Recent Advances in Asymmetric Catalytic Electrosynthesis


Abstract
{kind=link}
{kind=link}
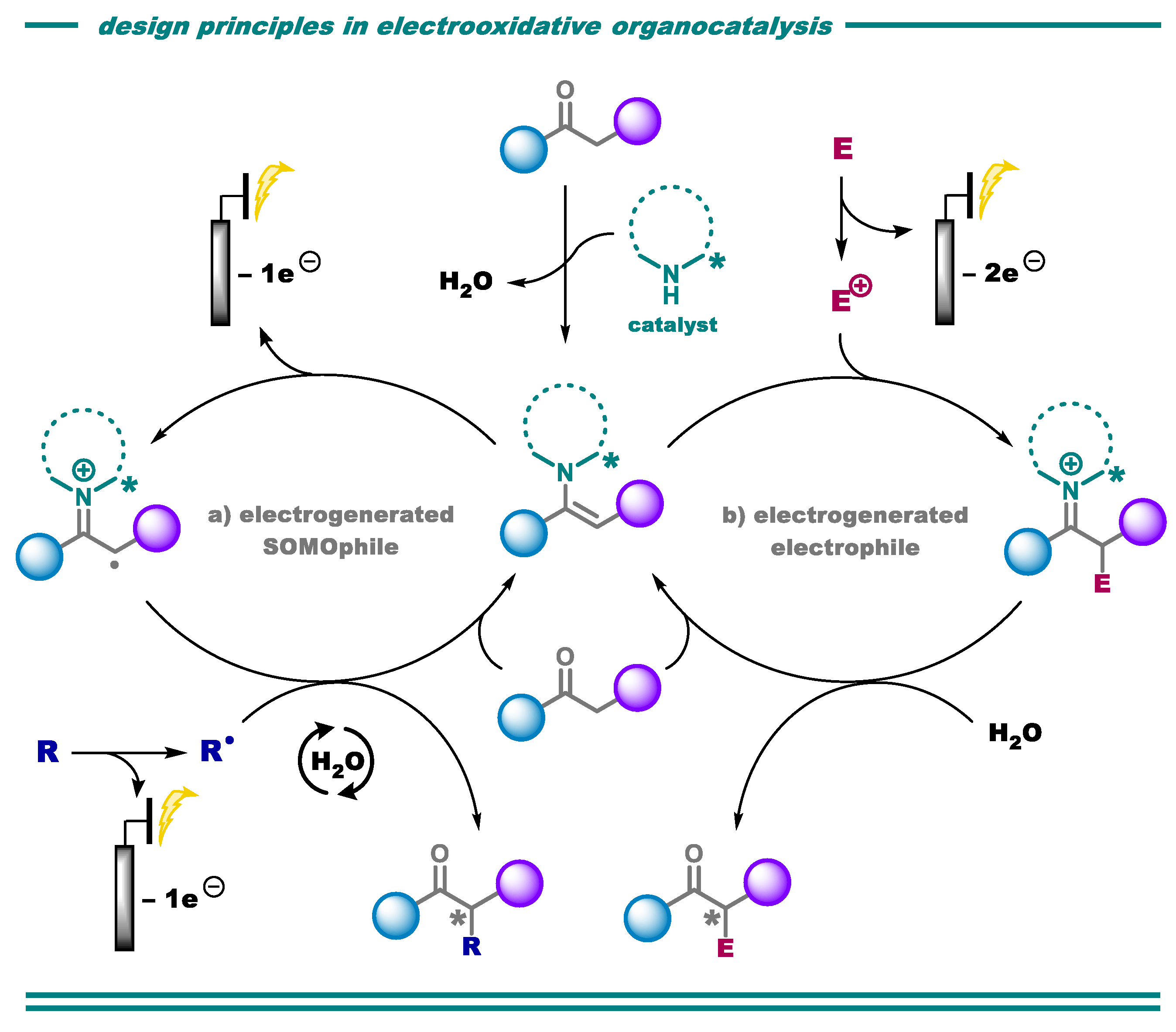{kind=link}
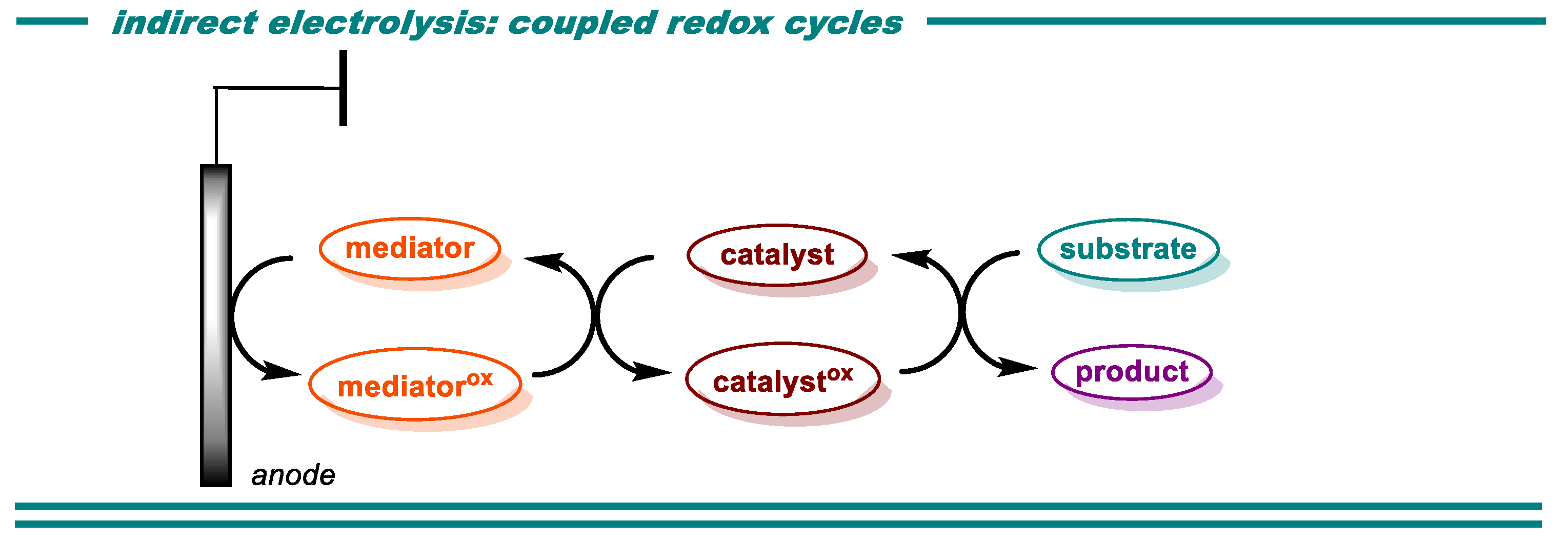{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Oxidative Transformations
2.1. C-H Functionalization
2.2. Alkene Functionalization
3. Reductive Transformations
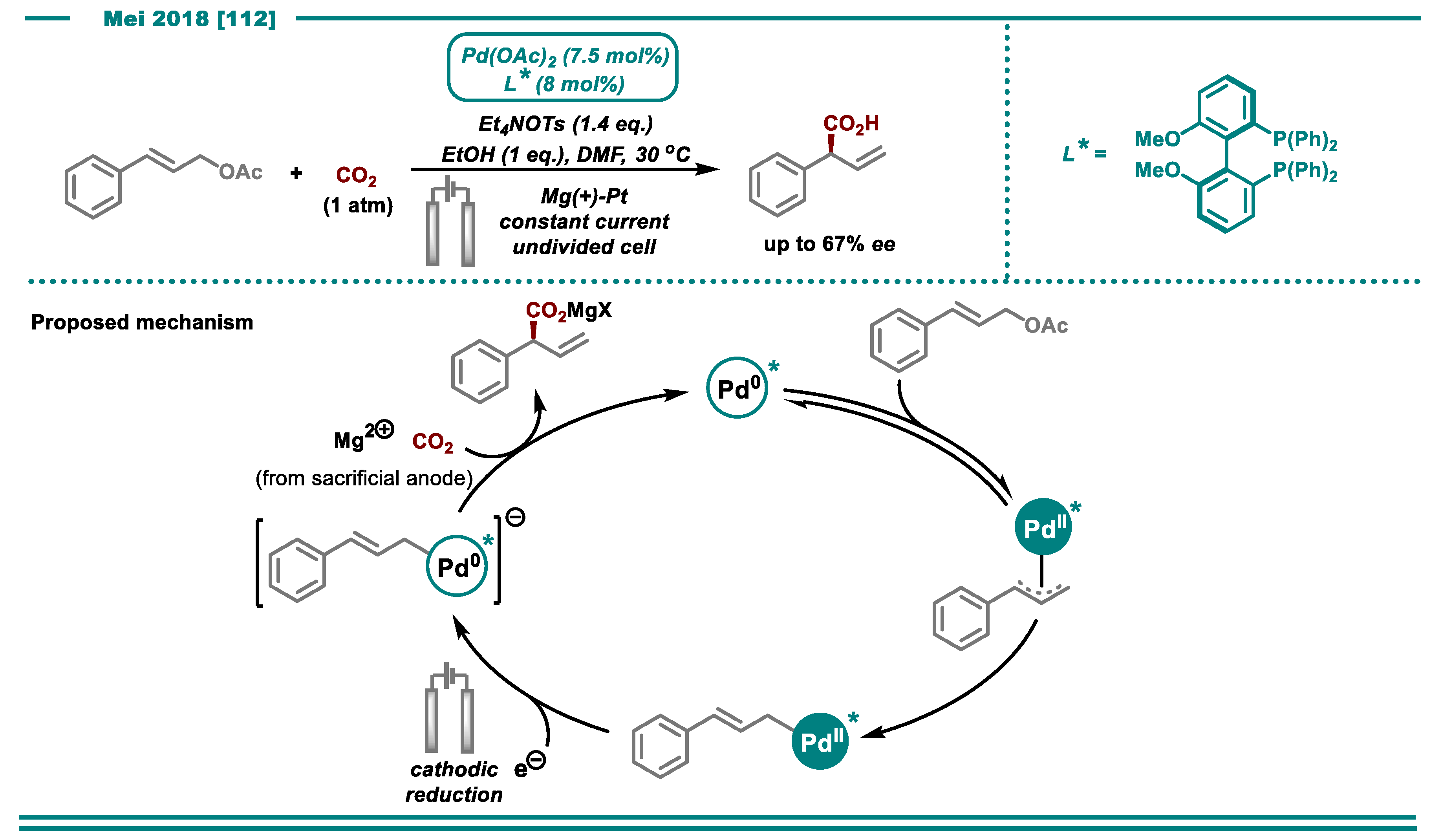
3.1. Carboxylation Using CO2
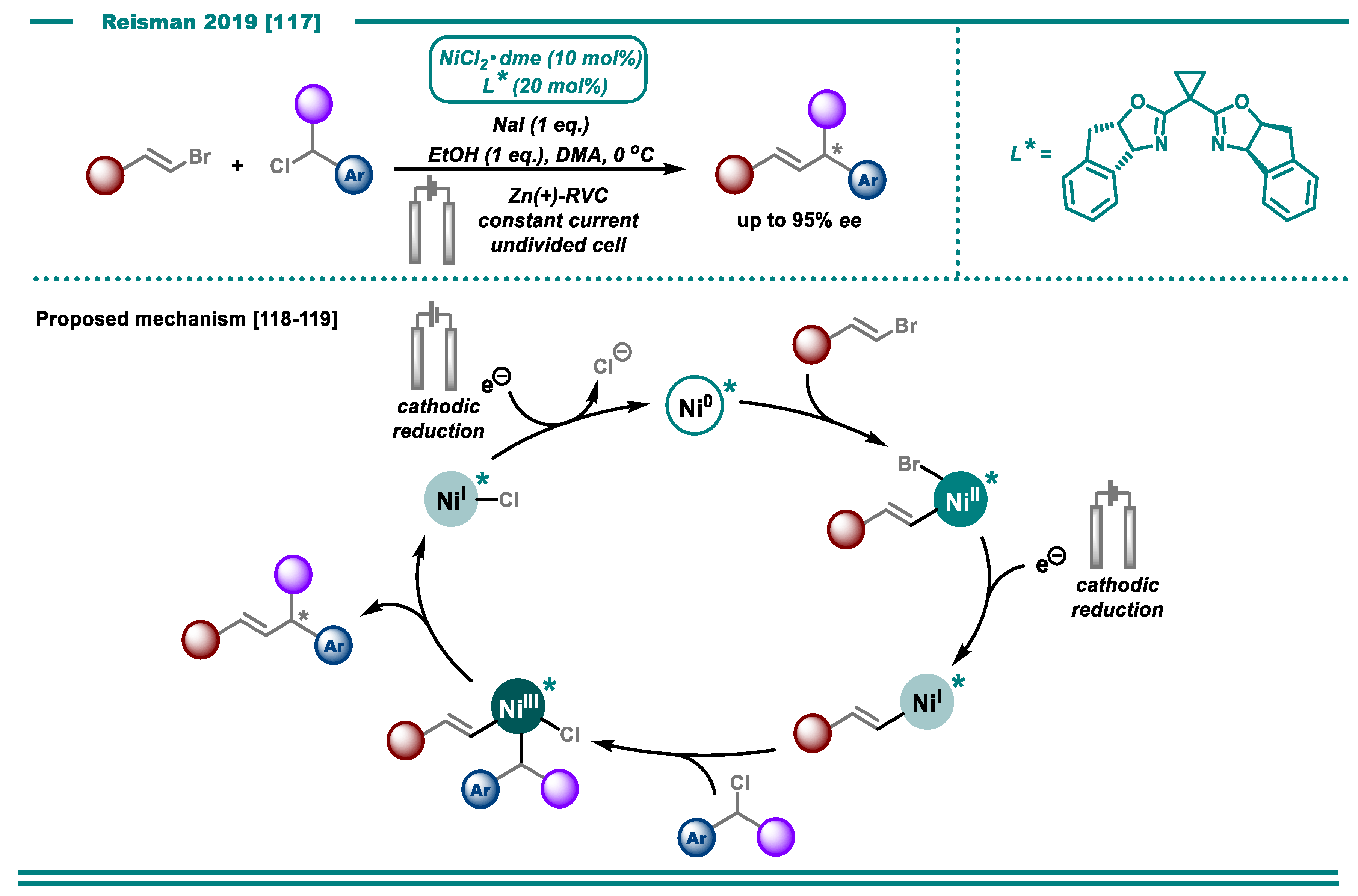
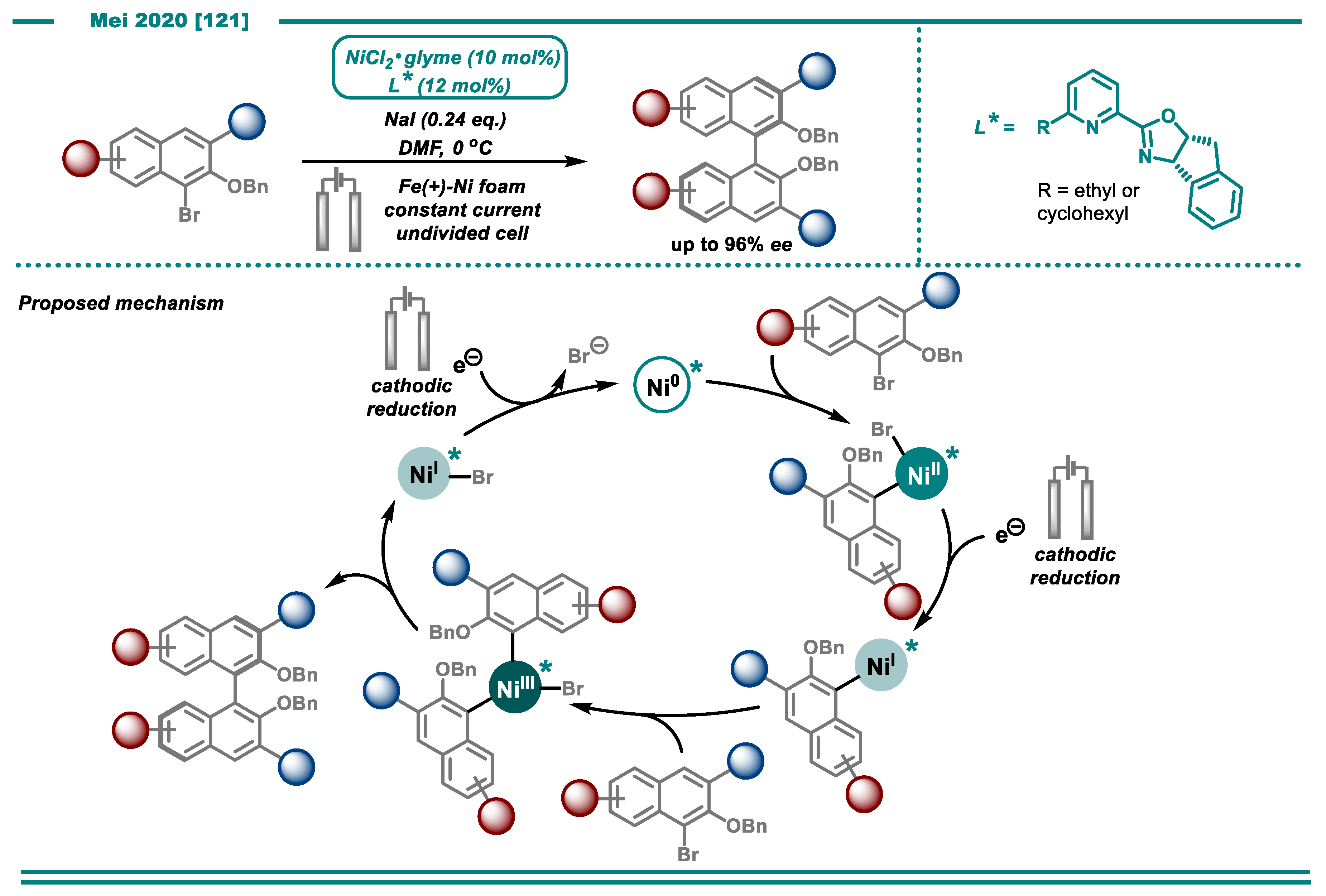
3.2. Cross-Electrophile Couplings
4. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Trost, B.M. Asymmetric catalysis: An enabling science. Proc. Nat. Acad. Sci. USA 2004, 101, 5348. [Google Scholar] [CrossRef]
- Ojima, I. Catalytic Asymmetric Synthesis, 3rd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Chang, X.; Zhang, Q.; Guo, C. Asymmetric Electrochemical Transformations. Angew. Chem. Int. Ed. 2020, 59, 12612–12622. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Li, L.; Luo, S. Asymmetric Electrochemical Catalysis. Chem. Eur. J. 2019, 25, 10033–10044. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Shinde, V.S.; Rueping, M. A review of asymmetric synthetic organic electrochemistry and electrocatalysis: Concepts, applications, recent developments and future directions. Beilstein J. Org. Chem. 2019, 15, 2710–2746. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.A.; Boydston, A.J. Recent Developments in Organocatalyzed Electroorganic Chemistry. Chem. Lett. 2015, 44, 10–16. [Google Scholar] [CrossRef]
- Wang, K.; Kong, W. Recent Advances in Transition Metal-Catalyzed Asymmetric Radical Reactions. Chin. J. Chem. 2018, 36, 247–256. [Google Scholar] [CrossRef]
- Saha, D. Catalytic Enantioselective Radical Transformations Enabled by Visible Light. Chem. Asian J. 2020, 15, 2129–2152. [Google Scholar] [CrossRef]
- Sibi, M.P.; Manyem, S.; Zimmerman, J. Enantioselective Radical Processes. Chem. Rev. 2003, 103, 3263–3296. [Google Scholar] [CrossRef]
- Jiang, C.; Chen, W.; Zheng, W.-H.; Lu, H. Advances in asymmetric visible-light photocatalysis. 2015–2019, Org. Biomol. Chem. 2019, 17, 8673–8689. [Google Scholar]
- Siu, J.C.; Fu, N.; Lin, S. Catalyzing Electrosynthesis: A Homogeneous Electrocatalytic Approach to Reaction Discovery. Acc. Chem. Res. 2020, 53, 547–560. [Google Scholar] [CrossRef]
- Francke, R. Integrating Catalytic Processes and Modern Electrolyte Concepts into Electrosynthesis. CHIMIA 2020, 74, 49–56. [Google Scholar] [CrossRef]
- Kärkäs, M.D. Electrochemical strategies for C–H functionalization and C–N bond formation. Chem. Soc. Rev. 2018, 47, 5786–5865. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230–13319. [Google Scholar] [CrossRef]
- Francke, R.; Little, R.D. Redox catalysis in organic electrosynthesis: Basic principles and recent developments. Chem. Soc. Rev. 2014, 43, 2492–2521. [Google Scholar] [CrossRef] [PubMed]
- Pollok, D.; Waldvogel, S.R. Electro-organic synthesis—A 21st century technique. Chem. Sci. 2020. [Google Scholar] [CrossRef]
- Krištofíková, D.; Modrocká, V.; Mečiarová, M.; Šebesta, R. Green Asymmetric Organocatalysis. ChemSusChem 2020, 13, 2828–2858. [Google Scholar] [CrossRef] [PubMed]
- Atobe, M. Organic electrosynthesis in flow microreactor. Curr. Opin. Electrochem. 2017, 2, 1–6. [Google Scholar] [CrossRef]
- Schäfer, H.J. Contributions of organic electrosynthesis to green chemistry. Comptes Rendus Chim. 2011, 14, 745–765. [Google Scholar] [CrossRef]
- Moeller, K.D. Using Physical Organic Chemistry To Shape the Course of Electrochemical Reactions. Chem. Rev. 2018, 118, 4817–4833. [Google Scholar] [CrossRef]
