
Roles of Nanostructured Bimetallic Supported on Alumina-Zeolite (AZ) in Light Cycle Oil (LCO) Upgrading



Abstract
:1. Introduction
2. Results and Discussion
2.1. Catalyst Characterization
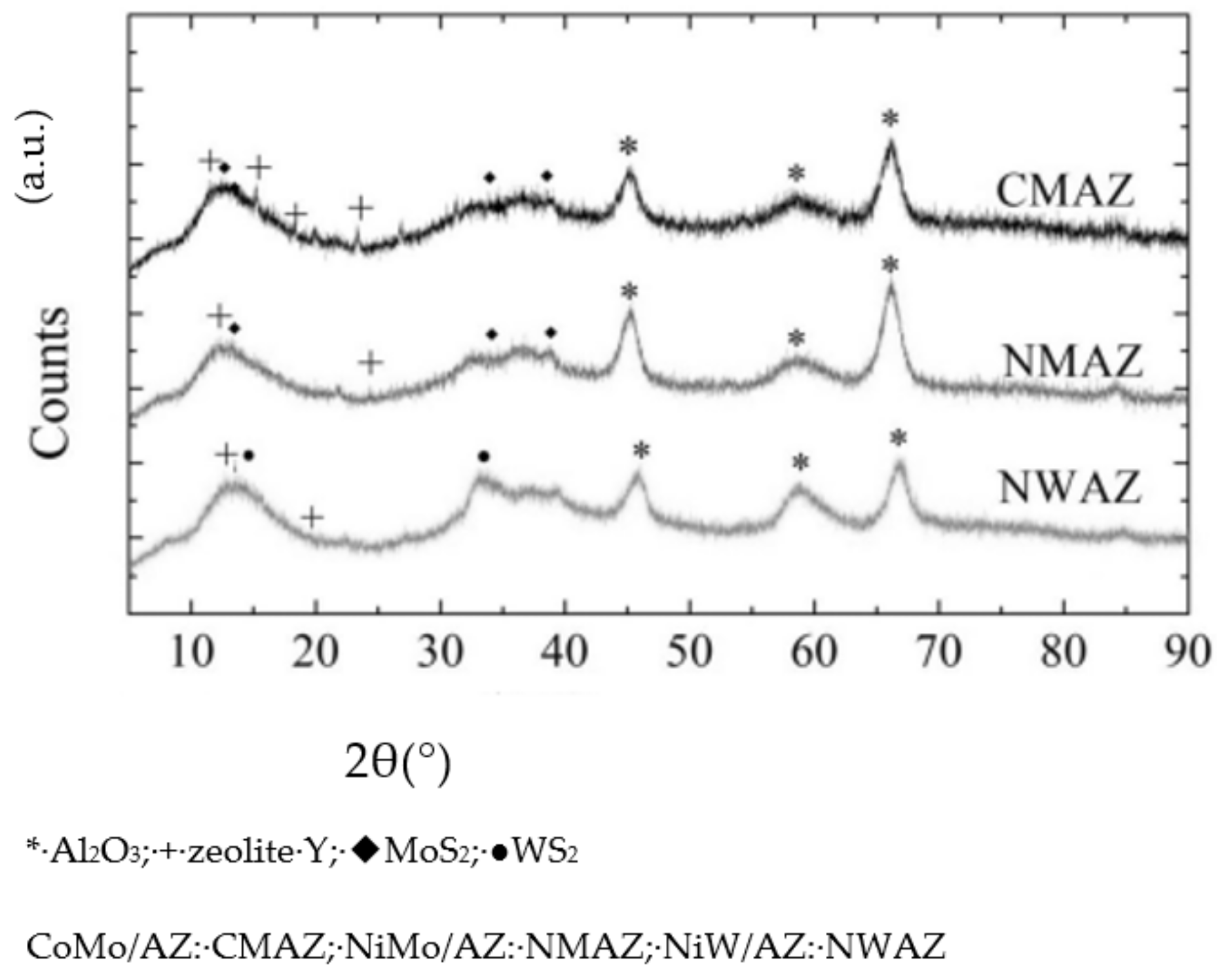
2.1.1. Texture Structures
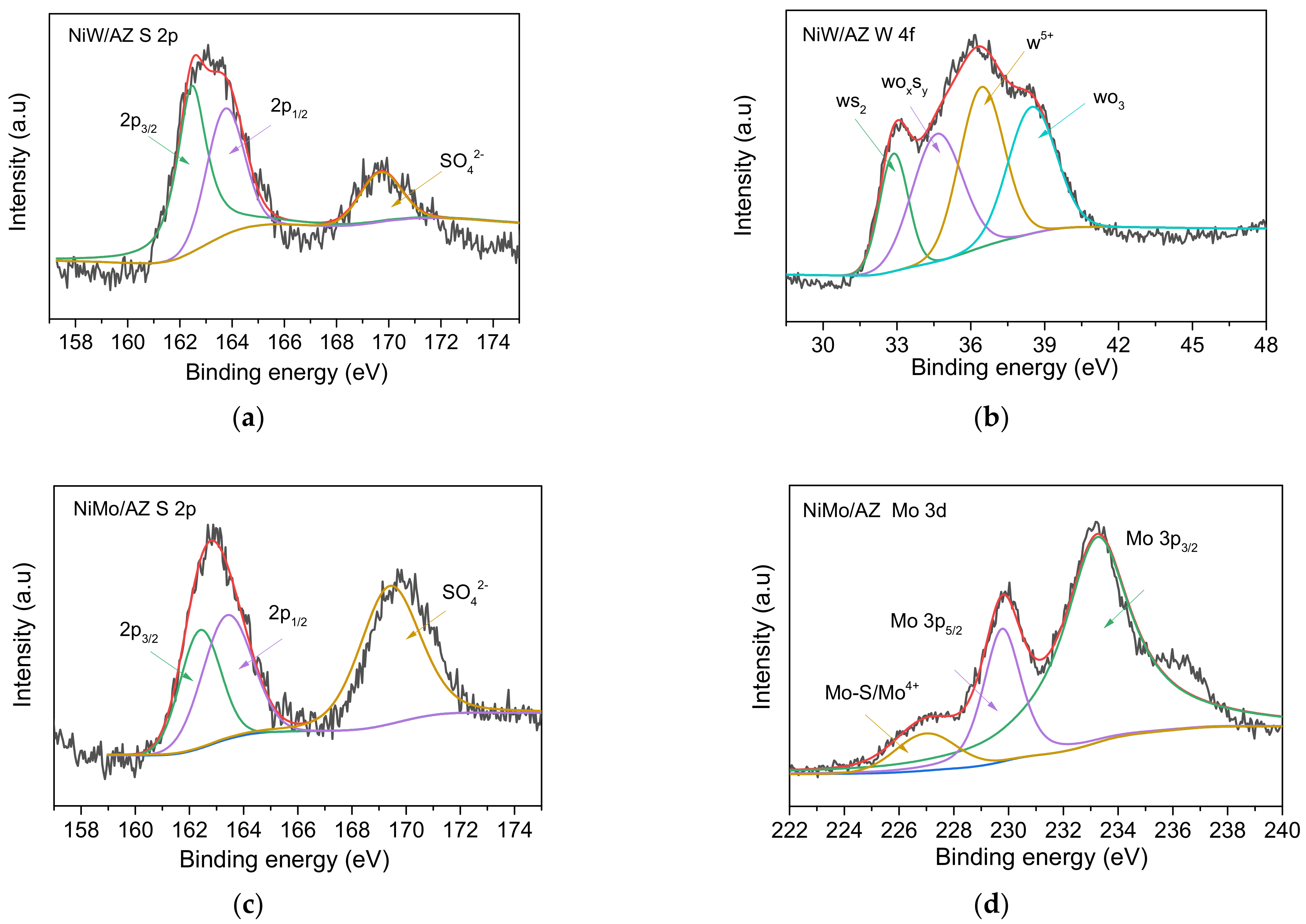
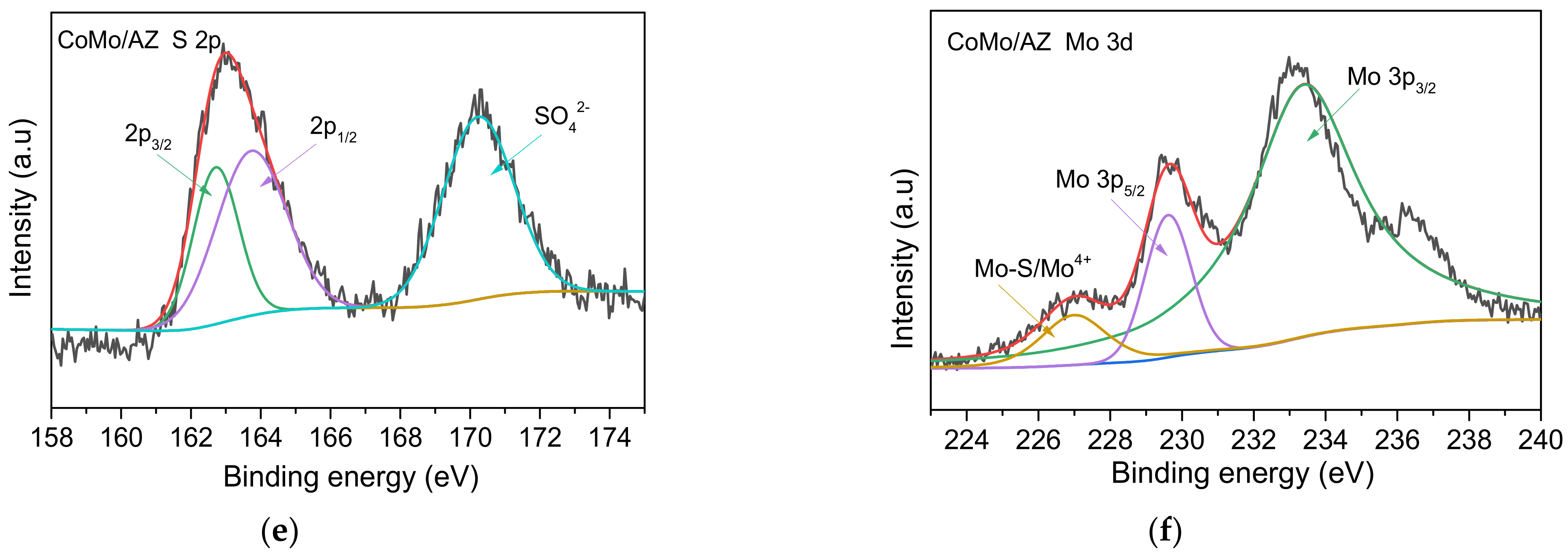
2.1.2. Morphology
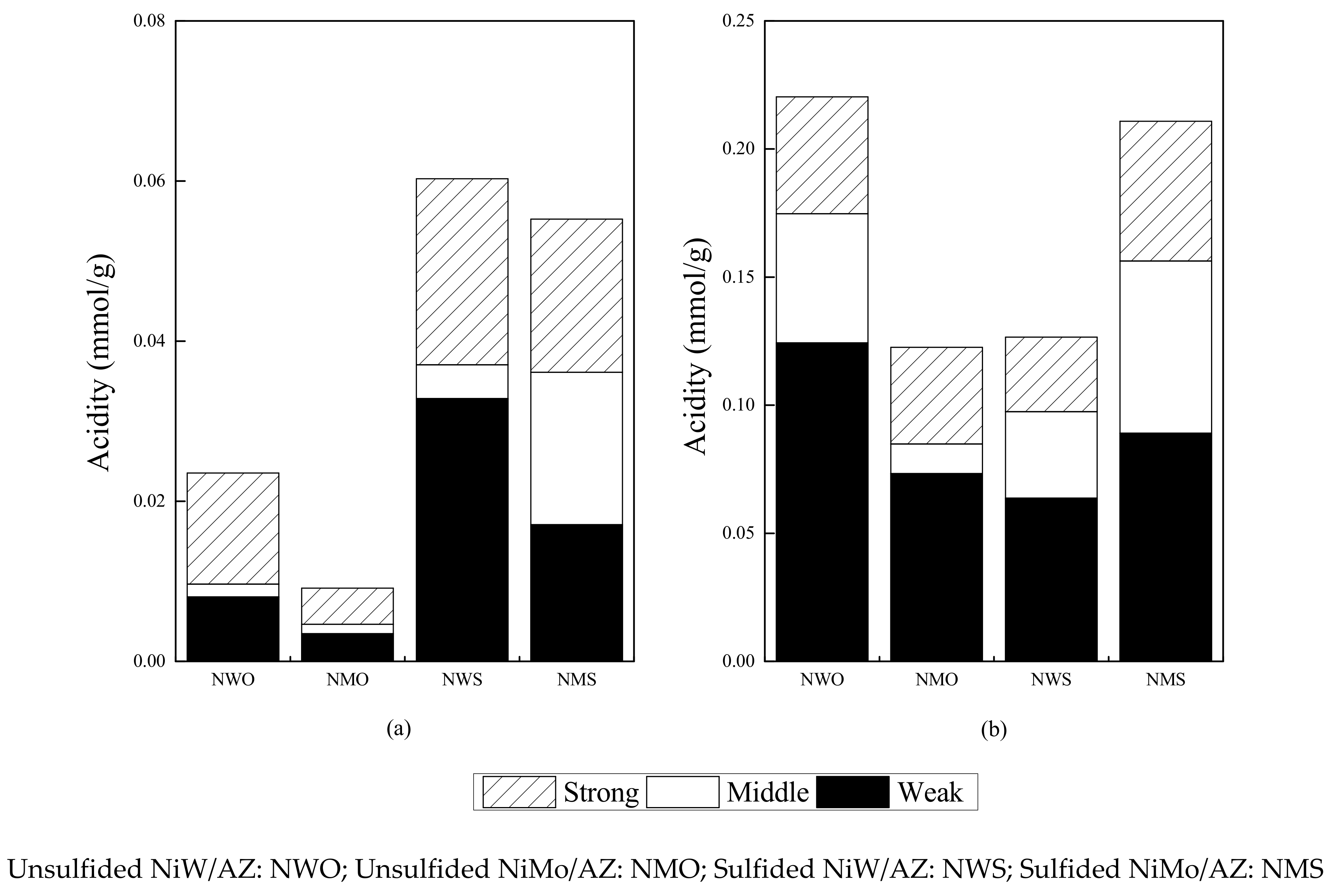
2.1.3. Acidity
2.2. Reaction Performances
2.2.1. Model Compounds as Feed
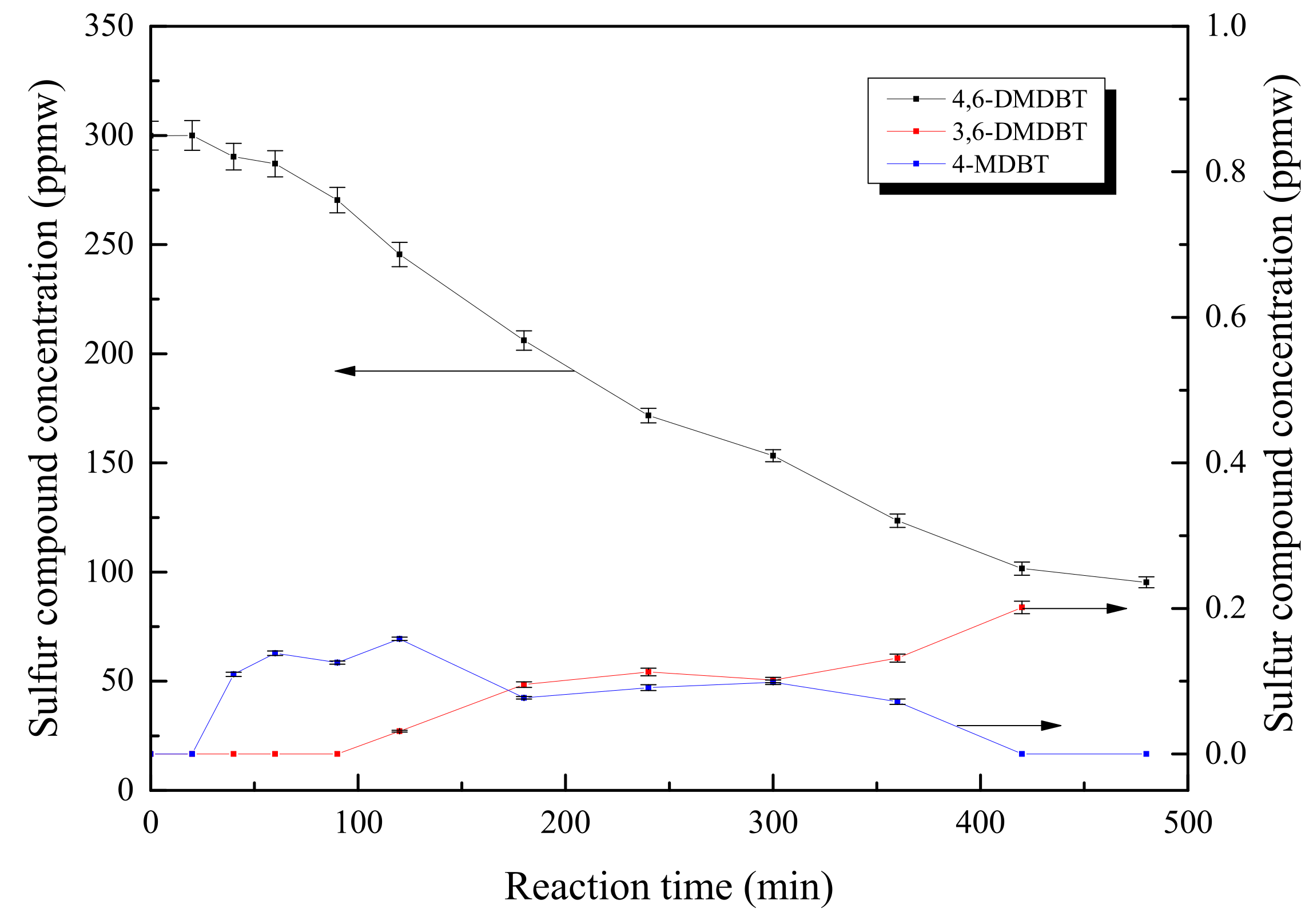
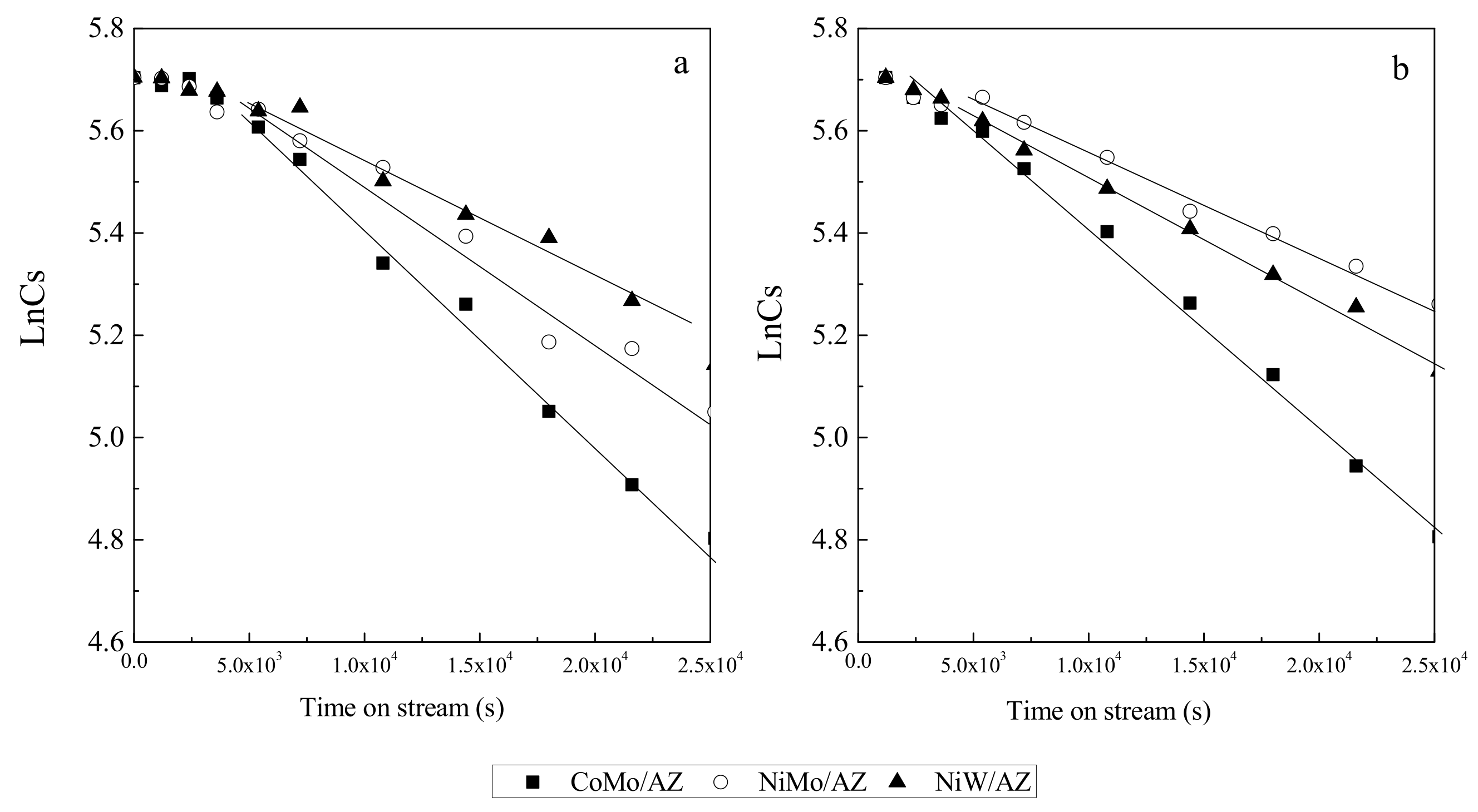
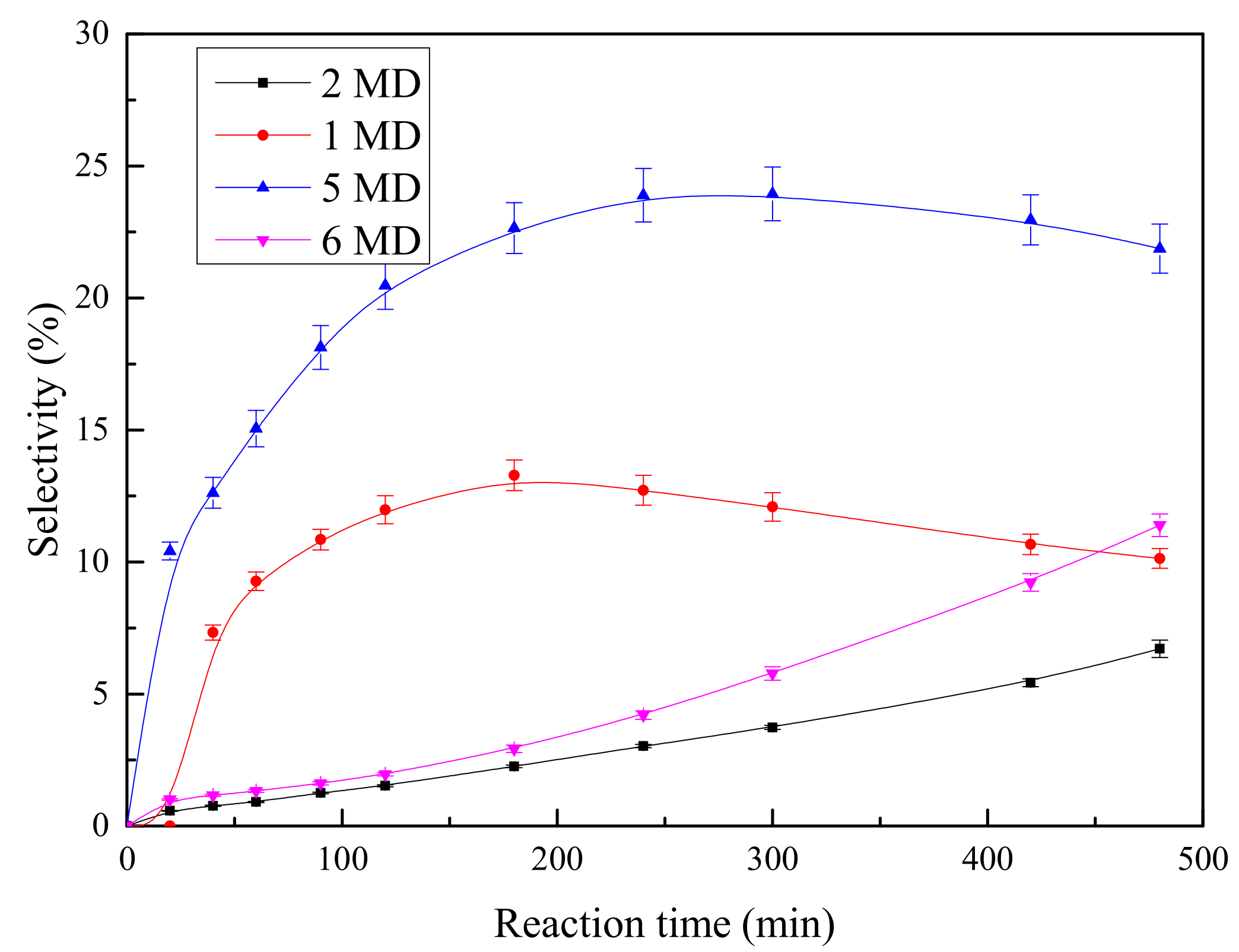
HDS Performance
Effects of Nitrogen Introduction on HDS Performance
HDA of 1-MN
Hydrogenation of 1-MN
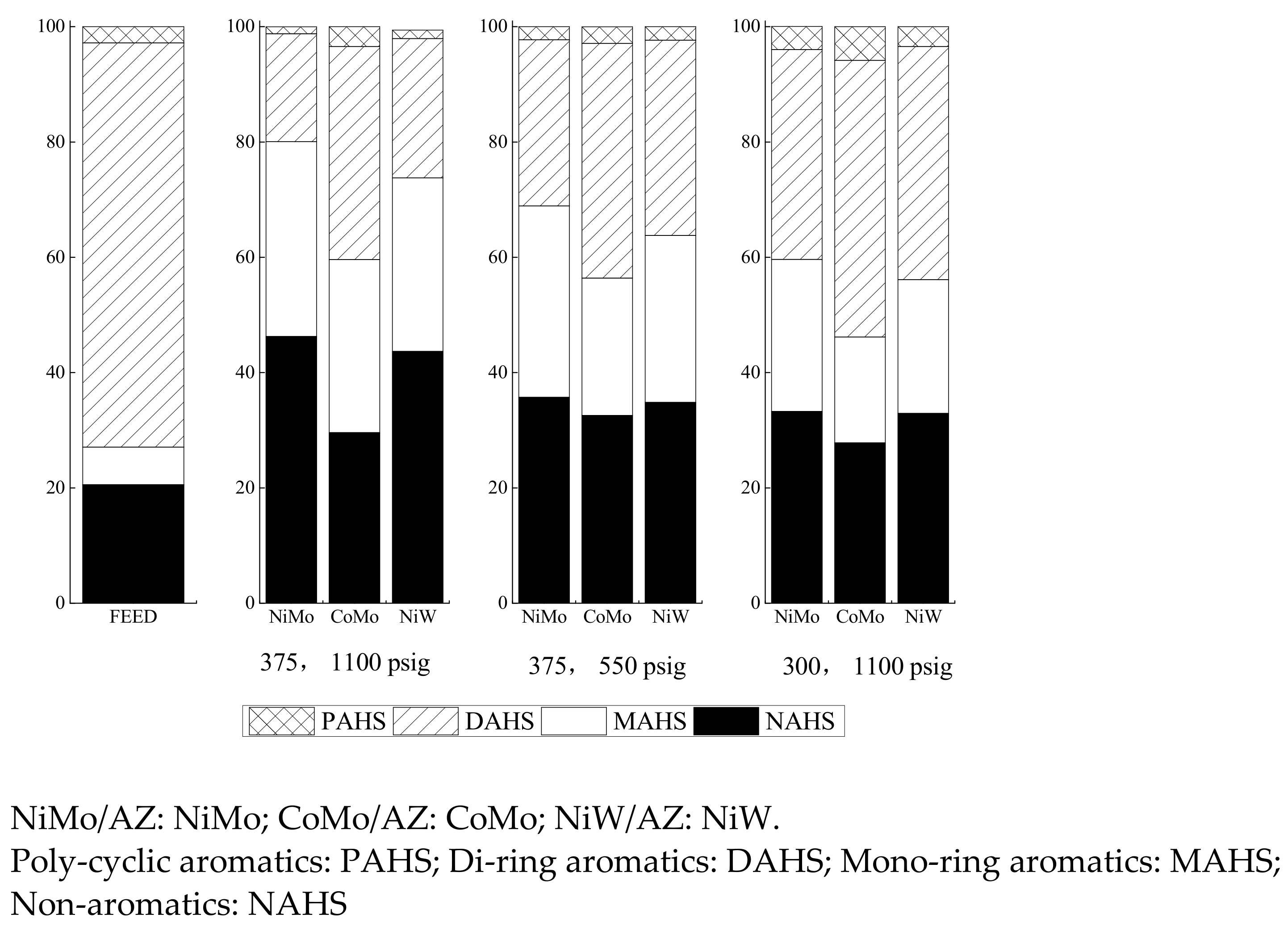
2.2.2. Real LCO as Feed
3. Experimental
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Catalyst Evaluation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LCO | Light cycle oil |
| CN | Cetane number |
| AZ | Alumina-zeolite |
| DDS | Direct desulfurization |
| HYD | hydrogenation |
| HDS | Hydrodesulfurization |
| HDN | Hydrodenitrogenation |
| HDA | Hydrodearomatization |
| FCC | Fluid catalytic cracking |
| PAH | Polycyclic aromatic hydrocarbons |
| GC | Gas chromatography |
| FID | Flame Ionization Detector |
| PFPD | Pulsed flame photometric detector |
| GC/MS | Gas chromatography and mass spectrometry |
| HPLC | High performance liquid chromatography |
| XRD | X-ray diffraction |
| FTIR | Fourier transform infrared spectroscopy |
| XPS | X-Ray Photoelectron Spectroscopy |
| TEM | Transmission electron microscopy |
| DMDBT | Dimethyl dibenzothiophene |
