
What Is the Real State of Single-Atom Catalysts under Electrochemical Conditions—From Adsorption to Surface Pourbaix Plots?

 ,
, 
Abstract
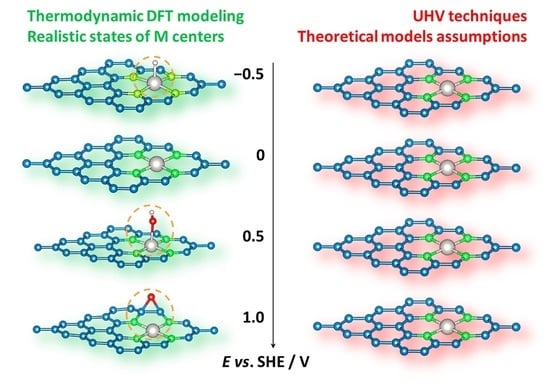
:
1. Introduction
2. Results
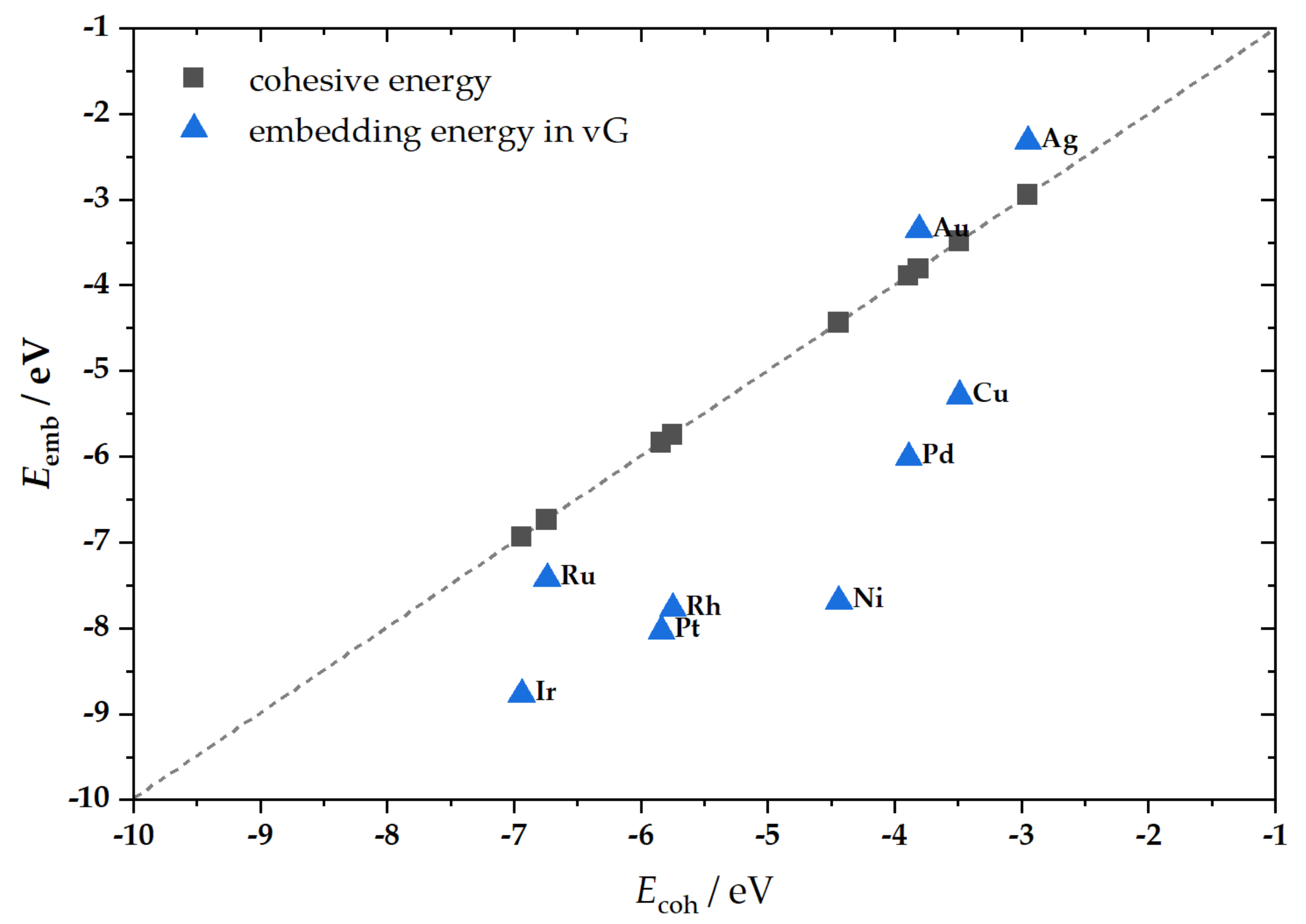
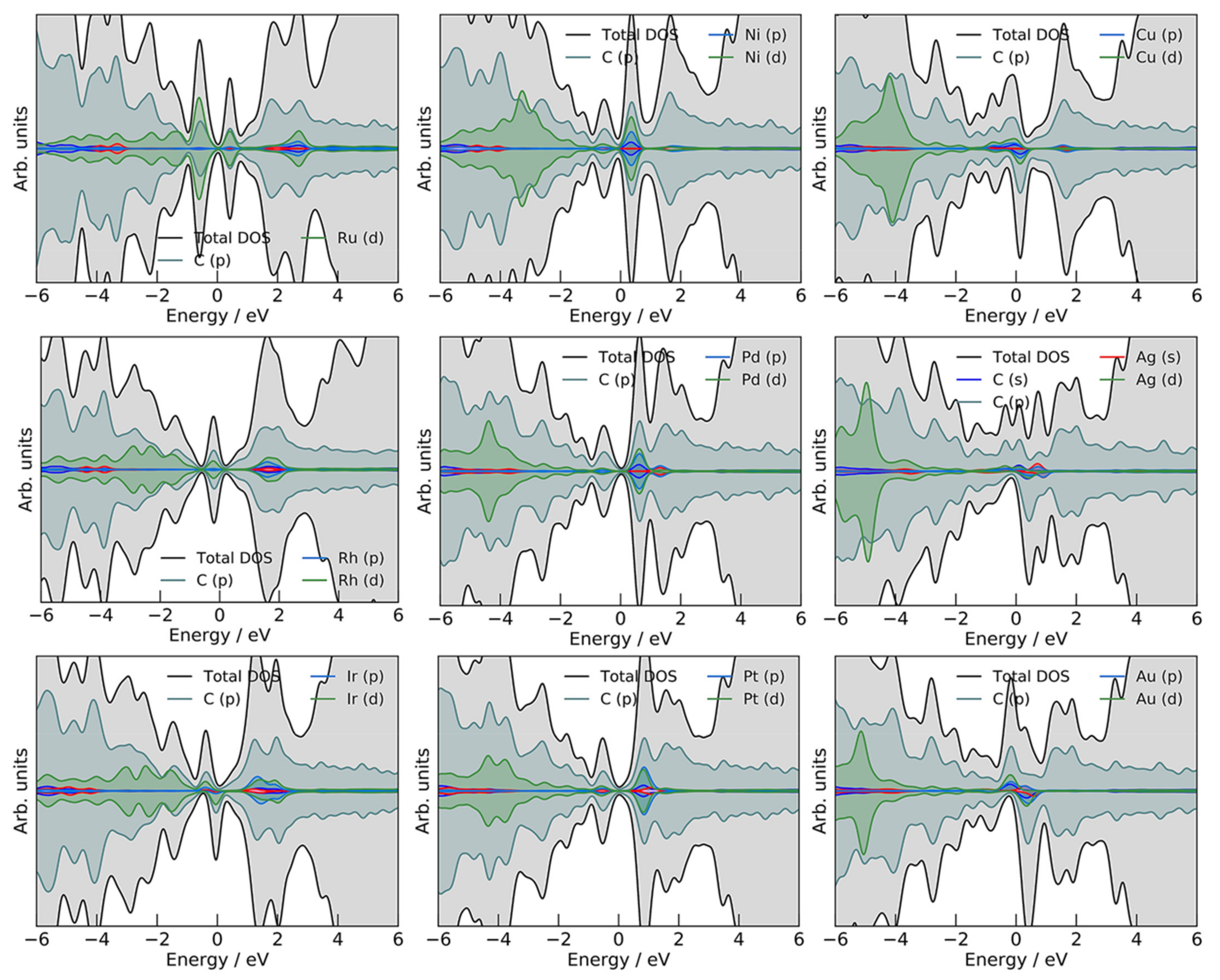
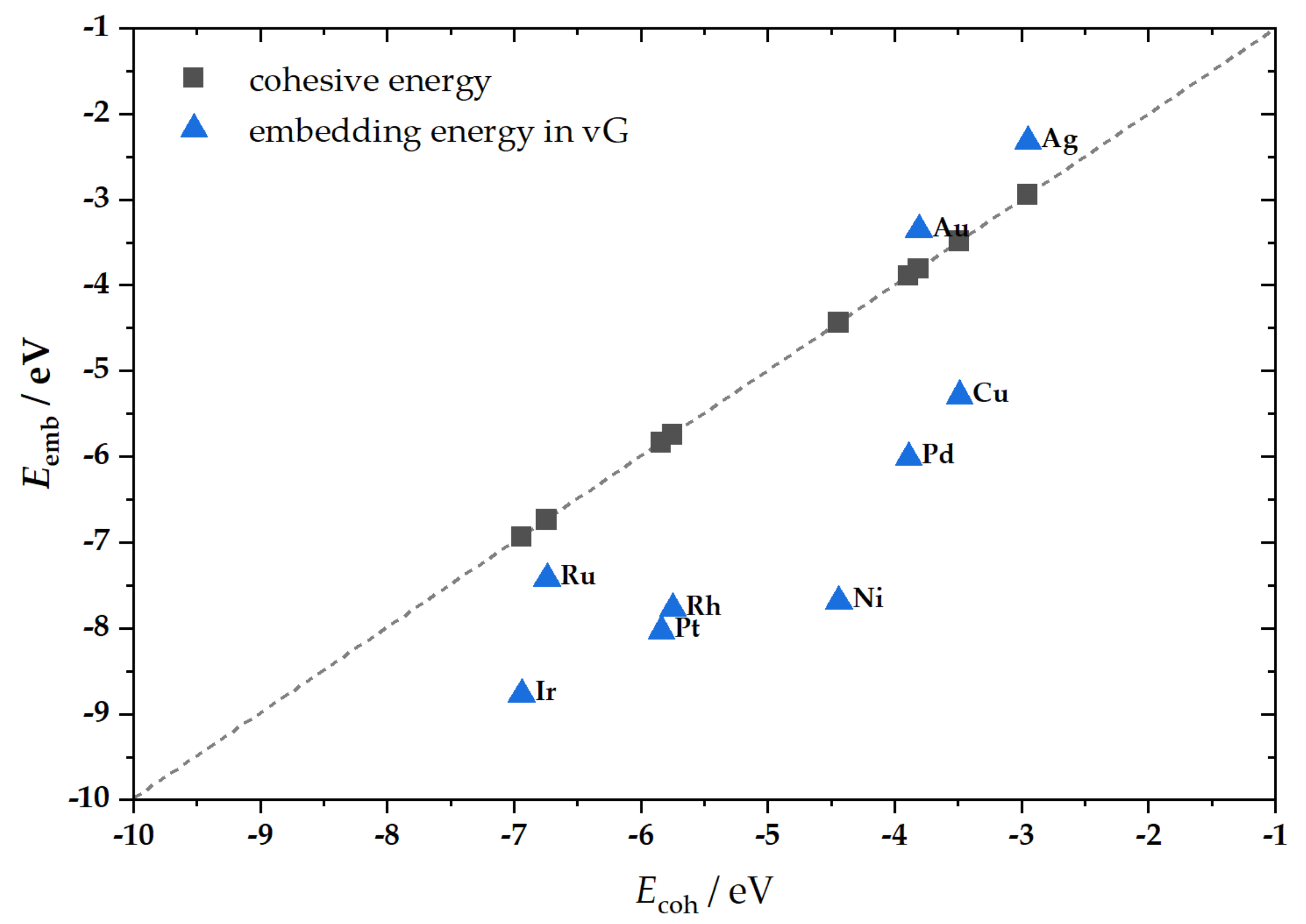
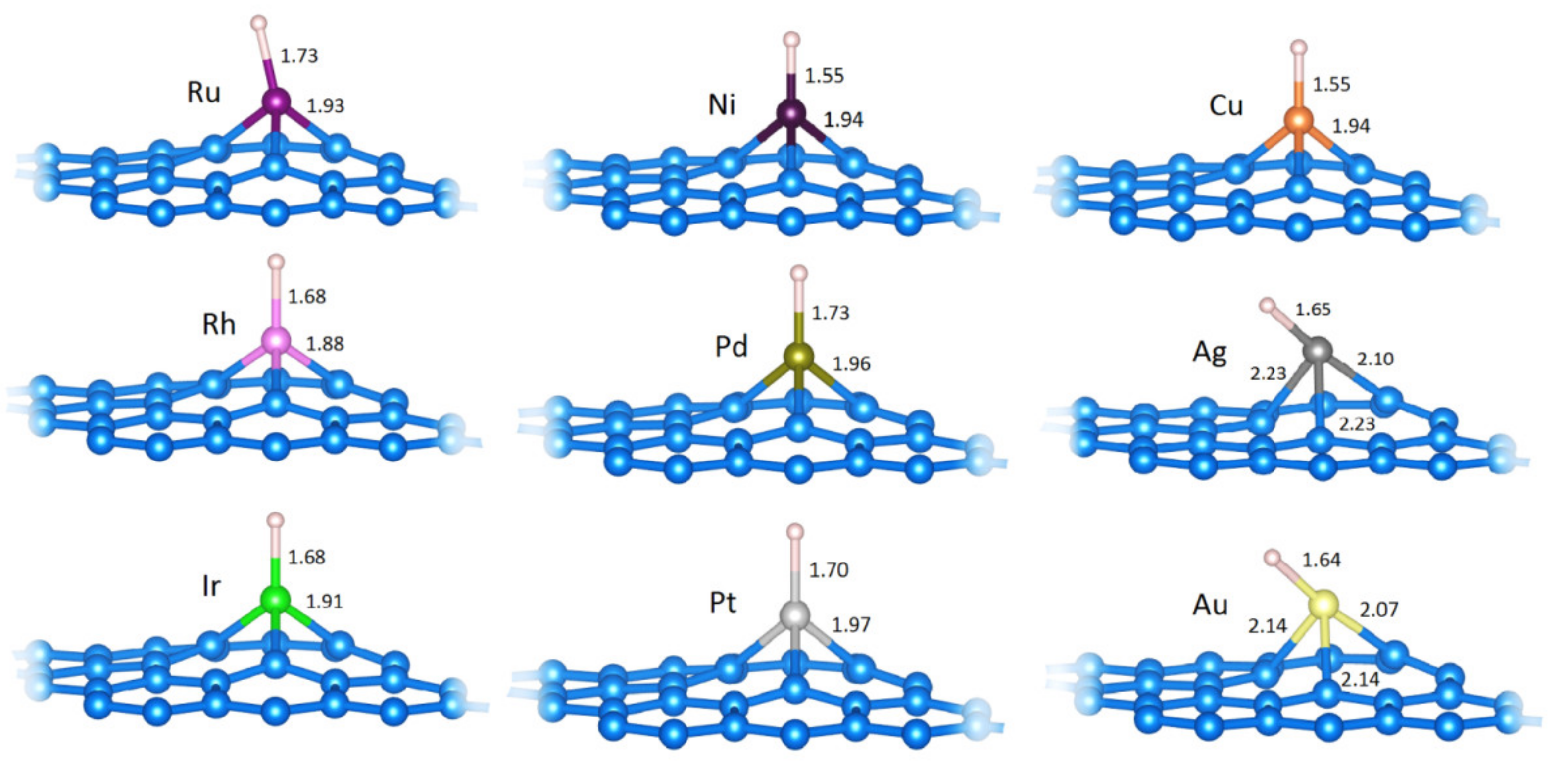
2.1. M@v-Graphene—Formation of SACs
2.2. A−M@v-Graphene
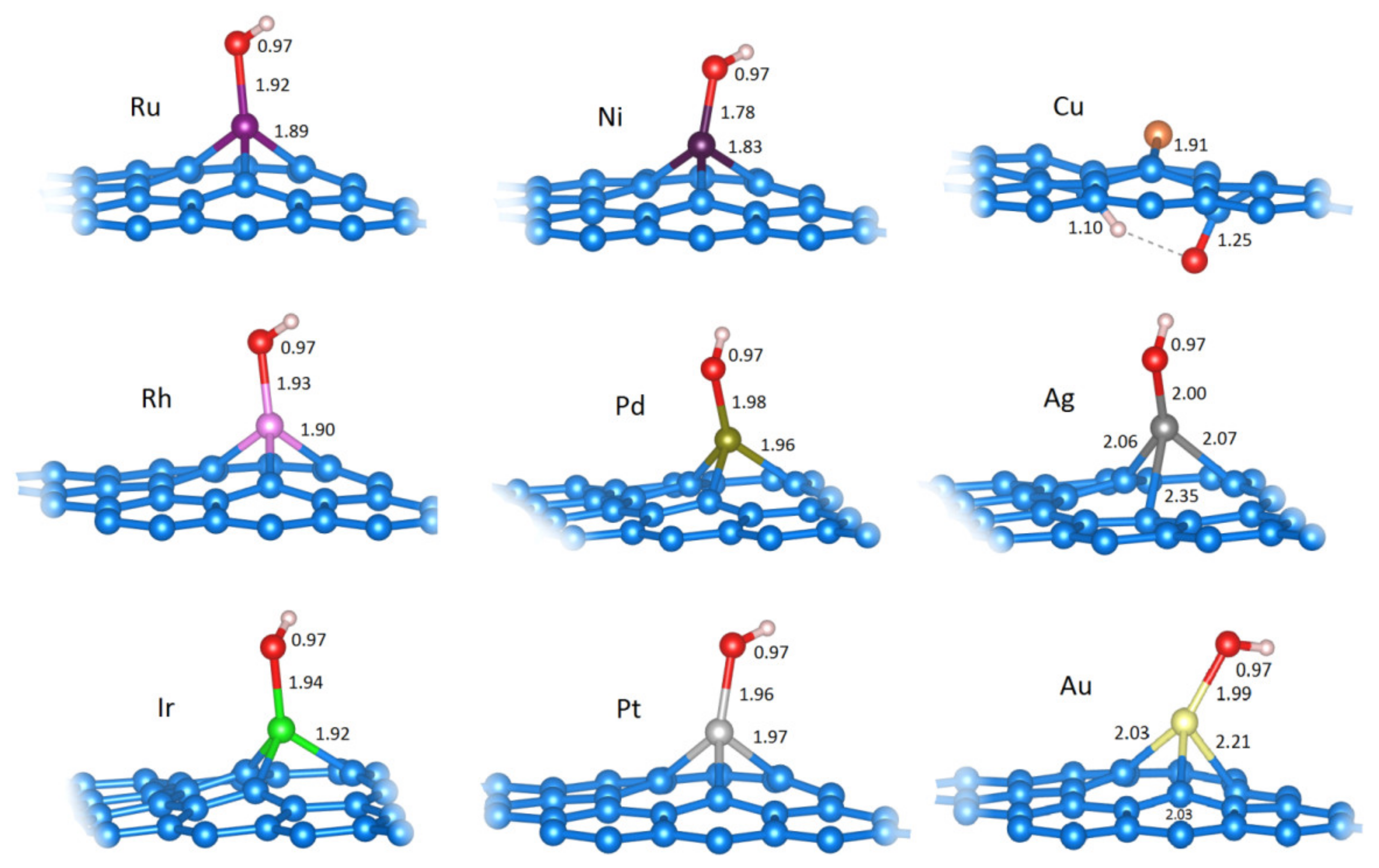
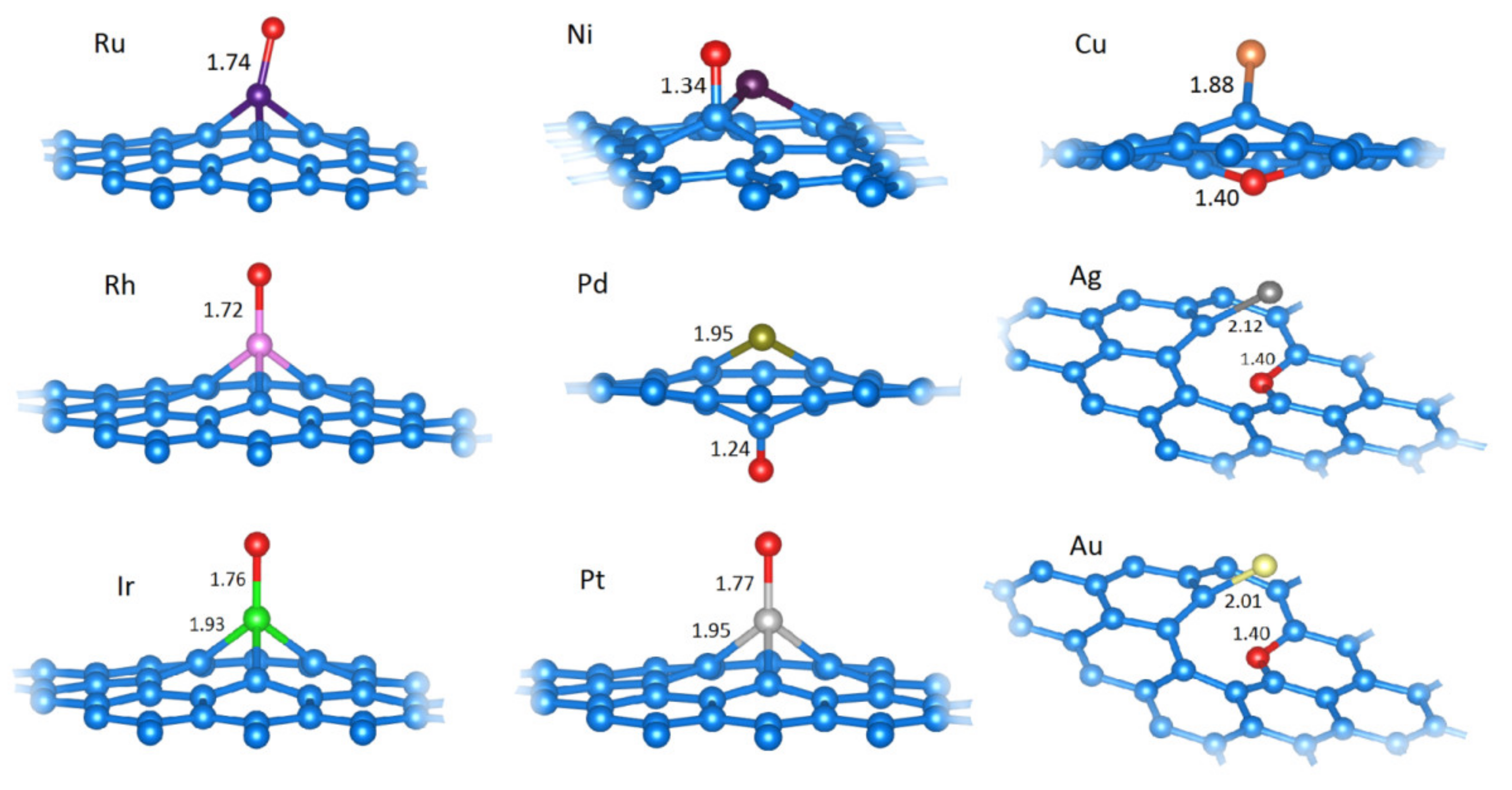
2.2.1. H Adsorption (H−M@vG)
2.2.2. OH Adsorption (OH−M@vG)
2.2.3. O Adsorption (O−M@vG)
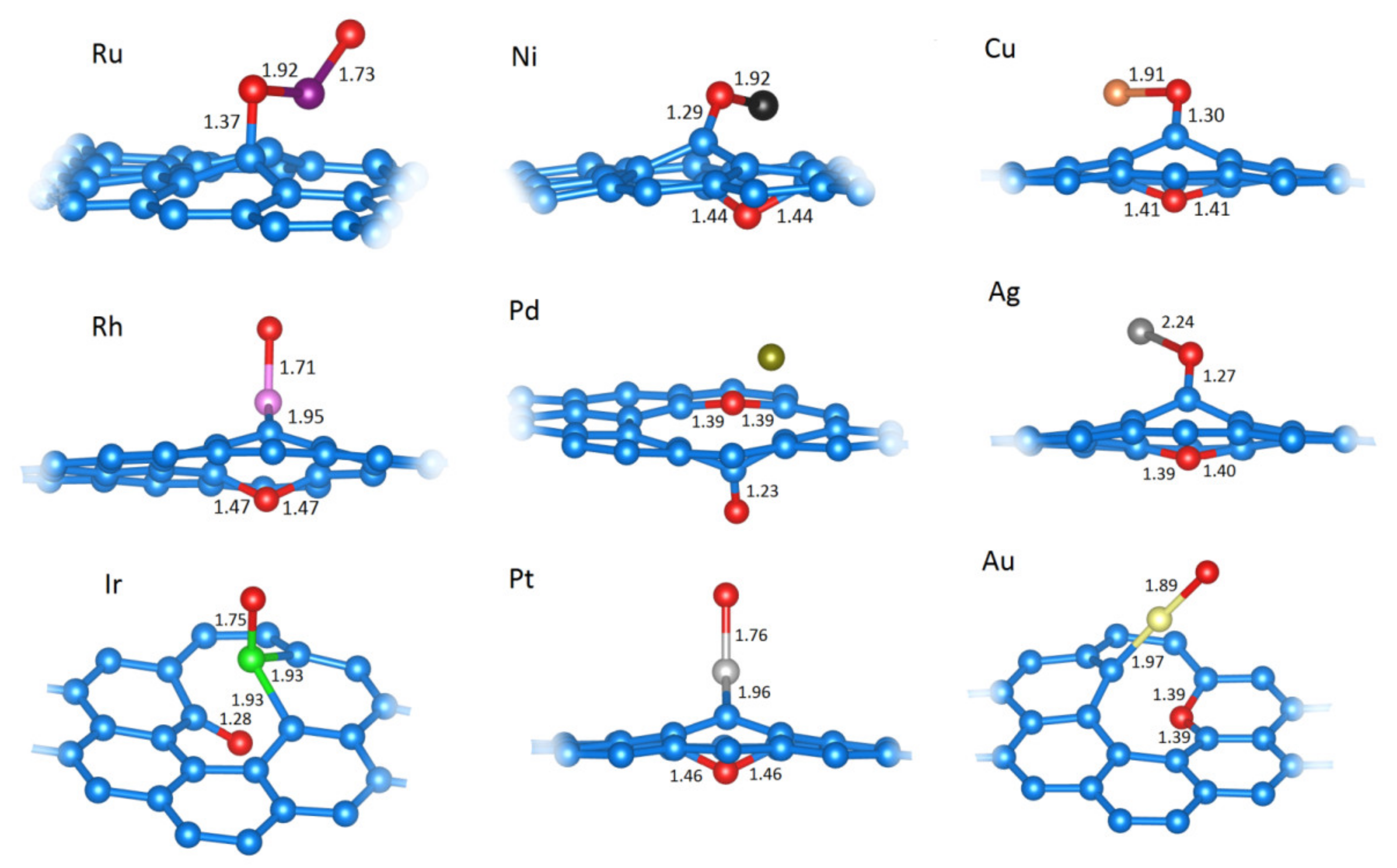
2.2.4. Extensive Oxidation of M@vG (2O−M@vG)
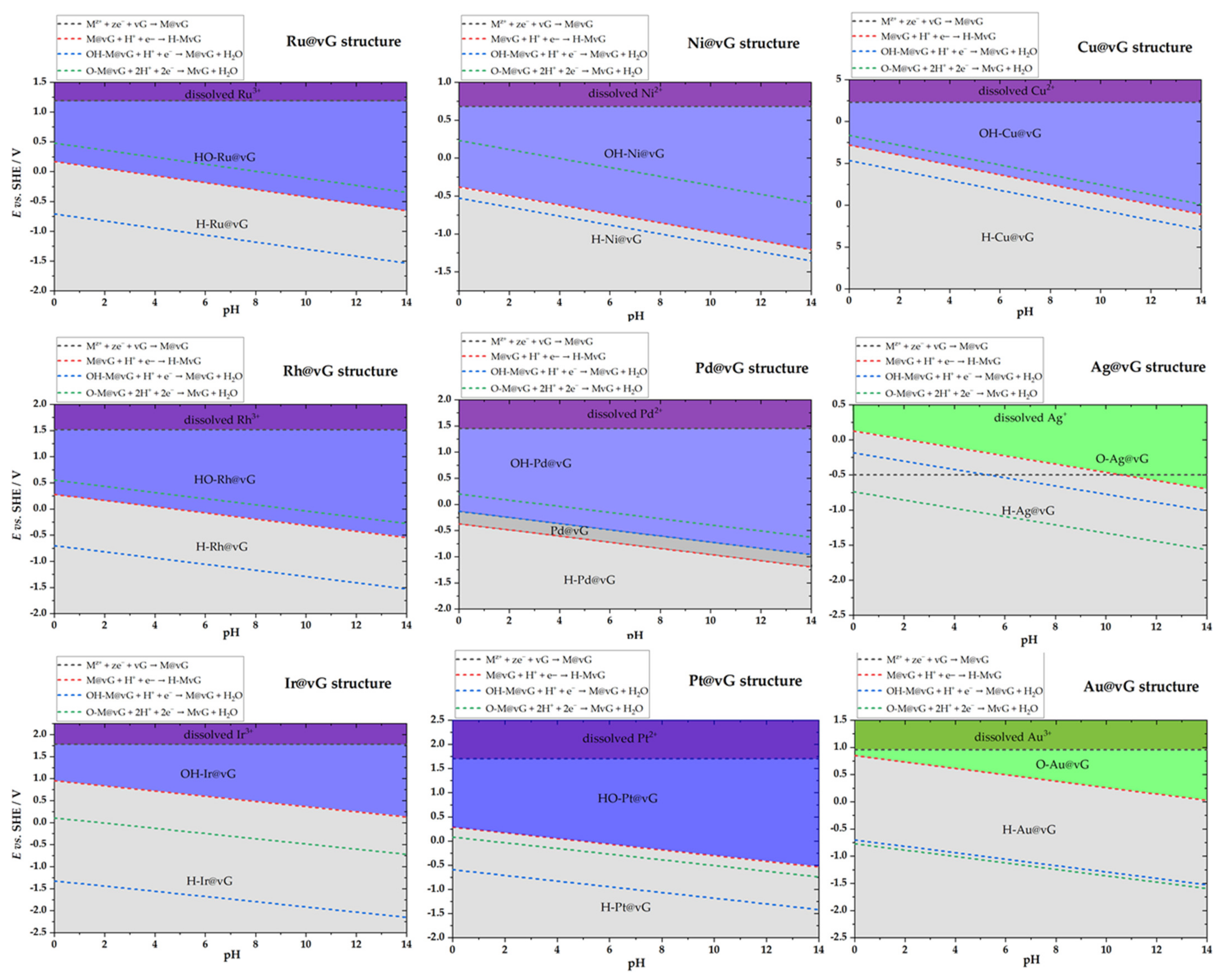
2.3. Surface Pourbaix Plots for M@vG Catalysts
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, G.; Shi, L.; Ye, J. Single-Atom Catalysts: Emerging Multifunctional Materials in Heterogeneous Catalysis. Adv. Energy Mater. 2018, 8, 1701343. [Google Scholar] [CrossRef]
- Liang, S.; Hao, C.; Shi, Y. The Power of Single-Atom Catalysis. ChemCatChem 2015, 7, 2559–2567. [Google Scholar] [CrossRef]
- Hossain, M.D.; Liu, Z.; Zhuang, M.; Yan, X.; Xu, G.L.; Gadre, C.A.; Tyagi, A.; Abidi, I.H.; Sun, C.J.; Wong, H.; et al. Rational Design of Graphene-Supported Single Atom Catalysts for Hydrogen Evolution Reaction. Adv. Energy Mater. 2019, 9, 1803689. [Google Scholar] [CrossRef]
- Cheng, N.; Stambula, S.; Wang, D.; Banis, M.N.; Liu, J.; Riese, A.; Xiao, B.; Li, R.; Sham, T.K.; Liu, L.M.; et al. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef]