- Heard, D.; Lennox, A. Electrode Materials in Modern Organic Electrochemistry. Angew. Chem. Int. Ed. 2020, accepted. [Google Scholar] [CrossRef]
- Sandford, C.; Edwards, M.A.; Klunder, K.J.; Hickey, D.P.; Li, M.; Barman, K.; Sigman, M.S.; White, H.S.; Minteer, S.D. A synthetic chemist’s guide to electroanalytical tools for studying reaction mechanisms. Chem. Sci. 2019, 10, 6404–6422. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Ma, C.; Zhu, Z. Enzymatic electrosynthesis as an emerging electrochemical synthesis platform. Curr. Opin. Electrochem. 2020, 19, 1–7. [Google Scholar] [CrossRef]
- Cadoux, C.; Milton, R.D. Recent Enzymatic Electrochemistry for Reductive Reactions. ChemElectroChem 2020, 7, 1974–1986. [Google Scholar] [CrossRef]
- Kohlmann, C.; Märkle, W.; Lütz, S. Electroenzymatic synthesis. J. Mol. Catal. B Enzym. 2008, 51, 57–72. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, D.; Jia, Z.; Lin, Q.; Huang, M.; Luo, S. Catalytic Asymmetric Oxidative Enamine Transformations. ACS Catal. 2018, 8, 5466–5484. [Google Scholar] [CrossRef]
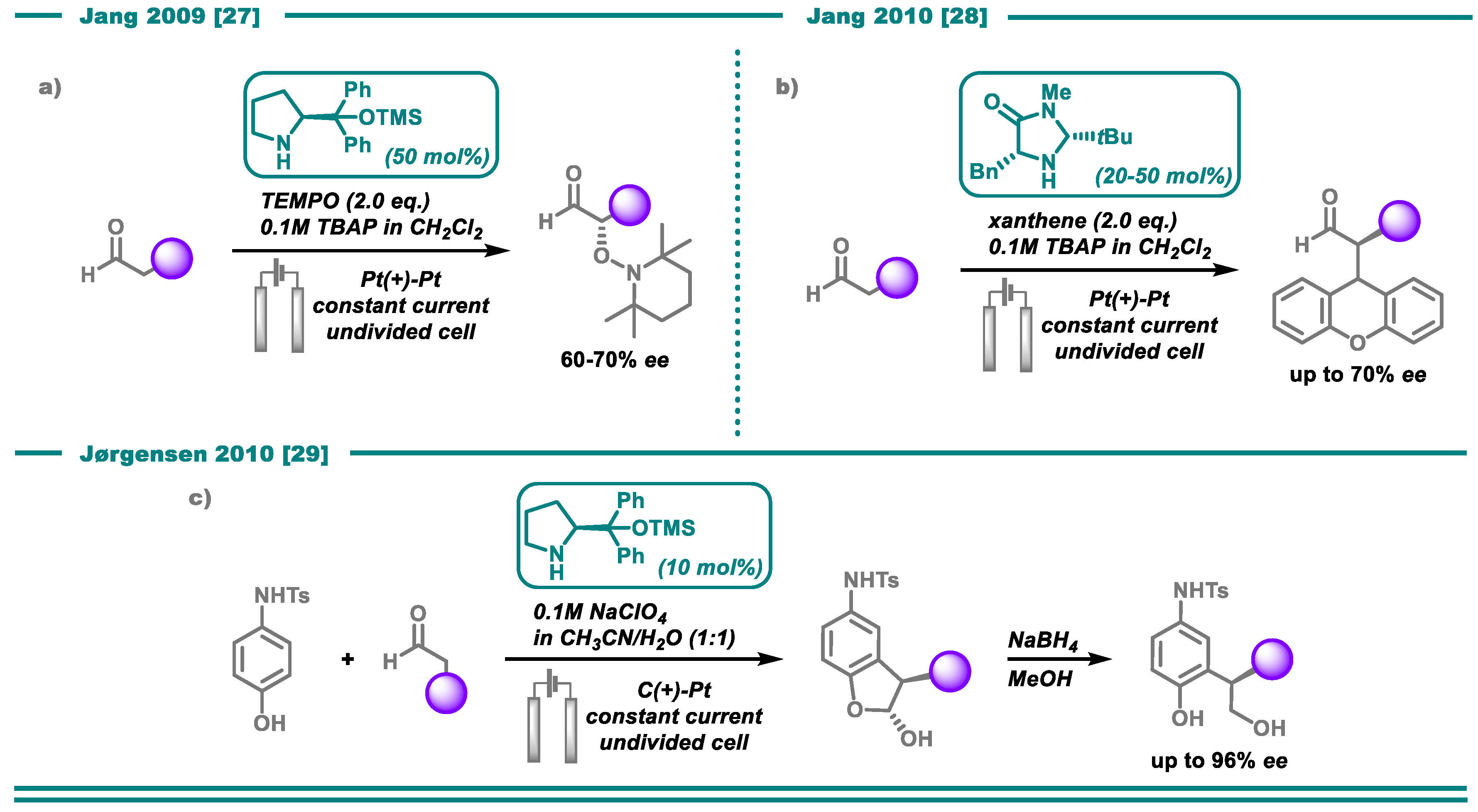
- Bui, N.-N.; Ho, X.-H.; Mho, S.-i.; Jang, H.-Y. Organocatalyzed α-Oxyamination of Aldehydes Using Anodic Oxidation. Eur. J. Org. Chem. 2009, 2009, 5309–5312. [Google Scholar] [CrossRef]
- Ho, X.-H.; Mho, S.-i.; Kang, H.; Jang, H.-Y. Electro-Organocatalysis: Enantioselective α-Alkylation of Aldehydes. Eur. J. Org. Chem. 2010, 2010, 4436–4441. [Google Scholar] [CrossRef]
- Jensen, K.L.; Franke, P.T.; Nielsen, L.T.; Daasbjerg, K.; Jørgensen, K.A. Anodic Oxidation and Organocatalysis: Direct Regio- and Stereoselective Access to meta-Substituted Anilines by α-Arylation of Aldehydes. Angew. Chem. Int. Ed. 2010, 49, 129–133. [Google Scholar] [CrossRef]
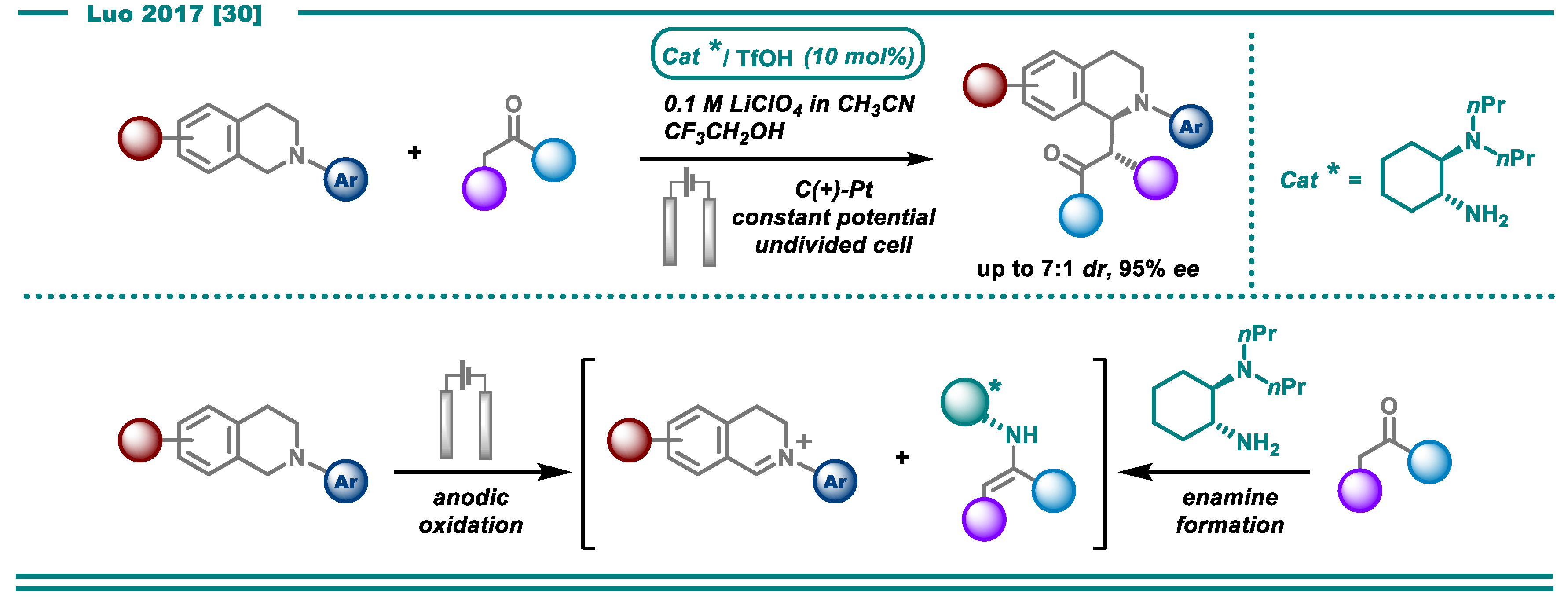
- Fu, N.; Li, L.; Yang, Q.; Luo, S. Catalytic Asymmetric Electrochemical Oxidative Coupling of Tertiary Amines with Simple Ketones. Org. Lett. 2017, 19, 2122–2125. [Google Scholar] [CrossRef]
- Osa, T.; Kashiwagi, Y.; Yanagisawa, Y.; Bobbitt, J.M. Enantioselective, electrocatalytic oxidative coupling of naphthol, naphthyl ether and phenanthrol on a TEMPO-modified graphite felt electrode in the presence of (–)-sparteine (TEMPO = 2,2,6,6-tetramethylpiperidin-1-yloxyl). J. Chem. Soc. Chem. Commun. 1994, 21, 2535–2537. [Google Scholar] [CrossRef]
- Kashiwagi, Y.; Yanagisawa, Y.; Kurashima, F.; Anzai, J.-i.; Osa, T.; Bobbitt, J.M. Enantioselective electrocatalytic oxidation of racemic alcohols on a TEMPO-modified graphite felt electrode by use of chiral base (TEMPO = 2,2,6,6-tetramethylpiperidin-1-yloxyl). Chem. Commun. 1996, 24, 2745–2746. [Google Scholar] [CrossRef]
- Yoshinori, Y.; Yoshitomo, K.; Futoshi, K.; Jun-ichi, A.; Tetsuo, O.; Enantioselective, B.J.M. Electrocatalytic Lactonization of Methyl-substituted Diols on a TEMPO-modified Graphite Felt Electrode in the Presence of (−)-Sparteine. Chem. Lett. 1996, 25, 1043–1044. [Google Scholar]
- Kashiwagi, Y.; Kurashima, F.; Kikuchi, C.; Anzai, J.-i.; Osa, T.; Bobbitt, J.M. Enantioselective electrocatalytic oxidation of racemic sec-alcohols using a chiral 1-azaspiro [5.5]undecane-N-oxyl radical. Tetrahedron Lett. 1999, 40, 6469–6472. [Google Scholar] [CrossRef]
- Kashiwagi, Y.; Kurashima, F.; Kikuchi, C.; Anzai, J.-i.; Osa, T.; Bobbitt, J.M. Enantioselective electrocatalytic oxidation of racemic amines using a chiral 1-azaspiro[5.5]undecane N-oxyl radical. Chem. Commun. 1999, 1983–1984. [Google Scholar] [CrossRef]