| LHSV | Liquid hourly space velocity |
| 1-MN | 1-methylnaphthlene |
| S | Sulfur |
| N | Nitrogen |
| MDBT | Methyl dibenzothiophene |
| 4,6-DMTHDBT | 4,6-dimethyltetrahydrodibenzothiophere |
| 4,6-DMHHDBT | 4,6-dimethylhexahydrodibenzothiophere |
| 3,3′-DMBiP | 3,3-dimethylbiphenyl |
| 3,6-DMDBT | 3,6-dimethyldibenzothiophere |
| 3,4′-DMBiP | 3,4′-dimethylbiphenyl |
| 3,3′-MCHT | 3,3′-methylcyclohexy toluene |
| IS | Initial state |
| TS | Transition state |
| FS | final states |
| EB | Energy barrier |
| Cs | Concentration of sulfur |
| MT | Methyltetralins |
| PAHS | Poly cyclic aromatics |
| DAHS | Di-ring aromatics |
| MAHS | Mono-ring aromatics |
| NAHS | Non-aromatics |
References
- Shi, Q.; Zhao, S.; Zhou, Y.; Gao, J.; Xu, C. Development of heavy oil upgrading technologies in China. Rev. Chem. Eng. 2020, 36, 1–19. [Google Scholar] [CrossRef]
- Peng, C.; Fang, X.C.; Zeng, R.H.; Guo, R.; Hao, W.Y. Commercial analysis of catalytic hydroprocessing technologies in producing diesel and gasoline by light cycle oil. Catal. Today 2016, 276, 11–18. [Google Scholar] [CrossRef]
- Peng, C.; Yang, X.; Fang, X.; Huang, X.; Cheng, Z.; Zeng, R.; Guo, R. Development of Light Cycle Oil (LCO) Hydrocracking Technology over a Commercial W-Ni Based Catalyst. China Pet. Process. Petrochem. Technol. 2015, 17, 30–36. [Google Scholar]
- Zhang, Z.; Zhang, W.; Zhang, Y.; Ji, D.; Jin, H.; Wang, G.; Zhang, Z. Technical review on flexible processing middle distillate for achieving maximum profit in China. Appl. Petrochem. Res. 2017, 7, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, K.G.; Cooper, B.H.; Topsoe, H. Catalyst and process technologies for ultra low sulfur diesel. Appl. Catal. A Gen. 1999, 189, 205–215. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, H.; Wang, M.; Pu, J.; Rogers, K.; Wang, H.; Ng, S.; Xu, R. Hydro-upgrading of light cycle oil-synthesis of NiMo/SiO2-Al2O3-TiO2 porous catalyst. J. Porous Mater. 2021, 28, 867–874. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, Q.; Zhou, Y.; Wei, Q.; Du, L.; Ding, S.; Jiang, S.; Zhang, Y. Effects of Ga- and P-modified USY-based NiMoS catalysts on ultra-deep hydrodesulfurization for FCC diesels. Catal. Today 2018, 305, 171–181. [Google Scholar] [CrossRef]
- Naranov, E.R.; Sadovnikov, A.A.; Maximov, A.L.; Karakhanov, E.A. Development of micro-mesoporous materials with lamellar structure as the support of NiW catalysts. Microporous Mesoporous Mater. 2018, 263, 150–157. [Google Scholar] [CrossRef]
- Ding, L.; Zheng, Y.; Zhang, Z.; Ring, Z.; Chen, J. Hydrotreating of light cycled oil using WNi/Al2O3 catalysts containing zeolite beta and/or chemically treated zeolite Y. J. Catal. 2006, 241, 435–445. [Google Scholar] [CrossRef]
- Ding, L.; Zheng, Y.; Zhang, Z.; Ring, Z.; Chen, J. Hydrotreating of light cycle oil using WNi catalysts containing hydrothermally and chemically treated zeolite Y. Catal. Today 2007, 125, 229–238. [Google Scholar] [CrossRef]
- Ding, L.; Zheng, Y.; Yang, H.; Parviz, R. LCO hydrotreating with Mo-Ni and W-Ni supported on nano- and micro-sized zeolite beta. Appl. Catal. A Gen. 2009, 353, 17–23. [Google Scholar] [CrossRef]
- Ding, L.; Zheng, M.; Wang, A.; Zhang, T. A Novel Route to the Preparation of Carbon Supported Nickel Phosphide Catalysts by a Microwave Heating Process. Catal. Lett. 2010, 135, 305–311. [Google Scholar] [CrossRef]
