Bioalcohol Reforming: An Overview of the Recent Advances for the Enhancement of Catalyst Stability





Abstract
Summary
| 1 | Introduction | Pag. 2 |
| 2 | Bioethanol reforming | Pag. 3 |
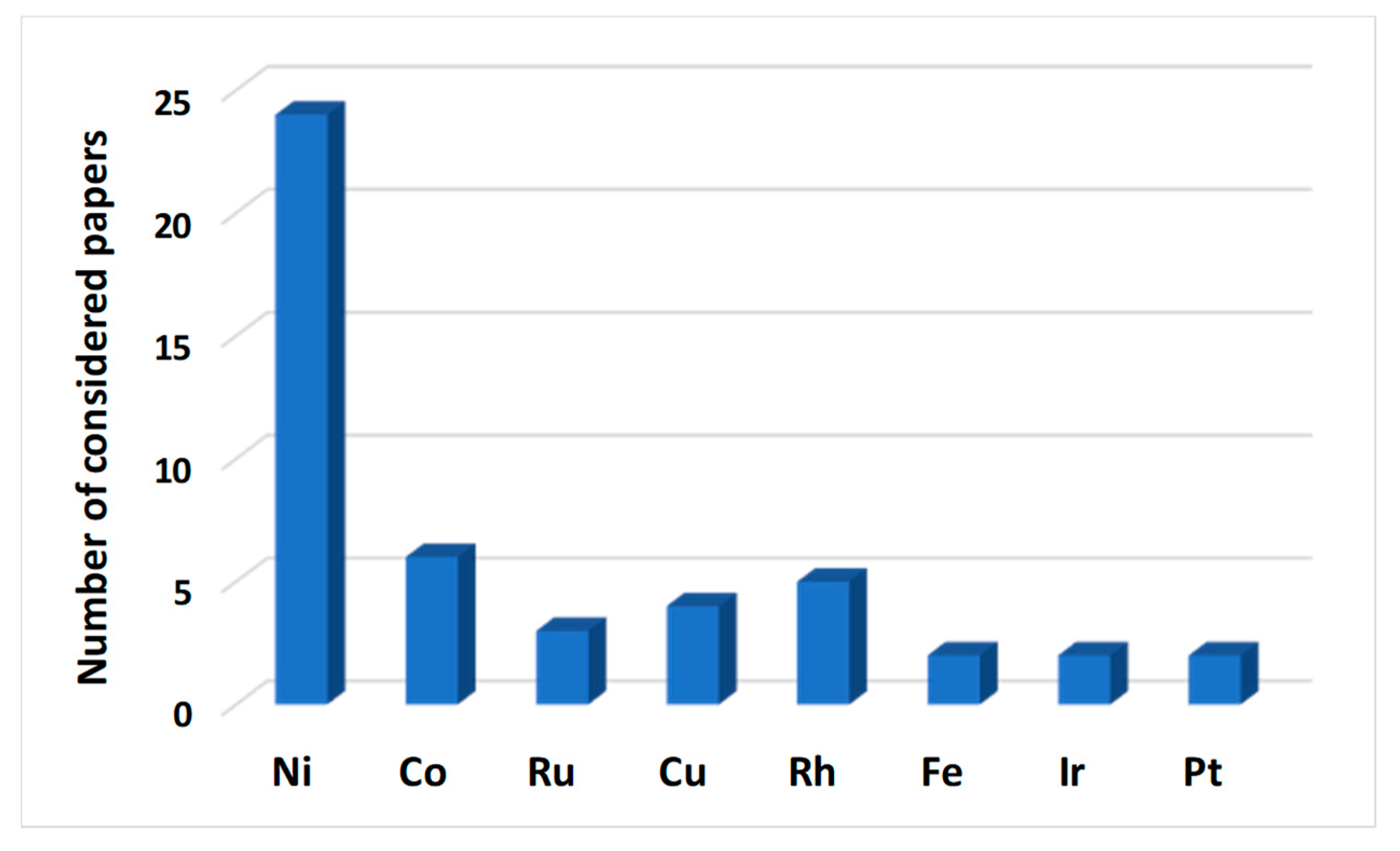
| 2.1 | The influence of the active phase | Pag. 8 |
| 2.2 | The role of the support | Pag. 12 |
| 2.3 | The effect of the addition of promoters | Pag. 13 |
| 3 | (Oxidative) Biomethanol steam reforming | Pag. 19 |
| 3.1 | The influence of the active phase | Pag. 20 |
| 3.2 | The role of the support | Pag. 25 |
| 3.3 | The effect of the addition of promoters | Pag. 27 |
| 3.4 | Unconventional reactor configuration, simulation, and theoretical studies | Pag. 32 |
| 3.5 | Oxidative steam reforming of methanol | Pag. 36 |
| 4 | Bioglycerol reforming | Pag. 39 |
| 4.1 | The influence of the active phase | Pag. 42 |
| 4.2 | The role of the support | Pag. 43 |
| 4.3 | The effect of the addition of promoters | Pag. 46 |
| 5 | Other bioalcohol reforming | Pag. 50 |
| 5.1 | The influence of the active phase | Pag. 51 |
| 5.2 | The role of the support | Pag. 53 |
| 5.3 | The effect of the addition of promoters | Pag. 54 |
| 6 | Conclusions | Pag. 54 |
1. Introduction
2. Bioethanol Reforming
2.1. The Influence of the Active Phase
2.2. The Role of the Support
2.3. The Effect of the Addition of Promoters
3. Oxidative Biomethanol Steam Reforming
3.1. The Influence of the Active Phase
3.2. The Role of the Support
3.3. The Effect of the Addition of Promoters
3.4. Unconventional Reactor Configuration, Simulation, and Theoretical Studies
3.5. Oxidative Steam Reforming of Methanol
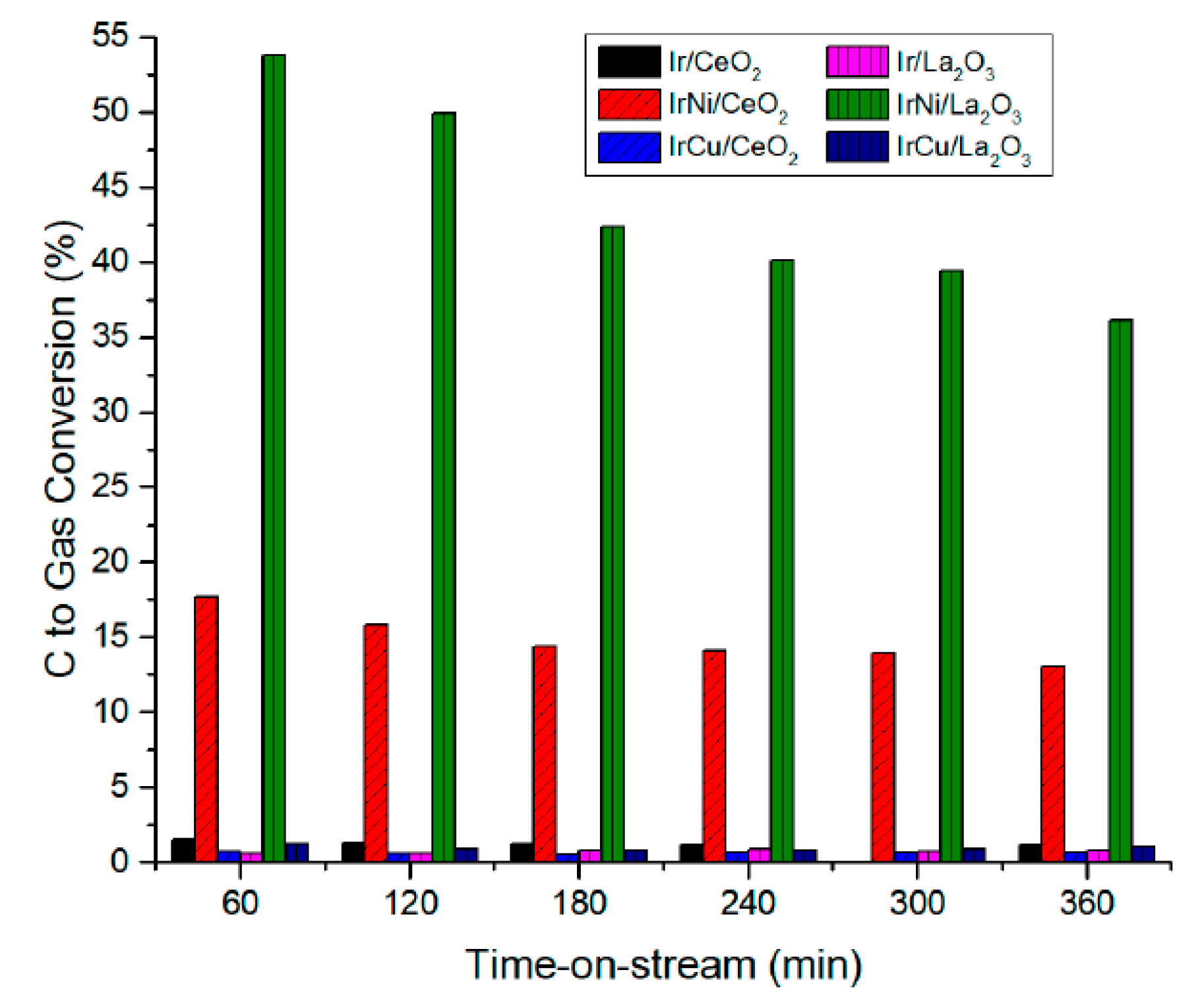
4. Bioglycerol Reforming
4.1. The Influence of the Active Phase
4.2. The Role of the Support
4.3. The Effect of the Addition of Promoters
5. Other Bioalcohol Reforming
5.1. The Influence of the Active Phase
5.2. The Role of the Support
5.3. The Effect of the Addition of Promoters
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APR | Aqueous-phase reforming |
| APRE | Aqueous-phase reforming of ethanol |
| ASRM | Autothermal steam reforming of methanol |
| ATR | Autothermal reforming |
| BET | Brunauer–Emmett–Teller surface area measurements |
| BSR | Butanol steam reforming |
| CCR | Carbon capture and recycling |
| CFD | Computational fluid dynamics |
| CFR | Carbon formation rate |
| CNF | Carbon nanofibers |
| CNT | Carbon nanotubes |
| DFT | Discrete Fourier transform |
| DRIFT | Diffuse reflectance infrared Fourier transform spectroscopy |
| DSC | Differential scanning calorimeter |
| EDR | Ethanol dry reforming |
| EDS | Energy-dispersive X-ray spectrometry |
| EDX | Energy-dispersive X-ray analysis |
| GAPR | Aqueous-phase reforming of glycerol |
| GDR | Glycerol dry reforming |
| GHSV | Gas hourly space velocity |
| GSR | Glycerol steam reforming |
| LHSV | Liquid hourly space velocity |
| MSR | Methanol steam reforming |
| MMT | Montmorillonite |
| OSRE | Oxidative steam reforming of ethanol |
| OSRM | Oxidative steam reforming of methanol |
| OSRB | Oxidative steam reforming of butanol |
| PEM | Polymer electrolyte membrane |
| PO | Partial oxidation |
| SEM | Scanning electron microscopy |
| TEM | Transmission electron microscopy |
| TGA | Thermogravimetric analysis |
| TOF | Turnover frequencies |
| TOS | Time-on-stream |
| TPD | Temperature-programmed desorption |
| TPH | Temperature-programmed hydrogenation |
| TPO | Temperature-programmed oxidation |
| TPR | Temperature-programmed reduction |
| WGS | Water–gas shift |
| WHSV | Weight hourly space velocity |
| X | Alcohol conversion |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction |
References
- Susmozas, A.; Iribarren, D.; Dufour, J. Assessing the life-cycle performance of hydrogen production via biofuel reforming in Europe. Resources 2015, 4, 398–411. [Google Scholar] [CrossRef]
- Al-Maamary, H.M.S.; Kazem, H.A.; Chaichan, M.T. The impact of oil price fluctuations on common renewable energies in GCC countries. Renew. Sustain. Energy Rev. 2017, 75, 989–1007. [Google Scholar] [CrossRef]
- Calles, J.A.; Carrero, A.; Vizcaíno, A.J.; García-Moreno, L.; Megía, P.J. Steam reforming of model Bio-Oil aqueous fraction using Ni-(Cu, Co, Cr)/SBA-15 catalysts. Int. J. Mol. Sci. 2019, 20, 512. [Google Scholar] [CrossRef] [PubMed]
- Colmenares, J.C.; Colmenares Quintero, R.F.; Pieta, I.S. Catalytic dry reforming for biomass-based fuels processing: Progress and future perspectives. Energy Technol. 2016, 4, 881–890. [Google Scholar] [CrossRef]
- Lisý, M.; Lisá, H.; Jecha, D.; Baláš, M.; Križan, P. Characteristic properties of alternative biomass fuels. Energies 2020, 13, 1448. [Google Scholar] [CrossRef]
- Palma, V.; Barba, D.; Meloni, E.; Ruocco, C.; Martino, M. Chapter 2—Membrane reactors for H2 and energy production. In Current Trends and Future Developments on (Bio-) Membranes; Basile, A., Napporn, T.W., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 33–56. [Google Scholar] [CrossRef]
- Alique, D.; Bruni, G.; Sanz, R.; Calles, J.A.; Tosti, S. Ultra-Pure hydrogen via co-valorization of olive mill wastewater and bioethanol in pd-membrane reactors. Processes 2020, 8, 219. [Google Scholar] [CrossRef]
- Frusteri, F.; Bonura, G. 5-Hydrogen production by reforming of bio-alcohols. In Compendium of Hydrogen Energy; Subramani, V., Basile, A., Veziroğlu, T.N., Eds.; Woodhead Publishing: Oxford, UK, 2015; pp. 109–136. [Google Scholar]
- Melikoglu, M.; Singh, V.; Leu, S.Y.; Webb, C.; Lin, C.S.K. Biochemical production of bioalcohols. In Handbook of Biofuels Production, 2nd ed.; Luque, R., Lin, C.S.K., Wilson, K., Clark, J., Eds.; Woodhead Publishing: Oxford, UK, 2016; pp. 237–258. [Google Scholar]
- Datta, A.; Hossain, A.; Roy, S. An overview on biofuels and their advantages and disadvantages. Asian J. Chem. 2019, 31, 1851–1858. [Google Scholar] [CrossRef]
- Ail, S.S.; Dasappa, S. Biomass to liquid transportation fuel via Fischer Tropsch synthesis—Technology review and current scenario. Renew. Sustain. Energy Rev. 2016, 58, 267–286. [Google Scholar] [CrossRef]
- Behera, S.; Singh, R.; Arora, R.; Sharma, N.K.; Shukla, M.; Kumar, S. Scope of algae as third generation biofuels. Front. Bioeng. Biotechnol. 2015, 2. [Google Scholar] [CrossRef]
- Seretis, A.; Tsiakaras, P. Crude bio-glycerol aqueous phase reforming and hydrogenolysis over commercial SiO2Al2O3 nickel catalyst. Renew. Energy 2016, 97, 373–379. [Google Scholar] [CrossRef]
- Coronado, C.J.R. Glycerol: Production, consumption, prices, characterization and new trends in combustion. Renew. Sustain. Energy Rev. 2013, 27, 475–493. [Google Scholar] [CrossRef]
- Liu, Z.; Peng, W.; Motahari-Nezhad, M.; Shahraki, S.; Beheshti, M. Circulating fluidized bed gasification of biomass for flexible end-use of syngas: A micro and nano scale study for production of bio-methanol. J. Clean. Prod. 2016, 129, 249–255. [Google Scholar] [CrossRef]
- Iaquaniello, G.; Centi, G.; Salladini, A.; Palo, E.; Perathoner, S.; Spadaccini, L. Waste-to-methanol: Process and economics assessment. Bioresour. Technol. 2017, 243, 611–619. [Google Scholar] [CrossRef]
- Anitha, M.; Kamarudin, S.K.; Shamsul, N.S.; Kofli, N.T. Determination of bio-methanol as intermediate product of anaerobic co-digestion in animal and agriculture wastes. Int. J. Hydrogen Energy 2015, 40, 11791–11799. [Google Scholar] [CrossRef]
- Bonfim-Rocha, L.; Gimenes, M.L.; Bernardo de Faria, S.H.; Silva, R.O.; Esteller, L.J. Multi-objective design of a new sustainable scenario for bio-methanol production in Brazil. J. Clean. Prod. 2018, 187, 1043–1056. [Google Scholar] [CrossRef]
- Hartley, U.W.; Amornraksa, S.; Kim-Lohsoontorn, P.; Laosiripojana, N. Thermodynamic analysis and experimental study of hydrogen production from oxidative reforming of n-butanol. Chem. Eng. J. 2015, 278, 2–12. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Meloni, E.; Ricca, A. Highly active and stable Pt-Ni/CeO2-SiO2 catalysts for ethanol reforming. J. Clean. Prod. 2017, 166, 263–272. [Google Scholar] [CrossRef]
- Bepari, S.; Kuila, D. Steam reforming of methanol, ethanol and glycerol over nickel-based catalysts-A review. Int. J. Hydrogen Energy 2019. [Google Scholar] [CrossRef]
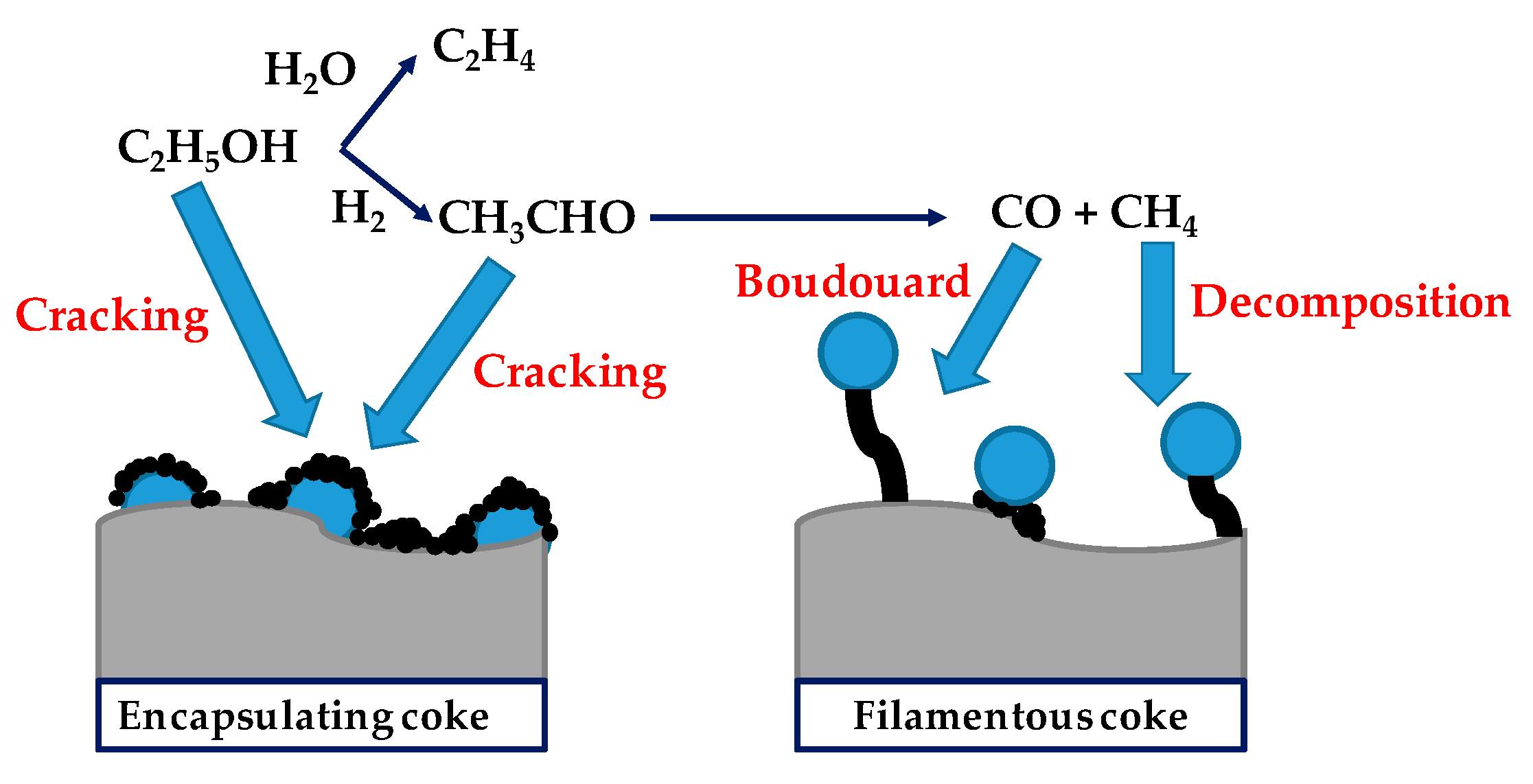
- Montero, C.; Remiro, A.; Valle, B.; Oar-Arteta, L.; Bilbao, J.; Gayubo, A.G. Origin and nature of coke in ethanol steam reforming and its role in deactivation of Ni/La2O3–αAl2O3 catalyst. Ind. Eng. Chem. Res. 2019, 58, 14736–14751. [Google Scholar] [CrossRef]
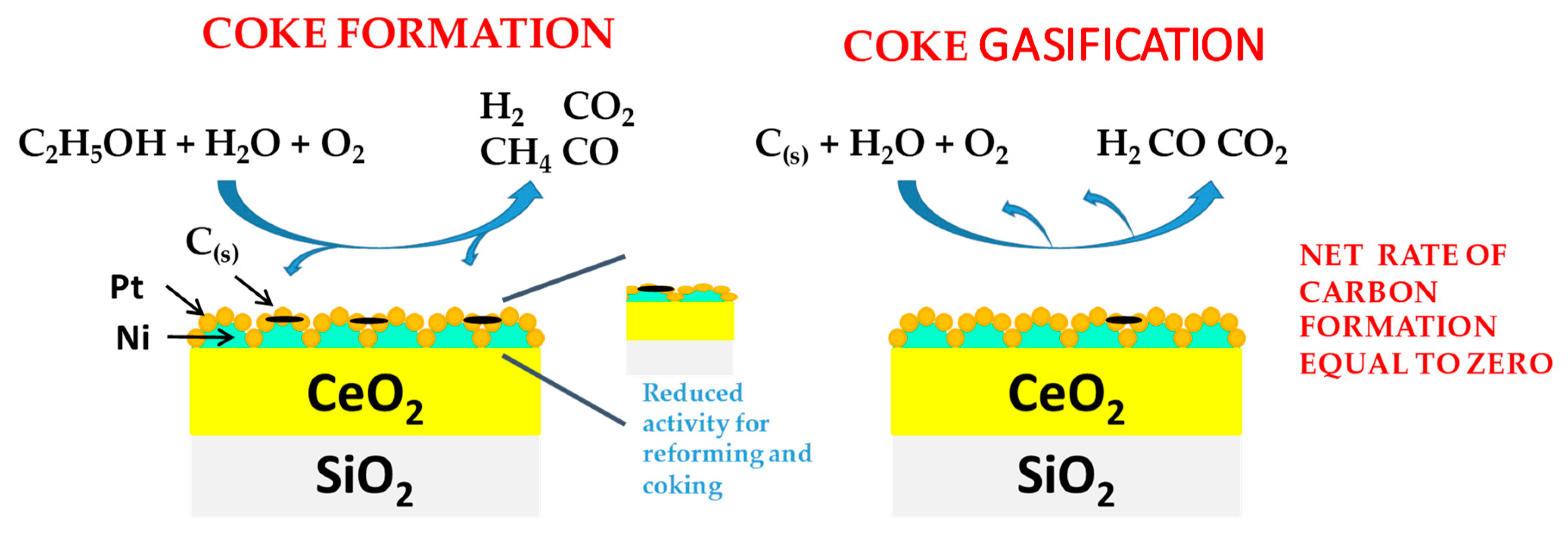
- Palma, V.; Ruocco, C.; Meloni, E.; Ricca, A. Influence of Catalytic Formulation and Operative Conditions on Coke Deposition over CeO2-SiO2 Based Catalysts for Ethanol Reforming. Energies 2017, 10, 1030. [Google Scholar] [CrossRef]
- Horng, R.-F.; Lai, M.-P.; Chiu, W.-C.; Huang, W.-C. Thermodynamic analysis of syngas production and carbon formation on oxidative steam reforming of butanol. Int. J. Hydrogen Energy 2016, 41, 889–896. [Google Scholar] [CrossRef]