- Kuroboshi, M.; Yoshihisa, H.; Cortona, M.N.; Kawakami, Y.; Gao, Z.; Tanaka, H. Electro-oxidative kinetic resolution of sec-alcohols by using an optically active N-oxyl mediator. Tetrahedron Lett. 2000, 41, 8131–8135. [Google Scholar] [CrossRef]
- Tanaka, H.; Kawakami, Y.; Goto, K.; Kuroboshi, M. An aqueous silica gel disperse electrolysis system. N-Oxyl-mediated electrooxidation of alcohols. Tetrahedron Lett. 2001, 42, 445–448. [Google Scholar] [CrossRef]
- Shiigi, H.; Mori, H.; Tanaka, T.; Demizu, Y.; Onomura, O. Chiral azabicyclo-N-oxyls mediated enantioselective electrooxidation of sec-alcohols. Tetrahedron Lett. 2008, 49, 5247–5251. [Google Scholar] [CrossRef]
- Wang, F.; Stahl, S.S. Electrochemical Oxidation of Organic Molecules at Lower Overpotential: Accessing Broader Functional Group Compatibility with Electron−Proton Transfer Mediators. Acc. Chem. Res. 2020, 53, 561–574. [Google Scholar] [CrossRef]
- Minato, D.; Arimoto, H.; Nagasue, Y.; Demizu, Y.; Onomura, O. Asymmetric electrochemical oxidation of 1,2-diols, aminoalcohols, and aminoaldehydes in the presence of chiral copper catalyst. Tetrahedron 2008, 64, 6675–6683. [Google Scholar] [CrossRef]
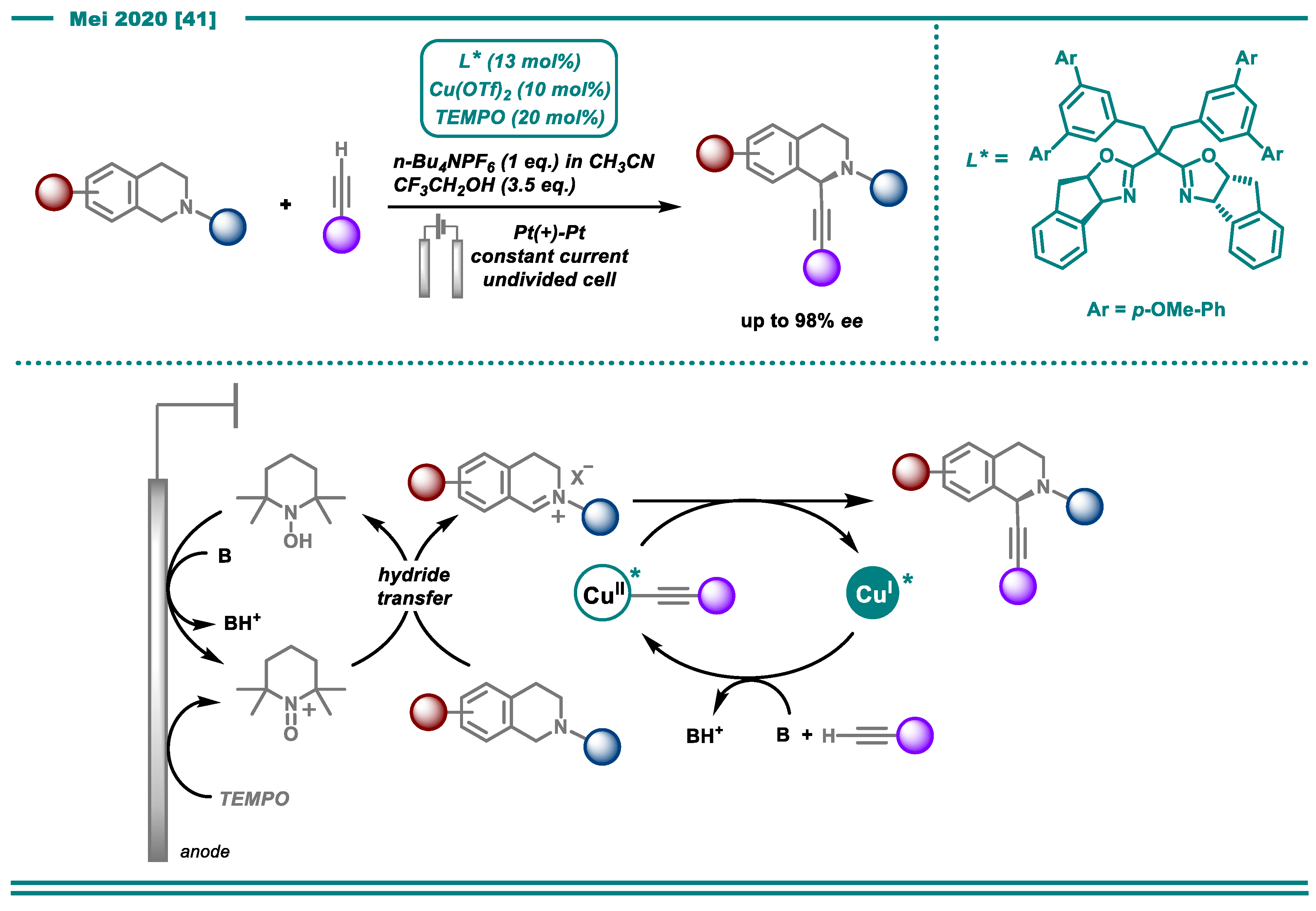
- Gao, P.-S.; Weng, X.-J.; Wang, Z.-H.; Zheng, C.; Sun, B.; Chen, Z.-H.; You, S.-L.; Mei, T.-S. CuII/TEMPO-Catalyzed Enantioselective C(sp3)–H Alkynylation of Tertiary Cyclic Amines through Shono-Type Oxidation. Angew. Chem. Int. Ed. 2020, 59, 15254–15259. [Google Scholar] [CrossRef]
- Shono, T.; Hamaguchi, H.; Matsumura, Y. Electroorganic chemistry. XX. Anodic oxidation of carbamates. J. Am. Chem. Soc. 1975, 97, 4264–4268. [Google Scholar] [CrossRef]
- Lu, F.-Y.; Chen, Y.-J.; Chen, Y.; Ding, X.; Guan, Z.; He, Y.-H. Highly enantioselective electrosynthesis of C2-quaternary indolin-3-ones. Chem. Commun. 2020, 56, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef] [PubMed]
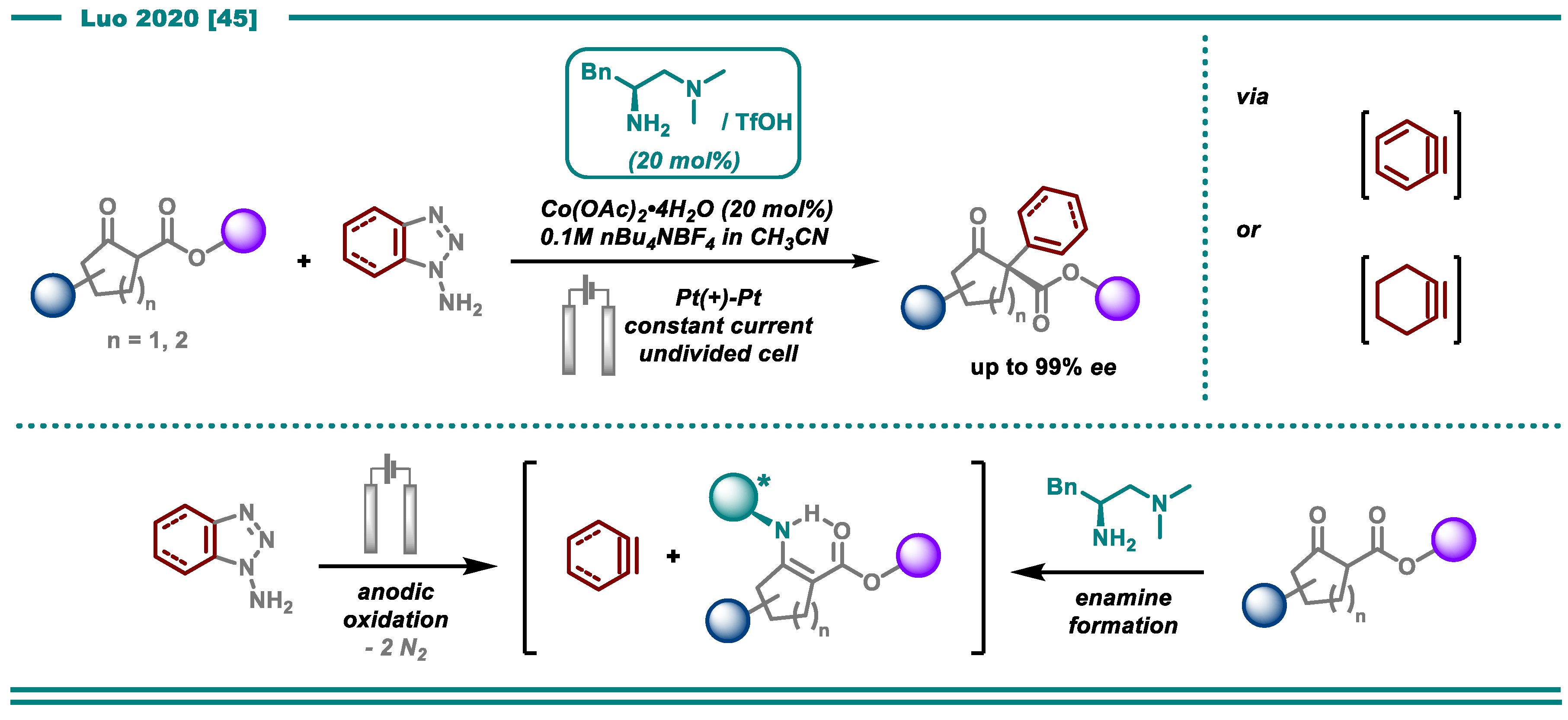
- Li, L.; Li, Y.; Fu, N.; Zhang, L.; Luo, S. Catalytic Asymmetric Electrochemical α-Arylation of Cyclic β-Ketocarbonyls with Anodic Benzyne Intermediates. Angew. Chem. Int. Ed. 2020, 59, 14347–14351. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.D.; Rees, C.W. Reactive intermediates. Part I. Synthesis and oxidation of 1- and 2-aminobenzotriazole. J. Chem. Soc. C 1969, 742–747. [Google Scholar] [CrossRef]
- Kato, H.; Nakazawa, S.; Kiyosawa, T.; Hirakawa, K. Heterocycles by cycloaddition. Part II. Cycloaddition–extrusion reactions of five-membered mesoionic compounds with benzyne: Prepartion of benz[c]azole and benzo[c]thiophen derivatives. J. Chem. Soc. Perkin Trans. 1 1976, 672–675. [Google Scholar] [CrossRef]
- Rigby, J.H.; Holsworth, D.D.; James, K. Vinyl isocyanates in synthesis. [4 + 2] Cycloaddition reactions with benzyne addends. J. Org. Chem. 1989, 54, 4019–4020. [Google Scholar] [CrossRef]
- Sakurai, H.; Sakaba, H.; Nakadaira, Y. Facile preparation of 2,3-benzo-1,4-diphenyl-7-silanorbornadiene derivatives and the first clear evidence of silylene-to-disilene thermal rearrangement. J. Am. Chem. Soc. 1982, 104, 6156–6158. [Google Scholar] [CrossRef]
- Cresp, T.M.; Wege, D. The addition of benzyne to azulene. Tetrahedron 1986, 42, 6713–6718. [Google Scholar] [CrossRef]
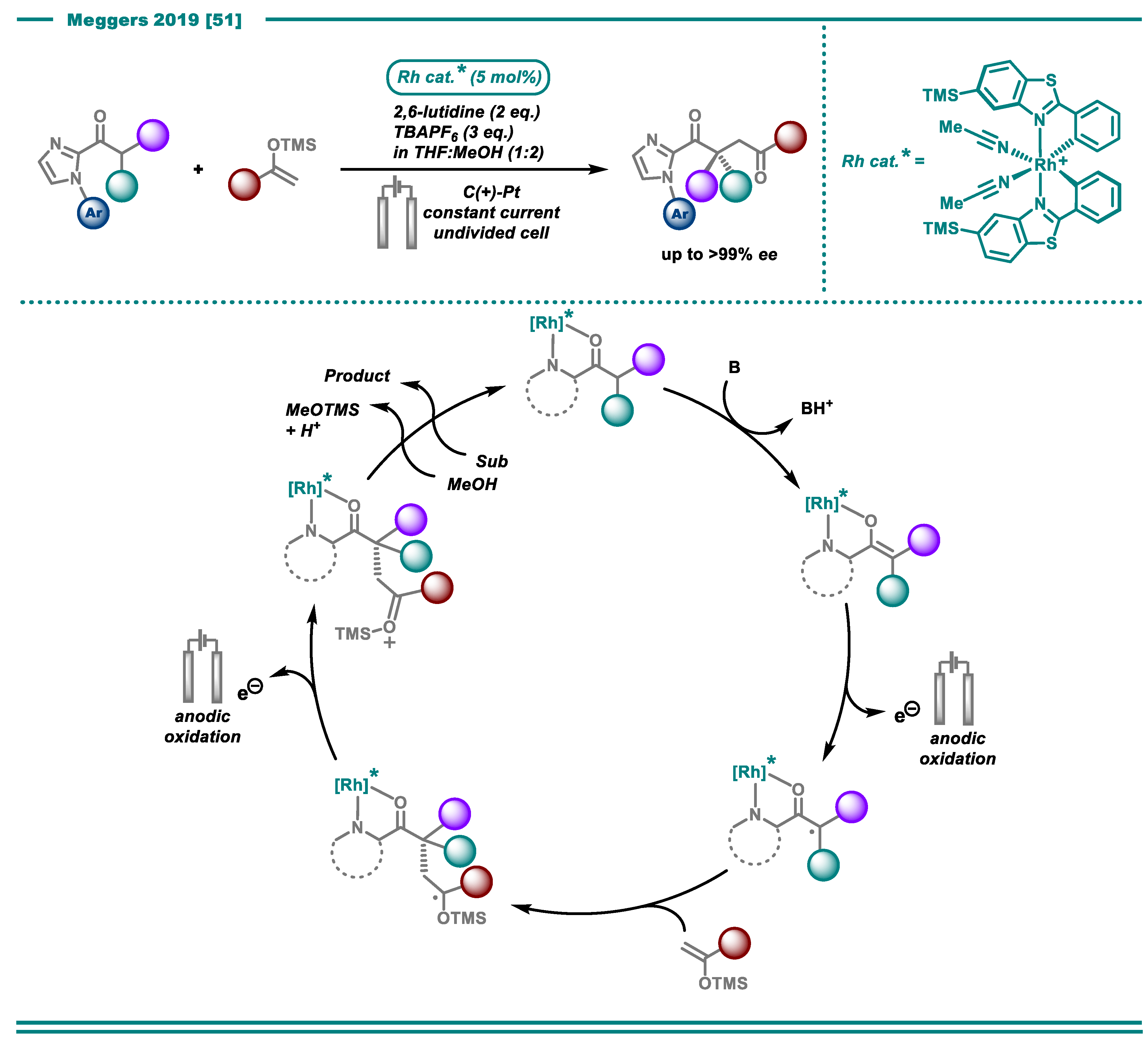
- Huang, X.; Zhang, Q.; Lin, J.; Harms, K.; Meggers, E. Electricity-driven asymmetric Lewis acid catalysis. Nat. Catal. 2019, 2, 34–40. [Google Scholar] [CrossRef]
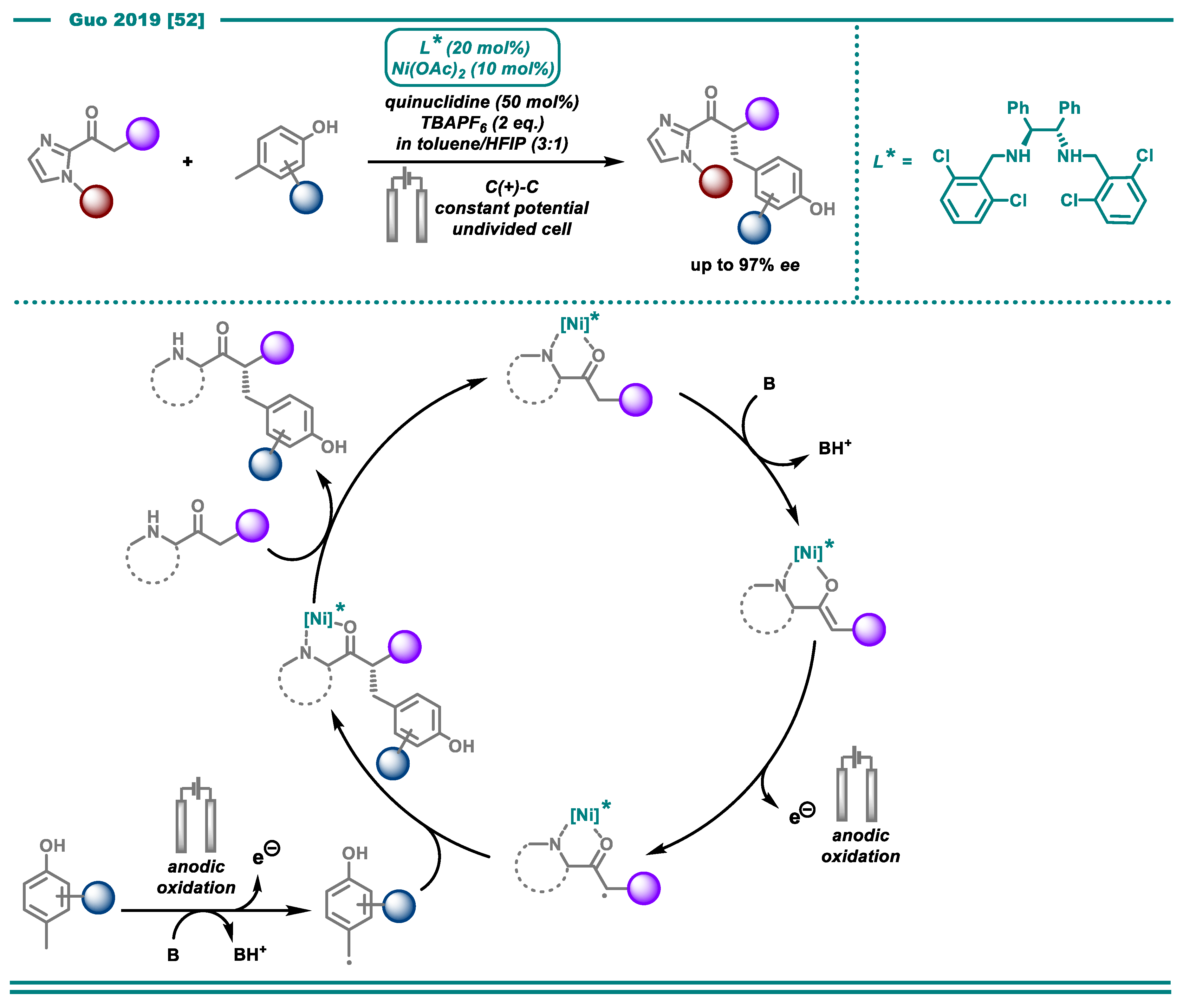
