Biocatalyzed Reactions towards Functional Food Components 4-Alkylcatechols and Their Analogues


 , and
, and
Abstract
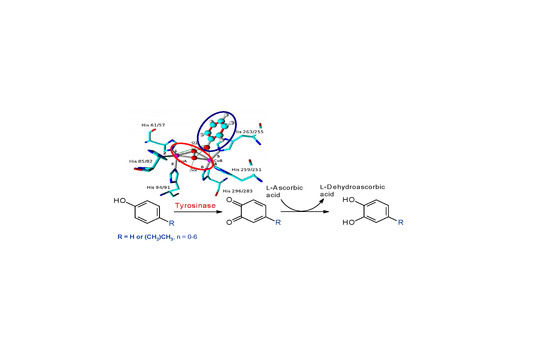
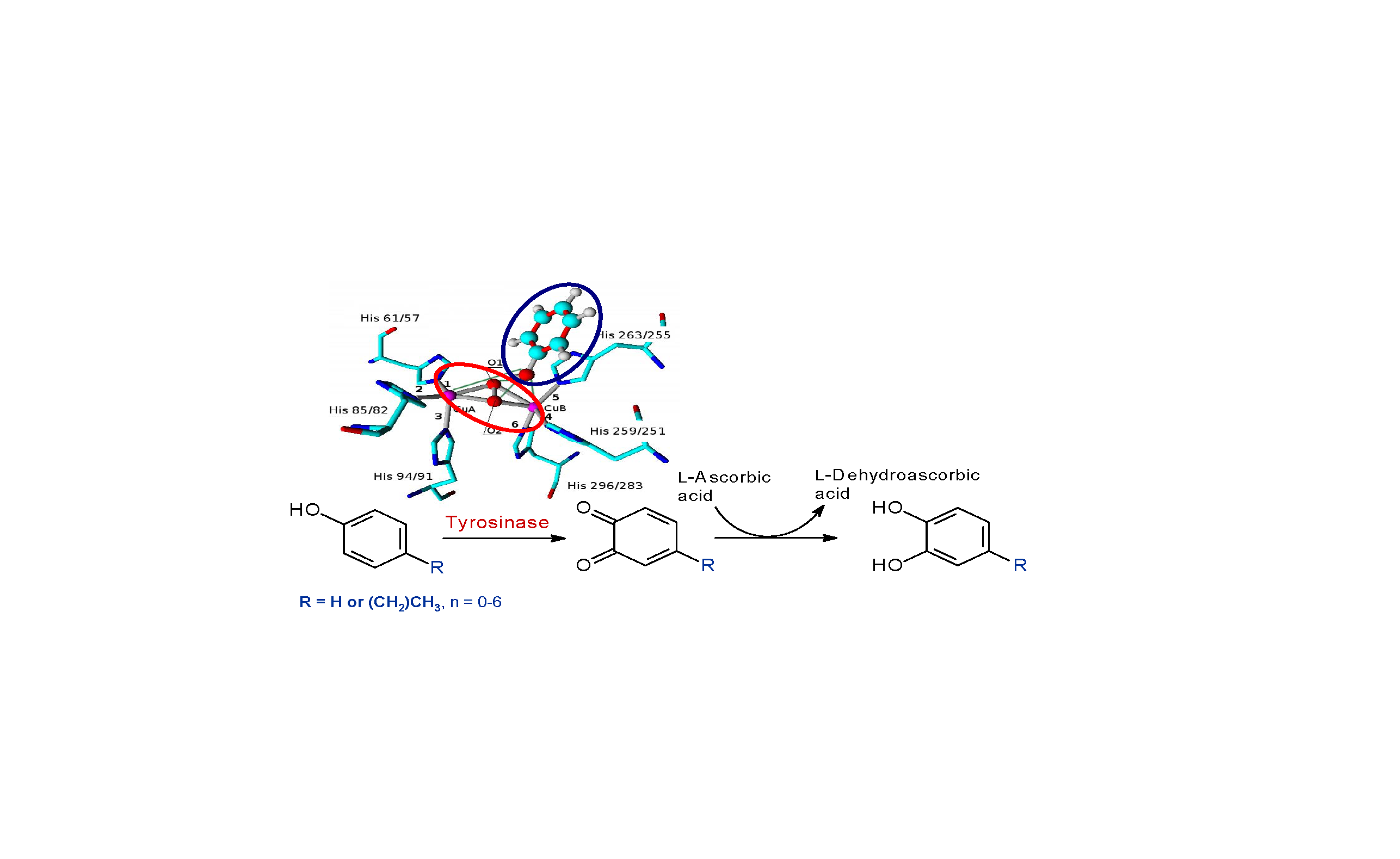
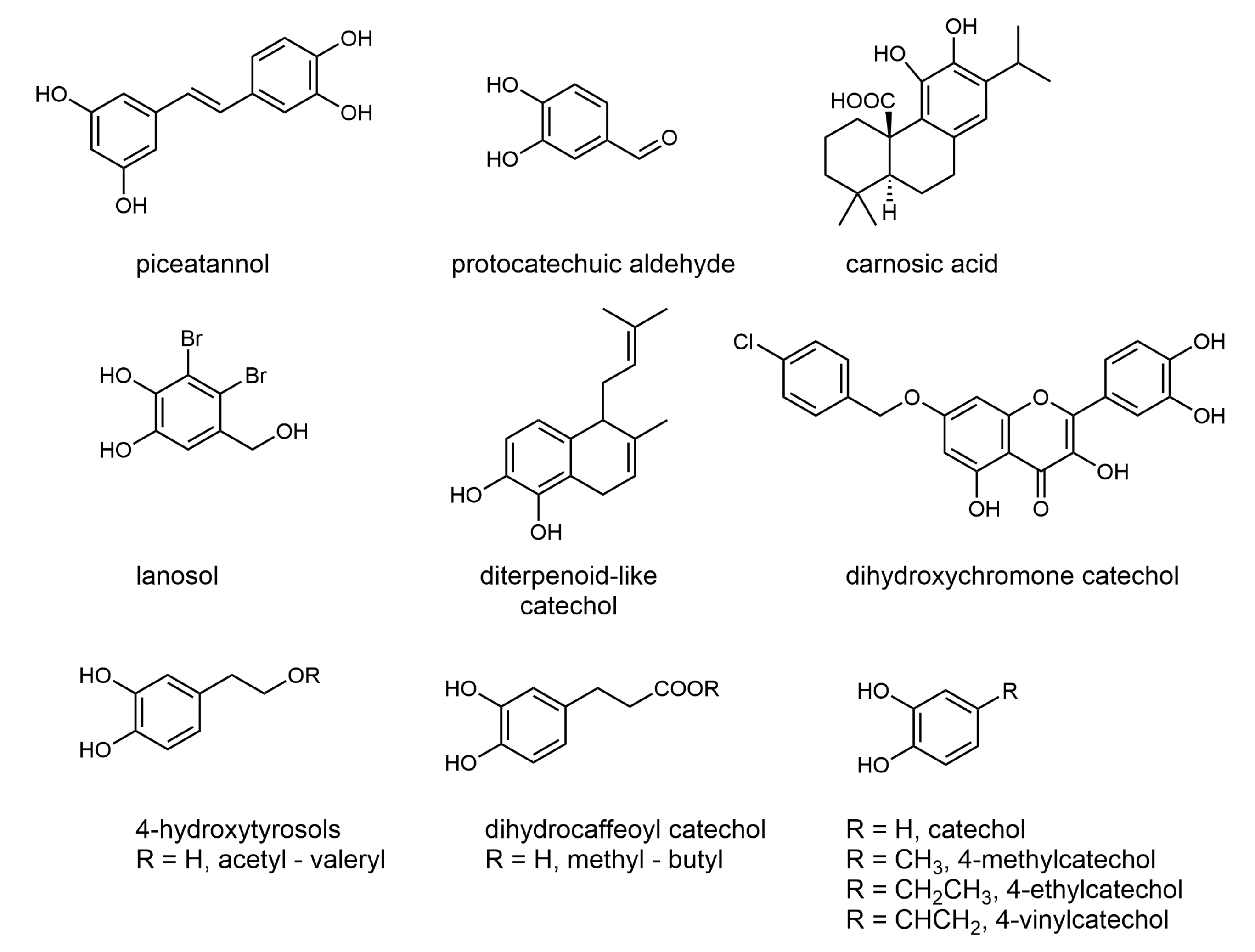
1. Introduction
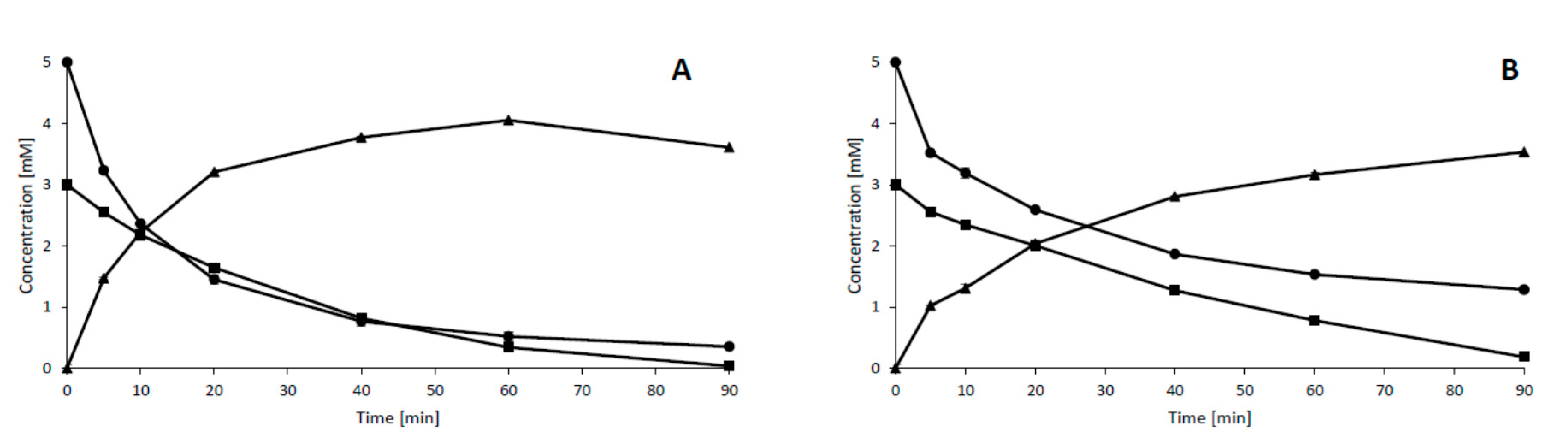
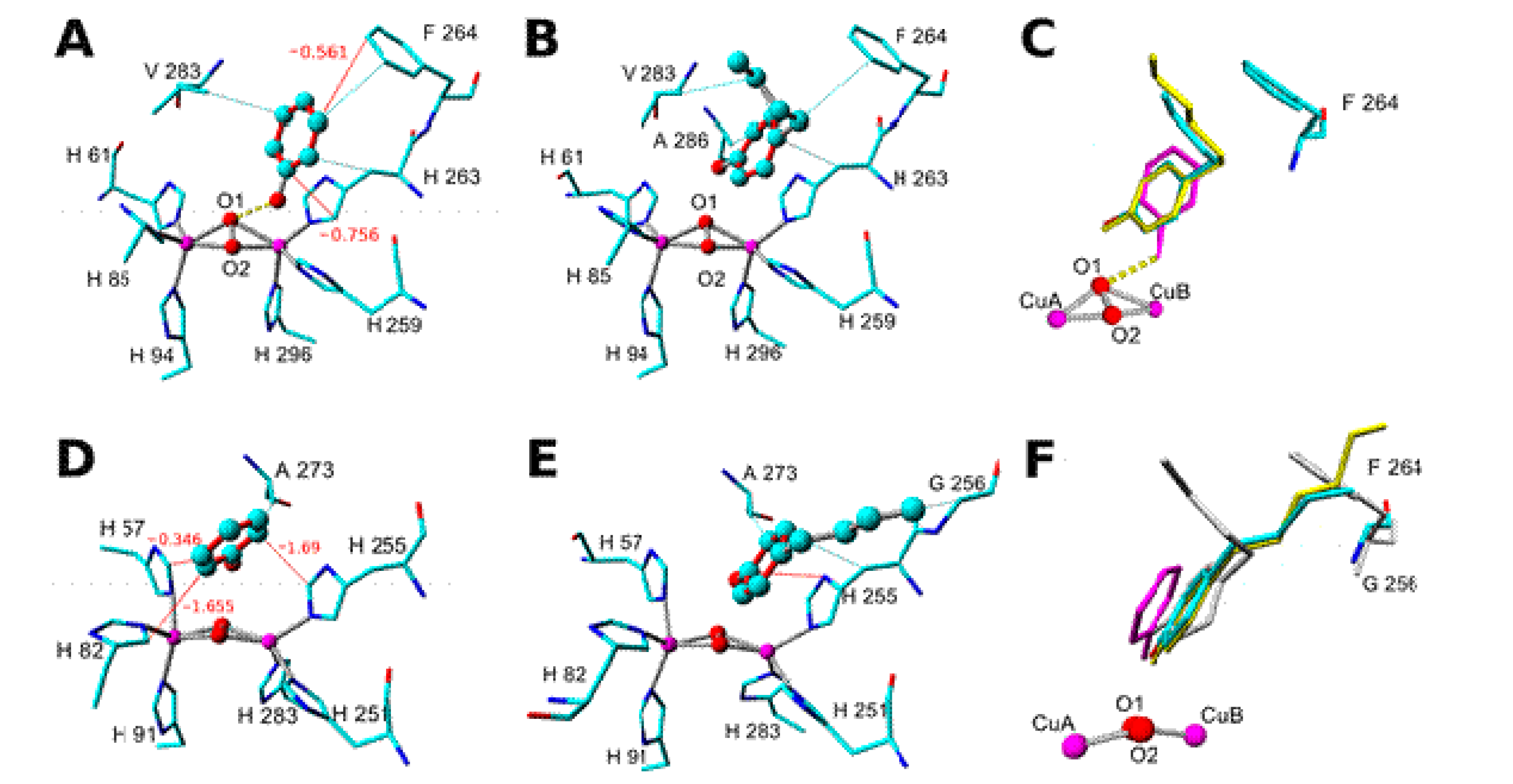
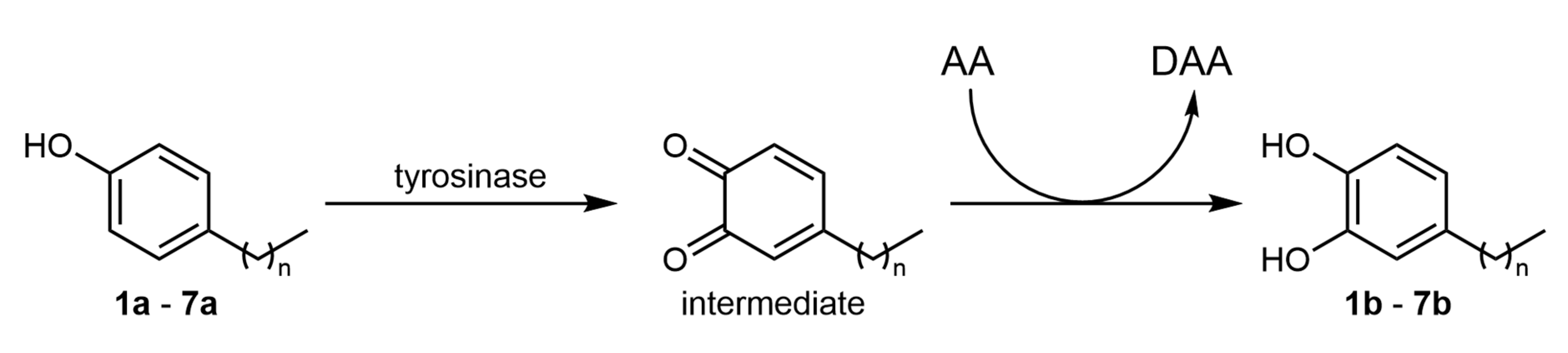
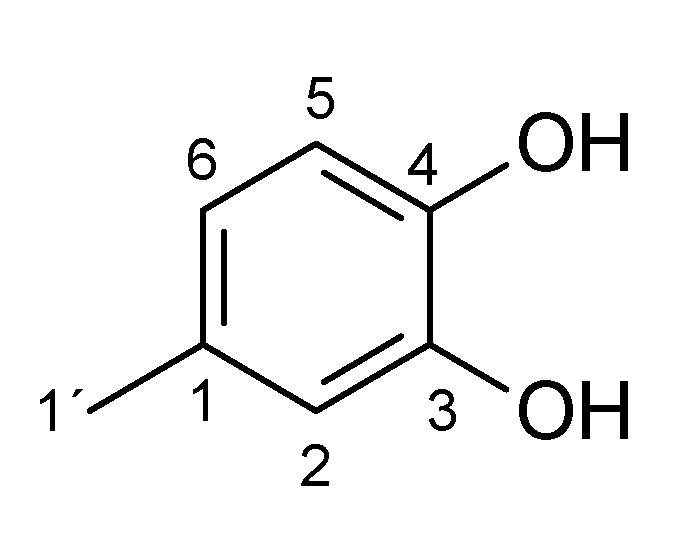
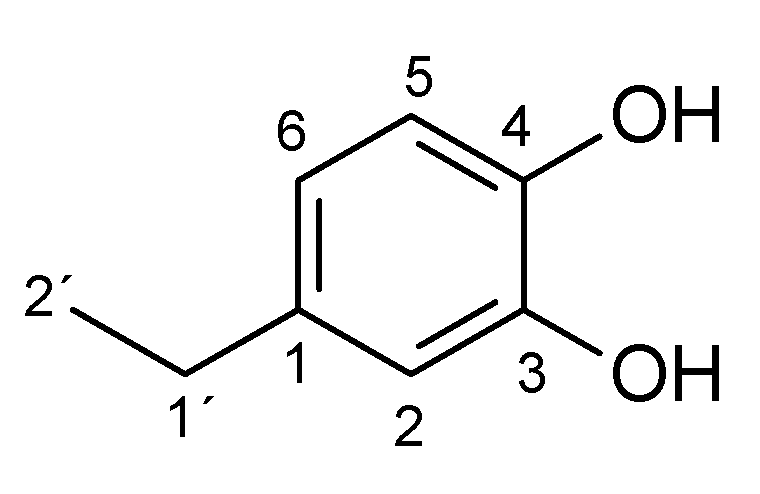
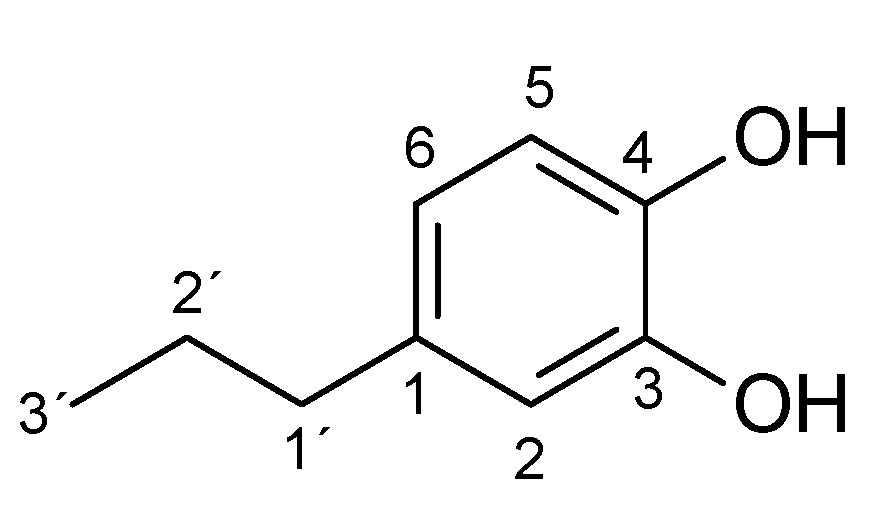
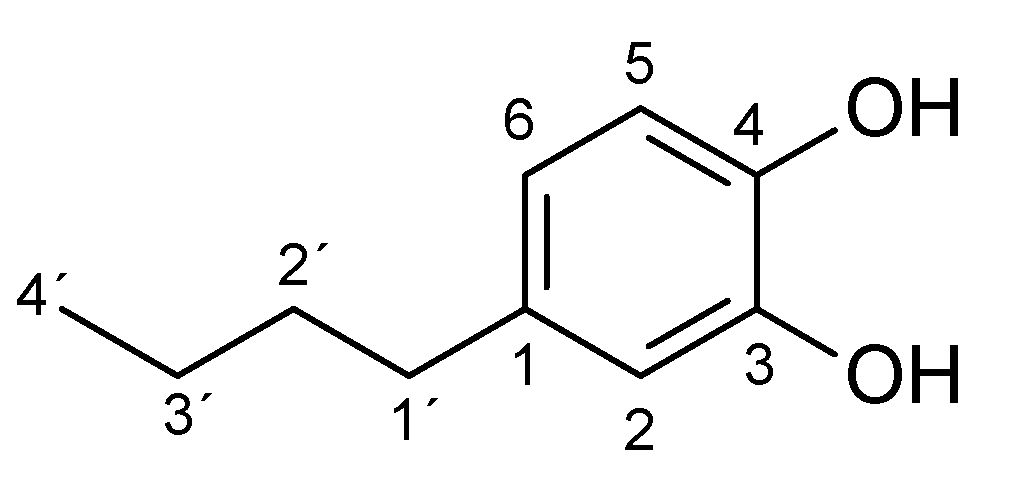
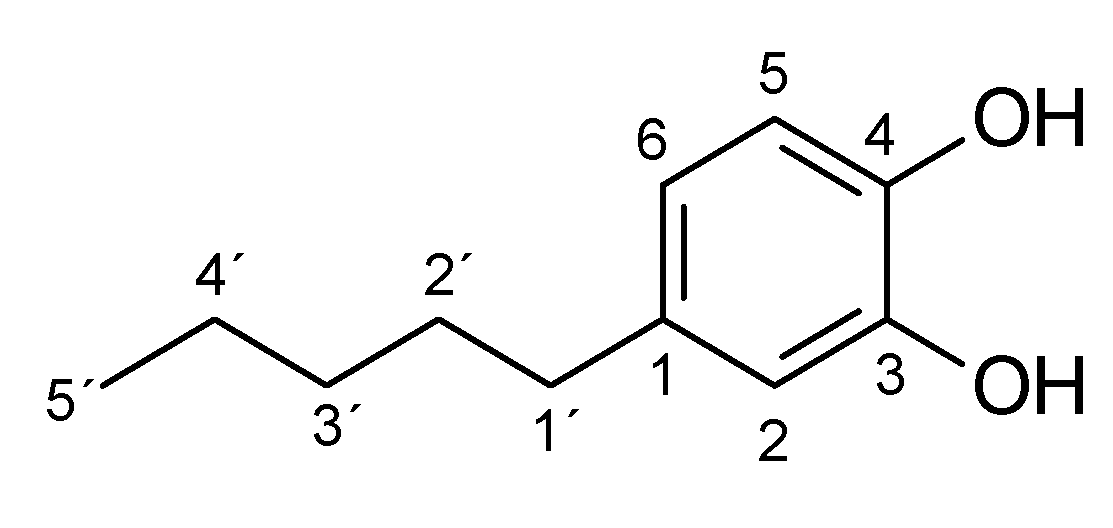
2. Results and Discussion
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Activity Assays
3.3. Biocatalyzed Reactions
3.3.1. Reactions on the Analytical Scale
3.3.2. Reactions on the Preparative Scale
3.3.3. Product Isolation
3.3.4. Preparative HPLC
3.4. Analytical Methods
3.4.1. Analytical HPLC
3.4.2. LC–MS
3.4.3. NMR
3.5. Molecular Modeling and Ligand Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A
Appendix A.1. 1 H and 13C NMR Spectrum of the Products





Appendix A.2. Effect of Substrate and Ascorbic Acid Concentrations on the Biocatalyzed Reactions
Appendix A.3. Effect of Cosolvents on Tyrosinase Activity and the Biocatalyzed Reactions
References
- Botta, G.; Bizzarri, B.M.; Garozzo, A.; Timpanaro, R.; Bisignano, B.; Amatore, D.; Palamara, A.T.; Nencioni, L.; Saladino, R. Carbon nanotubes supported tyrosinase in the synthesis of lipophilic hydroxytyrosol and dihydrocaffeoyl catechols with antiviral activity against DNA and RNA viruses. Bioorg. Med. Chem. 2015, 23, 5345–5351. [Google Scholar] [CrossRef] [PubMed]