- Pu, Z.; Amiinu, I.S.; Cheng, R.; Wang, P.; Zhang, C.; Mu, S.; Zhao, W.; Su, F.; Zhang, G.; Liao, S.; et al. Single-Atom Catalysts for Electrochemical Hydrogen Evolution Reaction: Recent Advances and Future Perspectives. Nano-Micro Lett. 2020, 12, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kim, H.-E.; Lee, H. Single-Atom Catalysts of Precious Metals for Electrochemical Reactions. ChemSusChem 2018, 11, 104–113. [Google Scholar] [CrossRef]
- Zhu, C.; Shi, Q.; Feng, S.; Du, D.; Lin, Y. Single-Atom Catalysts for Electrochemical Water Splitting. ACS Energy Lett. 2018, 3, 1713–1721. [Google Scholar] [CrossRef]
- Gutić, S.J.; Dobrota, A.S.; Fako, E.; Skorodumova, N.V.; López, N.; Pašti, I.A. Hydrogen Evolution Reaction-From Single Crystal to Single Atom Catalysts. Catalysts 2020, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Luo, M.; Liu, X.; Xing, W.; Xu, W.; Jiang, Z.; Gu, L. Zn Single Atom Catalyst for Highly Efficient Oxygen Reduction Reaction. Adv. Funct. Mater. 2017, 27, 1700802. [Google Scholar] [CrossRef]
- Cui, L.; Cui, L.; Li, Z.; Zhang, J.; Wang, H.; Lu, S.; Xiang, Y. A copper single-atom catalyst towards efficient and durable oxygen reduction for fuel cells. J. Mater. Chem. A 2019, 7, 16690–16695. [Google Scholar] [CrossRef]
- Yang, L.; Xu, H.; Liu, H.; Zeng, X.; Cheng, D.; Huang, Y.; Zheng, L.; Cao, R.; Cao, D. Oxygen-Reconstituted Active Species of Single-Atom Cu Catalysts for Oxygen Reduction Reaction. Research 2020, 2020, 7593023. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Xi, L.; Yu, Y.; Chen, N.; Sun, S.; Wang, W.; Lange, K.M.; Zhang, B. Single-Atom Au/NiFe Layered Double Hydroxide Electrocatalyst: Probing the Origin of Activity for Oxygen Evolution Reaction. J. Am. Chem. Soc. 2018, 140, 3876–3879. [Google Scholar] [CrossRef]
- Zhang, Z.; Feng, C.; Li, X.; Liu, C.; Wang, D.; Si, R.; Yang, J.; Zhou, S.; Zeng, J. In-Situ Generated High-Valent Iron Single-Atom Catalyst for Efficient Oxygen Evolution. Nano Lett. 2021, 21, 4795–4801. [Google Scholar] [CrossRef]