- Zhang, Q.; Chang, X.; Peng, L.; Guo, C. Asymmetric Lewis Acid Catalyzed Electrochemical Alkylation. Angew. Chem. Int. Ed. 2019, 58, 6999–7003. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, J.L.; Terrett, J.A.; MacMillan, D.W.C. O–H hydrogen bonding promotes H-atom transfer from α C–H bonds for C-alkylation of alcohols. Science 2015, 349, 1532–1536. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.H.; Shurtleff, V.W.; Terrett, J.A.; Cuthbertson, J.D.; MacMillan, D.W.C. Native functionality in triple catalytic cross-coupling: sp3 C–H bonds as latent nucleophiles. Science 2016, 352, 1304. [Google Scholar] [CrossRef] [PubMed]
- Le, C.; Liang, Y.; Evans, R.W.; Li, X.; MacMillan, D.W.C. Selective sp 3 C-H alkylation via polarity-match-based cross-coupling. Nature 2017, 547, 79–83. [Google Scholar] [CrossRef]
- Zhang, X.; MacMillan, D.W.C. Direct aldehyde C–H arylation and alkylation via the combination of nickel, hydrogen atom transfer, and photoredox catalysis. J. Am. Chem. Soc. 2017, 139, 11353–11356. [Google Scholar] [CrossRef]
- Twilton, J.; Christensen, M.; DiRocco, D.A.; Ruck, R.T.; Davies, I.W.; MacMillan, D.W.C. Selective Hydrogen Atom Abstraction through Induced Bond Polarization: Direct a-Arylation of Alcohols through Photoredox, HAT, and Nickel Catalysis. Angew. Chem. Int. Ed. 2018, 57, 5369–5373. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-B.; Feceu, A.; Martin, D.B.C. Catalyst-Controlled C–H Functionalization of Adamantanes Using Selective H-Atom Transfer. ACS Catal. 2019, 9, 5708–5715. [Google Scholar] [CrossRef]
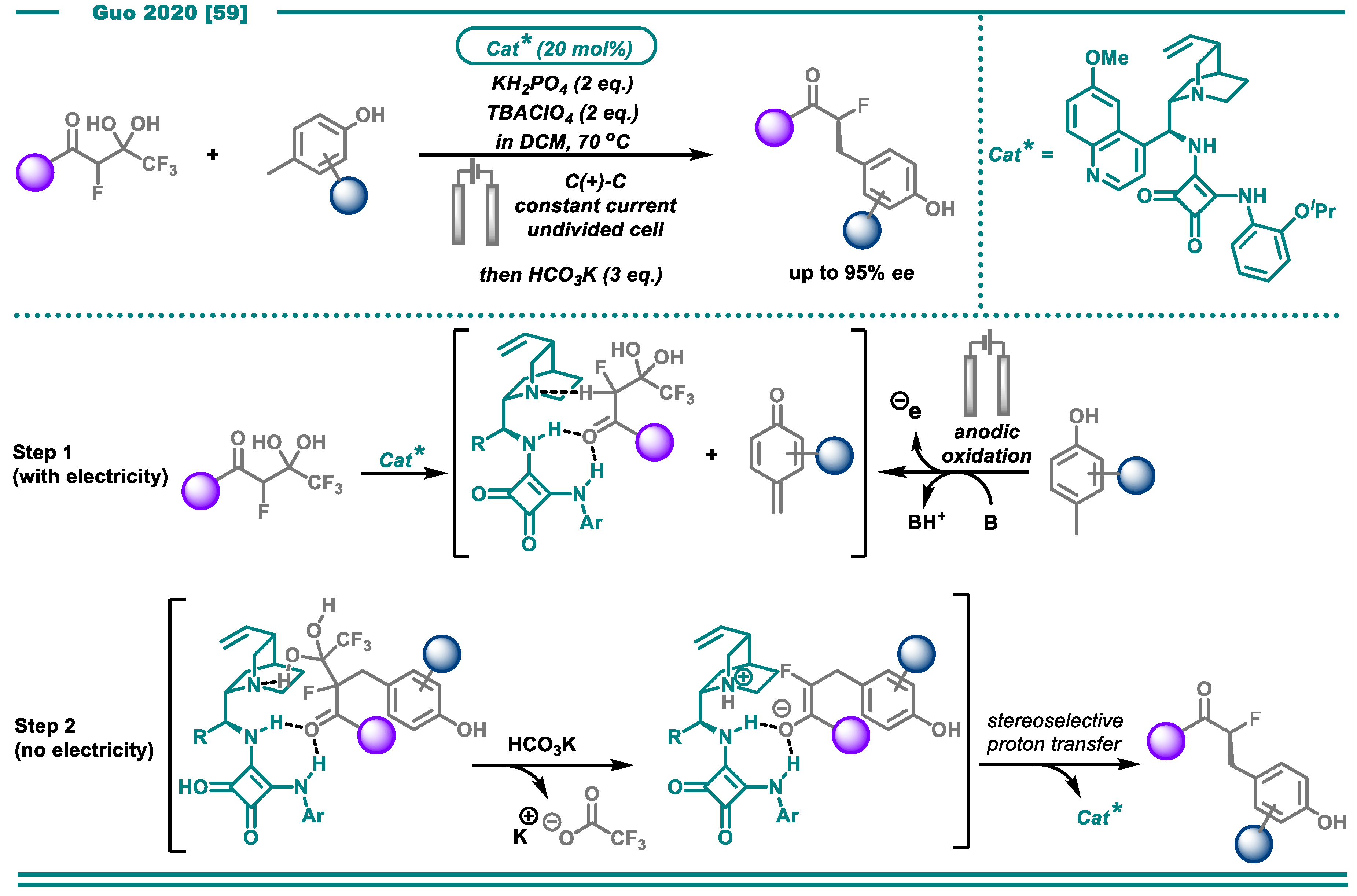
- Chang, X.; Zhang, J.; Zhang, Q.; Guo, C. Merging Electrosynthesis and Bifunctional Squaramide Catalysis in the Asymmetric Detrifluoroacetylative Alkylation Reactions. Angew. Chem. Int. Ed. 2020, accepted. [Google Scholar] [CrossRef]
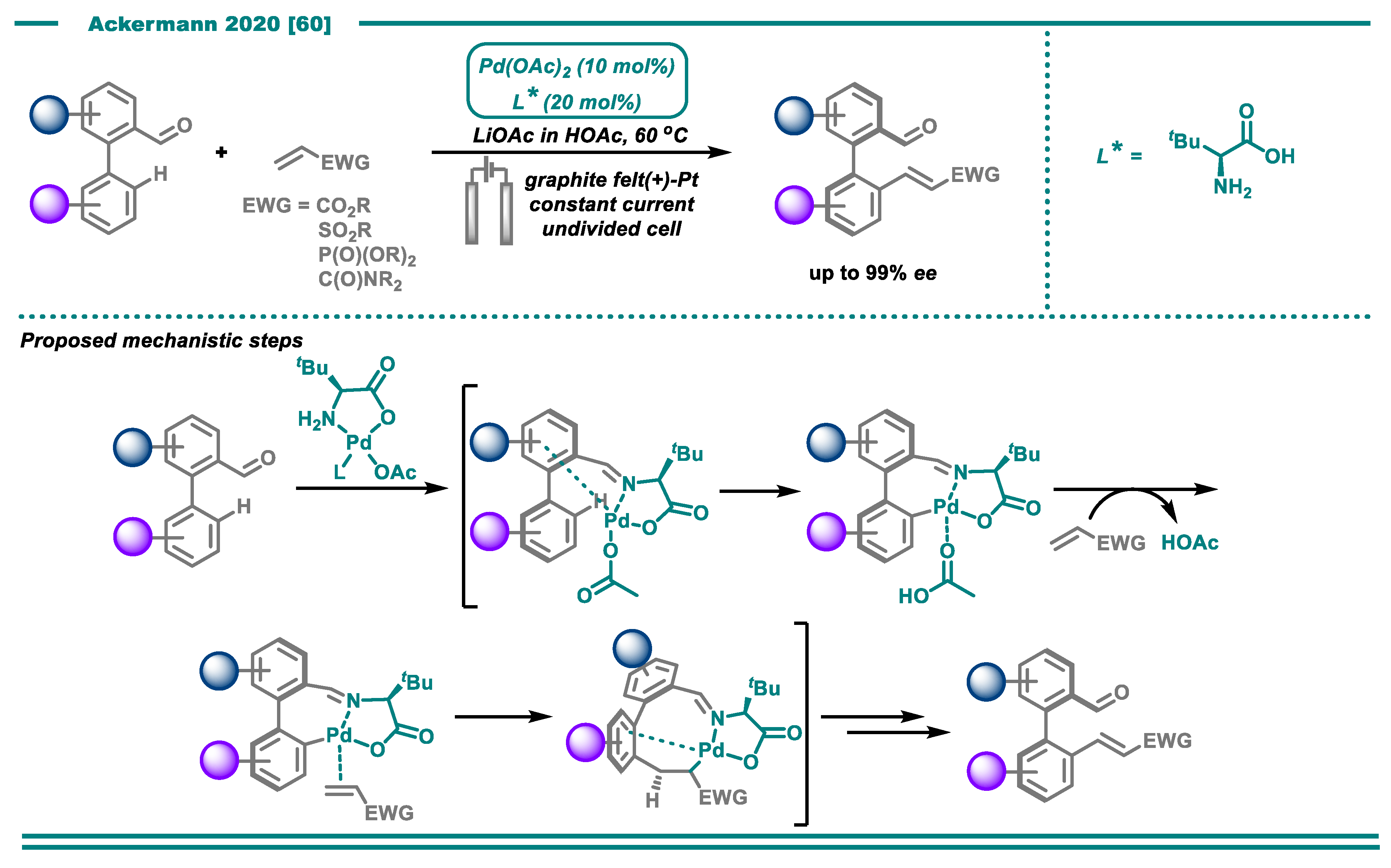
- Dhawa, U.; Tian, C.; Wdowik, T.; Oliveira, J.C.A.; Hao, J.; Ackermann, L. Enantioselective Palladaelectro-Catalyzed C–H Activations by Transient Directing Groups: Expedient Access to Helicenes. Angew. Chem. Int. Ed. 2020, 59, 13451–13457. [Google Scholar] [CrossRef]
- Hentges, S.G.; Sharpless, K.B. Asymmetric induction in the reaction of osmium tetroxide with olefins. J. Am. Chem. Soc. 1980, 102, 4263–4265. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Amberg, W.; Bennani, Y.L.; Crispino, G.A.; Hartung, J.; Jeong, K.S.; Kwong, H.L.; Morikawa, K.; Wang, Z.M. The osmium-catalyzed asymmetric dihydroxylation: A new ligand class and a process improvement. J. Org. Chem. 1992, 57, 2768–2771. [Google Scholar] [CrossRef]
- Xu, D.; Crispino, G.A.; Sharpless, K.B. Selective asymmetric dihydroxylation (AD) of dienes. J. Am. Chem. Soc. 1992, 114, 7570–7571. [Google Scholar] [CrossRef]
- Hoi-Lun, K.; Sorato, C.; Ogino, Y.; Hou, C.; Sharpless, K.B. Preclusion of the “second cycle” in the osmium-catalyzed asymmetric dihydroxylation of olefins leads to a superior process. Tetrahedron Lett. 1990, 31, 2999–3002. [Google Scholar] [CrossRef]
- Minato, M.; Yamamoto, K.; Tsuji, J. Osmium tetraoxide catalyzed vicinal hydroxylation of higher olefins by using hexacyanoferrate(III) ion as a cooxidant. J. Org. Chem. 1990, 55, 766–768. [Google Scholar] [CrossRef]
- Ogino, Y.; Chen, H.; Kwong, H.-L.; Sharpless, K.B. On the timing of hydrolysis / reoxidation in the osmium-catalyzed asymmetric dihydroxylation of olefins using potassium ferricyanide as the reoxidant. Tetrahedron Lett. 1991, 32, 3965–3968. [Google Scholar] [CrossRef]
- Amundsen, A.R.; Balko, E.N. Preparation of chiral diols by the osmium-catalysed, indirect anodic oxidation of olefins. J. Appl. Electrochem. 1992, 22, 810–816. [Google Scholar] [CrossRef]
- Torii, S.; Liu, P.; Tanaka, H. Electrochemical Os-Catalyzed Asymmetric Dihydroxylation of Olefins with Sharpless’ Ligand. Chem. Lett. 1995, 24, 319–320. [Google Scholar] [CrossRef]