- Valle, B.; Aramburu, B.; Olazar, M.; Bilbao, J.; Gayubo, A.G. Steam reforming of raw bio-oil over Ni/La2O3-αAl2O3: Influence of temperature on product yields and catalyst deactivation. Fuel 2018, 216, 463–474. [Google Scholar] [CrossRef]
- Coll, R.; Salvadó, J.; Farriol, X.; Montane, D. Steam reforming model compounds of biomass gasification tars: Conversion at different operating conditions and tendency towards coke formation. Fuel Process. Technol. 2001, 74, 19–31. [Google Scholar] [CrossRef]
- Larmier, K.; Chizallet, C.; Cadran, N.; Maury, S.; Abboud, J.; Lamic-Humblot, A.-F.; Marceau, E.; Lauron-Pernot, H. Mechanistic investigation of isopropanol conversion on alumina catalysts: Location of active sites for alkene/ether production. ACS Catal. 2015, 5, 4423–4437. [Google Scholar] [CrossRef]
- De Rezende, S.M.; Franchini, C.A.; Dieuzeide, M.L.; De Farias, A.M.D.; Amadeo, N.; Fraga, M. Glycerol steam reforming over layered double hydroxide-supported Pt catalysts. Chem. Eng. J. 2015, 272, 108–118. [Google Scholar] [CrossRef]
- Ochoa, A.; Aramburu, B.; Valle, B.; Resasco, D.E.; Bilbao, J.; Gayubo, A.G.; Castaño, P. Role of oxygenates and effect of operating conditions in the deactivation of a Ni supported catalyst during the steam reforming of bio-oil. Green Chem. 2017, 19, 4315–4333. [Google Scholar] [CrossRef]
- Sánchez, N.; Encinar, J.M.; Nogales, S.; González, J.F. Lanthanum Effect on Ni/Al2O3 as a Catalyst Applied in Steam Reforming of Glycerol for Hydrogen Production. Processes 2019, 7, 449. [Google Scholar] [CrossRef]
- Harun, N.; Abidin, S.Z.; Osazuwa, O.; Taufiq-Yap, Y.H.; Azizan, M.T. Hydrogen production from glycerol dry reforming over Ag-promoted Ni/Al2O3. Int. J. Hydrogen Energy 2019, 44, 213–225. [Google Scholar] [CrossRef]
- Gharahshiran, V.S.; Yousefpour, M.; Amini, V. A comparative study of zirconia and yttria promoted mesoporous carbon-nickel-cobalt catalysts in steam reforming of ethanol for hydrogen production. Mol. Catal. 2020, 484, 110767. [Google Scholar] [CrossRef]
- Ramesh, S.; Yang, E.-H.; Jung, J.-S.; Moon, D.J. Copper decorated perovskite an efficient catalyst for low temperature hydrogen production by steam reforming of glycerol. Int. J. Hydrogen Energy 2015, 40, 11428–11435. [Google Scholar] [CrossRef]
- Shao, J.; Zeng, G.; Li, Y. Effect of Zn substitution to a LaNiO3−δ perovskite structured catalyst in ethanol steam reforming. Int. J. Hydrogen Energy 2017, 42, 17362–17375. [Google Scholar] [CrossRef]
- Sun, C.; Zheng, Z.; Wang, S.; Li, X.; Wu, X.; An, X.; Xie, X. Yolk-shell structured Pt-CeO2 @Ni-SiO2 as an efficient catalyst for enhanced hydrogen production from ethanol steam reforming. Ceram. Int. 2018, 44, 1438–1442. [Google Scholar] [CrossRef]
- He, S.; Mei, Z.; Liu, N.; Zhang, L.; Lu, J.; Li, X.; Wang, J.; He, D.; Luo, Y. Ni/SBA-15 catalysts for hydrogen production by ethanol steam reforming: Effect of nickel precursor. Int. J. Hydrogen Energy 2017, 42, 14429–14438. [Google Scholar] [CrossRef]
- Ruocco, C.; Palma, V.; Ricca, A. Hydrogen production by oxidative reforming of ethanol in a fluidized bed reactor using a Pt Ni/CeO2SiO2 catalyst. Int. J. Hydrogen Energy 2019, 44, 12661–12670. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, L.; Deng, J.; Zhang, J.; Han, B.; Wang, Y.; Li, Z.; Yu, H.; Cai, W.; Deng, Z. Embedded Ni catalysts in Ni-O-Ce solid solution for stable hydrogen production from ethanol steam reforming reaction. Fuel Process. Technol. 2019, 193, 94–101. [Google Scholar] [CrossRef]
- Ricca, A.; Palma, V.; Martino, M.; Meloni, E. Innovative catalyst design for methane steam reforming intensification. Fuel 2017, 198, 175–182. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Castaldo, F.; Ricca, A.; Boettge, D. Ethanol steam reforming over bimetallic coated ceramic foams: Effect of reactor configuration and catalytic support. Int. J. Hydrogen Energy 2015, 40, 12650–12662. [Google Scholar] [CrossRef]
- Koc, S.; Avci, A.K. Reforming of glycerol to hydrogen over Ni-based catalysts in a microchannel reactor. Fuel Process. Technol. 2017, 156, 357–365. [Google Scholar] [CrossRef]
- Montero, C.; Ochoa, A.; Castaño, P.; Bilbao, J.; Gayubo, A.G. Monitoring Ni 0 and coke evolution during the deactivation of a Ni/La2O3–αAl2O3 catalyst in ethanol steam reforming in a fluidized bed. J. Catal. 2015, 331, 181–192. [Google Scholar] [CrossRef]
- Mondal, T.; Pant, K.K.; Dalai, A.K. Catalytic oxidative steam reforming of bio-ethanol for hydrogen production over Rh promoted Ni/CeO2–ZrO2 catalyst. Int. J. Hydrogen Energy 2015, 40, 2529–2544. [Google Scholar] [CrossRef]
- Li, M.-R.; Wang, G.-C. The mechanism of ethanol steam reforming on the Co0 and Co2+ sites: A DFT study. J. Catal. 2018, 365, 391–404. [Google Scholar] [CrossRef]
- Grewal, T.P.K.; Roy, S. Modeling the effect of coke deposition in a heat integrated ethanol reformer. Int. J. Hydrogen Energy 2016, 41, 19863–19880. [Google Scholar] [CrossRef]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous catalyst deactivation and regeneration: A review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Trevisanut, C.; Mari, M.; Millet, J.-M.M.; Cavani, F. Chemical-loop reforming of ethanol over metal ferrites: An analysis of structural features affecting reactivity. Int. J. Hydrogen Energy 2015, 40, 5264–5271. [Google Scholar] [CrossRef]
- Sharma, Y.C.; Kumar, A.; Prasad, R.; Upadhyay, S.N. Ethanol steam reforming for hydrogen production: Latest and effective catalyst modification strategies to minimize carbonaceous deactivation. Renew. Sustain. Energy Rev. 2017, 74, 89–103. [Google Scholar] [CrossRef]
- Li, D.; Li, X.; Gong, J. Catalytic reforming of oxygenates: State of the art and future prospects. Chem. Rev. 2016, 116, 11529–11653. [Google Scholar] [CrossRef] [PubMed]
- Lazar, M.D.; Senila, L.; Dan, M.; Mihet, M. Chapter 10—Crude bioethanol reforming process: The advantage of a biosource exploitation. In Ethanol; Basile, A., Iulianelli, A., Dalena, F., Veziroğlu, T.N., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 257–288. [Google Scholar]
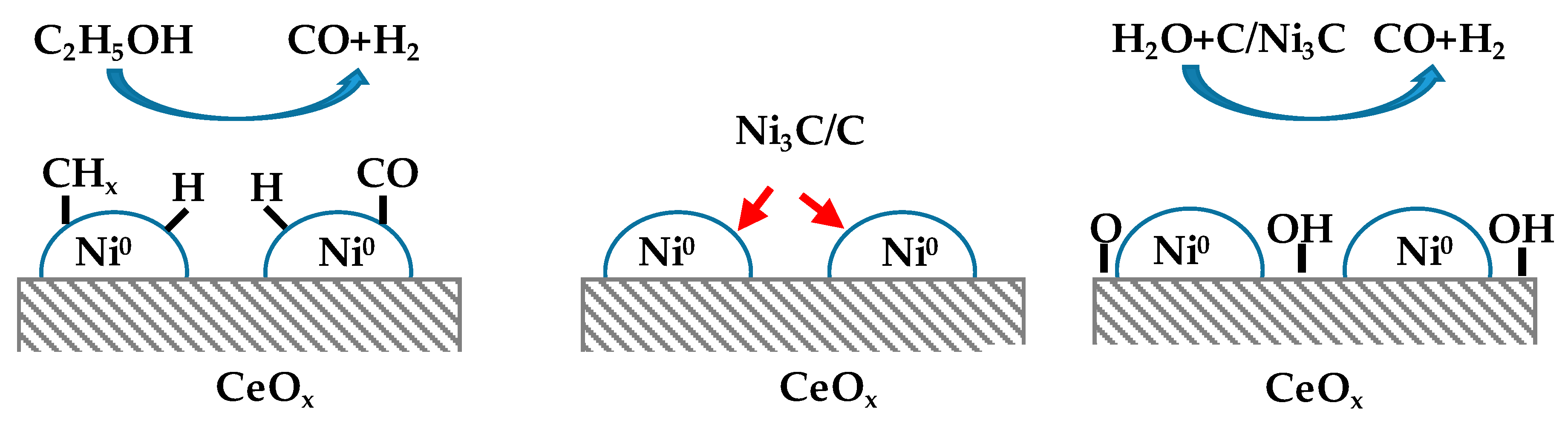
- Liu, Z.; Duchoň, T.; Wang, H.; Peterson, E.W.; Zhou, Y.; Luo, S.; Zhou, J.; Matolin, V.; Stacchiola, D.J.; Rodríguez, J.A.; et al. Mechanistic Insights of Ethanol Steam Reforming over Ni–CeOx(111): The Importance of Hydroxyl Groups for Suppressing Coke Formation. J. Phys. Chem. C 2015, 119, 18248–18256. [Google Scholar] [CrossRef]
- Gonçalves, A.A.S.; Faustino, P.B.; Assaf, J.M.; Jaroniec, M. One-Pot Synthesis of Mesoporous Ni–Ti–Al Ternary Oxides: Highly Active and Selective Catalysts for Steam Reforming of Ethanol. ACS Appl. Mater. Interfaces 2017, 9, 6079–6092. [Google Scholar] [CrossRef]
- Osorio-Vargas, P.; Flores-González, N.A.; Navarro, R.M.; Fierro, J.L.; Campos, C.H.; Reyes, P. Improved stability of Ni/Al2O3 catalysts by effect of promoters (La2O3, CeO2) for ethanol steam-reforming reaction. Catal. Today 2016, 259, 27–38. [Google Scholar] [CrossRef]
- Jo, S.W.; Kwak, B.S.; Kim, K.M.; Do, J.Y.; Park, N.-K.; Lee, T.J.; Lee, S.-T.; Kang, M. Reasonable harmony of Ni and Mn in core@shell-structured NiMn@SiO2 catalysts prepared for hydrogen production from ethanol steam reforming. Chem. Eng. J. 2016, 288, 858–868. [Google Scholar] [CrossRef]
- Chen, M.; Wang, C.; Wang, Y.; Tang, Z.; Yang, Z.; Zhang, H.; Wang, J. Hydrogen production from ethanol steam reforming: Effect of Ce content on catalytic performance of Co/Sepiolite catalyst. Fuel 2019, 247, 344–355. [Google Scholar] [CrossRef]
- Passos, A.R.; Martins, L.; Pulcinelli, S.H.; Santilli, C.; Briois, V. Correlation of Sol-Gel Alumina-Supported Cobalt Catalyst Processing to Cobalt Speciation, Ethanol Steam Reforming Activity, and Stability. ChemCatChem 2017, 9, 3918–3929. [Google Scholar] [CrossRef]
- Spallina, V.; Matturro, G.; Ruocco, C.; Meloni, E.; Palma, V.; Fernandez, E.; Melendez, J.; Tanaka, A.P.; Sole, J.V.; Annaland, M.V.S.; et al. Direct route from ethanol to pure hydrogen through autothermal reforming in a membrane reactor: Experimental demonstration, reactor modelling and design. Energy 2018, 143, 666–681. [Google Scholar] [CrossRef]
- Muñoz, M.; Moreno, S.; Molina, R. Oxidative steam reforming of ethanol (OSRE) over stable NiCo–MgAl catalysts by microwave or sonication assisted coprecipitation. Int. J. Hydrogen Energy 2017, 42, 12284–12294. [Google Scholar] [CrossRef]
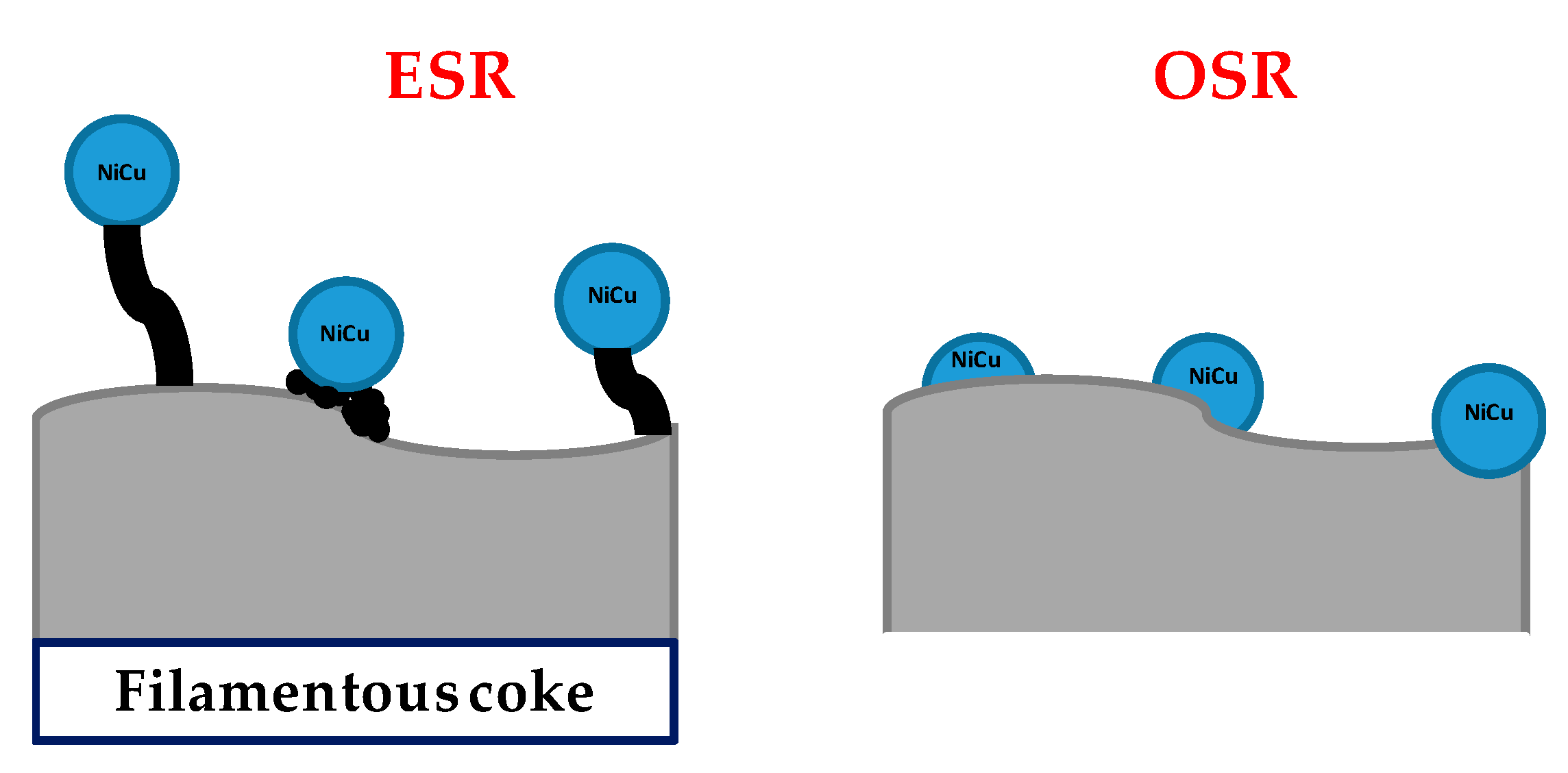
- Passos, A.R.; Pulcinelli, S.H.; Santilli, C.V.; Briois, V. Operando monitoring of metal sites and coke evolution during non-oxidative and oxidative ethanol steam reforming over Ni and NiCu ex-hydrotalcite catalysts. Catal. Today 2019, 336, 122–130. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Meloni, E.; Ricca, A. Oxidative steam reforming of ethanol on mesoporous silica supported PtNi/CeO2 catalysts. Int. J. Hydrogen Energy 2017, 42, 1598–1608. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Meloni, E.; Ricca, A. Oxidative reforming of ethanol over CeO2 -SiO2 based catalysts in a fluidized bed reactor. Chem. Eng. Process. Process. Intensif. 2018, 124, 319–327. [Google Scholar] [CrossRef]
- Yu, J.; Odriozola, J.; Reina, T.R. Dry reforming of ethanol and glycerol: Mini-review. Catalsts 2019, 9, 1015. [Google Scholar] [CrossRef]