- Yao, S.; Zheng, Y.; Ding, L.; Ng, S.; Yang, H. Co-promotion of fluorine and boron on NiMo/Al2O3 for hydrotreating light cycle oil. Catal. Sci. Technol. 2012, 2, 1925–1932. [Google Scholar] [CrossRef]
- Yao, S.; Zheng, Y.; Ng, S.; Ding, L.; Yang, H. The role of nanobeta zeolite in NiMo hydrotreating catalysts. Appl. Catal. A Gen. 2012, 435, 61–67. [Google Scholar] [CrossRef]
- Ding, L. Hydrotreating Catalyst Supported on Modified Al2O3 and Zeolites for Hydroupgrading Light Cycle Oil. Ph.D. Thesis, University of New Brunswick, Fredericton, NB, Canada, 2006. [Google Scholar]
- Zhang, H. Synthesis of Highly Active Unsupported Molybdenum Sulfide Catalysts for Hydrodesulfurization and Hydrodeoxygenation. Ph.D. Thesis, University of New Brunswick, Fredericton, NB, Canada, 2014. [Google Scholar]
- Wang, H. Integration of Catalytic Cracking and Hydrotreating Technology for Triglyceride Deoxygenation. Ph.D. Thesis, University of New Brunswick, Fredericton, NB, Canada, 2016. [Google Scholar]
- Zheng, P.; Li, T.; Chi, K.; Xiao, C.; Wang, X.; Fan, J.; Duan, A.; Xu, C. DFT insights into the direct desulfurization pathways of DBT and 4,6-DMDBT catalyzed by Co-promoted and Ni-promoted MoS2 corner sites. Chem. Eng. Sci. 2019, 206, 249–260. [Google Scholar] [CrossRef]
- Wu, T.; Chen, S.-L.; Yuan, G.-M.; Pan, X.; Du, J.; Zhang, Y.; Zhang, N. High Metal-Acid Balance and Selective Hydrogenation Activity Catalysts for Hydrocracking of 1-Methylnaphthalene to Benzene, Toluene, and Xylene. Ind. Eng. Chem. Res. 2020, 59, 5546–5556. [Google Scholar] [CrossRef]