- Wang, K.; Wang, X.; Liang, X. Synthesis of High Metal Loading Single Atom Catalysts and Exploration of the Active Center Structure. ChemCatChem 2021, 13, 28–58. [Google Scholar] [CrossRef]
- Speck, F.D.; Paul, M.T.Y.; Ruiz-Zepeda, F.; Gatalo, M.; Kim, H.; Kwon, H.C.; Mayrhofer, K.J.J.; Choi, M.; Choi, C.H.; Hodnik, N.; et al. Atomistic Insights into the Stability of Pt Single-Atom Electrocatalysts. J. Am. Chem. Soc. 2020, 142, 15496–15504. [Google Scholar] [CrossRef]
- Stamenkovic, V.R.; Fowler, B.; Mun, B.S.; Wang, G.; Ross, P.N.; Lucas, C.A.; Markovic, N.M. Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability. Science 2007, 315, 493–497. [Google Scholar] [CrossRef] [Green Version]
- Stamenkovic, V.R.; Mun, B.S.; Arenz, M.; Mayrhofer, K.J.J.; Lucas, C.A.; Wang, G.; Ross, P.N.; Markovic, N.M. Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces. Nat. Mater. 2007, 6, 241–247. [Google Scholar] [CrossRef]
- Stamenkovic, V.; Mun, B.S.; Mayrhofer, K.J.J.; Ross, P.N.; Markovic, N.M.; Rossmeisl, J.; Greeley, J.; Nørskov, J.K. Changing the Activity of Electrocatalysts for Oxygen Reduction by Tuning the Surface Electronic Structure. Angew. Chem. 2006, 118, 2963–2967. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Markovic, N.M.; Gasteiger, H.A.; Ross, P.N. Oxygen reduction on platinum low-index single-crystal surfaces in sulfuric acid solution. Rotating ring—Pt(hkl) disk studies. J. Phys. Chem. 1995, 99, 3411–3415. [Google Scholar] [CrossRef]
- Marković, N.M.; Gasteiger, H.A.; Ross, P.N. Oxygen reduction on platinum low-index single-crystal surfaces in alkaline solution: Rotating ring disk Pt(hkl) studies. J. Phys. Chem. 1996, 100, 6715–6721. [Google Scholar] [CrossRef]