- Torii, S.; Liu, P.; Bhuvaneswari, N.; Amatore, C.; Jutand, A. Chemical and Electrochemical Asymmetric Dihydroxylation of Olefins in I2−K2CO3−K2OsO2(OH)4 and I2−K3PO4/K2HPO4−K2OsO2(OH)4 Systems with Sharpless’ Ligand. J. Org. Chem. 1996, 61, 3055–3060. [Google Scholar] [CrossRef]
- Tanaka, H.; Kuroboshi, M.; Takeda, H.; Kanda, H.; Torii, S. Electrochemical asymmetric epoxidation of olefins by using an optically active Mn-salen complex. J. Electroanal. Chem. 2001, 507, 75–81. [Google Scholar] [CrossRef]
- Irie, R.; Noda, K.; Ito, Y.; Matsumoto, N.; Katsuki, T. Catalytic asymmetric epoxidation of unfunctionalized olefins. Tetrahedron Lett. 1990, 31, 7345–7348. [Google Scholar] [CrossRef]
- Zhang, W.; Loebach, J.L.; Wilson, S.R.; Jacobsen, E.N. Enantioselective epoxidation of unfunctionalized olefins catalyzed by salen manganese complexes. J. Am. Chem. Soc. 1990, 112, 2801–2803. [Google Scholar] [CrossRef]
- Katsuki, T. Mn-salen catalyst, competitor of enzymes, for asymmetric epoxidation. J. Mol. Catal. A 1996, 113, 87–107. [Google Scholar] [CrossRef]
- Page, P.C.B.; Marken, F.; Williamson, C.; Chan, Y.; Buckley, B.R.; Bethell, D. Enantioselective Organocatalytic Epoxidation Driven by Electrochemically Generated Percarbonate and Persulfate. Adv. Synth. Catal. 2008, 350, 1149–1154. [Google Scholar] [CrossRef]
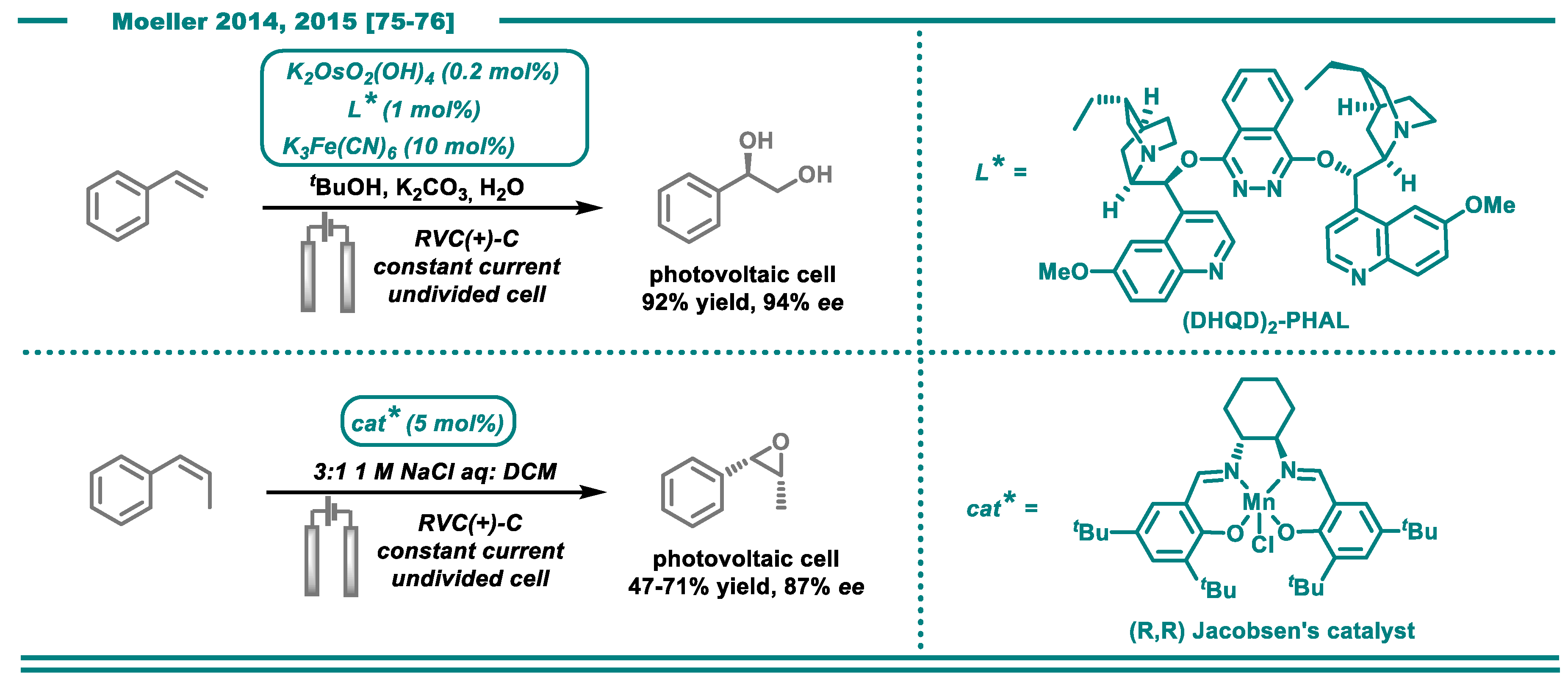
- Nguyen, B.H.; Perkins, R.J.; Smith, J.A.; Moeller, K.D. Photovoltaic-driven organic electrosynthesis and efforts toward more sustainable oxidation reactions. Beilstein J. Org. Chem. 2015, 11, 280–287. [Google Scholar] [CrossRef]
- Nguyen, B.H.; Redden, A.; Moeller, K.D. Sunlight, electrochemistry, and sustainable oxidation reactions. Green Chem. 2014, 16, 69–72. [Google Scholar] [CrossRef]
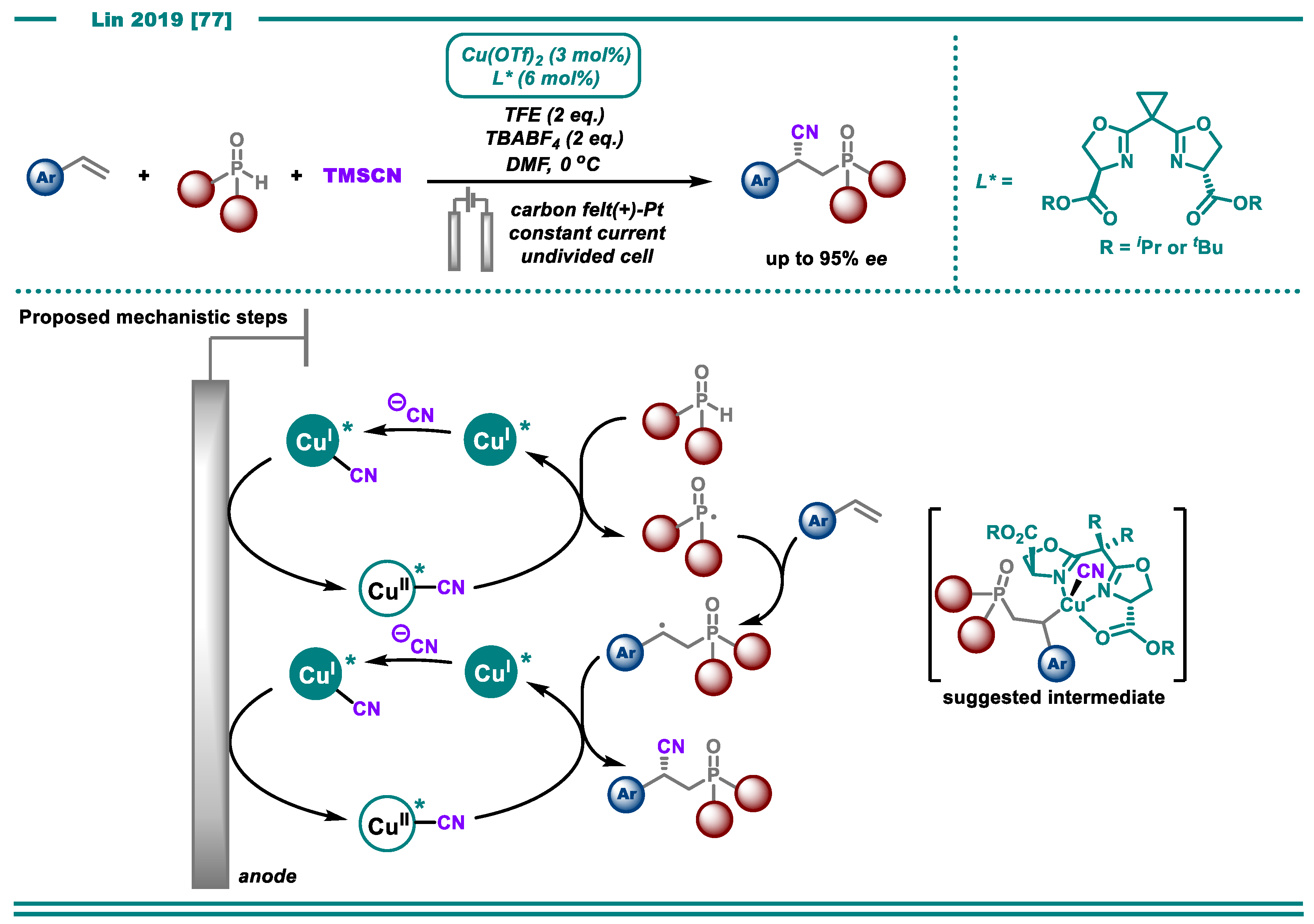
- Fu, N.; Song, L.; Liu, J.; Shen, Y.; Siu, J.C.; Lin, S. New Bisoxazoline Ligands Enable Enantioselective Electrocatalytic Cyanofunctionalization of Vinylarenes. J. Am. Chem. Soc. 2019, 141, 14480–14485. [Google Scholar] [CrossRef]
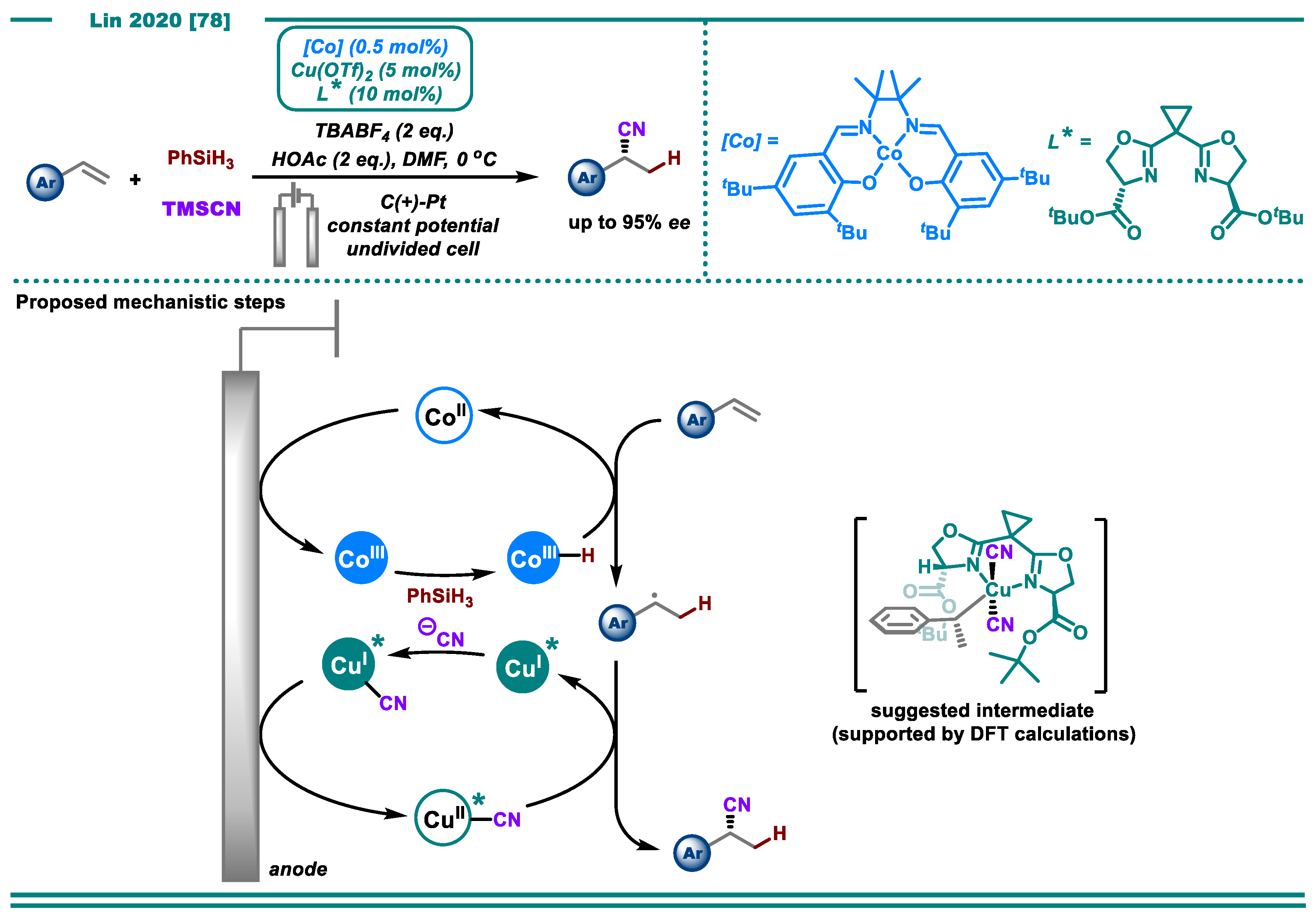
- Song, L.; Fu, N.; Ernst, B.G.; Lee, W.H.; Frederick, M.O.; DiStasio, R.A.; Lin, S. Dual Electrocatalysis Enables Enantioselective Hydrocyanation of Conjugated Alkenes. Nat. Chem. 2020, 12, 747–754. [Google Scholar] [CrossRef]
- Gourley, R.N.; Grimshaw, J.; Millar, P.G. Electrochemical reduction in the presence of tertiary amines: An asymmetric synthesis of 3, 4-dihydro-4-methylcoumarin. Chem. Commun. 1967, 1278–1279. [Google Scholar] [CrossRef]
- Gourley, R.N.; Grimshaw, J.; Millar, P.G. Electrochemical reactions. Part VIII. Asymmetric induction during the reduction of coumarins modified by the presence of tertiary amines. J. Chem. Soc. C 1970, 2318–2323. [Google Scholar] [CrossRef]
- Höweler, U.; Schoo, N.; Schäfer, H.-J. Enantioselective cathodic reduction of 4-substituted coumarins with alkaloids as catalysts, 2. AM1 and force-field study of the transition-state model. Liebigs Ann. Chem. 1993, 1993, 609–614. [Google Scholar] [CrossRef]
- Schoo, N.; Schäfer, H.-J. Electroorganic Synthesis, 54. Enantioselective Cathodic Reduction of 4-Substituted Coumarins with Alkaloids as Catalysts, 1. Liebigs Ann. Chem. 1993, 1993, 601–607. [Google Scholar] [CrossRef]
- Nielsen, M.F.; Batanero, B.; Löhl, T.; Schäfer, H.J.; Würthwein, E.-U.; Fröhlich, R. Enantioselective Cathodic Reduction of 4-Methylcoumarin: Dependence of Selectivity on Reaction Conditions and Investigation of the Mechanism. Chem. Eur. J. 1997, 3, 2011–2024. [Google Scholar] [CrossRef]