- Wang, H.; Li, G.; Rogers, K.; Lin, H.; Zheng, Y.; Ng, S. Hydrotreating of waste cooking oil over supported CoMoS catalyst Catalyst-deactivation mechanism study. Mol. Catal. 2017, 443, 228–240. [Google Scholar] [CrossRef]
- Cortes, J.C.; Rodriguez, C.; Molina, R.; Moreno, S. Hydrocracking of 1-methylnaphtalene (1MN) over modified clays-supported NiMoS and NiWS catalyst. Fuel 2021, 295, 120612. [Google Scholar] [CrossRef]
- Dik, P.P.; Golubev, I.S.; Kazakov, M.O.; Pereyma, V.Y.; Smirnova, M.Y.; Prosvirin, I.P.; Gerasimov, E.Y.; Kondrashev, D.O.; Golovachev, V.A.; Kleimenov, A.V.; et al. Influence of zeolite content in NiW/Y-ASA-Al2O3 catalyst for second stage hydrocracking. Catal. Today 2021, 377, 50–58. [Google Scholar] [CrossRef]
- Salam, M.A.; Cheah, Y.W.; Ho, P.H.; Olsson, L.; Creaser, D. Hydrotreatment of lignin dimers over NiMoS-USY: Effect of silica/alumina ratio. Sustain. Energy Fuels 2021, 5, 3445–3457. [Google Scholar] [CrossRef]
- Santos, B.M.; Zhao, W.; Zotin, J.L.; da Silva, M.A.P.; Oliviero, L.; Mauge, F. Impact of proximity between NiMoS and zeolitic HY sites on cyclohexene hydroconversion: An infrared operando study of sulfide catalysts. J. Catal. 2021, 396, 92–103. [Google Scholar] [CrossRef]
- Hu, E.; Yao, Z.; Zhao, L.; Wu, J.; Meng, H.; Huo, L.; Li, Y. Characteristics of zeolite-modified NiMo/Al2O3 catalysts and their hydrotreating performance for light cycled oil. Can. J. Chem. Eng. 2019, 97, 1107–1113. [Google Scholar] [CrossRef]
- Laredo, G.C.; Merino, P.M.V.; Hernandez, P.S. Light Cycle Oil Upgrading to High Quality Fuels and Petrochemicals: A Review. Ind. Eng. Chem. Res. 2018, 57, 7315–7321. [Google Scholar] [CrossRef]