- Zhao, S.; Cai, W.; Li, Y.; Yu, H.; Zhang, S.; Cui, L. Syngas production from ethanol dry reforming over Rh/CeO2 catalyst. J. Saudi Chem. Soc. 2018, 22, 58–65. [Google Scholar] [CrossRef]
- Nozawa, T.; Yoshida, A.; Hikichi, S.; Naito, S. Effects of Re addition upon aqueous phase reforming of ethanol over TiO2 supported Rh and Ir catalysts. Int. J. Hydrogen Energy 2015, 40, 4129–4140. [Google Scholar] [CrossRef]
- Xiong, H.; DeLaRiva, A.; Datye, A.K.; Wang, Y. Low-temperature aqueous-phase reforming of ethanol on bimetallic PdZn catalysts. Catal. Sci. Technol. 2015, 5, 254–263. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, L.; Tan, Q.; Yang, F.; Faria, J.; Resasco, D.E. Synergistic bimetallic Ru-Pt catalysts for the low-temperature aqueous phase reforming of ethanol. AIChE J. 2018, 65, 151–160. [Google Scholar] [CrossRef]
- Gogoi, P.; Nagpure, A.S.; Kandasamy, P.; Satyanarayana, C.V.V.; Raja, T.; Kandasamy, P.; Thirumalaiswamy, R. Insights into the catalytic activity of Ru/NaY catalysts for efficient H2 production through aqueous phase reforming. Sustain. Energy Fuels 2020, 4, 678–690. [Google Scholar] [CrossRef]
- Mulewa, W.; Tahir, M.; Amin, N.A.S. MMT-supported Ni/TiO2 nanocomposite for low temperature ethanol steam reforming toward hydrogen production. Chem. Eng. J. 2017, 326, 956–969. [Google Scholar] [CrossRef]
- Nejat, T.; Jalalinezhad, P.; Hormozi, F.; Bahrami, Z. Hydrogen production from steam reforming of ethanol over Ni-Co bimetallic catalysts and MCM-41 as support. J. Taiwan Inst. Chem. Eng. 2019, 97, 216–226. [Google Scholar] [CrossRef]
- Di Michele, A.; Dell’Angelo, A.; Tripodi, A.; Bahadori, E.; Sànchez, F.; Motta, D.; Dimitratos, N.; Rossetti, I.; Ramis, G.; Sanchez, F. Steam reforming of ethanol over Ni/MgAl2O4 catalysts. Int. J. Hydrogen Energy 2019, 44, 952–964. [Google Scholar] [CrossRef]
- Carvalho, F.L.; Asencios, Y.J.O.; Bellido, J.D.; Assaf, J.M. Bio-ethanol steam reforming for hydrogen production over Co3O4/CeO2 catalysts synthesized by one-step polymerization method. Fuel Process. Technol. 2016, 142, 182–191. [Google Scholar] [CrossRef]
- Konsolakis, M.; Ioakimidis, Z.; Kraia, T.; Marnellos, G.E. Hydrogen Production by Ethanol Steam Reforming (ESR) over CeO2 Supported Transition Metal (Fe, Co, Ni, Cu) Catalysts: Insight into the Structure-Activity Relationship. Catalsts 2016, 6, 39. [Google Scholar] [CrossRef]
- Greluk, M.; Słowik, G.; Rotko, M.; Machocki, A. Steam reforming and oxidative steam reforming of ethanol over PtKCo/CeO2 catalyst. Fuel 2016, 183, 518–530. [Google Scholar] [CrossRef]
- Riani, P.; Garbarino, G.; Canepa, F.; Busca, G. Cobalt nanoparticles mechanically deposited on α-Al2O3: A competitive catalyst for the production of hydrogen through ethanol steam reforming. J. Chem. Technol. Biotechnol. 2019, 94, 538–546. [Google Scholar] [CrossRef]
- Mhadmhan, S.; Natewong, P.; Prasongthum, N.; Samart, C.; Reubroycharoen, P. Investigation of Ni/SiO2 fiber catalysts prepared by different methods on hydrogen production from ethanol steam reforming. Catalysts 2018, 8, 319. [Google Scholar] [CrossRef]
- Rodrigues, T.S.; De Moura, A.B.; Silva, F.; Candido, E.G.; Da Silva, A.G.; De Oliveira, D.C.; Quiroz, J.; Camargo, P.H.; Bergamaschi, V.S.; Ferreira, J.C.; et al. Ni supported Ce0.9Sm0.1O2-δ nanowires: An efficient catalyst for ethanol steam reforming for hydrogen production. Fuel 2019, 237, 1244–1253. [Google Scholar] [CrossRef]
- Roy, B.; Leclerc, C. Study of preparation method and oxidization/reduction effect on the performance of nickel-cerium oxide catalysts for aqueous-phase reforming of ethanol. J. Power Sources 2015, 299, 114–124. [Google Scholar] [CrossRef]
- Sohn, H.; Ozkan, U.S. Cobalt-Based Catalysts for Ethanol Steam Reforming: An Overview. Energy Fuels 2016, 30, 5309–5322. [Google Scholar] [CrossRef]
- Yoshida, H.; Yamaoka, R.; Arai, M. Stable Hydrogen Production from Ethanol through Steam Reforming Reaction over Nickel-Containing Smectite-Derived Catalyst. Int. J. Mol. Sci. 2014, 16, 350–362. [Google Scholar] [CrossRef]
- Ogo, S.; Sekine, Y. Recent progress in ethanol steam reforming using non-noble transition metal catalysts: A review. Fuel Process. Technol. 2020, 199, 106238. [Google Scholar] [CrossRef]
- Hou, T.; Yu, B.; Zhang, S.; Xu, T.; Wang, D.; Cai, W. Hydrogen production from ethanol steam reforming over Rh/CeO2 catalyst. Catal. Commun. 2015, 58, 137–140. [Google Scholar] [CrossRef]
- Hou, T.; Lei, Y.; Zhang, S.; Zhang, J.; Cai, W. Ethanol dry reforming for syngas production over Ir/CeO2 catalyst. J. Rare Earths 2015, 33, 42–45. [Google Scholar] [CrossRef]
- Moraes, T.S.; Neto, R.C.R.; Ribeiro, M.; Mattos, L.V.; Kourtelesis, M.; Ladas, S.; Verykios, X.; Noronha, F.B. The study of the performance of PtNi/CeO2–nanocube catalysts for low temperature steam reforming of ethanol. Catal. Today 2015, 242, 35–49. [Google Scholar] [CrossRef]
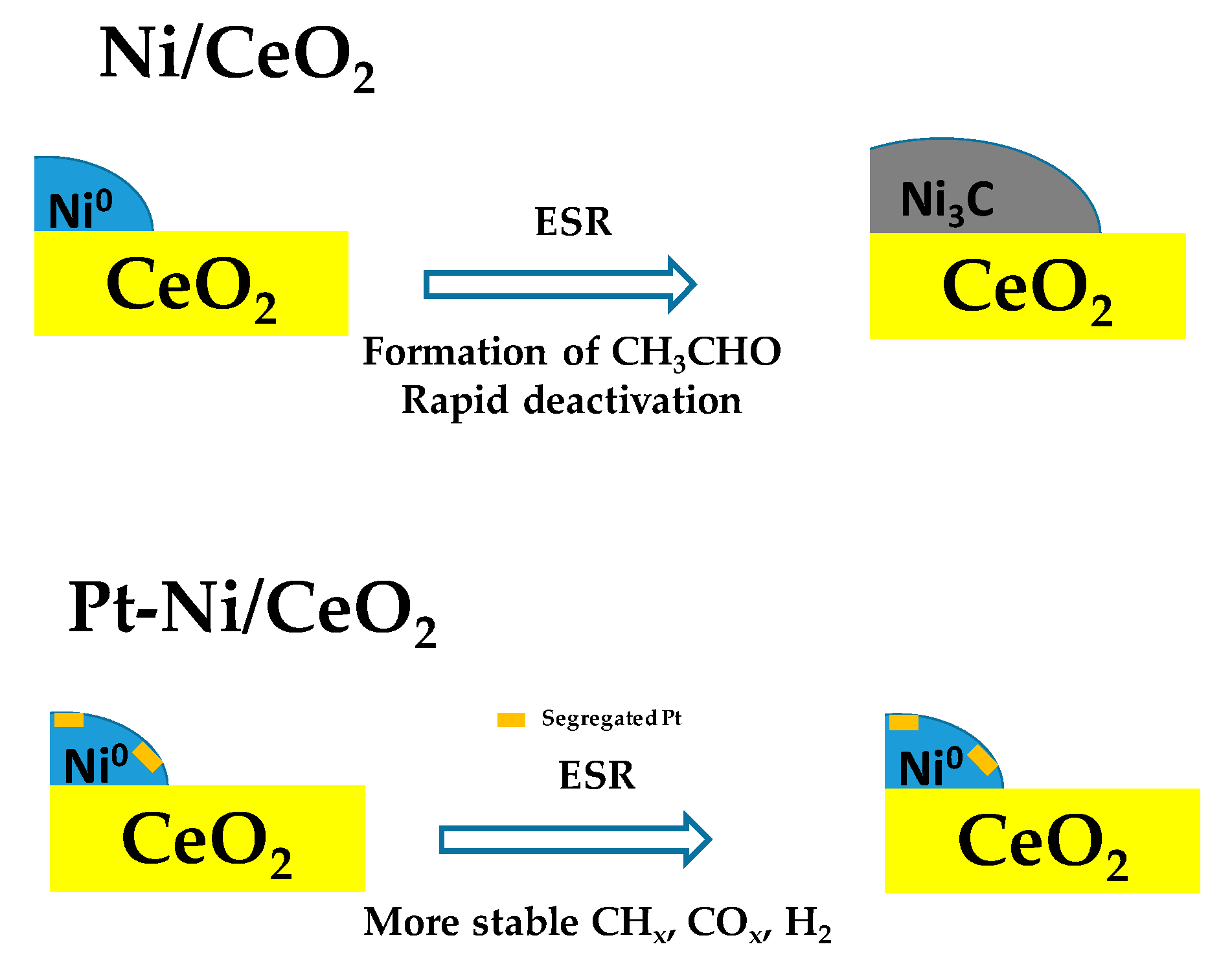
- Moraes, T.S.; Neto, R.C.R.; Ribeiro, M.; Mattos, L.V.; Kourtelesis, M.; Ladas, S.; Verykios, X.; Noronha, F.B. Ethanol conversion at low temperature over CeO2—Supported Ni-based catalysts. Effect of Pt addition to Ni catalyst. Appl. Catal. B Environ. 2016, 181, 754–768. [Google Scholar] [CrossRef]
- Cifuentes, B.; Valero, M.F.; Conesa, J.A.; Cobo, M. Hydrogen Production by Steam Reforming of Ethanol on Rh-Pt Catalysts: Influence of CeO2, ZrO2, and La2O3 as Supports. Catalsts 2015, 5, 1872–1896. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Meloni, E.; Ricca, A. Renewable hydrogen from ethanol reforming over CeO2-SiO2 based catalysts. Catalsts 2017, 7, 226. [Google Scholar] [CrossRef]
- Campos, C.H.; Pecchi, G.; Fierro, J.L.G.; Osorio-Vargas, P. Enhanced bimetallic Rh-Ni supported catalysts on alumina doped with mixed lanthanum-cerium oxides for ethanol steam reforming. Mol. Catal. 2019, 469, 87–97. [Google Scholar] [CrossRef]
- Sanchez, N.; Ruiz, R.; Cifuentes, B.; Cobo, M. Hydrogen from glucose: A combined study of glucose fermentation, bioethanol purification, and catalytic steam reforming. Int. J. Hydrogen Energy 2016, 41, 5640–5651. [Google Scholar] [CrossRef]
- Bilal, M.; Jackson, S.D. Ethanol steam reforming over Pt/Al2O3 and Rh/Al2O3 catalysts: The effect of impurities on selectivity and catalyst deactivation. Appl. Catal. A Gen. 2017, 529, 98–107. [Google Scholar] [CrossRef]
- Iulianelli, A.; Palma, V.; Bagnato, G.; Ruocco, C.; Huang, Y.; Veziroglu, N.T.; Basile, A. From bioethanol exploitation to high grade hydrogen generation: Steam reforming promoted by a Co-Pt catalyst in a Pd-based membrane reactor. Renew. Energy 2018, 119, 834–843. [Google Scholar] [CrossRef]
- Nichele, V.; Signoretto, M.; Pinna, F.; Ghedini, E.; Compagnoni, M.; Rossetti, I.; Cruciani, G.; Di Michele, A. Bimetallic Ni–Cu catalysts for the low-temperature ethanol steam reforming: Importance of metal–support interactions. Catal. Lett. 2014, 145, 549–558. [Google Scholar] [CrossRef]
- Rodriguez-Gomez, A.; Caballero, A. Bimetallic Ni-Co/SBA-15 catalysts for reforming of ethanol: How cobalt modifies the nickel metal phase and product distribution. Mol. Catal. 2018, 449, 122–130. [Google Scholar] [CrossRef]
- Chen, L.-C.; Cheng, H.; Chiang, C.-W.; Lin, S.D. Sustainable hydrogen production by ethanol steam reforming using a partially reduced copper-nickel oxide catalyst. ChemSusChem 2015, 8, 1787–1793. [Google Scholar] [CrossRef]
- Braga, A.H.; Ribeiro, M.; Noronha, F.B.; Galante, D.; Bueno, J.M.C.; Santos, J.B.O. Effects of Co addition to supported Ni catalysts on hydrogen production from oxidative steam reforming of ethanol. Energy Fuels 2018, 32, 12814–12825. [Google Scholar] [CrossRef]
- Baruah, R.; Dixit, M.; Parejiya, A.; Basarkar, P.; Bhargav, A.; Sharma, S. Oxidative steam reforming of ethanol on rhodium catalyst—I: Spatially resolved steady-state experiments and microkinetic modeling. Int. J. Hydrogen Energy 2017, 42, 10184–10198. [Google Scholar] [CrossRef]
- Weng, S.-F.; Hsieh, H.-C.; Lee, C.-S. Hydrogen production from oxidative steam reforming of ethanol on nickel-substituted pyrochlore phase catalysts. Int. J. Hydrogen Energy 2017, 42, 2849–2860. [Google Scholar] [CrossRef]
- Hsieh, H.-C.; Chang, Y.-C.; Tsai, P.-W.; Lin, Y.-Y.; Chuang, Y.-C.; Sheu, H.-S.; Lee, C.-S. Metal substituted pyrochlore phase LixLa2−xCe1.8Ru0.2O7−δ (x = 0.0–0.6) as an effective catalyst for oxidative and auto-thermal steam reforming of ethanol. Catal. Sci. Technol. 2019, 9, 1406–1419. [Google Scholar] [CrossRef]
- Cao, D.; Zeng, F.; Zhao, Z.; Cai, W.; Li, Y.; Yu, H.; Zhang, S.; Qu, F. Cu based catalysts for syngas production from ethanol dry reforming: Effect of oxide supports. Fuel 2018, 219, 406–416. [Google Scholar] [CrossRef]
- Cao, N.; Cai, W.; Li, Y.; Li, C.; Yu, H.; Zhang, S.; Qu, F. Syngas Production from Ethanol Dry Reforming over Cu/Ce0.8Zr0.2O2 Catalyst. Catal. Lett. 2017, 147, 2929–2939. [Google Scholar] [CrossRef]
- Chen, Q.; Cai, W.; Liu, Y.; Zhang, S.; Li, Y.; Huang, D.; Wang, T.; Li, Y. Synthesis of Cu-Ce0.8Zr0.2O2 catalyst by ball milling for CO2 reforming of ethanol. J. Saudi Chem. Soc. 2019, 23, 111–117. [Google Scholar] [CrossRef]
