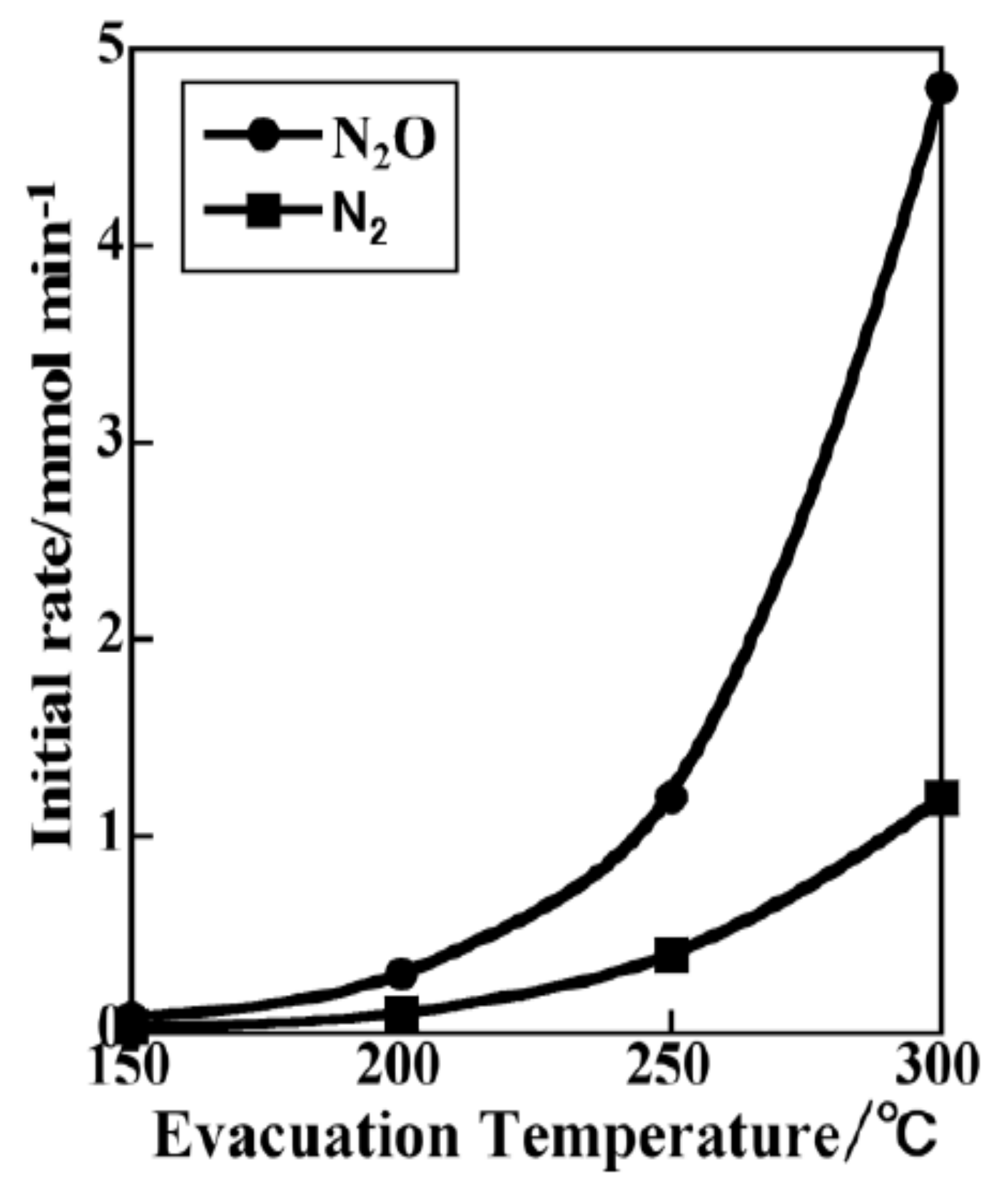
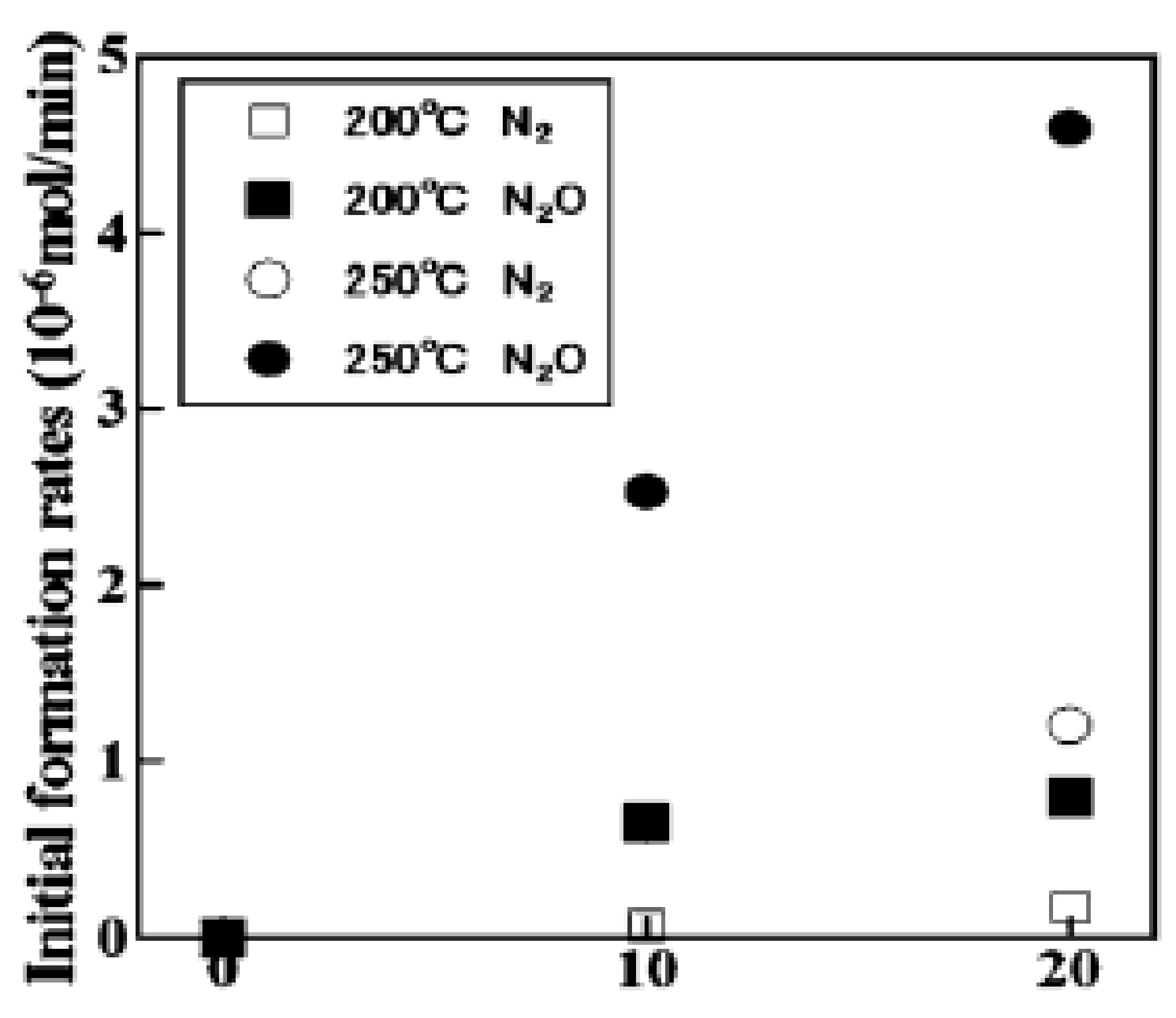
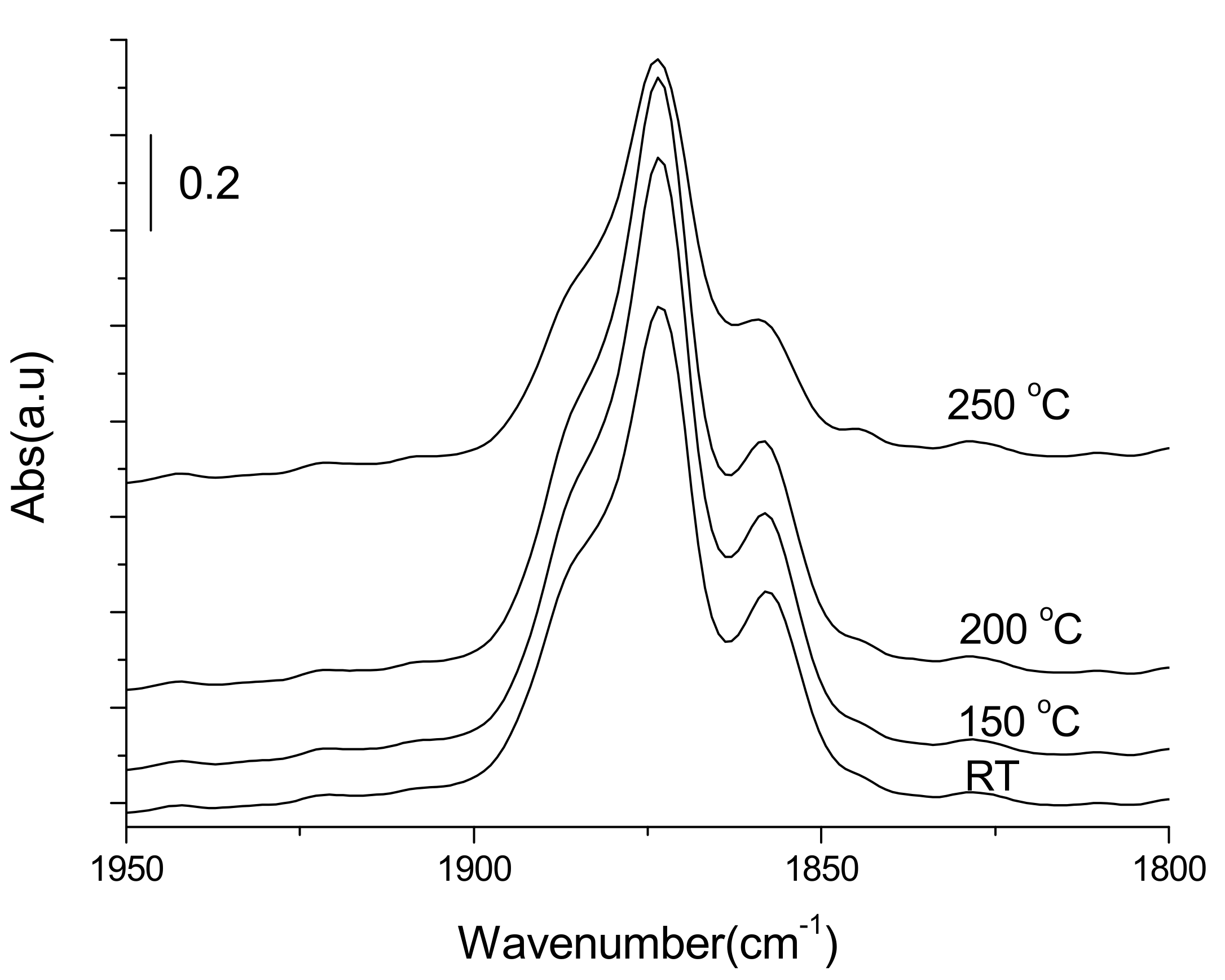
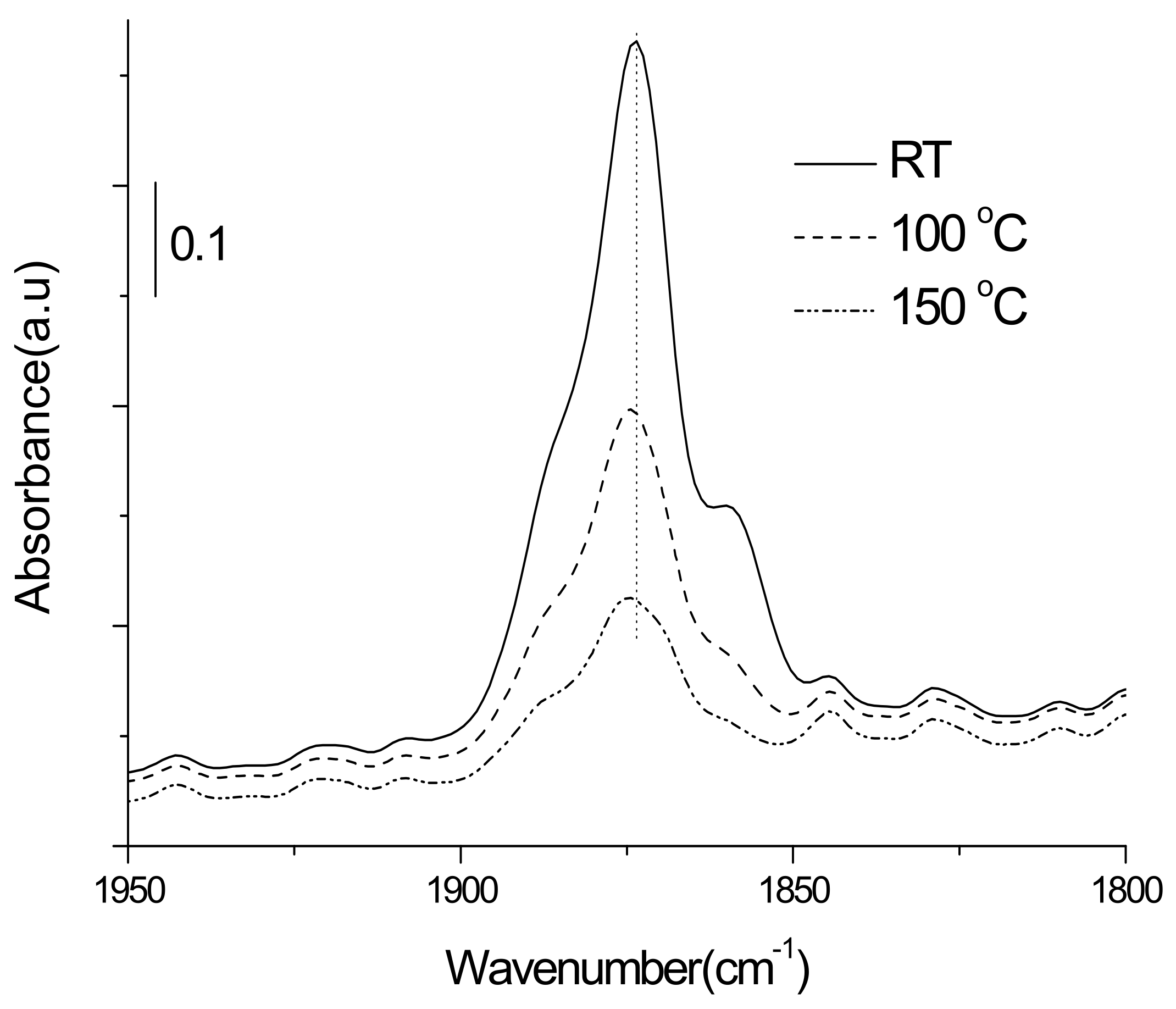
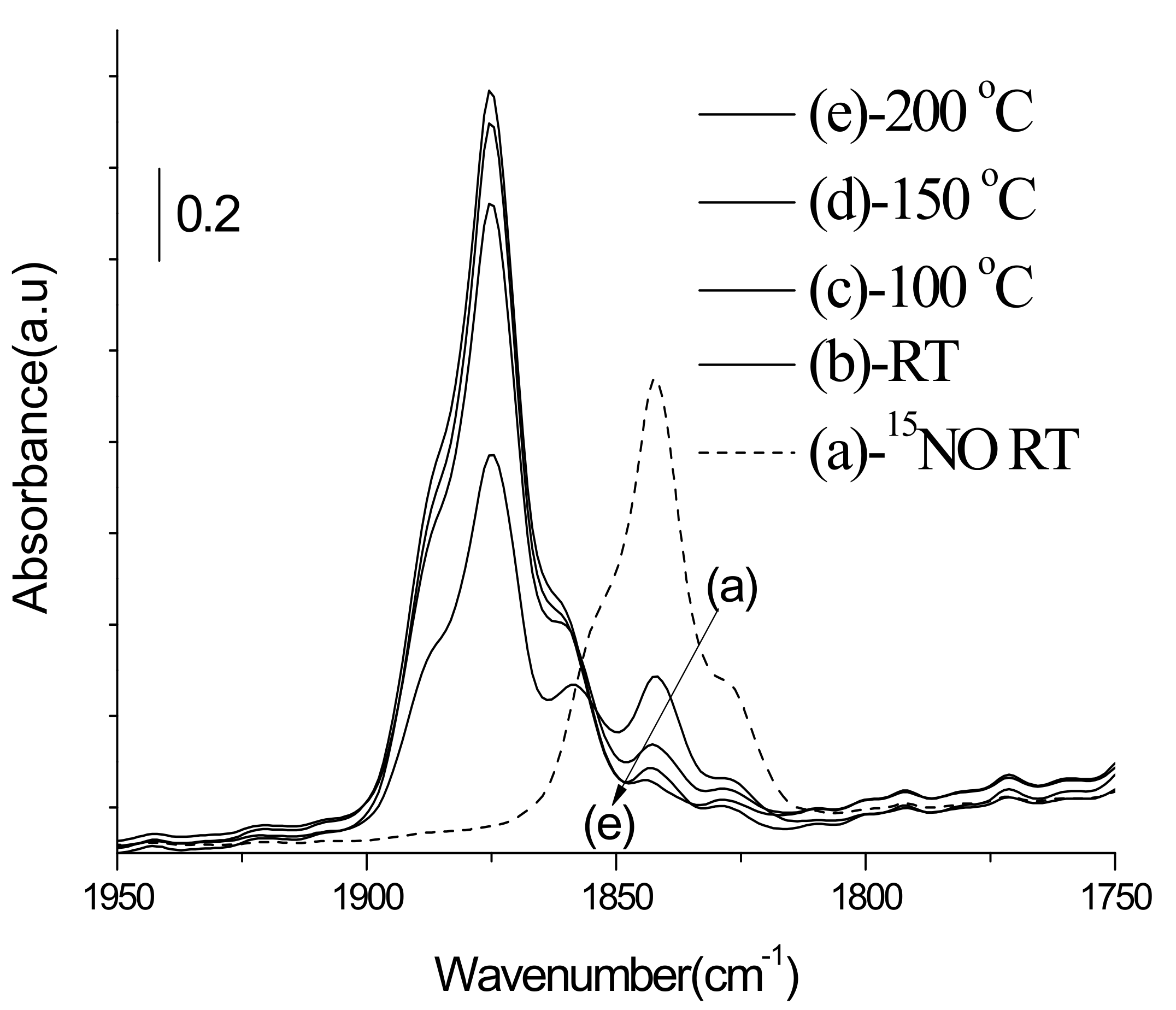
3.3.1. Adsorption-Desorption Behavior of NO and CO on Nanoporous VSB-5
3.3.2. Isotopic 15NO Adsorption
3.3.3. CO Adsorption
3.3.4. Co-Adsorption of NO and CO
4. Experimental
4.1. Catalyst Preparation
4.2. Catalyst Characterization
4.3. Procedure for NOx Storage/Reduction
5. Conclusions and Perspective
- (1)
- Although there is a wide general agreement concerning the mechanism of NSR processes, diversified and sometimes contradictory results have also been reported, mainly due to different experimental