- Kariv, E.; Terni, H.A.; Gileadi, E. The role of quinidine in induction of asymmetric synthesis at mercury cathodes. Electrochim. Acta 1973, 18, 433–441. [Google Scholar] [CrossRef]
- Chen, B.-L.; Xiao, Y.; Xu, X.-M.; Yang, H.-P.; Wang, H.; Lu, J.-X. Alkaloid induced enantioselective electroreduction of acetophenone. Electrochim. Acta 2013, 107, 320–326. [Google Scholar] [CrossRef]
- Kariv, E.; Terni, H.A.; Gileadi, E. Asymmetric Induction by Alkaloids in Electrolytic Reductions. J. Electrochem. Soc. 1973, 120, 639. [Google Scholar] [CrossRef]
- Hermolin, J.; Kopilov, J.; Gileadi, E. Asymmetric reduction of 2- and 4-acetylpyridine in the presence of strychnine. J. Electroanal. Chem. 1976, 71, 245–248. [Google Scholar] [CrossRef]
- Kopilov, J.; Shatzmiller, S.; Kariv, E. Asymmetric induction in reduction of acetyl pyridines at a mercury cathode. Electrochim. Acta 1976, 21, 535–536. [Google Scholar] [CrossRef]
- Kopilov, J.; Kariv, E.; Miller, L.L. Asymmetric, cathodic reduction of acetylpyridines. J. Am. Chem. Soc. 1977, 99, 3450–3454. [Google Scholar] [CrossRef]
- Yadav, A.K.; Singh, A. Enantioselective Cathodic Reduction of Some Prochiral Ketones in the Presence of (1R,2S)-(-)-N,N-Dimethylephedrinium Tetrafluoroborate at a Mercury Pool Cathode. Bull. Chem. Soc. Jpn. 2002, 75, 587–588. [Google Scholar] [CrossRef]
- Yadav, A.K.; Manju, M.P.; Chhinpa, R. Enantioselective cathodic reduction of some prochiral ketones in the presence of (−)-N,N′-dimethylquininium tetrafluoroborate at mercury cathode. Tetrahedron Asymmetry 2003, 14, 1079–1081. [Google Scholar] [CrossRef]
- Vago, M.; Williams, F.J.; Calvo, E.J. Enantioselective electrocatalytic hydrogenation of ethyl pyruvate on carbon supported Pd electrodes. Electrochem. Commun. 2007, 9, 2725–2728. [Google Scholar] [CrossRef]
- Jubault, M.; Raoult, E.; Armand, J.; Boulares, L. Effect of cathodic potential on the electrochemical synthesis of optically active amino-acids. J. Chem. Soc. Chem. Commun. 1977, 250–251. [Google Scholar] [CrossRef]
- Jubault, M.; Raoult, E.; Peltier, D. Preprotonation of the substrate by protonated alkaloid in asymmetric electrosynthesis. J. Chem. Soc. Chem. Commun. 1979, 232–233. [Google Scholar] [CrossRef]
- Jubault, M. Effect of alkaloid concentration in asymmetric electrosynthesis. J. Chem. Soc. Chem. Commun. 1980, 953–954. [Google Scholar] [CrossRef]
- Park, J.W.; Choi, M.H.; Park, K.K. Electrocatalytic reduction of benzoylformic acid mediated by methyl viologen. Tetrahedron Lett. 1995, 36, 2637–2638. [Google Scholar] [CrossRef]
- Hazard, R.; Jaouannet, S.; Tallec, A. Stereochemistry of electroreductions of bromocyclopropanes: 1-asymmetric electrochemical synthesis by reduction at a mercury cathode in the presence of adsorbed alkaloids. Tetrahedron 1982, 38, 93–102. [Google Scholar] [CrossRef]
- Moutet, J.-C.; Cho, L.Y.; Duboc-Toia, C.; Ménage, S.; Riesgo, E.C.; Thummel, R.P. Heterogeneous and homogeneous asymmetric electrocatalytic hydrogenation with rhodium(III) complexes containing chiral polypyridyl ligands. New J. Chem. 1999, 23, 939–944. [Google Scholar] [CrossRef]
- Ohno, T.; Nishioka, T.; Hisaeda, Y.; Murakami, Y. Hydrophobic vitamin B12: Part 13. Asymmetric reaction of hydrophobic vitamin B12 under electrochemical conditions and rationalization of enantioselectivity based on conformational analysis. J. Mol. Struct. 1994, 308, 207–218. [Google Scholar] [CrossRef]
- Hisaeda, Y.; Nishioka, T.; Inoue, Y.; Asada, K.; Hayashi, T. Electrochemical reactions mediated by vitamin B12 derivatives in organic solvents. Coord. Chem. Rev. 2000, 198, 21–37. [Google Scholar] [CrossRef]