- Dang, C.; Wu, S.; Yang, G.; Cao, Y.; Wang, H.; Peng, F.; Yu, H. Syngas production by dry reforming of the mixture of glycerol and ethanol with CaCO3. J. Energy Chem. 2020, 43, 90–97. [Google Scholar] [CrossRef]
- Da Costa-Serra, J.F.; Chica, A. Catalysts based on Co-Birnessite and Co-Todorokite for the efficient production of hydrogen by ethanol steam reforming. Int. J. Hydrogen Energy 2018, 43, 16859–16865. [Google Scholar] [CrossRef]
- Wei, Y.; Cai, W.; Deng, S.; Li, Z.; Yu, H.; Zhang, S.; Yu, Z.; Cui, L.; Qu, F. Efficient syngas production via dry reforming of renewable ethanol over Ni/KIT-6 nanocatalysts. Renew. Energy 2020, 145, 1507–1516. [Google Scholar] [CrossRef]
- Chai, R.; Li, Y.; Zhang, Q.; Zhao, G.; Liu, Y.; Lu, Y. Monolithic Ni–MOx/Ni-foam (M = Al, Zr or Y) catalysts with enhanced heat/mass transfer for energy-efficient catalytic oxy-methane reforming. Catal. Commun. 2015, 70, 1–5. [Google Scholar] [CrossRef]
- Moraes, T.S.; Borges, L.E.P.; Farrauto, R.; Noronha, F.B. Steam reforming of ethanol on Rh/SiCeO2 washcoated monolith catalyst: Stable catalyst performance. Int. J. Hydrogen Energy 2018, 43, 115–126. [Google Scholar] [CrossRef]
- Gaudillere, C.; González, J.J.; Chica, A.; Serra, J.M. YSZ monoliths promoted with Co as catalysts for the production of H2 by steam reforming of ethanol. Appl. Catal. A Gen. 2017, 538, 165–173. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Ricca, A. Ceramic foams coated with PtNi/CeO2ZrO2 for bioethanol steam reforming. Int. J. Hydrogen Energy 2016, 41, 11526–11536. [Google Scholar] [CrossRef]
- Turczyniak, S.; Greluk, M.; Słowik, G.; Gac, W.; Zafeiratos, S.; Machocki, A. Surface state and catalytic performance of ceria-supported cobalt catalysts in the steam reforming of ethanol. ChemCatChem 2017, 9, 782–797. [Google Scholar] [CrossRef]
- Greluk, M.; Rybak, P.; Słowik, G.; Rotko, M.; Machocki, A. Comparative study on steam and oxidative steam reforming of ethanol over 2KCo/ZrO2 catalyst. Catal. Today 2015, 242, 50–59. [Google Scholar] [CrossRef]
- Greluk, M.; Rotko, M.; Turczyniak-Surdacka, S. Enhanced catalytic performance of La2O3 promoted Co/CeO2 and Ni/CeO2 catalysts for effective hydrogen production by ethanol steam reforming. Renew. Energy 2020, 155, 378–395. [Google Scholar] [CrossRef]
- Cerdá-Moreno, C.; Da Costa-Serra, J.F.; Chica, A. Co and La supported on Zn-Hydrotalcite-derived material as efficient catalyst for ethanol steam reforming. Int. J. Hydrogen Energy 2019, 44, 12685–12692. [Google Scholar] [CrossRef]
- Kim, D.; Kwak, B.S.; Park, N.-K.; Han, G.B.; Kang, M. Dynamic hydrogen production from ethanol steam-reforming reaction on NixMoy/SBA-15 catalytic system. Int. J. Energy Res. 2015, 39, 279–292. [Google Scholar] [CrossRef]
- Kwak, B.S.; Lee, G.; Park, S.-M.; Kang, M. Effect of MnOx in the catalytic stabilization of Co2MnO4 spinel during the ethanol steam reforming reaction. Appl. Catal. A Gen. 2015, 503, 165–175. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, W.; Yao, S.; Johnson-Peck, A.C.; Zhao, F.; Michorczyk, P.; Kubacka, A.; Stach, E.A.; Fernández-García, M.; Senanayake, S.D.; et al. Superior performance of Ni–W–Ce mixed-metal oxide catalysts for ethanol steam reforming: Synergistic effects of W- and Ni-dopants. J. Catal. 2015, 321, 90–99. [Google Scholar] [CrossRef]
- Li, D.; Zeng, L.; Li, X.; Wang, X.; Ma, H.; Assabumrungrat, S.; Gong, J. Ceria-promoted Ni/SBA-15 catalysts for ethanol steam reforming with enhanced activity and resistance to deactivation. Appl. Catal. B Environ. 2015, 176, 532–541. [Google Scholar] [CrossRef]
- Du, Y.-L.; Wu, X.; Cheng, Q.; Huang, Y.-L.; Huang, W. Development of Ni-Based Catalysts Derived from Hydrotalcite-Like Compounds Precursors for Synthesis Gas Production via Methane or Ethanol Reforming. Catalysts 2017, 7, 70. [Google Scholar] [CrossRef]
- Marinho, A.L.A.; Rabelo-Neto, R.C.; Noronha, F.B.; Mattos, L.V. Steam reforming of ethanol over Ni-based catalysts obtained from LaNiO3 and LaNiO3/CeSiO2 perovskite-type oxides for the production of hydrogen. Appl. Catal. A Gen. 2016, 520, 53–64. [Google Scholar] [CrossRef]
- Dan, M.; Senila, L.; Roman, M.; Mihet, M.; Lazar, M.D. From wood wastes to hydrogen—Preparation and catalytic steam reforming of crude bio-ethanol obtained from fir wood. Renew. Energy 2015, 74, 27–36. [Google Scholar] [CrossRef]
- He, S.; He, S.; Zhang, L.; Li, X.; Wang, J.; He, D.; Lu, J.; Luo, Y. Hydrogen production by ethanol steam reforming over Ni/SBA-15 mesoporous catalysts: Effect of Au addition. Catal. Today 2015, 258, 162–168. [Google Scholar] [CrossRef]
- Mondal, T.; Pant, K.K.; Dalai, A.K. Oxidative and non-oxidative steam reforming of crude bio-ethanol for hydrogen production over Rh promoted Ni/CeO2-ZrO2 catalyst. Appl. Catal. A Gen. 2015, 499, 19–31. [Google Scholar] [CrossRef]
- Bahari, M.B.; Phuc, N.H.H.; Abdullah, B.; Alenazey, F.; Vo, D.-V.N. Ethanol dry reforming for syngas production over Ce-promoted Ni/Al2O3 catalyst. J. Environ. Chem. Eng. 2016, 4, 4830–4838. [Google Scholar] [CrossRef]
- Bahari, M.B.; Phuc, N.H.H.; Alenazey, F.; Vu, K.B.; Ainirazali, N.; Vo, D.-V.N. Catalytic performance of La-Ni/Al2O3 catalyst for CO2 reforming of ethanol. Catal. Today 2017, 291, 67–75. [Google Scholar] [CrossRef]
- Fayaz, F.; Danh, H.T.; Nguyen-Huy, C.; Vu, K.B.; Abdullah, B.; Vo, D.-V.N. Promotional Effect of Ce-dopant on Al2O3-supported Co Catalysts for Syngas Production via CO2 Reforming of Ethanol. Procedia Eng. 2016, 148, 646–653. [Google Scholar] [CrossRef]
- Fayaz, F.; Bach, L.-G.; Bahari, M.B.; Nguyen, D.T.; Vu, K.B.; Kanthasamy, R.; Samart, C.; Nguyen-Huy, C.; Vo, D.-V.N. Stability evaluation of ethanol dry reforming on Lanthania-doped cobalt-based catalysts for hydrogen-rich syngas generation. Int. J. Energy Res. 2018, 43, 405–416. [Google Scholar] [CrossRef]
- Shafiqah, M.-N.N.; Nguyen, T.D.; Jun, L.N.; Bahari, M.B.; Phuong, P.T.; Abdullah, B.; Vo, D.-V.N. Production of Syngas from Ethanol CO2 Reforming on La-Doped Cu/Al2O3: Impact of Promoter Loading; AIP Publishing LLC: Melville, NY, USA, 2019; p. 020011. [Google Scholar]
- Słowik, G.; Greluk, M.; Rotko, M.; Machocki, A. Evolution of the structure of unpromoted and potassium-promoted ceria-supported nickel catalysts in the steam reforming of ethanol. Appl. Catal. B Environ. 2018, 221, 490–509. [Google Scholar] [CrossRef]
- Cifuentes, B.; Hernández, M.; Monsalve, S.; Cobo, M. Hydrogen production by steam reforming of ethanol on a RhPt/CeO2/SiO2 catalyst: Synergistic effect of the Si:Ce ratio on the catalyst performance. Appl. Catal. A Gen. 2016, 523, 283–293. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, L.; Zhu, J.; Han, B.; Zhao, L.; Yu, H.; Deng, Z.; Shi, W. Study on different CeO2 structure stability during ethanol steam reforming reaction over Ir/CeO2 nanocatalysts. Appl. Catal. A Gen. 2018, 564, 226–233. [Google Scholar] [CrossRef]
- Osorio-Vargas, P.; Campos, C.H.; Navarro, R.M.; Fierro, J.L.; Reyes, P. Rh/Al2O3–La2O3 catalysts promoted with CeO2 for ethanol steam reforming reaction. J. Mol. Catal. A Chem. 2015, 407, 169–181. [Google Scholar] [CrossRef]
- Gunduz, S.; Doğu, T. Hydrogen by steam reforming of ethanol over Co–Mg incorporated novel mesoporous alumina catalysts in tubular and microwave reactors. Appl. Catal. B Environ. 2015, 168, 497–508. [Google Scholar] [CrossRef]
- Zhao, L.; Han, T.; Wang, H.; Zhang, L.; Liu, Y. Ni-Co alloy catalyst from LaNi1−xCoxO3 perovskite supported on zirconia for steam reforming of ethanol. Appl. Catal. B Environ. 2016, 187, 19–29. [Google Scholar] [CrossRef]
- Bepari, S.; Basu, S.; Pradhan, N.C.; Dalai, A.K. Steam reforming of ethanol over cerium-promoted Ni-Mg-Al hydrotalcite catalysts. Catal. Today 2017, 291, 47–57. [Google Scholar] [CrossRef]
- Prasongthum, N.; Xiao, R.; Zhang, H.; Tsubaki, N.; Natewong, P.; Reubroycharoen, P. Highly active and stable Ni supported on CNTs-SiO2 fiber catalysts for steam reforming of ethanol. Fuel Process. Technol. 2017, 160, 185–195. [Google Scholar] [CrossRef]
- Dai, R.; Zheng, Z.; Sun, C.; Li, X.; Wang, S.; Wu, X.; An, X.; Xie, X. Pt nanoparticles encapsulated in a hollow zeolite microreactor as a highly active and stable catalyst for low-temperature ethanol steam reforming. Fuel 2018, 214, 88–97. [Google Scholar] [CrossRef]
- Dobosz, J.; Małecka, M.; Zawadzki, M. Hydrogen generation via ethanol steam reforming over Co/HAp catalysts. J. Energy Inst. 2018, 91, 411–423. [Google Scholar] [CrossRef]
- Chen, M.; Wang, Y.; Yang, Z.; Liang, T.; Liu, S.; Zhou, Z.; Li, X. Effect of Mg-modified mesoporous Ni/Attapulgite catalysts on catalytic performance and resistance to carbon deposition for ethanol steam reforming. Fuel 2018, 220, 32–46. [Google Scholar] [CrossRef]
- Xiao, Z.; Li, Y.; Hou, F.; Wu, C.; Pan, L.; Zou, J.; Wang, L.; Zhang, X.; Liu, G.; Li, G. Engineering oxygen vacancies and nickel dispersion on CeO2 by Pr doping for highly stable ethanol steam reforming. Appl. Catal. B Environ. 2019, 258, 117940. [Google Scholar] [CrossRef]