- Sueishi, Y.; Nii, R.; Kakizaki, N. Resveratrol analogues like piceatannol are potent antioxidants as quantitatively demonstrated through the high scavenging ability against reactive oxygen species and methyl radical. Bioorg. Med. Chem. Lett. 2017, 27, 5203–5206. [Google Scholar] [CrossRef] [PubMed]
- Yum, S.; Doh, H.-J.; Hong, S.; Jeong, S.; Kim, D.-D.; Park, M.; Jung, Y. Piceatannol, a hydroxystilbene natural product, stabilizes HIF-1α protein by inhibiting HIF prolyl hydroxylase. Eur. J. Pharmacol. 2013, 699, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ji, Y.; Kang, Z.; Lv, C.; Jiang, W. Protocatechuic aldehyde ameliorates experimental pulmonary fibrosis by modulating HMGB1/RAGE pathway. Toxicol. Appl. Pharmacol. 2015, 283, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Gigante, B.; Santos, C.; Silva, A.M.; Curto, M.J.M.; Nascimento, M.S.J.; Pinto, E.; Pedro, M.; Cerqueira, F.; Pinto, M.M.; Duarte, M.P.; et al. Catechols from abietic acid: Synthesis and evaluation as bioactive compounds. Bioorg. Med. Chem. 2003, 11, 1631–1638. [Google Scholar] [CrossRef]
- Park, S.-H.; Song, J.-H.; Kim, T.; Shin, W.-S.; Park, G.M.; Lee, S.; Kim, Y.-J.; Choi, P.; Kim, H.; Kim, H.-S.; et al. Anti-human rhinoviral activity of polybromocatechol compounds isolated from the Rhodophyta, Neorhodomela aculeata. Mar. Drugs 2012, 10, 2222–2233. [Google Scholar] [CrossRef]