- Minguzzi, A.; Fan, F.R.F.; Vertova, A.; Rondinini, S.; Bard, A.J. Dynamic potential–pH diagrams application to electrocatalysts for water oxidation. Chem. Sci. 2011, 3, 217–229. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Chan, M.K.Y.; Zhao, Z.-J.; Kubal, J.; Fan, D.; Greeley, J. Towards First Principles-Based Prediction of Highly Accurate Electrochemical Pourbaix Diagrams. J. Phys. Chem. C 2015, 119, 18177–18187. [Google Scholar] [CrossRef]
- Hansen, H.A.; Rossmeisl, J.; Nørskov, J.K. Surface Pourbaix diagrams and oxygen reduction activity of Pt, Ag and Ni(111) surfaces studied by DFT. Phys. Chem. Chem. Phys. 2008, 10, 3722–3730. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Majumdar, A.; Lenz, M.; Cramer, C.J.; Truhlar, D.G. Construction of Pourbaix Diagrams for Ruthenium-Based Water-Oxidation Catalysts by Density Functional Theory. Angew. Chem. Int. Ed. 2012, 51, 12810–12814. [Google Scholar] [CrossRef]
- Gossenberger, F.; Juarez, F.; Groß, A. Sulfate, Bisulfate, and Hydrogen Co-adsorption on Pt(111) and Au(111) in an Electrochemical Environment. Front. Chem. 2020, 8, 634. [Google Scholar] [CrossRef]
- Zagal, J.H.; Koper, M.T.M. Reactivity Descriptors for the Activity of Molecular MN4 Catalysts for the Oxygen Reduction Reaction. Angew. Chem. Int. Ed. 2016, 55, 14510–14521. [Google Scholar] [CrossRef]
- Krasheninnikov, A.V.; Lehtinen, P.O.; Foster, A.S.; Pyykkö, P.; Nieminen, R.M. Embedding transition-metal atoms in graphene: Structure, bonding, and magnetism. Phys. Rev. Lett. 2009, 102, 126807. [Google Scholar] [CrossRef] [Green Version]
- Santos, E.J.G.; Ayuela, A.; Sanchez-Portal, D. First-Principles Study of Substitutional Metal Impurities in Graphene: Structural, Electronic and Magnetic Properties. New J. Phys. 2010, 12, 053012. [Google Scholar] [CrossRef] [Green Version]