- Lee, J.H.; Do, J.Y.; Park, N.-K.; Ryu, H.-J.; Seo, M.W.; Kang, M. Hydrogen production on Pd0.01Zn0.29Mg0.7Al2O4 spinel catalyst by low temperature ethanol steam reforming reaction. J. Energy Inst. 2019, 92, 1064–1076. [Google Scholar] [CrossRef]
- Yang, P.; Li, N.; Teng, J.; Wu, J.; Ma, H. Effect of template on catalytic performance of La0.7Ce0.3Ni0.7Fe0.3O3 for ethanol steam reforming reaction. J. Rare Earths 2019, 37, 594–601. [Google Scholar] [CrossRef]
- Dai, R.; Zheng, Z.; Yan, W.; Lian, C.; Wu, X.; An, X.; Xie, X. Dragon fruit-like Pt-Cu@mSiO2 nanocomposite as an efficient catalyst for low-temperature ethanol steam reforming. Chem. Eng. J. 2020, 379, 122299. [Google Scholar] [CrossRef]
- Zhurka, M.D.; Lemonidou, A.A.; Kechagiopoulos, P.N. Elucidation of metal and support effects during ethanol steam reforming over Ni and Rh based catalysts supported on (CeO2)-ZrO2-La2O3. Catal. Today 2020. [Google Scholar] [CrossRef]
- Morales, M.; Segarra, M. Steam reforming and oxidative steam reforming of ethanol over La0.6Sr0.4CoO3− perovskite as catalyst precursor for hydrogen production. Appl. Catal. A Gen. 2015, 502, 305–311. [Google Scholar] [CrossRef]
- Fang, W.; Pirez, C.; Paul, S.; Jiménez-Ruiz, M.; Jobic, H.; Dumeignil, F.; Jalowiecki-Duhamel, L. Advanced functionalized Mg2AlNiXHZOY nano-oxyhydrides ex-hydrotalcites for hydrogen production from oxidative steam reforming of ethanol. Int. J. Hydrogen Energy 2016, 41, 15443–15452. [Google Scholar] [CrossRef]
- Muñoz, M.; Moreno, S.; Molina, R. Promoter effect of Ce and Pr on the catalytic stability of the Ni-Co system for the oxidative steam reforming of ethanol. Appl. Catal. A Gen. 2016, 526, 84–94. [Google Scholar] [CrossRef]
- Hsieh, H.-C.; Tsai, P.-W.; Chang, Y.-C.; Weng, S.-F.; Sheu, H.-S.; Chuang, Y.-C.; Lee, C.-S. Oxidative steam reforming of ethanol over MxLa2−xCe1.8Ru0.2O7−δ (M = Mg, Ca) catalysts: Effect of alkaline earth metal substitution and support on stability and activity. RSC Adv. 2019, 9, 39932–39944. [Google Scholar] [CrossRef]
- Bej, B.; Bepari, S.; Pradhan, N.C.; Neogi, S. Production of hydrogen by dry reforming of ethanol over alumina supported nano-NiO/SiO2 catalyst. Catal. Today 2017, 291, 58–66. [Google Scholar] [CrossRef]
- Samsudeen, K.; Ahmed, A.F.; Yahya, M.; Ahmed, A.; Anis, F. Effect of Calcination Temperature on Hydrogen Production via Ethanol Dry Reforming Over Ni/Al2O3 Catalyst. Int. J. Res. Sci. 2018, 4, 5–9. [Google Scholar] [CrossRef]
- Usman, M.; Daud, W.M.A.W. Recent advances in the methanol synthesis via methane reforming processes. RSC Adv. 2015, 5, 21945–21972. [Google Scholar] [CrossRef]
- Kothandaraman, J.; Kar, S.; Goeppert, A.; Sen, R.; Prakash, G.K.S. Advances in Homogeneous Catalysis for Low Temperature Methanol Reforming in the Context of the Methanol Economy. Top. Catal. 2018, 61, 542–559. [Google Scholar] [CrossRef]
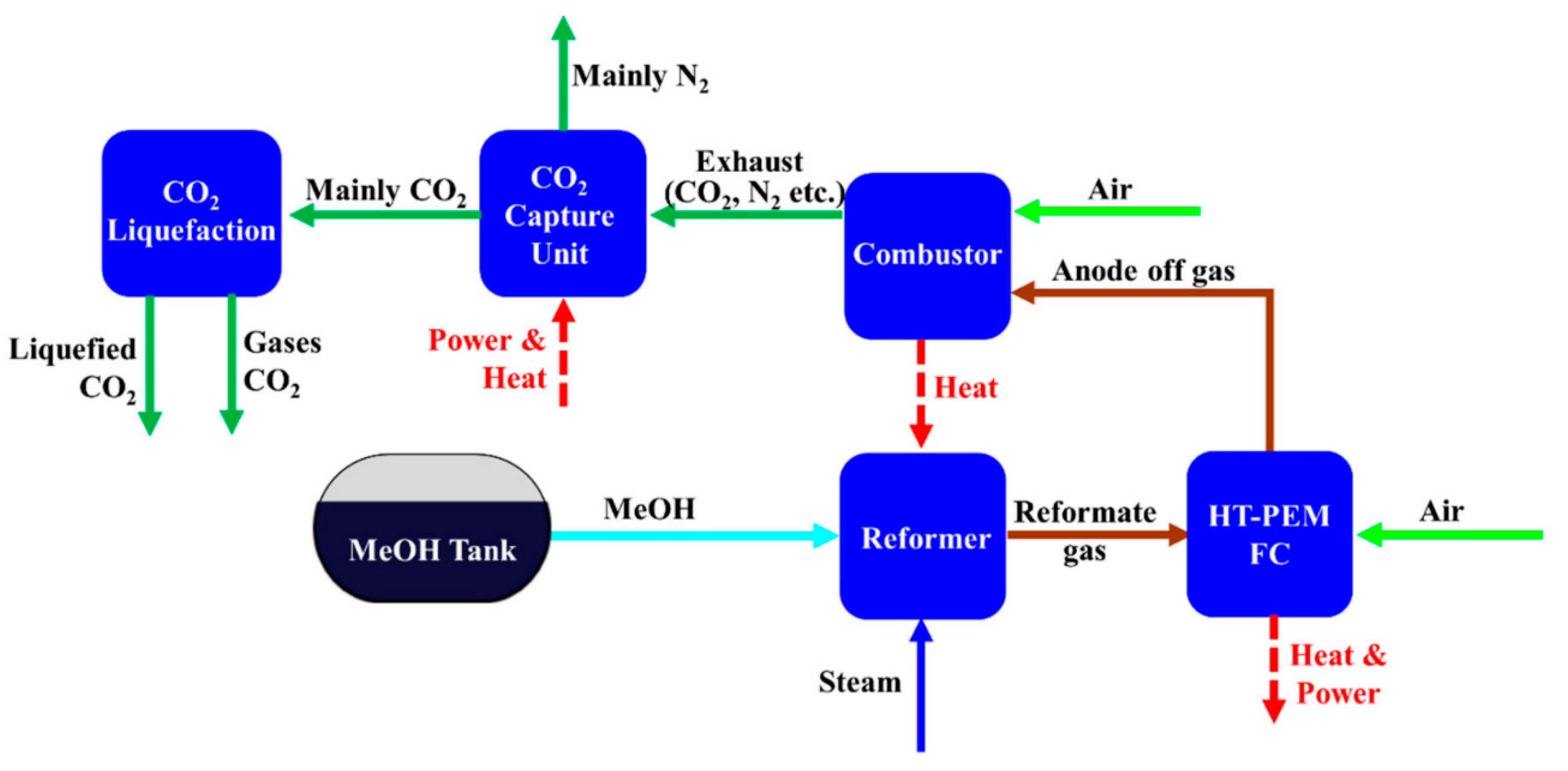
- Lee, H.; Jung, I.; Roh, G.; Na, Y.; Kang, H. Comparative Analysis of On-Board Methane and Methanol Reforming Systems Combined with HT-PEM Fuel Cell and CO2 Capture/Liquefaction System for Hydrogen Fueled Ship Application. Energies 2020, 13, 224. [Google Scholar] [CrossRef]
- Araya, S.S.; Liso, V.; Cui, X.; Li, N.; Zhu, J.; Sahlin, S.L.; Jensen, S.; Nielsen, M.; Kær, S. A Review of The Methanol Economy: The Fuel Cell Route. Energies 2020, 13, 596. [Google Scholar] [CrossRef]
- Iulianelli, A.; Ghasemzadeh, K.; Basile, A. Progress in Methanol Steam Reforming Modelling via Membrane Reactors Technology. Membranes 2018, 8, 65. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Martino, M.; Meloni, E.; Ricca, A. Bimetallic supported catalysts for hydrocarbons and alcohols reforming reactions. In Hydrogen Production, Separation and Purification for Energy; Institution of Engineering and Technology: London, UK, 2017; pp. 39–70. [Google Scholar]
- Xu, X.; Shuai, K.; Xu, B. Review on copper and palladium based catalysts for methanol steam reforming to produce hydrogen. Catalysts 2017, 7, 183. [Google Scholar] [CrossRef]
- Iulianelli, A.; Ribeirinha, P.; Mendes, A.; Basile, A. Methanol steam reforming for hydrogen generation via conventional and membrane reactors: A review. Renew. Sustain. Energy Rev. 2014, 29, 355–368. [Google Scholar] [CrossRef]
- Tonelli, F.; Gorriz, O.; Tarditi, A.M.; Cornaglia, L.; Arrúa, L.; Abello, M.C. Activity and stability of a CuO/CeO2 catalyst for methanol steam reforming. Int. J. Hydrogen Energy 2015, 40, 13379–13387. [Google Scholar] [CrossRef]
- Das, D.; Llorca, J.; Domínguez, M.; Colussi, S.; Trovarelli, A.; Gayen, A. Methanol steam reforming behavior of copper impregnated over CeO2–ZrO2 derived from a surfactant assisted coprecipitation route. Int. J. Hydrogen Energy 2015, 40, 10463–10479. [Google Scholar] [CrossRef]
- Deshmane, V.G.; Abrokwah, R.Y.; Kuila, D. Synthesis of stable Cu-MCM-41 nanocatalysts for H2 production with high selectivity via steam reforming of methanol. Int. J. Hydrogen Energy 2015, 40, 10439–10452. [Google Scholar] [CrossRef]
- Xu, T.; Zou, J.; Tao, W.; Zhang, S.; Cui, L.; Zeng, F.; Wang, D.; Cai, W. Co-nanocasting synthesis of Cu based composite oxide and its promoted catalytic activity for methanol steam reforming. Fuel 2016, 183, 238–244. [Google Scholar] [CrossRef]
- Thattarathody, R.; Sheintuch, M. Kinetics and dynamics of methanol steam reforming on CuO/ZnO/alumina catalyst. Appl. Catal. A Gen. 2017, 540, 47–56. [Google Scholar] [CrossRef]
- Bagherzadeh, S.B.; Haghighi, M. Plasma-enhanced comparative hydrothermal and coprecipitation preparation of CuO/ZnO/Al2O3 nanocatalyst used in hydrogen production via methanol steam reforming. Energy Convers. Manag. 2017, 142, 452–465. [Google Scholar] [CrossRef]
- Ajamein, H.; Haghighi, M.; Alaei, S. The role of various fuels on microwave-enhanced combustion synthesis of CuO/ZnO/Al2O3 nanocatalyst used in hydrogen production via methanol steam reforming. Energy Convers. Manag. 2017, 137, 61–73. [Google Scholar] [CrossRef]
- Abrokwah, R.Y.; Deshmane, V.G.; Kuila, D. Comparative performance of M-MCM-41 (M: Cu, Co, Ni, Pd, Zn and Sn) catalysts for steam reforming of methanol. J. Mol. Catal. A Chem. 2016, 425, 10–20. [Google Scholar] [CrossRef]
- Papadopoulou, E.; Ioannides, T. Methanol Reforming over Cobalt Catalysts Prepared from Fumarate Precursors: TPD Investigation. Catalysts 2016, 6, 33. [Google Scholar] [CrossRef]
- Li, J.; Mei, X.; Zhang, L.; Yu, Z.; Liu, Q.; Wei, T.; Wu, W.; Dong, D.; Xu, L.; Hu, X. A comparative study of catalytic behaviors of Mn, Fe, Co, Ni, Cu and Zn–Based catalysts in steam reforming of methanol, acetic acid and acetone. Int. J. Hydrogen Energy 2020, 45, 3815–3832. [Google Scholar] [CrossRef]
- Maiti, S.; Llorca, J.; Dominguez, M.; Colussi, S.; Trovarelli, A.; Priolkar, K.R.; Aquilanti, G.; Gayen, A. Combustion synthesized copper-ion substituted FeAl2O4 (Cu0.1Fe0.9Al2O4): A superior catalyst for methanol steam reforming compared to its impregnated analogue. J. Power Sources 2016, 304, 319–331. [Google Scholar] [CrossRef]
- Qing, S.-J.; Hou, X.-N.; Liu, Y.-J.; Wang, L.; Li, L.-D.; Gao, Z.-X. Catalytic performance of Cu-Ni-Al spinel for methanol steam reforming to hydrogen. J. Fuel Chem. Technol. 2018, 46, 1210–1217. [Google Scholar] [CrossRef]
- Luo, X.; Hong, Y.; Wang, F.; Hao, S.; Pang, C.H.; Lester, E.H.; Wu, T. Development of nano NixMgyO solid solutions with outstanding anti-carbon deposition capability for the steam reforming of methanol. Appl. Catal. B Environ. 2016, 194, 84–97. [Google Scholar] [CrossRef]
- Zeng, Z.; Liu, G.; Geng, J.; Jing, D.; Hong, X.; Guo, L. A high-performance PdZn alloy catalyst obtained from metal-organic framework for methanol steam reforming hydrogen production. Int. J. Hydrogen Energy 2019, 44, 24387–24397. [Google Scholar] [CrossRef]
- Claudio-Piedras, A.; Ramírez-Zamora, R.M.; Alcántar-Vázquez, B.C.; Gutiérrez-Martínez, A.; Mondragón-Galicia, G.; Morales-Anzures, F.; Pérez-Hernández, R.; Modragón-Galicia, G. One dimensional Pt/CeO2-NR catalysts for hydrogen production by steam reforming of methanol: Effect of Pt precursor. Catal. Today 2019. [Google Scholar] [CrossRef]
- Barrios, C.; Bosco, M.V.; Baltanás, M.A.; Bonivardi, A.L. Hydrogen production by methanol steam reforming: Catalytic performance of supported-Pd on zinc–cerium oxides’ nanocomposites. Appl. Catal. B Environ. 2015, 179, 262–275. [Google Scholar] [CrossRef]
- Ajamein, H.; Haghighi, M.; Shokrani, R.; Abdollahifar, M. On the solution combustion synthesis of copper based nanocatalysts for steam methanol reforming: Effect of precursor, ultrasound irradiation and urea/nitrate ratio. J. Mol. Catal. A Chem. 2016, 421, 222–234. [Google Scholar] [CrossRef]
- Lin, L.; Zhou, W.; Gao, R.; Yao, S.; Zhang, X.; Xu, W.; Zheng, S.; Jiang, Z.; Yu, Q.; Li, Y.-W.; et al. Low-temperature hydrogen production from water and methanol using Pt/α-MoC catalysts. Nature 2017, 544, 80–83. [Google Scholar] [CrossRef]
- Cai, F.; Ibrahim, J.J.; Fu, Y.; Kong, W.; Zhang, J.; Sun, Y. Low-temperature hydrogen production from methanol steam reforming on Zn-modified Pt/MoC catalysts. Appl. Catal. B Environ. 2020, 264, 118500. [Google Scholar] [CrossRef]