conditions used by the various researchers, such as different lean and regeneration periods, different reducing gases, different catalyst preparation and pretreatment, and different mass- and heat-transfer conditions. NOx storage is a sequential process (initially, nitrite and eventually, nitrate formation), which still does not clearly state the nature of the Ba species, nor does it account for the NOx slip during the storage process. The NOx reduction mechanism and the product selectivity under rich conditions are dependent on the reductants. NH3, which has recently been considered to be an intermediate reductant, has not been universally observed and accepted by researchers.
- (2)
- The noble metal Pt is widely used by researchers for the oxidation of NO and the subsequent reduction of stored NOx. However, the nature of the Pt particles, such as the loading, dispersion, size and the morphology, and metal-support interaction influencing the catalytic activity are still the subject of debate. There is still no conclusive understanding of the optimum loading, shape, and dispersion of Pt particles for the best catalytic activity. Investigations have been carried out with other noble metals, such as Pd and Rh. Furthermore, the potential of metal alloys needs to be explored.
- (3)
- The storage component best investigated and commonly used is Ba; however, the nature of the storage component is still not clear. According to some researchers, BaCO3 is the dominant storage component under real conditions, whereas others claim that BaO is a better potential storage component than BaCO3. Researchers have revealed that the proximity of Ba and the noble metal is important in the storage process; however, systematic investigations are still lacking regarding this aspect. Overall, it is accepted that BaAl2 O4 is formed at higher temperatures during the NSR operation, although the exact role of BaAl2 O4 is unknown. Storage materials, other than Ba, such as K, Mg, and Ca showed promising performance in the storage step, as well as less sulfur poisoning, which needs to be explored more profoundly.