- Pawar, R.; Das, T.; Mishra, S.; Nutan; Pancholi, B.; Gupta, S.K.; Bhat, S.V. Synthesis, anti-HIV activity, integrase enzyme inhibition and molecular modeling of catechol, hydroquinone and quinol labdane analogs. Bioorg. Med. Chem. Lett. 2014, 24, 302–307. [Google Scholar] [CrossRef]
- Lee, C.; Lee, J.M.; Lee, N.-R.; Kim, D.-E.; Jeong, Y.-J.; Chong, Y. Investigation of the pharmacophore space of Severe Acute Respiratory Syndrome coronavirus (SARS-CoV) NTPase/helicase by dihydroxychromone derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 4538–4541. [Google Scholar] [CrossRef]
- Senger, D.R.; Li, D.; Jaminet, S.-C.; Cao, S. Activation of the Nrf2 cell defense pathway by ancient foods: Disease prevention by important molecules and microbes lost from the modern western diet. PLoS ONE 2016, 11, e148042. [Google Scholar] [CrossRef]
- Espín, J.C.; Soler-Rivas, C.; Cantos, E.; Tomás-Barberán, F.A.; Wichers, H.J. Synthesis of the antioxidant hydroxytyrosol using tyrosinase as biocatalyst. J. Agric. Food Chem. 2001, 49, 1187–1193. [Google Scholar] [CrossRef]
- Xu, D.-Y.; Chen, J.-Y.; Yang, Z. Use of cross-linked tyrosinase aggregates as catalyst for synthesis of L–DOPA. Biochem. Eng. J. 2012, 63, 88–94. [Google Scholar] [CrossRef]
- Min, K.; Park, K.; Park, D.-H.; Yoo, Y.J. Overview on the biotechnological production of L-DOPA. Appl. Microbiol. Biotechnol. 2015, 99, 575–584. [Google Scholar] [CrossRef]