- Pašti, I.A.; Jovanović, A.; Dobrota, A.S.; Mentus, S.V.; Johansson, B.; Skorodumova, N.V. Atomic adsorption on graphene with a single vacancy: Systematic DFT study through the periodic table of elements. Phys. Chem. Chem. Phys. 2018, 20, 858–865. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Siahrostami, S.; Zheng, T.; Hu, Y.; Hwang, S.; Stavitski, E.; Peng, Y.; Dynes, J.; Gangisetty, M.; Su, D.; et al. Isolated Ni single atoms in graphene nanosheets for high-performance CO2 reduction. Energy Environ. Sci. 2018, 11, 893–903. [Google Scholar] [CrossRef]
- Jiang, K.; Siahrostami, S.; Akey, A.J.; Li, Y.; Lu, Z.; Lattimer, J.; Hu, Y.; Stokes, C.; Gangishetty, M.; Chen, G.; et al. Transition-Metal Single Atoms in a Graphene Shell as Active Centers for Highly Efficient Artificial Photosynthesis. Chem 2017, 3, 950–960. [Google Scholar] [CrossRef] [Green Version]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; John Wiley & Sons: New York, NY, USA, 1996; ISBN 047141526X. [Google Scholar]
- Jovanović, A.Z.; Mentus, S.V.; Skorodumova, N.V.; Pašti, I.A. Reactivity Screening of Single Atoms on Modified Graphene Surface: From Formation and Scaling Relations to Catalytic Activity. Adv. Mater. Interfaces 2021, 8, 2001814. [Google Scholar] [CrossRef]
- Ganose, A.M.; Jackson, A.J.; Scanlon, D.O. sumo: Command-line tools for plotting and analysis of periodic ab initio calculations. J. Open Source Softw. 2018, 3, 717. [Google Scholar] [CrossRef]
- Marković, N.M.; Ross, P.N. Surface science studies of model fuel cell electrocatalysts. Surf. Sci. Rep. 2002, 45, 117–229. [Google Scholar] [CrossRef]