- (4)
- Besides stabilizing the dispersed noble metal particles, the support material can also affect the oxidation state of the noble metal and the spillover process, which has an effect on the oxidation, storage, and regeneration (reduction) steps, and thus, the overall performance of the NSR catalyst. For Ba-based lean traps, NOx storage capacity seems to follow the basicity of the oxide support, while the rate of NOx reduction tends to decrease with stronger basicity of the support. An open question is the role of the support in the spillover processes occurring in the NSR system. Furthermore, for a deeper understanding of the surface processes during NSR, the structure and chemistry of the interfaces of the support with catalytic (noble metals) and storage components (alkali and alkaline earth metals) need to be investigated more thoroughly, particularly under dynamic conditions.
Funding
Data Availability Statement
Conflicts of Interest
References
- Roy, S.; Baiker, A. NOx Storage-Reduction Catalysis: From Mechanism and Materials Properties to Storage-Reduction Performance. Chem. Rev. 2009, 109, 4054–4091. [Google Scholar] [CrossRef]
- Centi, G.; Arena, G.E.; Perathoner, S. Nanostructured Catalysts for NOx Storage-Reduction and N2O decomposition. J. Catal. 2003, 216, 443–454. [Google Scholar] [CrossRef]
- Kaspar, J.; Fornasiero, N.P.; Hicky, N. Automotive Catalytic Converters: Current Status and Some Perspectives. Catal. Today 2003, 77, 419–449. [Google Scholar] [CrossRef]
- Takahashi, N.; Shinjoh, H.; Suzuki, T.; Yamazaki, K.; Yokota, K.; Suzuki, H.; Miyoshi, N.; Matsumoto, H.; Tanizawa, T.; Tanaka, T.; et al. The New Concept 3-way Catalyst for Automotive Lean-burn Engine: NOx Storage and Reduction Cataltst. Catal. Today 1996, 27, 63–69. [Google Scholar] [CrossRef]
- Matsumoto, S. Denox catalyst for automotive lean-burn engine. Catal. Today 1996, 29, 43–45. [Google Scholar] [CrossRef]
- Matsumoto, S.; Ikeda, Y.; Suzuki, H.; Ogai, M.; Miyoshi, N. NOx Storage-Reduction Catalyst for Automotive Exhaust with Improved Torerance against Sulfur Poisoning. Appl. Catal. B Environ. 2000, 25, 115–124. [Google Scholar] [CrossRef]
- Yamazaki, K.; Suzuki, T.; Takahashi, N.; Yokota, K.; Sugiura, M. Effect of the Addition of Transition Metals to Pt/Ba/Al2O3 Catalyst on the NOx Storage-Reduction Catalysis under Oxidizing Conditions in the Presence of SO2. Appl. Catal. B Environ. 2001, 30, 459–468. [Google Scholar] [CrossRef]
- Forzatti, P. Present Status and Perspectives in De-NOx SCR Catalysis. Appl. Catal. A General. 2001, 222, 221–236. [Google Scholar] [CrossRef]
- Busca, G.; Lietti, L.; Ramis, G.; Berti, F. Chemical and mechanistic aspects of the selective catalytic reduction of NOx by ammonia over oxide catalysts: A review. Appl. Catal. B Environ. 1998, 18, 1–36. [Google Scholar] [CrossRef]
- Iwamoto, M.; Yahiro, H.; Shundo, S.; Yu-u, Y.; Mizuno, N. Influence of Sulfur Dioxide on Catalytic Removal of Nitric Oxide over Copper Ion-Exchanged ZSM-5 Zeolite. Appl. Catal. 1991, 69, L15–L19. [Google Scholar] [CrossRef]
- Theinnoi, K.; Sitshebo, S.; Houel, V.; Rajaram, R.R.; Tsolakis, A. Hydrogen Promotion of Low-Temperature Passive Hydrocarbon-Selective Catalytic Reduction (SCR) over a Silver Catalyst. Energy Fuels 2008, 22, 4109–4114. [Google Scholar] [CrossRef]
- Burch, R.; Breen, J.P.; Hill, C.J.; Krutzsch, B.; Konrad, B.; Jobson, E.; Cider, L.; Eranen, K.; Klingstedt, F.; Lindfors, L.-E. Exceptional Activity for NOx Reduction at Low Temperatures Using Combinations of Hydrogen and Higher Hydrocarbons on Ag/Al2O3 Catalysts. Top. Catal. 2004, 30–31, 19–25. [Google Scholar] [CrossRef]
- Frola, F.; Manzoli, M.; Prinetto, F.; Ghiotti, G. Pt−Ba/Al2O3 NSR Catalysts at Different Ba Loading: Characterization of Morphological, Structural, and Surface Properties. J. Phys. Chem. C 2008, 112, 12869–12878. [Google Scholar] [CrossRef]