- Guazzaroni, M.; Crestini, C.; Saladino, R. Layer-by-Layer coated tyrosinase: An efficient and selective synthesis of catechols. Biorg. Med. Chem. 2012, 20, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Guazzaroni, M.; Pasqualini, M.; Botta, G.; Saladino, R. A novel synthesis of bioactive catechols by layer-by-layer immobilized tyrosinase in an organic solvent medium. ChemCatChem 2012, 4, 89–99. [Google Scholar] [CrossRef]
- Bozzini, T.; Botta, G.; Delfino, M.; Onofri, S.; Saladino, R.; Amatore, D.; Sgarbanti, R.; Nencioni, L.; Palamara, A.T. Tyrosinase and Layer-by-Layer supported tyrosinases in the synthesis of lipophilic catechols with antiinfluenza activity. Bioorg. Med. Chem. 2013, 21, 7699–7708. [Google Scholar] [CrossRef] [PubMed]
- Botta, G.; Delfino, M.; Guazzaroni, M.; Crestini, C.; Onofri, S.; Saladino, R. Selective Synthesis of DOPA and DOPA peptides by native and immobilized tyrosinase in organic solvent. ChemPlusChem 2013, 78, 325–330. [Google Scholar] [CrossRef]
- Ramsden, C.A.; Riley, P.A. Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg. Med. Chem. 2014, 22, 2388–2395. [Google Scholar] [CrossRef]
- Deri-Zenaty, B.; Bachar, S.; Rebroš, M.; Fishman, A. A coupled enzymatic reaction of tyrosinase and glucose dehydrogenase for the production of hydroxytyrosol. Appl. Microbiol. Biotechnol. 2020, 104, 4945–4955. [Google Scholar] [CrossRef]
- Yamada, K.; Akiba, Y.; Shibuya, T.; Kashiwada, A.; Matsuda, K.; Hirata, M. Water purification through bioconversion of phenol compounds by tyrosinase and chemical adsorption by chitosan beads. Biotechnol. Prog. 2005, 21, 823–829. [Google Scholar] [CrossRef]