- Gabrielli, C.; Grand, P.P.; Lasia, A.; Perrot, H. Investigation of Hydrogen Adsorption-Absorption into Thin Palladium Films: I. Theory. J. Electrochem. Soc. 2004, 151, A1925. [Google Scholar] [CrossRef]
- Łosiewicz, B.; Jurczakowski, R.; Lasia, A. Kinetics of hydrogen underpotential deposition at iridium in sulfuric and perchloric acids. Electrochim. Acta 2017, 225, 160–167. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, J23–J26. [Google Scholar] [CrossRef] [Green Version]
- Nordlander, P.; Holloway, S.; Nørskov, J.K. Hydrogen adsorption on metal surfaces. Surf. Sci. 1984, 136, 59–81. [Google Scholar] [CrossRef]
- Park, J.; Elmlund, H.; Ercius, P.; Yuk, J.M.; Limmer, D.T.; Chen, Q.; Kim, K.; Han, S.H.; Weitz, D.A.; Zettl, A.; et al. 3D structure of individual nanocrystals in solution by electron microscopy. Science 2015, 349, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Jia, Q.; Ramaswamy, N.; Hafiz, H.; Tylus, U.; Strickland, K.; Wu, G.; Barbiellini, B.; Bansil, A.; Holby, E.F.; Zelenay, P.; et al. Experimental Observation of Redox-Induced Fe–N Switching Behavior as a Determinant Role for Oxygen Reduction Activity. ACS Nano 2015, 9, 12496–12505. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, A.; Pettersson, L.G.M.; Hammer, B.; Bligaard, T.; Christensen, C.H.; Nørskov, J.K. The electronic structure effect in heterogeneous catalysis. Catal. Lett. 2005, 100, 111–114. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M | Mtot/μB | Eemb(M)/eV | Eembref(M)/eV | d(C−M)/Å | h(M)/Å | Δq(M)/e |
|---|---|---|---|---|---|---|
| Ni | 0.00 | −6.77 | −6.64 1,*; −6.89 1,# −6.78 1,†; −5.72 1,‡ | 1.79 | 1.19 | −0.42 |
| Cu | 0.85 | −3.75 | −3.61 1,*; −3.87 1,# −3.75 1,†; −2.89 1,‡; −3.69 2,* | 1.88 | 1.35 | −0.52 |
| Ru | 0.00 | −8.98 | −8.81 1,*; −9.16 1,# −8.99 1,†; −7.67 1,‡ | 1.88 | 1.47 | −0.54 |