- Happel, H.; Desikusumastuti, A.; Sobota, M.; Laurin, M.; Libuda, J. Impact of Sulfur Poisoning on the Uptake of a NOx Storage Reduction (NSR) Model Catalyst. J. Phys. Chem. C 2010, 114, 4568–4575. [Google Scholar] [CrossRef]
- Lietti, L.; Forzatti, P.; Nova, I.; Tronconi, E. NOx Storage Reduction over Pt@Ba/γ-Al2O3. J. Catal. 2001, 204, 175–191. [Google Scholar] [CrossRef]
- Rankovic, N.; Nicolle, A.; Costa, P.D. Detailed Kinetic Modeling Study of NOx Oxidation and Storage and Their Interactions over Pt/Ba/Al2O3 Monolith Catalysts. J. Phys. Chem. C 2010, 114, 7102–7111. [Google Scholar] [CrossRef]
- Lesage, T.; Saussey, J.; Malo, S.; Hervieu, M.; Hedouin, C.; Blanchard, G.; Daturi, M. Operando FTIR study of NOx storage over a Pt/K/Mn/Al2O3-CeO2 catalyst. Appl. Catal. B Environ. 2007, 72, 166–177. [Google Scholar] [CrossRef]
- Liu, Y.; Meng, M.; Zou, X.; Li, Z.; Zha, Y. In Situ DRIFTS Investigation on the NOx Storage Mechanisms over Pt/K/TiO2–ZrO2. Catalyst Catal. Commun. 2008, 10, 173–177. [Google Scholar] [CrossRef]
- de Lucas, A.; Caravaca, A.; Sanchez, P.; Dorado, F.; Valverde, J.L. A New Improvement of Catalysis by Solid-State Electrochemistry:An Electrochemically Assisted NOx Storage/Reduction catalyst. J. Catal. 2008, 259, 54–65. [Google Scholar]
- Yamamoto, K.; Kikuchi, R.; Takeguchi, T.; Eguchi, K. Development of NO Sorbents Tolerant to Sulfur Oxides. J. Catal. 2006, 238, 449–457. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Z.; Chung, J.S. Molecular NO2 Induced K2Ti2O5–K2Ti6O13 Structure Switching in the Dry Gas Phase: Lattice Potassium Reactivity. Chem. Commun. 2009, 35, 5284–5286. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Sohn, J.H.; Chung, J.S. Thermally Stable Pt/K2Ti2O5 as High-Temperature NOx Storage and Reduction Catalyst. Appl. Catal. B Environ. 2009, 89, 97–103. [Google Scholar] [CrossRef]
- Sun, X.; Li, Y. Synthesis and Characterization of Ion-Exchangeable Titanate Nanotubes. Chem. Europian. J. 2003, 9, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Gao, X.; Zhu, H.; Zheng, Z.; Yan, T.; Wu, F.; Ringer, S.; Song, D. Titanate Nanotubes and Nanorods Prepared from Rutile Powder. Adv. Funct. Mater. 2005, 15, 1310–1318. [Google Scholar] [CrossRef]
- Tsai, C.; Teng, H. Nanotube Formation from a Sodium Titanate Powder via Low-Temperature Acid Treatment. Langmuir 2008, 24, 3434–3438. [Google Scholar] [CrossRef]
- Bavykin, D.V.; Friedrich, J.M.; Lapkin, A.A.; Walsh, F.C. Stability of Aqueous Suspensions of Titanate Nanotubes. Chem. Mater. 2006, 18, 1124–11129. [Google Scholar] [CrossRef]
- Sun, X.; Chen, X.; Li, Y. Large-Scale Synthesis of Sodium and Potassium Titanate Nanobelts. Inorg. Chem. 2002, 41, 4996–4998. [Google Scholar] [CrossRef]
- Idakiev, V.; Yuan, Z.-Y.; Tabakova, T.; Su, B.-L. Titanium Oxide Nanotubes as Supports of Nano-sized Gold Catalysts for Low Temperature Water-gas Shift Reaction. Appl. Catal. A Gen. 2005, 281, 149–155. [Google Scholar] [CrossRef]
- Torrente-Murciano, L.; Lapkin, A.A.; Bavykin, D.V.; Walsh, F.C.; Wilson, K. Highly selective Pd/titanate nanotube catalysts for the double-bond migration reaction. J. Catal. 2007, 245, 272. [Google Scholar] [CrossRef]
- Ntho, T.A.; Anderson, J.A.; Scurrell, M.S. CO Oxidation over Titanate Nanotube Supported Au: Deactivation due to Bicarbonate. J. Catal. 2009, 261, 94. [Google Scholar] [CrossRef]
- Shen, W.; Nitta, A.; Chen, Z.; Eda, T.; Yoshida, A.; Naito, S. NOx storage and reduction over potassium titanate nnanobelt-based catalyst with high storage capacity. J. Catal. 2011, 280, 161–167. [Google Scholar] [CrossRef]