- Yamada, K.; Inoue, T.; Akiba, Y.; Kashiwada, A.; Matsuda, K.; Hirata, M. Removal of p-alkylphenols from aqueous solutions by combined use of mushroom tyrosinase and chitosan beads. Biosci. Biotechnol. Biochem. 2006, 70, 2467–2475. [Google Scholar] [CrossRef][Green Version]
- Flurkey, A.; Cooksey, J.; Reddy, A.; Spoonmore, K.; Rescigno, A.; Inlow, J.; Flurkey, W.H. Enzyme, protein, carbohydrate, and phenolic contaminants in commercial tyrosinase preparations: Potential problems affecting tyrosinase activity and inhibition studies. J. Agric. Food Chem. 2008, 56, 4760–4768. [Google Scholar] [CrossRef] [PubMed]
- Inlow, J.K. Homology models of four Agaricus bisporus tyrosinases. Int. J. Biol. Macromol. 2012, 50, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Ismaya, W.T.; Rozeboom, H.J.; Weijn, A.; Mes, J.J.; Fussetti, F.; Wichers, H.J.; Dijkstra, B.W. Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry 2011, 50, 5477–5486. [Google Scholar] [CrossRef] [PubMed]
- Mauracher, S.G.; Molitor, C.; Al-Oweini, R.; Kortz, U.; Rompel, A. Latent and active abPPO4 mushroom tyrosinase cocrystallized with hexatungstotellurate (VI) in a single crystal. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 2301–2315. [Google Scholar] [CrossRef] [PubMed]
- Matoba, Y.; Kihara, S.; Bando, N.; Yoshitsu, H.; Sakaguchi, M.; Kayama, K.; Yamagisawa, S.; Ogura, T.; Sugiyama, M. Catalytic mechanism of the tyrosinase reaction toward the Tyr98 residue in the caddie protein. PLoS Biol. 2018, 16, e3000077. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Honda, K.; Uchimaru, T.; Mikami, M.; Tanabe, K. The magnitude of the CH/π Interaction between benzene and some model hydrocarbons. J. Am. Chem. Soc. 2000, 122, 3746–3753. [Google Scholar] [CrossRef]