- Su, H.; Walder, L.; Zhang, Z.-d.; Scheffold, R. Asymmetric Catalysis by Vitamin B12. The isomerization of achiral epoxides to optically active allylic alcohols. Helv. Chim. Acta 1988, 71, 1073–1078. [Google Scholar] [CrossRef]
- Franco, D.; Riahi, A.; Hénin, F.; Muzart, J.; Duñach, E. Electrochemical Reduction of a Racemic Allyl β-Keto Ester Catalyzed by Nickel Complexes: Asymmetric Induction. Eur. J. Org. Chem. 2002, 2002, 2257–2259. [Google Scholar] [CrossRef]
- Hori, Y. Modern Aspects of Electrochemistry; Vayenas, C.G., White, R.E., Gamboa-Aldeco, M.E., Eds.; Springer: New York, NY, USA, 2008; pp. 89–189. [Google Scholar]
- Liu, Y.; Li, F.; Zhang, X.; Ji, X. Recent progress on electrochemical reduction of CO2 to methanol. Curr. Opin. Green Sustain. Chem. 2020, 23, 10–17. [Google Scholar] [CrossRef]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.-J.; Jouny, M.; Luc, W.; Zhu, W.; Zhu, J.-J.; Jiao, F. A Highly Porous Copper Electrocatalyst for Carbon Dioxide Reduction. Adv. Mater. 2018, 30, 1803111. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Kattel, S.; Jiao, F.; Chen, J.G. Shape-Controlled CO2 Electrochemical Reduction on Nanosized Pd Hydride Cubes and Octahedra. Adv. Energy Mater. 2019, 9, 1802840. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, H.; Zhao, S.-F.; Niu, D.-F.; Lu, J.-X. Asymmetric electrochemical carboxylation of prochiral acetophenone: An efficient route to optically active atrolactic acid via selective fixation of carbon dioxid. J. Electroanal. Chem. 2009, 630, 35–41. [Google Scholar] [CrossRef]
- Zhao, S.-F.; Zhu, M.-X.; Zhang, K.; Wang, H.; Lu, J.-X. Alkaloid induced asymmetric electrocarboxylation of 4-methylpropiophenone. Tetrahedron Lett. 2011, 52, 2702–2705. [Google Scholar] [CrossRef]
- Chen, B.-L.; Tu, Z.-Y.; Zhu, H.-W.; Sun, W.-W.; Wang, H.; Lu, J.-X. CO2 as a C1-organic building block: Enantioselective electrocarboxylation of aromatic ketones with CO2 catalyzed by cinchona alkaloids under mild conditions. Electrochim. Acta 2014, 116, 475–483. [Google Scholar] [CrossRef]
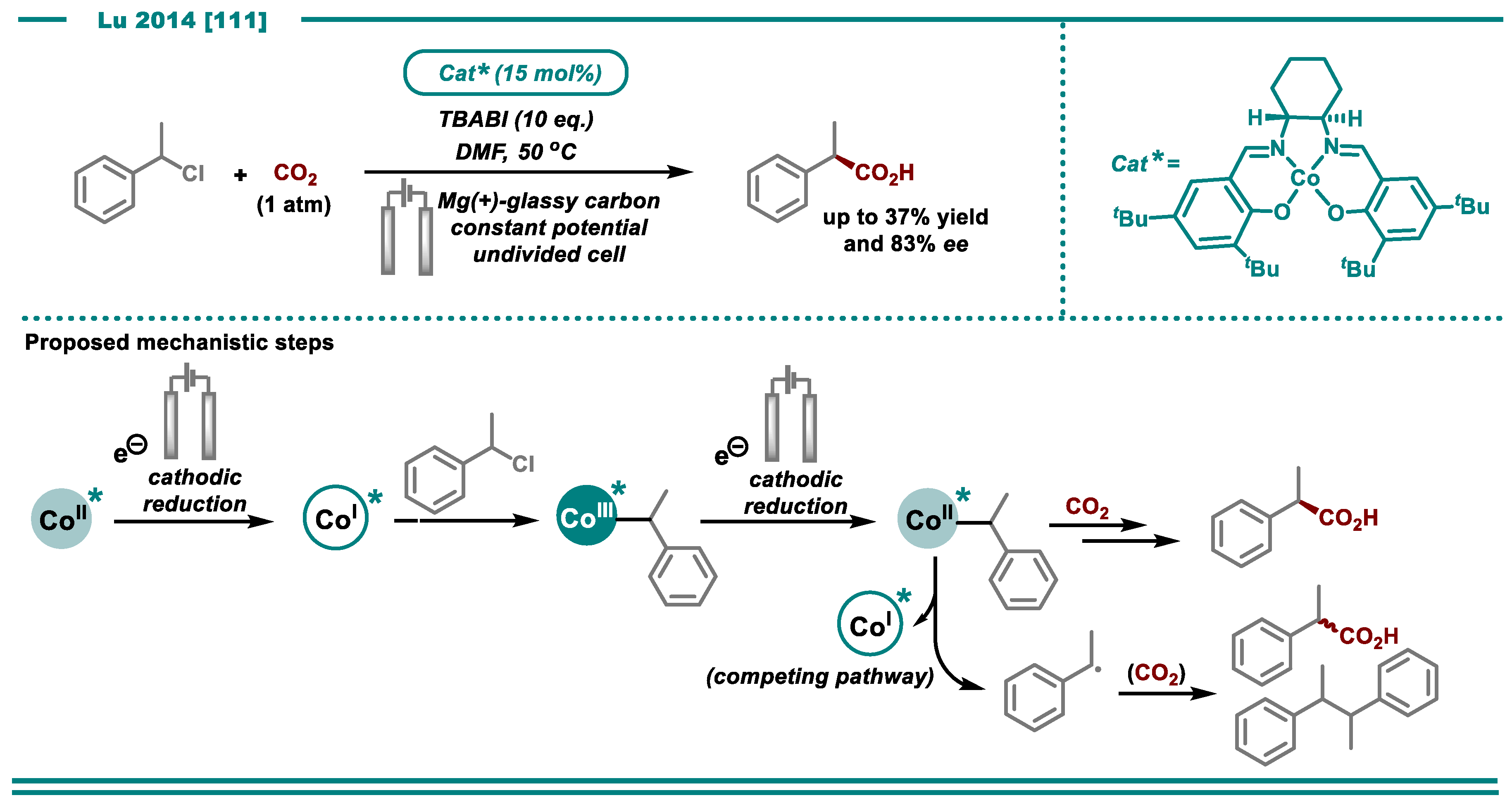
- Chen, B.-L.; Zhu, H.-W.; Xiao, Y.; Sun, Q.-L.; Wang, H.; Lu, J.-X. Asymmetric electrocarboxylation of 1-phenylethyl chloride catalyzed by electrogenerated chiral [CoI(salen)]−complex. Electrochem. Commun. 2014, 42, 55–59. [Google Scholar] [CrossRef]
- Jiao, K.-J.; Li, Z.-M.; Xu, X.-T.; Zhang, L.-P.; Li, Y.-Q.; Zhang, K.; Mei, T.-S. Palladium-catalyzed reductive electrocarboxylation of allyl esters with carbon dioxide. Org. Chem. Front. 2018, 5, 2244–2248. [Google Scholar] [CrossRef]
- Shatskiy, A.; Lundberg, H.; Kärkäs, M.D. Organic Electrosynthesis: Applications in Complex Molecule Synthesis. ChemElectroChem 2019, 6, 4067–4092. [Google Scholar] [CrossRef]
- Everson, D.A.; Weix, D.J. Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem. 2014, 79, 4793–4798. [Google Scholar] [CrossRef] [PubMed]
- Goldfogel, M.J.; Huang, L.; Weix, D.J. Cross-Electrophile Coupling: Principles and New Reactions. In Nickel Catalysis in Synthesis: Methods and Reactions; Ogoshi, S., Ed.; Wiley-VCH: Weinheim, Germany, 2020; Volume 352. [Google Scholar]
- Truesdell, B.L.; Hamby, T.B.; Sevov, C.S. General C(sp2)–C(sp3) Cross-Electrophile Coupling Reactions Enabled by Overcharge Protection of Homogeneous Electrocatalysts. J. Am. Chem. Soc. 2020, 142, 5884–5893. [Google Scholar] [CrossRef] [PubMed]
- DeLano, T.J.; Reisman, S.E. Enantioselective Electroreductive Coupling of Alkenyl and Benzyl Halides via Nickel Catalysis. ACS Catal. 2019, 9, 6751–6754. [Google Scholar] [CrossRef]