- Chen, X.; Dong, Y.; Yu, X.; Wang, Z.; Liu, Y.; Liu, J.; Yao, S. Steric Hindrance of Methyl Group on the Reaction Pathway of Hydrodesulfurization in the Presence of Quinoline. Catal. Lett. 2021, 151, 194–211. [Google Scholar] [CrossRef]
- Vega-Merino, P.M.; Quintana-Solorzano, R.; Laredo-Sanchez, G.C.; Arzate-Barbosa, E.; Olmos-Cerda, E.H. Impact of variables on the naphthalene hydrogenation for the tetralin formation towards BTX production. Int. J. Oil Gas Coal Technol. 2020, 23, 504–517. [Google Scholar] [CrossRef]
- Cao, Z.; Zhang, X.; Xu, C.; Huang, X.; Wu, Z.; Peng, C.; Duan, A. Selective hydrocracking of light cycle oil into high-octane gasoline over bi-functional catalysts. J. Energy Chem. 2021, 52, 41–50. [Google Scholar] [CrossRef]
- Tung, N.T.; Shnzaki, A.; Qian, E.W. Hydrodesulfurization, Hydrodenitrogenation and Hydrodearomatization over CoMo/SAPO-11-Al2O3 Catalysts. J. Jpn. Pet. Inst. 2017, 60, 301–310. [Google Scholar]
- Peng, C.; Zhou, Z.; Cheng, Z.; Fang, X. Upgrading of Light Cycle Oil to High-Octane Gasoline through Selective Hydrocracking over Non-Noble Metal Bifunctional Catalysts. Energy Fuels 2019, 33, 1090–1097. [Google Scholar] [CrossRef]
- Ward, J.W. Nature of active sites on zeolite. Rare earth Y zeolite. J. Catal. 1969, 13, 321–327. [Google Scholar]
- Wu, X. Acidity and Catalytic Activity of Zeolite Catalysts Bound with Silica and Alumina; Texas A&M University: College Station, TX, USA, 2003. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | NiMo/AZ | CoMo/AZ | NiW/AZ |
|---|---|---|---|