- Liu, X.; Men, Y.; Wang, J.; He, R.; Wang, Y. Remarkable support effect on the reactivity of Pt/In2O3/MOx catalysts for methanol steam reforming. J. Power Sources 2017, 364, 341–350. [Google Scholar] [CrossRef]
- Diaz-Perez, M.A.; Moya, J.; Serrano-Ruiz, J.C.; Faria, J. Interplay of support chemistry and reaction conditions on copper catalyzed methanol steam reforming. Ind. Eng. Chem. Res. 2018, 57, 15268–15279. [Google Scholar] [CrossRef] [PubMed]
- Tahay, P.; Khani, Y.; Jabari, M.; Bahadoran, F.; Safari, N.; Zamanian, A. Synthesis of cubic and hexagonal ZnTiO3 as catalyst support in steam reforming of methanol: Study of physical and chemical properties of copper catalysts on the H2 and CO selectivity and coke formation. Int. J. Hydrogen Energy 2020, 45, 9484–9495. [Google Scholar] [CrossRef]
- Talkhoncheh, S.K.; Minaei, S.; Ajamein, H.; Haghighi, M.; Abdollahifar, M. Synthesis of CuO/ZnO/Al2O3 /ZrO2/CeO2 nanocatalysts via homogeneous precipitation and combustion methods used in methanol steam reforming for fuel cell grade hydrogen production. RSC Adv. 2016, 6, 57199–57209. [Google Scholar] [CrossRef]
- Taghizadeh, M.; Akhoundzadeh, H.; Rezayan, A.; Sadeghian, M. Excellent catalytic performance of 3D-mesoporous KIT-6 supported Cu and Ce nanoparticles in methanol steam reforming. Int. J. Hydrogen Energy 2018, 43, 10926–10937. [Google Scholar] [CrossRef]
- Phongboonchoo, Y.; Thouchprasitchai, N.; Pongstabodee, S. Hydrogen production with a low carbon monoxide content via methanol steam reforming over CuxCeyMgz/Al2O3 catalysts: Optimization and stability. Int. J. Hydrogen Energy 2017, 42, 12220–12235. [Google Scholar] [CrossRef]
- Hou, X.; Qing, S.; Liu, Y.; Li, L.; Gao, Z.; Qin, Y. Enhancing effect of MgO modification of Cu–Al spinel oxide catalyst for methanol steam reforming. Int. J. Hydrogen Energy 2020, 45, 477–489. [Google Scholar] [CrossRef]
- Liu, X.; Toyir, J.; De La Piscina, P.R.; Homs, N. Hydrogen production from methanol steam reforming over Al2O3- and ZrO2- modified CuOZnOGa2O3 catalysts. Int. J. Hydrogen Energy 2017, 42, 13704–13711. [Google Scholar] [CrossRef]
- Mohtashami, Y.; Taghizadeh, M. Performance of the ZrO2 promoted Cu ZnO catalyst supported on acetic acid-treated MCM-41 in methanol steam reforming. Int. J. Hydrogen Energy 2019, 44, 5725–5738. [Google Scholar] [CrossRef]
- Lu, J.; Li, X.; He, S.; Han, C.; Wan, G.; Lei, Y.; Chen, R.; Liu, P.; Chen, K.; Zhang, L.; et al. Hydrogen production via methanol steam reforming over Ni-based catalysts: Influences of Lanthanum (La) addition and supports. Int. J. Hydrogen Energy 2017, 42, 3647–3657. [Google Scholar] [CrossRef]
- Azenha, C.; Mateos-Pedrero, C.; Queirós, S.; Concepción, P.; Mendes, A. Innovative ZrO2-supported CuPd catalysts for the selective production of hydrogen from methanol steam reforming. Appl. Catal. B Environ. 2017, 203, 400–407. [Google Scholar] [CrossRef]
- Liu, D.; Men, Y.; Wang, J.; Kolb, G.; Liu, X.; Wang, Y.; Sun, Q. Highly active and durable Pt/In2O3/Al2O3 catalysts in methanol steam reforming. Int. J. Hydrogen Energy 2016, 41, 21990–21999. [Google Scholar] [CrossRef]
- Martinelli, M.; Jacobs, G.; Graham, U.; Crocker, M. Methanol Steam Reforming: Na Doping of Pt/YSZ Provides Fine Tuning of Selectivity. Catalysts 2017, 7, 148. [Google Scholar] [CrossRef]
- Zhang, R.; Huang, C.; Zong, L.; Lu, K.; Wang, X.; Cai, J. Hydrogen Production from Methanol Steam Reforming over TiO2 and CeO2 Pillared Clay Supported Au Catalysts. Appl. Sci. 2018, 8, 176. [Google Scholar] [CrossRef]
- Lytkina, A.A.; Orekhova, N.V.; Ermilova, M.; Yaroslavtsev, A.B. The influence of the support composition and structure (MXZr1-XO2-δ) of bimetallic catalysts on the activity in methanol steam reforming. Int. J. Hydrogen Energy 2018, 43, 198–207. [Google Scholar] [CrossRef]
- Lu, P.-J.; Cai, F.-F.; Zhang, J.; Liu, Y.; Sun, Y.-H. Hydrogen production from methanol steam reforming over B-modified CuZnAlO catalysts. J. Fuel Chem. Technol. 2019, 47, 791–798. [Google Scholar] [CrossRef]
- Maiti, S.; Das, D.; Pal, K.; Llorca, J.; Soler, L.; Colussi, S.; Trovarelli, A.; Priolkar, K.R.; Sarode, P.; Asakura, K.; et al. Methanol steam reforming behavior of sol-gel synthesized nanodimensional CuxFe1-xAl2O4 hercynites. Appl. Catal. A Gen. 2019, 570, 73–83. [Google Scholar] [CrossRef]
- Song, Q.; Men, Y.; Wang, J.; Liu, S.; Chai, S.; An, W.; Wang, K.; Li, Y.; Tang, Y. Methanol steam reforming for hydrogen production over ternary composite ZnyCe1Zr9Ox catalysts. Int. J. Hydrogen Energy 2020, 45, 9592–9602. [Google Scholar] [CrossRef]
- Mateos-Pedrero, C.; Silva, H.; Tanaka, D.P.; Liguori, S.; Iulianelli, A.; Basile, A.; Mendes, A. CuO/ZnO catalysts for methanol steam reforming: The role of the support polarity ratio and surface area. Appl. Catal. B Environ. 2015, 174, 67–76. [Google Scholar] [CrossRef]
- Kim, S.; Yun, S.-W.; Lee, B.; Heo, J.; Kim, K.; Kim, Y.-T.; Lim, H. Steam reforming of methanol for ultra-pure H2 production in a membrane reactor: Techno-economic analysis. Int. J. Hydrogen Energy 2019, 44, 2330–2339. [Google Scholar] [CrossRef]
- Köpfle, N.; Mayr, L.; Schmidmair, D.; Bernardi, J.; Knop-Gericke, A.; Hävecker, M.; Klötzer, B.; Penner, S. A Comparative Discussion of the Catalytic Activity and CO2-Selectivity of Cu-Zr and Pd-Zr (Intermetallic) Compounds in Methanol Steam Reforming. Catalysts 2017, 7, 53. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, Y.; Pei, P.; Yu, W.; Chu, X.; Li, S.; Yang, K. Hydrogen production from cylindrical methanol steam reforming microreactor with porous Cu-Al fiber sintered felt. Int. J. Hydrogen Energy 2018, 43, 3643–3654. [Google Scholar] [CrossRef]
- Ke, Y.; Zhou, W.; Chu, X.; Yuan, D.; Wan, S.; Yu, W.; Liu, Y. Porous copper fiber sintered felts with surface microchannels for methanol steam reforming microreactor for hydrogen production. Int. J. Hydrogen Energy 2019, 44, 5755–5765. [Google Scholar] [CrossRef]
- Tajrishi, O.Z.; Taghizadeh, M.; Kiadehi, A.D. Methanol steam reforming in a microchannel reactor by Zn-, Ce- and Zr- modified mesoporous Cu/SBA-15 nanocatalyst. Int. J. Hydrogen Energy 2018, 43, 14103–14120. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, W.; Lin, Y.; Chen, L.; Chu, X.; Zheng, T.; Wan, S.; Lin, J. Novel copper foam with ordered hole arrays as catalyst support for methanol steam reforming microreactor. Appl. Energy 2019, 246, 24–37. [Google Scholar] [CrossRef]
- Sarafraz, M.; Safaei, M.R.; Goodarzi, M.; Arjomandi, M. Reforming of methanol with steam in a micro-reactor with Cu–SiO2 porous catalyst. Int. J. Hydrogen Energy 2019, 44, 19628–19639. [Google Scholar] [CrossRef]
- Shanmugam, V.; Neuberg, S.; Zapf, R.; Pennemann, H.; Kolb, G. Hydrogen production over highly active Pt based catalyst coatings by steam reforming of methanol: Effect of support and co-support. Int. J. Hydrogen Energy 2020, 45, 1658–1670. [Google Scholar] [CrossRef]
- Zhuang, X.; Xia, X.; Xu, X.; Li, L. Experimental investigation on hydrogen production by methanol steam reforming in a novel multichannel micro packed bed reformer. Int. J. Hydrogen Energy 2020, 45, 11024–11034. [Google Scholar] [CrossRef]
- Zhu, J.; Araya, S.S.; Cui, X.; Sahlin, S.L.; Kær, S.K. Modeling and design of a multi-tubular packed-bed reactor for methanol steam reforming over a Cu/ZnO/Al2O3 catalyst. Energies 2020, 13, 610. [Google Scholar] [CrossRef]
- Ke, C.; Lin, Z. Density functional theory based micro-and macro-kinetic studies of Ni-catalyzed methanol steam reforming. Catalysts 2020, 10, 349. [Google Scholar] [CrossRef]
- Wang, G.; Wang, F.; Chen, B. Performance study on methanol steam reforming rib micro-reactor with waste heat recovery. Energies 2020, 13, 1564. [Google Scholar] [CrossRef]
- Udani, P.P.C.; Gunawardana, P.V.D.S.; Lee, H.C.; Kim, D.H. Steam reforming and oxidative steam reforming of methanol over CuO–CeO2 catalysts. Int. J. Hydrogen Energy 2009, 34, 7648–7655. [Google Scholar] [CrossRef]
- Kim, J.H.; Jang, Y.S.; Kim, D.H. Multiple steady states in the oxidative steam reforming of methanol. Chem. Eng. J. 2018, 338, 752–763. [Google Scholar] [CrossRef]
- Turco, M.; Bagnasco, G.; Costantino, U.; Marmottini, F.; Montanari, T.; Ramis, G.; Busca, G. Production of hydrogen from oxidative steam reforming of methanol: I. Preparation and characterization of Cu/ZnO/Al2O3 catalysts from a hydrotalcite-like LDH precursor. J. Catal. 2004, 228, 43–55. [Google Scholar] [CrossRef]
- Pojanavaraphan, C.; Satitthai, U.; Luengnaruemitchai, A.; Gulari, E. Activity and stability of Au/CeO2–Fe2O3 catalysts for the hydrogen production via oxidative steam reforming of methanol. J. Ind. Eng. Chem. 2015, 22, 41–52. [Google Scholar] [CrossRef]
- Pérez-Hernández, R.; Gutiérrez-Martínez, A.; Espinosa-Pesqueira, M.; Estanislao, M.L.; Palacios, J. Effect of the bimetallic Ni/Cu loading on the ZrO2 support for H2 production in the autothermal steam reforming of methanol. Catal. Today 2015, 250, 166–172. [Google Scholar] [CrossRef]