- Goldfeder, M.; Kanteev, M.; Isaschar-Ovdat, S.; Adir, N.; Fishman, A. Determination of tyrosinase substrate-binding modes reveals mechanistic differences between type-3 copper proteins. Nat. Commun. 2014, 5, 4505. [Google Scholar] [CrossRef]
- Romanovskaya, I.I.; Shesterenko, Y.A.; Sevastyanov, O.V. Elimination of phenol with the use of tyrosinase of fungi. J. Water Chem. Technol. 2009, 31, 135–138. [Google Scholar] [CrossRef]
- Selinheimo, E.; Gasparetti, C.; Mattinen, M.-L.; Steffensen, C.L.; Buchert, J.; Kruus, K. Comparison of substrate specificity of tyrosinases from Trichoderma reesei and Agaricus bisporus. Enzym. Microb. Technol. 2009, 44, 1–10. [Google Scholar] [CrossRef]
- Guo, M.; Lu, F.; Du, L.; Pu, J.; Bai, D. Optimization of the expression of a laccase gene from Trametes versicolor in Pichia methanolica. Appl. Microbiol. Biotechnol. 2006, 71, 848–852. [Google Scholar] [CrossRef]
- Rucká, L.; Chmátal, M.; Kulik, N.; Petrásková, L.; Pelantová, H.; Novotný, P.; Příhodová, R.; Pátek, M.; Martínková, L. Genetic and functional diversity of nitrilases in Agaricomycotina. Int. J. Mol. Sci. 2019, 20, 5990. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Matoba, Y.; Kumagai, T.; Yamamoto, A.; Yoshitsu, H.; Sugiyama, M. Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. J. Biol. Chem. 2006, 281, 8981–8990. [Google Scholar] [CrossRef]