| SBET, m2/g | 141 | 185 | 149 |
| Total pore volume, cm3/g | 0.44 | 0.49 | 0.39 |
| Micropore volume, cm3/g | 0.061 | 0.084 | 0.066 |
| Average layer length, nm | 5.10 | 6.11 | 8.40 |
| Average layer number | 1.74 | 1.79 | 1.52 |
| Catalysts | B Acids, ×10−3 mmol/g | L Acids, mmol/g | Ratio of B/L | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Weak | Middle | Strong | Total | Weak | Middle | Strong | Total | Weak | Middle | Strong | Total | |
| NWS | 32.8 | 4.2 | 23.3 | 60.3 | 63.7 | 33.7 | 29.1 | 126.5 | 0.52 | 0.13 | 0.80 | 0.48 |
| NMS | 17.1 | 19.0 | 19.1 | 55.2 | 89.1 | 67.2 | 54.5 | 210.8 | 0.19 | 0.28 | 0.35 | 0.26 |
| CMS | 18.0 | 16.0 | 13.8 | 47.8 | 21.0 | 15.2 | 9.2 | 45.4 | 0.86 | 1.05 | 1.50 | 1.05 |
| k1 × 10−5, s−1·gcat−1 | CoMo/AZ | NiMo/AZ | NiW/AZ |
|---|---|---|---|
| Model 1 without N | 4.37 | 3.07 | 2.48 |
| Model 2 with N | 3.87 | 2.09 | 2.25 |
| Compositions, % | Model 1 without Nitrogen | Model 2 with Nitrogen | ||||
|---|---|---|---|---|---|---|
| CoMo/AZ | NiMo/AZ | NiW/AZ | CoMo/AZ | NiMo/AZ | NiW/AZ | |
| 1-MT | 10.1 | 14.1 | 9.7 | 16.3 | 17.9 | 15.5 |
| 2-MT | 6.7 | 4.4 | 7.6 | 3.3 | 1.7 | 3.9 |
| 5-MT | 21.9 | 27.4 | 22.1 | 30.3 | 33.9 | 29.4 |
| 6-MT | 11.4 | 6.3 | 12.6 | 2.4 | 4.2 | 4.9 |
| 4H-HC | - | - | - | - | - | - |
| decalin | 8.4 | 7.1 | 8.1 | 5.6 | 4.2 | 6.1 |
| single ring | 10.1 | 7.3 | 8.2 | 5.1 | 4.3 | 6.0 |
| C2 toluene | 4.9 | 5.6 | 5.2 | 5.1 | 4.6 | 5.0 |
| naphathalene | 3.9 | 3.1 | 4.0 | 2.7 | 2.5 | 2.9 |
| polymer | 1.5 | 0.6 | 1.4 | 1.1 | 0.9 | 1.2 |
| Feed | Catalyst | 375 °C 1100 psig | 375 °C, 550 psig | 300 °C, 1100 psig |
|---|---|---|---|---|