- Yoshida, A.; Shen, W.; Eda, T.; Watanabe, R.; Ito, T.; Naito, S. NOx storage/reduction over alkali-metal-nitrate impregnated titanate nanobelt catalysts and investigation of alkali metal cation migration using XPS. Catal. Today 2012, 184, 78–82. [Google Scholar] [CrossRef]
- Gopalakrishnan, R.; Bartholomew, C.H. Zeolite Selective Catalytic Reduction Catalysts for NOx Removal from Nuclear Waste Processing Plants. ACS Symp. Ser. 1995, 587, 56–70. [Google Scholar]
- Shelef, M. Selective catalytic reduction of NOx with N-free reductants. Chem. Rev. 1995, 95, 209–225. [Google Scholar] [CrossRef]
- Li, Y.; Armor, J.N. Selective catalytic reduction of NOx with methane over metal exchange zeolites. Appl. Catal. B Environ. 1993, 2, 239–256. [Google Scholar] [CrossRef]
- Korhonen, S.T.; Fickel, D.W.; Lobo, R.F.; Weckhuysen, B.M.; Beale, A.M. Isolated Cu2+ ions: Active sites for selective catalytic reduction of NO. Chem. Commun. 2011, 47, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Wang, X.; Qi, G.; Wang, J.; Shen, M.; Li, W. Characterization of copper species over Cu/SAPO-34 in selective catalytic reduction of NOx with ammonia: Relationships between active Cu sites and de-NOx performance at low temperature. J. Catal. 2013, 297, 56–64. [Google Scholar] [CrossRef]
- Burch, R.; Scire, R. An investigation of the mechanism of the selective catalytic reduction of NO on various metal/ZSM-5 catalysts: Reactions of H2/NO mixtures. Catal. Lett. 1994, 27, 177–186. [Google Scholar] [CrossRef]
- Yu, Q.; Richter, M.; Kong, F.; Li, L.; Wu, G.; Guan, N. Selective catalytic reduction of NO by hydrogen over Pt/ZSM-35. Catal. Today 2010, 158, 452–458. [Google Scholar] [CrossRef]
- Ravata, V.; Aghalayam, P. Effect of Noble Metals Deposition on the Catalytic Activity of MAPO-5 Catalysts for the reduction of NO by CO. Appl. Catal. A Gen. 2010, 389, 9–18. [Google Scholar] [CrossRef]
- Mihaylov, M.; Hadjiivanov, K. FTIR Study of CO and NO Adsorption and Coadsorption on Ni-ZSM-5 and Ni/SiO2. Langmuir 2002, 18, 4376–4383. [Google Scholar] [CrossRef]
- Mosqueda-Jiménez, B.I.; Jentys, A.; Seshan, K.; Lercher, J.A. Structure-activity Relationship for Ni-containing Zeolites during NO reduction 1. Influence of Acid Sites. J. Catal. 2003, 218, 348–353. [Google Scholar] [CrossRef]
- Mihaylova, A.; Hadjiivanov, K.; Dzwigaj, S.; Che, M. Remarkable Effect of the Preparation Technique on the State of Cobalt Ions Zeolites Evidenced by FTIR Spectroscopy of Adsorbed CO and NO, TPR and XRD. J. Phys. Chem. B 2006, 110, 19530–19536. [Google Scholar] [CrossRef] [PubMed]
- Hadjiivanov, K.; Kno1zinger, H.; Mihaylov, M. FTIR study of CO adsorption on Ni−ZSM-5. J. Phys. Chem. B 2002, 106, 2618–2624. [Google Scholar] [CrossRef]
- Gianotti, E.; Vishnuvarthan, M.; Berlier, G.; Marchese, L.; Coluccia, S. FT-IR Study of Cobalt Containing Aluminophosphates with Chabasite Like Structure by Using CO and NO as Molecular Probes. Catal. Lett. 2009, 133, 27–32. [Google Scholar] [CrossRef]
- Iwasaki, M.; Shinjoh, H. NO Evolution Reaction with NO2 Adsorption over Fe/ZSM-5: In situ FT/IR Observation and Relationships with Fe Sites. J. Catal. 2010, 273, 29–38. [Google Scholar] [CrossRef]
- Chen, Z.; Zhou, D.; Gao, T.; Shen, W.; Dong, X.; Naito, S. Unusual adsorption and drsorption behaviors of NO and CO on nanoporous nickel phosphate VSB-5: In situ FT-IR and XRD study. Catal. Today 2015, 258, 199–204. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Surf. Area. (Pore vol.) | MeNO3 Loaded (wt%) | Pt Size TEM (nm) | Full Trapping Period (min) | NO Stored (mmol/g) | N2 Desorb. (mmol/g) |
|---|---|---|---|---|---|---|
| TiO2(A) [ST-01] | 275 m2/g (0.63 cm3/g) | K (20) | 2–3 | 2 | 0.92 | 0.45 |
| K (33) | 18 | 2.45 | 1.11 | |||
| TiO2(R) [MT150 A] | 101 m2/g (0.36 cm3/g) | K (20) | 3–4 | 5 | 0.77 | 0.40 |
| K (33) | 1 | 1.71 | 0.66 | |||