| Rh | 0.03 | −8.48 | −8.34 1,*; −8.69 1,# −8.49 1,†; −7.05 1,‡ | 1.89 | 1.44 | −0.35 |
| Pd | 0.00 | −5.43 | −5.27 1,*; −5.62 1,# −5.44 1,†; −4.30 1,‡ | 1.94 | 1.45 | −0.34 |
| Ag | 1.01 | −1.89 | −1.72 1,*; −2.11 1,# −1.89 1,†; −1.28 1,‡−1.76 2,* | 2.16 2.21 2.21 | 1.77 | −0.46 |
| Ir | 0.71 | −9.31 | −9.28 1,*; −9.77 1,# −9.45 1,†; −7.67 1,‡ | 1.90 | 1.50 | −0.59 |
| Pt | 0.00 | −7.34 | −7.08 1,*; −7.57 1,# −7.34 1,†; −6.02 1,‡ | 1.94 | 1.51 | −0.28 |
| Au | 0.99 | −2.60 | −2.40 1,*; −2.93 1,#; −2.60 1,†; −1.80 1,‡ −2.07 2,* | 2.082.082.09 | 1.65 | −0.22 |
| M | Mtot/μB | Eads(H)/eV | d(M−H)/Å | Δq(M)/e | Δq(H) */e |
|---|---|---|---|---|---|
| Ni | 0.00 | −1.89 | 1.55 | −0.10 | 0.41 |
| Cu | 1.67 | −1.99 | 1.55 | −0.05 | 0.34 |
| Ru | 0.96 | −2.44 | 1.73 | −0.60 | 0.23 |
| Rh | 0.00 | −2.55 | 1.68 | −0.17 | 0.27 |
| Pd | 0.00 | −1.90 | 1.73 | −0.05 | 0.29 |
| Ag | 0.00 | −2.40 | 1.65 | 0.06 | 0.29 |
| Ir | 0.00 | −3.22 | 1.68 | 0.11 | 0.23 |
| Pt | 0.00 | −2.56 | 1.70 | −0.10 | 0.28 |
| Au | 0.00 | −3.12 | 1.64 | −0.03 | 0.20 |
| M | Mtot/μB | Eads(OH)/eV | d(M/C−O)/Å | Δq(M)*/e | Δq(OH) */e |
|---|---|---|---|---|---|
| Ni | 0.01 | −3.61 | 1.78 | −0.35 | 0.50 |
| Cu | 0.00 | −3.55 | 1.25 | 0.05 | 0.54 |
| Ru | 0.00 | −3.79 | 1.92 | −0.44 | 0.53 |
| Rh | 0.00 | −3.78 | 1.93 | −0.37 | 0.50 |
| Pd | 0.00 | −3.21 | 1.98 | 0.08 | 0.54 |
| Ag | 0.10 | −3.27 | 2.00 | −0.19 | 0.49 |
| Ir | 0.00 | −4.40 | 1.94 | −0.16 | 0.49 |
| Pt | 0.00 | −3.67 | 1.96 | −0.33 | 0.49 |
| Au | 0.00 | −3.78 | 1.99 | −0.29 | 0.32 |
| M | Mtot/μB | Eads(O)/eV | d(M/C−O)/Å | Δq(M) */e | Δq(O) */e |
|---|---|---|---|---|---|
| Ni | 0.00 | −5.07 | 1.34 | −0.14 | 1.76 |
| Cu | −0.66 | −5.86 | 1.40 | 0.35 | 1.58 |
| Ru | 0.96 | −4.58 | 1.74 | −0.58 | 0.72 |