| LCO1 | CoMo/AZ | 877 | 1016 | 2845 |
| NiMo/AZ | 722 | 909 | 3198 | |
| NiW/AZ | 1121 | 1099 | 3538 | |
| LCO2 | CoMo/AZ | 586 | 1096 | 2334 |
| NiMo/AZ | 512 | 612 | 2430 | |
| NiW/AZ | 567 | 958 | 3022 |
| Feed | Catalyst | 375 °C, 1100 psig | 375 °C, 550 psig | 300 °C, 1100 psig |
|---|---|---|---|---|
| LCO1 | CoMo/AZ | 104.6 | 265.2 | 415.2 |
| NiMo/AZ | 55.0 | 192.4 | 327.4 | |
| NiW/AZ | 83.8 | 208.7 | 378.4 | |
| LCO2 | CoMo/AZ | 14.5 | 49.7 | 93.1 |
| NiMo/AZ | 5.4 | 17.6 | 57.3 | |
| NiW/AZ | 4.0 | 22.4 | 69.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pu, J.; Zhang, H.; Wang, M.; Rogers, K.; Wang, H.; Wang, H.; Ng, S.; Sun, P. Roles of Nanostructured Bimetallic Supported on Alumina-Zeolite (AZ) in Light Cycle Oil (LCO) Upgrading. Catalysts 2021, 11, 1277. https://doi.org/10.3390/catal11111277
Pu J, Zhang H, Wang M, Rogers K, Wang H, Wang H, Ng S, Sun P. Roles of Nanostructured Bimetallic Supported on Alumina-Zeolite (AZ) in Light Cycle Oil (LCO) Upgrading. Catalysts. 2021; 11(11):1277. https://doi.org/10.3390/catal11111277
Chicago/Turabian StylePu, Jianglong, Haiping Zhang, Min Wang, Kyle Rogers, Hongmei Wang, Hui Wang, Siauw Ng, and Ping Sun. 2021. "Roles of Nanostructured Bimetallic Supported on Alumina-Zeolite (AZ) in Light Cycle Oil (LCO) Upgrading" Catalysts 11, no. 11: 1277. https://doi.org/10.3390/catal11111277
APA StylePu, J., Zhang, H., Wang, M., Rogers, K., Wang, H., Wang, H., Ng, S., & Sun, P. (2021). Roles of Nanostructured Bimetallic Supported on Alumina-Zeolite (AZ) in Light Cycle Oil (LCO) Upgrading. Catalysts, 11(11), 1277. https://doi.org/10.3390/catal11111277