- Murphy, R.B.; Philipp, D.M.; Friesner, R.A. A mixed quantum mechanics/molecular mechanics (QM/MM) method for large-scale modeling of chemistry in protein environments. J. Comp. Chem. 2000, 21, 1442–1457. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, J.T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Vaught, A. Graphing with Gnuplot and Xmgr. Linux J. 1996, 1996, 7. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | logP 1 | Reaction Time [h] | Product (Amount; Isolated Yield) 2 |
|---|---|---|---|
| 4-methylphenol (1a) | 1.94 | 3 | 1b (45 mg; 45%) |
| 4-ethylphenol (2a) | 2.58 | 4 | 2b (65 mg; 59%) |
| 4-propylphenol (3a) | 3.20 | 4.5 | 3b (46 mg; 45%) |
| 4-butylphenol (4a) | 3.65 | 4.5 | 4b (34 mg; 28%) |
| 4-pentylphenol (5a) | 4.06 | 4.5 | 5b (15 mg; 10%) |
| Enzyme | Ligand | Binding SP Score (kcal/mol) | ||
|---|---|---|---|---|
| Before MDS 1 | After MDS | |||
| After 10 ns | After 20 ns | |||
| PPO3 | phenol | −6.4 | −5.359 | −5.360 |
| 2-methylphenol | −6.477 | −5.763 | −5.150 | |
| 3-methylphenol | −6.926 | −5.943 | −6.509 | |
| 4-methylphenol (1a) | −6.880 | −6.465 | −6.245 | |
| 4-ethylphenol (2a) | −6.188 | −6.256 | −6.533 | |
| 4-propylphenol (3a) | −6.120 | −6.02 | −6.05 | |
| 4-butylphenol (4a) | −5.719 | −5.883 | −5.999 | |
| 4-pentylphenol (5a) | −5.337 | −5.132 | −5.079 | |
| 4-hexylphenol (6a) | −5.394 | −5.615 | −5.251 | |
| 4-heptylphenol (7a) | −5.451 | −4.585 | −4.359 | |
| PPO4 | phenol | −5.407 | −5.352 | −5.33 |
| 2-methylphenol | −5.587 | −4.097 | n.a. 2 | |
| 3-methylphenol | −5.532 | −5.206 | −5.493 | |