- Zhang, S.-K.; Samanta, R.C.; del Vecchio, A.; Ackermann, L. Evolution of High-Valent Nickela-Electrocatalyzed C−H Activation: From Cross(-Electrophile)-Couplings to Electrooxidative C−H Transformations. Chem. Eur. J. 2020, 26, 10936–10947. [Google Scholar] [CrossRef]
- Poremba, K.E.; Dibrell, S.E.; Reisman, S.E. Nickel-Catalyzed Enantioselective Reductive Cross-Coupling Reactions. ACS Catal. 2020, 10, 8237–8246. [Google Scholar] [CrossRef]
- Cherney, A.H.; Reisman, S.E. Nickel-Catalyzed Asymmetric Reductive Cross-Coupling Between Vinyl and Benzyl Electrophiles. J. Am. Chem. Soc. 2014, 136, 14365–14368. [Google Scholar] [CrossRef]
- Qiu, H.; Shuai, B.; Wang, Y.-Z.; Liu, D.; Chen, Y.-G.; Gao, P.-S.; Ma, H.-X.; Chen, S.; Mei, T.-S. Enantioselective Ni-Catalyzed Electrochemical Synthesis of Biaryl Atropisomers. J. Am. Chem. Soc. 2020, 142, 9872–9878. [Google Scholar] [CrossRef]
- Mo, Y.; Lu, Z.; Rughoobur, G.; Patil, P.; Gershenfeld, N.; Akinwande, A.I.; Buchwald, S.L.; Jensen, K.F. Microfluidic electrochemistry for single-electron transfer redox-neutral reactions. Science 2020, 368, 1352–1357. [Google Scholar] [CrossRef]
- Leech, M.C.; Garcia, A.D.; Petti, A.; Dobbs, A.P.; Lam, K. Organic electrosynthesis: From academia to industry. React. Chem. Eng. 2020, 5, 977–990. [Google Scholar] [CrossRef]
- Anson, C.W.; Stahl, S.S. Mediated Fuel Cells: Soluble Redox Mediators and Their Applications to Electrochemical Reduction of O2 and Oxidation of H2, Alcohols, Biomass, and Complex Fuels. Chem. Rev. 2020, 120, 3749–3786. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.-C.; Xiong, Z.-Y.; Pirhaghani, S.; Wirth, T. Enantioselective Electrochemical Lactonization Using Chiral Iodoarenes as Mediators. Synthesis 2019, 51, 276–284. [Google Scholar] [CrossRef]
- Broese, T.; Francke, R. Electrosynthesis Using a Recyclable Mediator–Electrolyte System Based on Ionically Tagged Phenyl Iodide and 1,1,1,3,3,3-Hexafluoroisopropanol. Org. Lett. 2016, 18, 5896–5899. [Google Scholar] [CrossRef] [PubMed]
- Doobary, S.; Sedikides, A.T.; Caldora, H.P.; Poole, D.L.; Lennox, A.J.J. Electrochemical Vicinal Difluorination of Alkenes: Scalable and Amenable to Electron-Rich Substrates. Angew. Chem. Int. Ed. 2020, 59, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Francke, R. Electrogenerated hypervalent iodine compounds as mediators in organic synthesis. Curr. Opin. Electrochem. 2019, 15, 83–88. [Google Scholar] [CrossRef]
- Massignan, L.; Tan, X.; Meyer, T.H.; Kuniyil, R.; Messinis, A.M.; Ackermann, L. C−H Oxygenation Reactions Enabled by Dual Catalysis with Electrogenerated Hypervalent Iodine Species and Ruthenium Complexes. Angew. Chem. Int. Ed. 2020, 59, 3184–3189. [Google Scholar] [CrossRef]
- Liu, K.; Song, C.; Lei, A. Recent advances in iodine mediated electrochemical oxidative cross-coupling. Org. Biomol. Chem. 2018, 16, 2375–2387. [Google Scholar] [CrossRef]



















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Margarita, C.; Lundberg, H. Recent Advances in Asymmetric Catalytic Electrosynthesis. Catalysts 2020, 10, 982. https://doi.org/10.3390/catal10090982
Margarita C, Lundberg H. Recent Advances in Asymmetric Catalytic Electrosynthesis. Catalysts. 2020; 10(9):982. https://doi.org/10.3390/catal10090982
Chicago/Turabian StyleMargarita, Cristiana, and Helena Lundberg. 2020. "Recent Advances in Asymmetric Catalytic Electrosynthesis" Catalysts 10, no. 9: 982. https://doi.org/10.3390/catal10090982
APA StyleMargarita, C., & Lundberg, H. (2020). Recent Advances in Asymmetric Catalytic Electrosynthesis. Catalysts, 10(9), 982. https://doi.org/10.3390/catal10090982