| TiO2(A/R) [P-25] | 54 m2/g (0.10 cm3/g) | K (20) | 3–4 | 13 | 1.53 | 0.56 |
| K (33) | 8 | 1.28 | 0.67 | |||
| K2 Ti8 O17 [KTN] | 320 m2/g (0.01 cm3/g) | K (20) | 1–2 | 25 | 1.57 | 0.67 |
| K (33) | 34 | 2.53 | 1.10 | |||
| Na (28) | 2–3 | 23 | 1.50 | 0.71 | ||
| Rb (27) | 3–4 | 16 | 1.28 | 0.59 |
| Catalysts | Condition | Pt 4f7/2 | Ti 2p3/2 | K 2p3/2 | N 1s | O 1s |
|---|---|---|---|---|---|---|
| Pt-KNO3/KTN | NOx stored | — (0) | 457.7 (0.01) | 292.7 (2.3) | 407.1 | 532.8 (1) |
| H2 reduced | 69.2 (1) | 457.7 (1) | 292.2 (1) | — | 529.4 (1) | |
| Pt-KNO3/TiO2 (ST-01) | NOx stored | 73.7 (0.8) 70.5 (0.2) | 457.8 (1.1) | 292.4 (0.9) | 406.9 | 529.2 (0.8) 532.1 (0.2) |
| H2 reduced | 70.5 (1) | 458.0 (1) | 292.7 (1) | — | 530.0 (1) |
| Catalysts | Trapping Period | NOx Stored | N2 Desorbed |
|---|---|---|---|
| Pt/2.5 K/ZrO2 | 20.7 | 1.2 | 0.7 |
| Pt/3.5 K/ZrO2 | 18.3 | 1.3 | 0.8 |
| Pt/4.9 K/ZrO2 | 15.8 | 1.5 | 0.9 |
| Pt/2.5 K/CeO2 | 18.6 | 1.1 | 0.7 |
| Pt/3.5 K/CeO2 | 24.1 | 1.6 | 0.9 |
| Pt/4.9 K/CeO2 | 28.6 | 2.4 | 1.5 |
| Pt4 f7/2 | K2 p3/2 | Ce3 d5/2 | N 1s | O 1s | |
|---|---|---|---|---|---|
| Reference | 71.0 | 292.7 | 883.1 | 398.1 | 533.2 |
| NOx stored | 72.3 (5) | 292.7 (0.3) | 889.0 (1.2) | 407.1 | 532.6, 529.0 |
| H2 reduced | 70.6 (1) | 293.6 (1) | 885.6 (1) | - | 531.6, 529.0 |
| Catalysts | Trapping Period | NOx Stored | N2 Desorbed |
|---|---|---|---|
| Pt-2.5 K/CeO2 | 19 min | 1.1 | 0.6 |
| Pt-3.3 K/CeO2 | 24 min | 1.6 | 0.9 |
| Pt-4.9 K/CeO2 | 29 min | 2.4 | 1.1 |
| Cu/1.3 K/CeO2 | 16 min | 1.9 | 0.8 |
| Co/1.3 K/CeO2 | 15 min | 1.1 | 0.5 |
| Catalysts | Condition | Pt 4f7/2 | Cu 2p3/2 | K 2p3/2 | Ce 3d5/2 | N 1s | O 1s |
|---|---|---|---|---|---|---|---|
| reference | 71.0 | 932.7 | 292.7 | 883.1 | 398.1 | 533.2 | |
| Pt/KNO3 /CeO2 | NOx stored | 72.3 (5) | 292.7 (0.3) | 889.0 (1.2) | 407.1 | 532.6, 529.0 | |
| H2 reduced | 70.6 (1) | 293.6 (1) | 885.6 (1) | - | 531.6, 529.0 | ||
| Cu/KNO3 /CeO2 | NOxstored | 933.7 (10) | 292.7 (0.1) | 889.0 (2.5) | 406.9 | 532.5, 529.0 | |
| H2 reduced | 932.6 (1) | 293.6 (1) | 885.6 (1) | 531.7, 529.0 |
| Bases (Treat.) | S.A. | Ads.amt. (77 K) | NO(a).amt. (cm3 g−1) | SEM/ EDX | NO-CO Initial Rate (10−7 mol/min−1) | ||||
|---|---|---|---|---|---|---|---|---|---|
| m2 g−1 | N2 | CO | 293 K | 353 K | P/Ni | CO2 | N2 O | N2 | |
| VSB-5(TEA) | 15.0 | 2.5 | 3.8 | 4.7 | 4.4 | 0.595 | 1.4 | 2.5 | 1.0 |
| VSB-5(NH3) | 318.0 | 93.5 | 106.0 | 36.1 | 35.3 | 0.625 | 3.0 | 3.0 | 2.5 |
| TEA(NaOH treat.) | 28.4 | 5.8 | 13.5 | 16.0 | 15.0 | 0.718 | 14.0 | 13.0 | 2.5 |
| NH3(NaOH treat.) | 108.0 | 32.1 | 36.0 | 12.6 | 12.3 | 0.477 | 20.0 | 14.0 | 33.0 |
| Catalysts | S.A. | NO(a).amt. (cm3 g−1) | Initial Rates of Product Formation (10−7 mol/min−1) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| m2 g−1 | 293 K | 353 K | NO-H2 Reaction | NO-CO Reaction | NO-CH4 Reaction | ||||
| N2 | N2O | N2 | N2O | N2(N2O) | CH3CN | ||||
| VSB-5(TEA) | 15.0 | 4.7 | 4.4 | 14.3 | 2.3 | 3.5 | 0.8 | 0.6 | 0.2 |
| VSB-5(NH3) | 318.0 | 36.1 | 35.3 | 18.6 | 3.5 | 5.8 | 1.4 | 1.3 | 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naito, S. Ordered Nanostructure Catalysts Efficient for NOx Storage/Reduction (NSR) Processes. Catalysts 2021, 11, 1348. https://doi.org/10.3390/catal11111348
Naito S. Ordered Nanostructure Catalysts Efficient for NOx Storage/Reduction (NSR) Processes. Catalysts. 2021; 11(11):1348. https://doi.org/10.3390/catal11111348
Chicago/Turabian StyleNaito, Shuichi. 2021. "Ordered Nanostructure Catalysts Efficient for NOx Storage/Reduction (NSR) Processes" Catalysts 11, no. 11: 1348. https://doi.org/10.3390/catal11111348
APA StyleNaito, S. (2021). Ordered Nanostructure Catalysts Efficient for NOx Storage/Reduction (NSR) Processes. Catalysts, 11(11), 1348. https://doi.org/10.3390/catal11111348