- Mierczynski, P.; Vasilev, K.; Mierczynska, A.; Maniukiewicz, W.; Ciesielski, R.; Rogowski, J.; Szynkowska, I.M.; Trifonov, A.Y.; Dubkov, S.V.; Gromov, D.G. The effect of gold on modern bimetallic Au–Cu/MWCNT catalysts for the oxy-steam reforming of methanol. Catal. Sci. Technol. 2016, 6, 4168–4183. [Google Scholar] [CrossRef]
- Jampa, S.; Jamieson, A.M.; Chaisuwan, T.; Luengnaruemitchai, A.; Wongkasemjit, S. Achievement of hydrogen production from autothermal steam reforming of methanol over Cu-loaded mesoporous CeO2 and Cu-loaded mesoporous CeO2–ZrO2 catalysts. Int. J. Hydrogen Energy 2017, 42, 15073–15084. [Google Scholar] [CrossRef]
- Pu, Y.-C.; Li, S.-R.; Yan, S.; Huang, X.; Wang, D.; Ye, Y.-Y.; Liu, Y.-Q. An improved Cu/ZnO catalyst promoted by Sc2O3 for hydrogen production from methanol reforming. Fuel 2019, 241, 607–615. [Google Scholar] [CrossRef]
- Adeniyi, A.G.; Ighalo, J.O. A review of steam reforming of glycerol. Chem. Pap. 2019, 73, 2619–2635. [Google Scholar] [CrossRef]
- Roslan, N.A.; Abidin, S.Z.; Ideris, A.; Vo, D.-V.N. A review on glycerol reforming processes over Ni-based catalyst for hydrogen and syngas productions. Int. J. Hydrogen Energy 2019. [Google Scholar] [CrossRef]
- Bulutoglu, P.S.; Say, Z.; Bac, S.; Ozensoy, E.; Avci, A.K. Dry reforming of glycerol over Rh-based ceria and zirconia catalysts: New insights on catalyst activity and stability. Appl. Catal. A Gen. 2018, 564, 157–171. [Google Scholar] [CrossRef]
- Bagnato, G.; Iulianelli, A.; Sanna, A.; Basile, A. Glycerol Production and Transformation: A Critical Review with Particular Emphasis on Glycerol Reforming Reaction for Producing Hydrogen in Conventional and Membrane Reactors. Membranes 2017, 7, 17. [Google Scholar] [CrossRef]
- Silva, J.; Soria, M.A.; Madeira, L.M. Challenges and strategies for optimization of glycerol steam reforming process. Renew. Sustain. Energy Rev. 2015, 42, 1187–1213. [Google Scholar] [CrossRef]
- Schwengber, C.A.; Alves, H.J.; Schaffner, R.A.; Silva, F.A.; Sequinel, R.; Bach, V.R.; Ferracin, R.J. Overview of glycerol reforming for hydrogen production. Renew. Sustain. Energy Rev. 2016, 58, 259–266. [Google Scholar] [CrossRef]
- Seadira, T.; Sadanandam, G.; Ntho, T.A.; Lu, X.; Masuku, C.M.; Scurrell, M. Hydrogen production from glycerol reforming: Conventional and green production. Rev. Chem. Eng. 2018, 34, 695–726. [Google Scholar] [CrossRef]
- Charisiou, N.; Siakavelas, G.; Papageridis, K.; Baklavaridis, A.; Tzounis, L.; Polychronopoulou, K.; Goula, M. Hydrogen production via the glycerol steam reforming reaction over nickel supported on alumina and lanthana-alumina catalysts. Int. J. Hydrogen Energy 2017, 42, 13039–13060. [Google Scholar] [CrossRef]
- Gallegos-Suárez, E.; Guerrero-Ruiz, A.; Rodríguez-Ramos, I. Efficient hydrogen production from glycerol by steam reforming with carbon supported ruthenium catalysts. Carbon 2016, 96, 578–587. [Google Scholar] [CrossRef]
- Papageridis, K.; Siakavelas, G.; Charisiou, N.D.; Avraam, D.; Tzounis, L.; Kousi, K.; Goula, M. Comparative study of Ni, Co, Cu supported on γ-alumina catalysts for hydrogen production via the glycerol steam reforming reaction. Fuel Process. Technol. 2016, 152, 156–175. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Papageridis, K.; Siakavelas, G.; Tzounis, L.; Kousi, K.; Baker, M.A.; Hinder, S.J.; Sebastian, V.; Polychronopoulou, K.; Goula, M. Glycerol Steam Reforming for Hydrogen Production over Nickel Supported on Alumina, Zirconia and Silica Catalysts. Top. Catal. 2017, 60, 1226–1250. [Google Scholar] [CrossRef]
- Charisiou, N.; Siakavelas, G.; Tzounis, L.; Dou, B.; Sebastian, V.; Hinder, S.; Baker, M.; Polychronopoulou, K.; Goula, M. Ni/Y2O3–ZrO2 catalyst for hydrogen production through the glycerol steam reforming reaction. Int. J. Hydrogen Energy 2020, 45, 10442–10460. [Google Scholar] [CrossRef]
- Senseni, A.Z.; Rezaei, M.; Meshkani, F. Glycerol steam reforming over noble metal nanocatalysts. Chem. Eng. Res. Des. 2017, 123, 360–366. [Google Scholar] [CrossRef]
- Silva, J.; Ribeiro, L.; Órfão, J.; Soria, M.A.; Madeira, L.M. Low temperature glycerol steam reforming over a Rh-based catalyst combined with oxidative regeneration. Int. J. Hydrogen Energy 2019, 44, 2461–2473. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, C.; Wang, K.; Dou, B.; Song, Y.; Chen, H.; Xu, Y. Highly dispersed Ni/montmorillonite catalyst for glycerol steam reforming: Effect of Ni loading and calcination temperature. Appl. Therm. Eng. 2016, 109, 99–108. [Google Scholar] [CrossRef]
- Bepari, S.; Pradhan, N.C.; Dalai, A.K. Selective production of hydrogen by steam reforming of glycerol over Ni/Fly ash catalyst. Catal. Today 2017, 291, 36–46. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, M.; Yang, Z.; Liang, T.; Liu, S.; Zhou, Z.; Li, X. Bimetallic Ni-M (M = Co, Cu and Zn) supported on attapulgite as catalysts for hydrogen production from glycerol steam reforming. Appl. Catal. A Gen. 2018, 550, 214–227. [Google Scholar] [CrossRef]
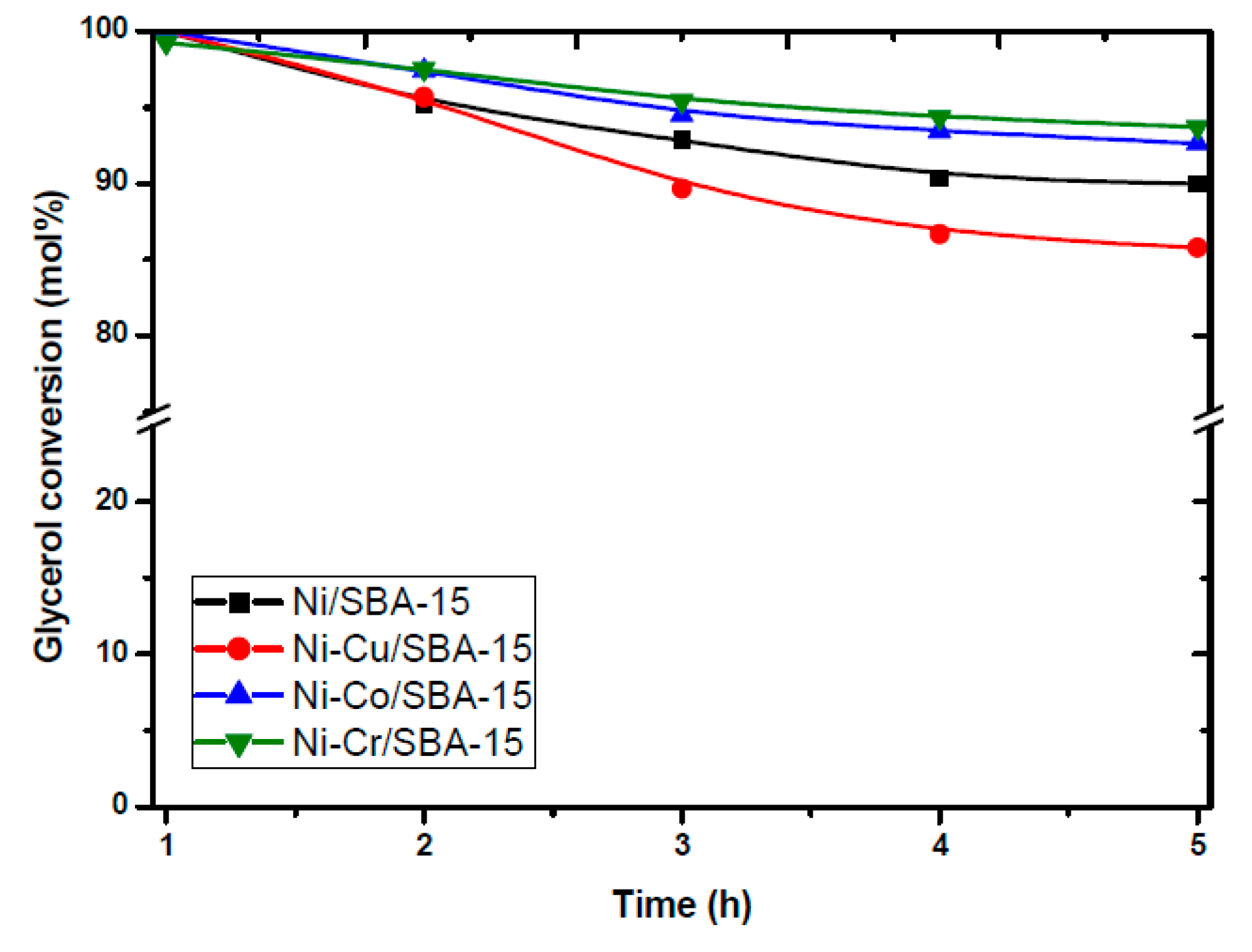
- Carrero, A.; Calles, J.; García-Moreno, L.; Vizcaíno, A. Production of Renewable Hydrogen from Glycerol Steam Reforming over Bimetallic Ni-(Cu,Co,Cr) Catalysts Supported on SBA-15 Silica. Catalysts 2017, 7, 55. [Google Scholar] [CrossRef]
- Wang, R.; Liu, S.; Liu, S.; Li, X.; Zhang, Y.; Xie, C.; Zhou, S.; Qiu, Y.; Luo, S.; Jing, F.; et al. Glycerol steam reforming for hydrogen production over bimetallic MNi/CNTs (M Co, Cu and Fe) catalysts. Catal. Today 2019. [Google Scholar] [CrossRef]
- Tavanarad, M.; Meshkani, F.; Rezaei, M. Production of syngas via glycerol dry reforming on Ni catalysts supported on mesoporous nanocrystalline Al2O3. J. CO2 Util. 2018, 24, 298–305. [Google Scholar] [CrossRef]
- Bac, S.; Say, Z.; Koçak, Y.; Ercan, K.E.; Harfouche, M.; Ozensoy, E.; Avci, A.K. Exceptionally active and stable catalysts for CO2 reforming of glycerol to syngas. Appl. Catal. B Environ. 2019, 256, 117808. [Google Scholar] [CrossRef]
- Larimi, A.; Kazemeini, M.; Khorasheh, F. Aqueous phase reforming of glycerol using highly active and stable Pt0.05CexZr0.95-xO2 ternary solid solution catalysts. Appl. Catal. A Gen. 2016, 523, 230–240. [Google Scholar] [CrossRef]
- Espinosa-Moreno, F.; Balla, P.; Shen, W.; Chavarría-Hernández, J.C.; Ruiz-Gómez, M.; Tlecuitl-Beristain, S. Ir-Based Bimetallic Catalysts for Hydrogen Production through Glycerol Aqueous-Phase Reforming. Catalysts 2018, 8, 613. [Google Scholar] [CrossRef]
- Kousi, K.; Chourdakis, N.; Matralis, H.; Kontarides, D.; Papadopoulou, C.; Verykios, X. Glycerol steam reforming over modified Ni-based catalysts. Appl. Catal. A Gen. 2016, 518, 129–141. [Google Scholar] [CrossRef]
- Cheng, C.-K.; Foo, S.Y.; Adesina, A.A. Glycerol Steam Reforming over Bimetallic Co−Ni/Al2O3. Ind. Eng. Chem. Res. 2010, 49, 10804–10817. [Google Scholar] [CrossRef]
- Zamzuri, N.H.; Mat, R.; Amin, N.A.S.; Talebian-Kiakalaieh, A. Hydrogen production from catalytic steam reforming of glycerol over various supported nickel catalysts. Int. J. Hydrogen Energy 2017, 42, 9087–9098. [Google Scholar] [CrossRef]
- Bobadilla, L.; Penkova, A.; Alvarez, A.; Domínguez, M.; Romero-Sarria, F.; Centeno, M.A.; Odriozola, J.; Leal, M.I.D. Glycerol steam reforming on bimetallic NiSn/CeO2–MgO–Al2O3 catalysts: Influence of the support, reaction parameters and deactivation/regeneration processes. Appl. Catal. A Gen. 2015, 492, 38–47. [Google Scholar] [CrossRef]
- Charisiou, N.; Papageridis, K.; Tzounis, L.; Sebastian, V.; Hinder, S.; Baker, M.; Alketbi, M.; Polychronopoulou, K.; Goula, M. Ni supported on CaO-MgO-Al2O3 as a highly selective and stable catalyst for H2 production via the glycerol steam reforming reaction. Int. J. Hydrogen Energy 2019, 44, 256–273. [Google Scholar] [CrossRef]
- Menezes, J.P.D.S.; Duarte, K.R.; Manfro, R.L.; Souza, M.M.V.M. Effect of niobia addition on cobalt catalysts supported on alumina for glycerol steam reforming. Renew. Energy 2020, 148, 864–875. [Google Scholar] [CrossRef]
- Arif, N.N.M.; Vo, D.-V.N.; Azizan, M.T.; Abidin, S.Z. Carbon Dioxide Dry Reforming of Glycerol for Hydrogen Production using Ni/ZrO2 and Ni/CaO as Catalysts. Bull. Chem. React. Eng. Catal. 2016, 11, 200. [Google Scholar] [CrossRef]
- Lee, H.-J.; Shin, G.S.; Kim, Y.-C. Characterization of supported Ni catalysts for aqueous-phase reforming of glycerol. Korean J. Chem. Eng. 2015, 32, 1267–1272. [Google Scholar] [CrossRef]
- Yancheshmeh, M.S.; Sahraei, O.A.; Aissaoui, M.; Iliuta, M.C. A novel synthesis of NiAl2O4 spinel from a Ni-Al mixed-metal alkoxide as a highly efficient catalyst for hydrogen production by glycerol steam reforming. Appl. Catal. B Environ. 2020, 265, 118535. [Google Scholar] [CrossRef]
- Lima, D.S.; Calgaro, C.O.; Perez-Lopez, O.W. Hydrogen production by glycerol steam reforming over Ni based catalysts prepared by different methods. Biomass Bioenergy 2019, 130, 105358. [Google Scholar] [CrossRef]
- Dieuzeide, M.; Laborde, M.; Amadeo, N.; Cannilla, C.; Bonura, G.; Frusteri, F. Hydrogen production by glycerol steam reforming: How Mg doping affects the catalytic behaviour of Ni/Al2O3 catalysts. Int. J. Hydrogen Energy 2016, 41, 157–166. [Google Scholar] [CrossRef]