| 4-methylphenol (1a) | −5.479 | −5.218 | −4.244 | |
| 4-ethylphenol (2a) | −5.874 | −5.816 | −5.760 | |
| 4-propylphenol (3a) | −5.303 | −5.39 | −4.88 | |
| 4-butylphenol (4a) | −4.874 | −4.732 | −4.758 | |
| 4-pentylphenol (5a) | −4.645 | −4.410 | −4.326 | |
| 4-hexylphenol (6a) | −4.156 | −4.24 | −4.19 | |
| 4-heptylphenol (7a) | −3.827 | −4.198 | −4.298 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínková, L.; Příhodová, R.; Kulik, N.; Pelantová, H.; Křístková, B.; Petrásková, L.; Biedermann, D. Biocatalyzed Reactions towards Functional Food Components 4-Alkylcatechols and Their Analogues. Catalysts 2020, 10, 1077. https://doi.org/10.3390/catal10091077
Martínková L, Příhodová R, Kulik N, Pelantová H, Křístková B, Petrásková L, Biedermann D. Biocatalyzed Reactions towards Functional Food Components 4-Alkylcatechols and Their Analogues. Catalysts. 2020; 10(9):1077. https://doi.org/10.3390/catal10091077
Chicago/Turabian StyleMartínková, Ludmila, Romana Příhodová, Natalia Kulik, Helena Pelantová, Barbora Křístková, Lucie Petrásková, and David Biedermann. 2020. "Biocatalyzed Reactions towards Functional Food Components 4-Alkylcatechols and Their Analogues" Catalysts 10, no. 9: 1077. https://doi.org/10.3390/catal10091077
APA StyleMartínková, L., Příhodová, R., Kulik, N., Pelantová, H., Křístková, B., Petrásková, L., & Biedermann, D. (2020). Biocatalyzed Reactions towards Functional Food Components 4-Alkylcatechols and Their Analogues. Catalysts, 10(9), 1077. https://doi.org/10.3390/catal10091077