| Rh | 0.00 | −4.43 | 1.72 | −0.63 | 0.84 |
| Pd | 0.00 | −5.14 | 1.24 | 0.05 | 1.73 |
| Ag | 0.80 | −7.01 | 1.40 | 0.28 | 1.49 |
| Ir | 0.00 | −5.32 | 1.76 | −0.34 | 0.78 |
| Pt | 0.00 | −5.37 | 1.77 | −0.53 | 0.77 |
| Au | 0.70 | −7.08 | 1.40 | 0.31 | 1.57 |
| M | Mtot/μB | Eadsdiff(O)/eV | Eadsint(O)/eV |
|---|---|---|---|
| Ni | 0.00 | −4.43 | −4.75 |
| Cu | 0.00 | −5.72 | −5.79 |
| Ru | 0.89 | −4.13 | −4.35 |
| Rh | 0.00 | −3.31 | −3.87 |
| Pd | 0.00 | −4.91 | −5.02 |
| Ag | 0.00 | −5.64 | −6.32 |
| Ir | 0.00 | −3.24 | −4.28 |
| Pt | 0.00 | −2.67 | −4.02 |
| Au | 1.00 | −3.34 | −5.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobrota, A.S.; Đokić, T.; Skorodumova, N.V.; Mentus, S.V.; Pašti, I.A. What Is the Real State of Single-Atom Catalysts under Electrochemical Conditions—From Adsorption to Surface Pourbaix Plots? Catalysts 2021, 11, 1207. https://doi.org/10.3390/catal11101207
Dobrota AS, Đokić T, Skorodumova NV, Mentus SV, Pašti IA. What Is the Real State of Single-Atom Catalysts under Electrochemical Conditions—From Adsorption to Surface Pourbaix Plots? Catalysts. 2021; 11(10):1207. https://doi.org/10.3390/catal11101207
Chicago/Turabian StyleDobrota, Ana S., Tanja Đokić, Natalia V. Skorodumova, Slavko V. Mentus, and Igor A. Pašti. 2021. "What Is the Real State of Single-Atom Catalysts under Electrochemical Conditions—From Adsorption to Surface Pourbaix Plots?" Catalysts 11, no. 10: 1207. https://doi.org/10.3390/catal11101207
APA StyleDobrota, A. S., Đokić, T., Skorodumova, N. V., Mentus, S. V., & Pašti, I. A. (2021). What Is the Real State of Single-Atom Catalysts under Electrochemical Conditions—From Adsorption to Surface Pourbaix Plots? Catalysts, 11(10), 1207. https://doi.org/10.3390/catal11101207