- Demsash, H.; Mohan, R. Steam reforming of glycerol to hydrogen over ceria promoted nickel–alumina catalysts. Int. J. Hydrogen Energy 2016, 41, 22732–22742. [Google Scholar] [CrossRef]
- Carrero, A.; Vizcaíno, A.; Calles, J.; García-Moreno, L. Hydrogen production through glycerol steam reforming using Co catalysts supported on SBA-15 doped with Zr, Ce and La. J. Energy Chem. 2017, 26, 42–48. [Google Scholar] [CrossRef]
- Veiga, S.; Bussi, J. Steam reforming of crude glycerol over nickel supported on activated carbon. Energy Convers. Manag. 2017, 141, 79–84. [Google Scholar] [CrossRef]
- Sahraei, O.A.Z.; Larachi, F.; Abatzoglou, N.; Iliuta, M. Hydrogen production by glycerol steam reforming catalyzed by Ni-promoted Fe/Mg-bearing metallurgical wastes. Appl. Catal. B Environ. 2017, 219, 183–193. [Google Scholar] [CrossRef]
- Dobosz, J.; Cichy, M.; Zawadzki, M.; Borowiecki, T. Glycerol steam reforming over calcium hydroxyapatite supported cobalt and cobalt-cerium catalysts. J. Energy Chem. 2018, 27, 404–412. [Google Scholar] [CrossRef]
- Siew, K.W.; Lee, H.C.; Gimbun, J.; Chin, S.Y.; Khan, M.R.; Taufiq-Yap, Y.H.; Cheng, C.-K. Syngas production from glycerol-dry(CO2 ) reforming over La-promoted Ni/Al2O3 catalyst. Renew. Energy 2015, 74, 441–447. [Google Scholar] [CrossRef]
- Reynoso, A.; Iriarte-Velasco, U.; Gutiérrez-Ortiz, M.A.; Ayastuy, J. Highly stable Pt/CoAl2O4 catalysts in Aqueous-Phase Reforming of glycerol. Catal. Today 2020. [Google Scholar] [CrossRef]
- Pendem, C.; Sarkar, B.; Siddiqui, N.; Konathala, L.N.S.K.; Baskar, C.; Bal, R. K-Promoted Pt-Hydrotalcite Catalyst for Production of H2 by Aqueous Phase Reforming of Glycerol. ACS Sustain. Chem. Eng. 2017, 6, 2122–2131. [Google Scholar] [CrossRef]
- Wang, M.; Au, C.T.; Lai, S.Y. H2 production from catalytic steam reforming of n-propanol over ruthenium and ruthenium-nickel bimetallic catalysts supported on ceria-alumina oxides with different ceria loadings. Int. J. Hydrogen Energy 2015, 40, 13926–13935. [Google Scholar] [CrossRef]
- Patel, R.; Patel, S. Process development for bio-butanol steam reforming for PEMFC application. Int. J. Eng. Technol. 2018, 7, 110–112. [Google Scholar] [CrossRef]
- Patel, R.; Patel, S. Effect of operating conditions on hydrogen production in butanol reforming: A review. Int. Conf. Multidiscip. Res. Pract. 2015, 145–150. [Google Scholar]
- Patel, R.; Patel, S. Renewable hydrogen production from butanol: A review. Clean Energy 2017, 1, 90–101. [Google Scholar] [CrossRef]
- Harju, H.; Lehtonen, J.; Lefferts, L. Steam reforming of n -butanol over Rh/ZrO2 catalyst: Role of 1-butene and butyraldehyde. Appl. Catal. B Environ. 2016, 182, 33–46. [Google Scholar] [CrossRef]
- Dhanala, V.; Maity, S.K.; Shee, D. Oxidative steam reforming of isobutanol over Ni/γ-Al2O3 catalysts: A comparison with thermodynamic equilibrium analysis. J. Ind. Eng. Chem. 2015, 27, 153–163. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, X.; Wang, Y. Catalytic performance of mesoporous MgO supported Ni catalyst in steam reforming of model compounds of biomass fermentation for hydrogen production. Int. J. Hydrogen Energy 2016, 41, 17846–17857. [Google Scholar] [CrossRef]
- Yadav, A.K.; Vaidya, P.D. Renewable hydrogen production by steam reforming of butanol over multiwalled carbon nanotube-supported catalysts. Int. J. Hydrogen Energy 2019, 44, 30014–30023. [Google Scholar] [CrossRef]
- Harju, H.; Lehtonen, J.; Lefferts, L. Steam- and autothermal-reforming of n-butanol over Rh/ZrO2 catalyst. Catal. Today 2015, 244, 47–57. [Google Scholar] [CrossRef]
- Harju, H.; Pipitone, G.; Lefferts, L. Influence of the catalyst particle size on the aqueous phase reforming of n-butanol over Rh/ZrO2. Front. Chem. 2020, 8, 17. [Google Scholar] [CrossRef]
- Yadav, A.K.; Vaidya, P.D. A study on the efficacy of noble metal catalysts for butanol steam reforming. Int. J. Hydrogen Energy 2019, 44, 25575–25588. [Google Scholar] [CrossRef]
- Bizkarra, K.; Barrio, V.; Yartu, A.; Requies, J.M.; Arias, P.L.; Cambra, J.F. Hydrogen production from n-butanol over alumina and modified alumina nickel catalysts. Int. J. Hydrogen Energy 2015, 40, 5272–5280. [Google Scholar] [CrossRef]
- Lobo, R.; Marshall, C.L.; Dietrich, P.J.; Ribeiro, F.H.; Akatay, C.; Stach, E.A.; Mane, A.; Lei, Y.; Elam, J.; Miller, J.T. Understanding the chemistry of H2 production for 1-propanol reforming: Pathway and support modification effects. ACS Catal. 2012, 2, 2316–2326. [Google Scholar] [CrossRef]
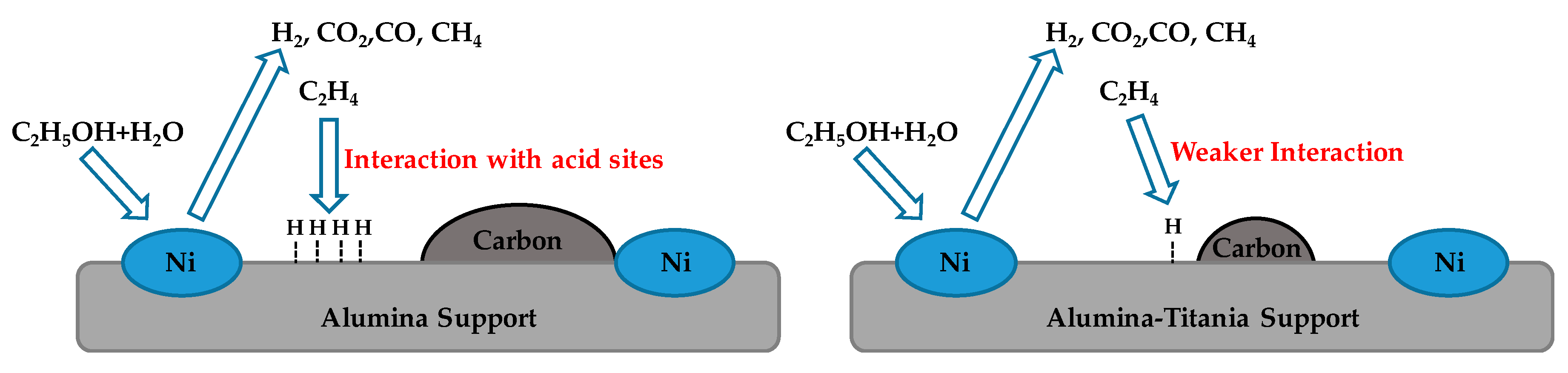
- Li, Y.; Zhang, L.; Zhang, Z.; Liu, Q.; Zhang, S.; Liu, Q.; Hu, G.; Wang, Y.; Hu, X. Steam reforming of the alcohols with varied structures: Impacts of acidic sites of Ni catalysts on coking. Appl. Catal. A Gen. 2019, 584, 117162. [Google Scholar] [CrossRef]
- Shejale, A.D.; Yadav, G.D. Noble metal promoted Ni–Cu/La2O3–MgO catalyst for renewable and enhanced hydrogen production via steam reforming of bio-based n-butanol: Effect of promotion with Pt, Ru and Pd on catalytic activity and selectivity. Clean Technol. Environ. Policy 2019, 21, 1323–1339. [Google Scholar] [CrossRef]
- Sharma, M.P.; Akyurtlu, J.F.; Akyurtlu, A. Autothermal reforming of isobutanol over promoted nickel xerogel catalysts for hydrogen production. Int. J. Hydrogen Energy 2015, 40, 13368–13378. [Google Scholar] [CrossRef]
- Lei, Y.; Lee, S.; Low, K.-B.; Marshall, C.L.; Elam, J.W. Combining Electronic and Geometric Effects of ZnO-Promoted Pt Nanocatalysts for Aqueous Phase Reforming of 1-Propanol. ACS Catal. 2016, 6, 3457–3460. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
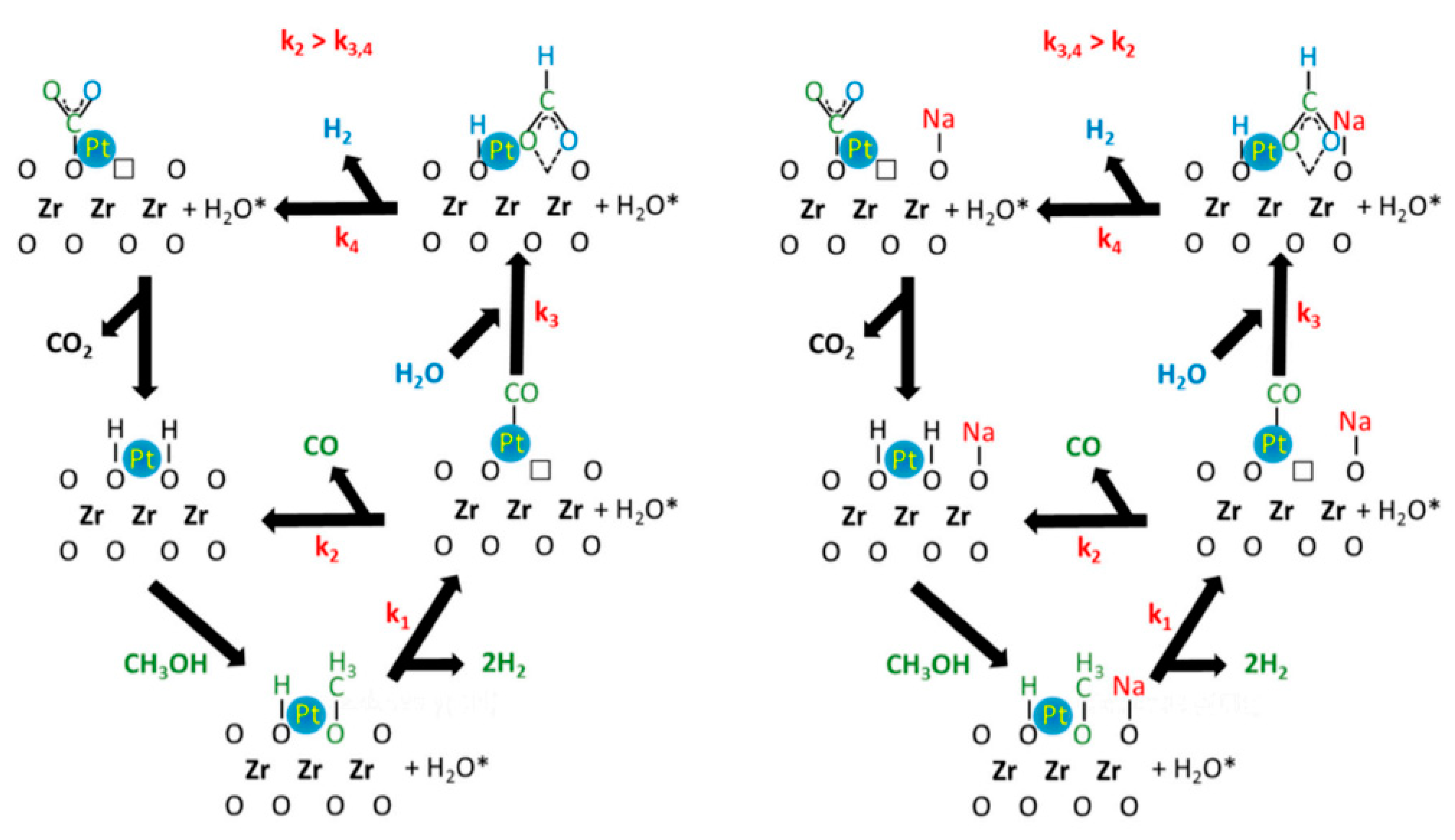{kind=link}
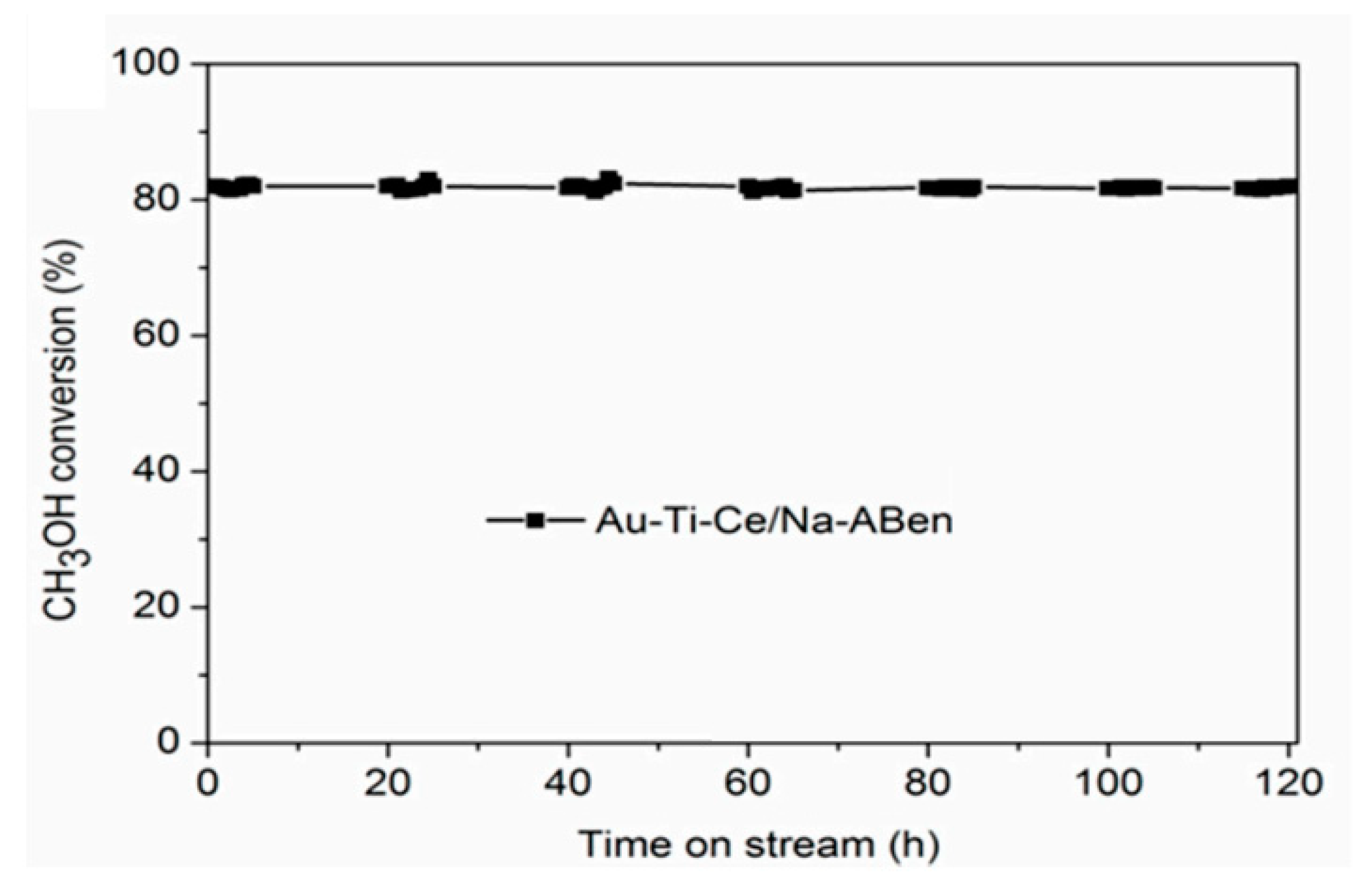{kind=link}
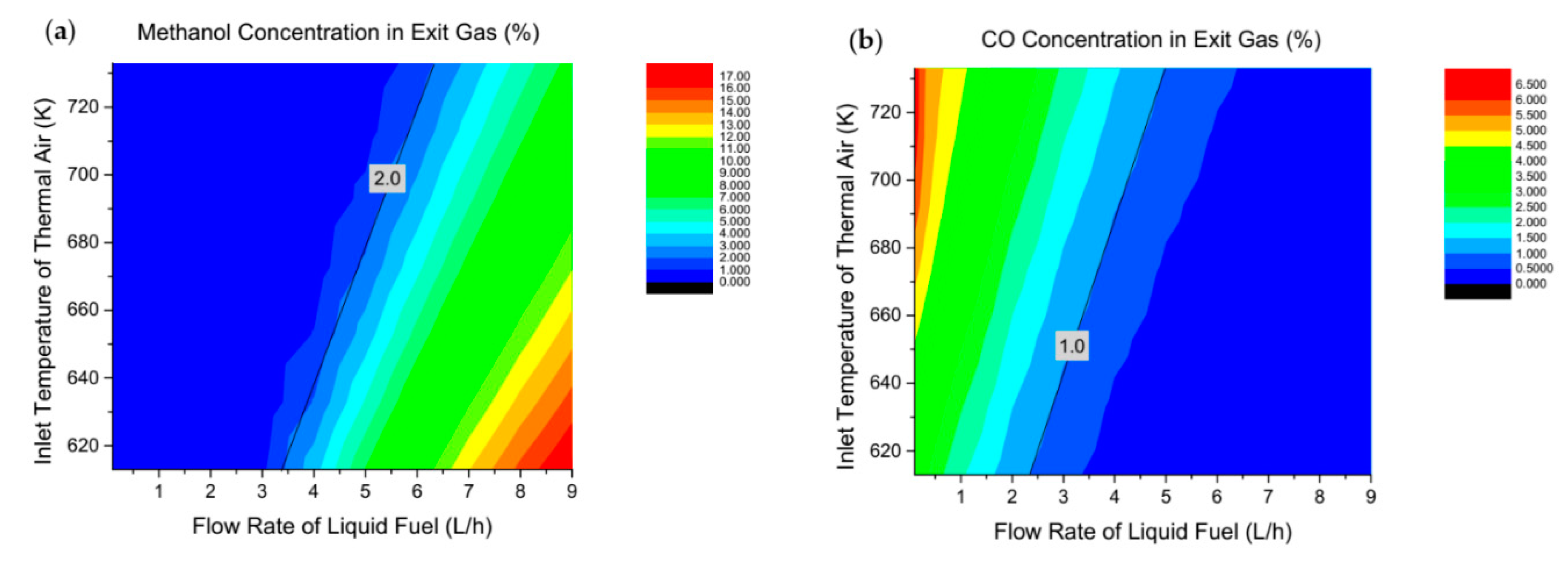{kind=link}
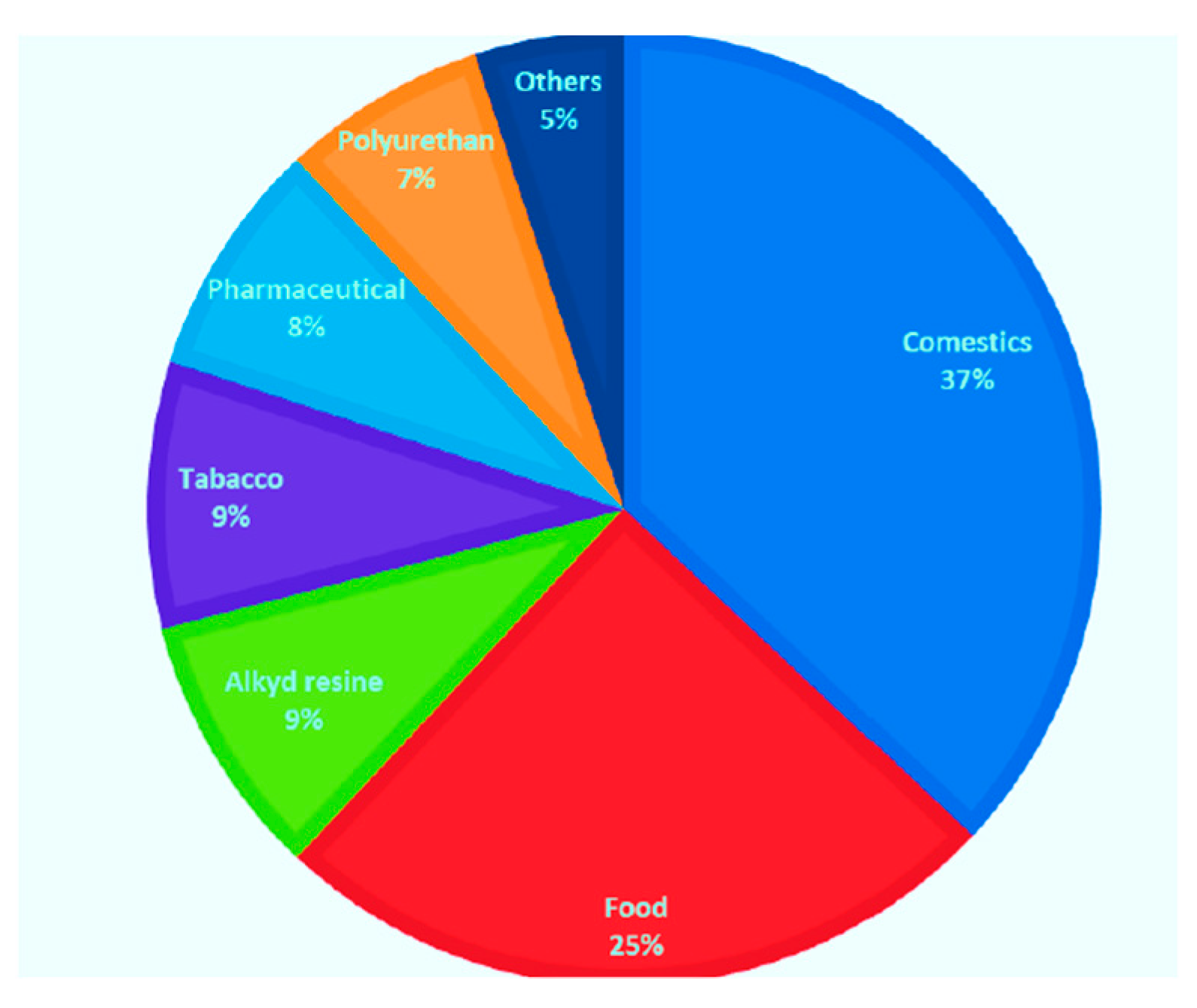{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst a | Operative Conditions b | WHSV c (h−1) | X EtOH (%) | Carbon Formation Rate d (Multiplied for 1000) | Ref. |
|---|---|---|---|---|---|
| Ethanol steam reforming | |||||
| 1Pt-3Ni/CeO2-SiO2 | T = 450 °C S/E = 4 %C2H5OH = 10% | 4.1 | 95% after 310 min | 3 | [23] |
| 0.5Rh-1Pt-3Ni/CeO2-SiO2 | T = 450 °C S/E = 4 %C2H5OH = 10% | 4.1 | 93% after 1300 min | 0.84 | |
| 1Rh-3Ni/CeO2-SiO2 | T = 450 °C S/E = 4 %C2H5OH = 10% | 4.1 | 91% after 4900 min | 0.065 | |
| 1Pt-3Ni-0.5K/CeO2-SiO2 | T = 450 °C S/E = 4 %C2H5OH = 10% | 4.1 | 92% after 200 min | 17 | |
| 1Pt-3Ni-0.5Cs/CeO2-SiO2 | T = 450 °C S/E = 4 %C2H5OH = 10% | 4.1 | 94% after 1600 min | 0.39 | |
| Ni-Co/mesoporous carbon (MC) | T = 375 °C S/E = 12 %C2H5OH = 4% | 1.2 | 0% after 700 min | 14 | [32] |
| Ni-Co/2Zr-MC | T = 375 °C S/E = 12 %C2H5OH = 4% | 1.2 | 77% after 700 min | 7.1 | |
| Ni-Co/2Y-MC | T = 375 °C S/E = 12 %C2H5OH = 4% | 1.2 | 90% after 700 min | 8.1 | |
| LaNi0.85Zn0.15O3-δ | T = 700 °C S/E = 3 %C2H5OH = 18.6% | 8.2 | 100% after 8 h | 2.7 | [34] |
| 10Ni/9La2O3-αAl2O3 | T = 500 °C S/E = 3 %C2H5OH = 8 % | 11.5 | 32 % after 20 h | 0.19 | [42] |
| 5Co/CeO2 | T = 500 °C S/E = 3 %C2H5OH = 25% | 6.8 | 94% after 6 h | 25 | [71] |
| 10Co/CeO2 | T = 500 °C S/E = 3 %C2H5OH = 25% | 6.8 | 98% after 6 h | 75 | |
| 20Co/CeO2 | T = 500 °C S/E = 3 %C2H5OH = 25% | 6.8 | 98% after 6 h | 58 | |
| 10Ni/CeO2 | T = 300 °C S/E = 3 %C2H5OH = 2.5% | 2.1 | 10% after 30 h | 0.15 | [84] |
| 1Pt10Ni/CeO2 | T = 300 °C S/E = 3 %C2H5OH = 2.5% | 2.1 | 33% after 30 h | 0.19 | [84] |
| 1Rh-10Ni/15La2O3-10CeO2-Al2O3 | T = 500 °C S/E = 3 %C2H5OH = 18.8% | 42.2 | 100% after 24 h | 0.00031 | [87] |
| Co-La/CeO2 La/Co mol ratio 0.1 | T = 420 °C S/E = 12 %C2H5OH = 7.7% | 9.5 | 60% after 21 h | 0.17 | [110] |
| Ni-La/CeO2 La/Ni molar ratio of 0.1 | T = 420 °C S/E = 12 %C2H5OH = 7.7% | 9.5 | 99% after 21 h | 19 | |
| 3Ni/SBA-15 | T = 650 °C S/E = 4 %C2H5OH = 4.5% | 25.7 | 70% after 50 h | 0.19 | [115] |
| 3NiCe/SBA-15 Ce/Ni molar ratio of 1:1 | T = 650 °C S/E = 4 %C2H5OH = 4.5% | 25.7 | 90% after 50 h | 0.045 | [115] |
| 0.4Pt-0.4Rh/CeO2-SiO2 Si/Ce molar ratio of 1:2 | T = 680 °C S/E = 3 %C2H5OH = 1.8% | 14.3 | 100% for 72 h | 0.16 | [127] |
| 2Ir/CeO2 nanoparticles | T = 650 °C S/E = 3 %C2H5OH = 25% | 9.23 | 80% after 45 h | 0.0083 | [128] |
| 2Ir/CeO2 nanoroads | T = 650 °C S/E = 3 %C2H5OH = 25% | 9.23 | 55% after 45 h | 0.0096 | [128] |
| 1Rh/Al2O3 | T = 500 °C S/E = 3 %C2H5OH = 14% | 40.7 | 80% after 45 h | 0.028 | [129] |
| 1Rh-15%La2O3-Al2O3 | T = 500 °C S/E = 3 %C2H5OH = 14% | 40.7 | 90% after 45 h | 0.0029 | [129] |
| 1Rh-15%La2O3-5%CeO2-Al2O3 | T = 500 °C S/E = 3 %C2H5OH = 14% | 40.7 | 97% after 45 h | 0.0016 | [129] |
| Co-Mg@mesoporous Al2O3 Co/Al molar ratio of 0.1:1 Mg/Al mol ratio 0.25:1 | T = 550 °C S/E = 5 %C2H5OH = 6.7% | 4.8 | 100% after 4 h | 9 | [130] |
| LaNiO3/ZrO2 | T = 650 °C S/E = 3 %C2H5OH = 5% | 27.1 | 80% after 50 h | 0.57 | [131] |
| LaNi0.7Co0.3O3/ZrO2 | T = 650 °C S/E = 3 %C2H5OH = 5% | 27.1 | 96% after 50 h | 0.36 | |
| LaCoO3/ZrO2 | T = 650 °C S/E = 3 %C2H5OH = 5% | 27.1 | 70% after 50 h | 0.68 | |
| 10Ce/Ni-Mg-Al | T = 540 °C S/E = 6 %C2H5OH = 14.3% | 2.1 | 83% after 10 h | 0.51 | [132] |
| 5Ni/CNTs-SiO2 fibers | T = 450 °C S/E = 9 %C2H5OH = 10% | 2.6 | 87% after 22 h | 1.2 | [133] |
| 10Ni/CNTs-SiO2 fibers | T = 450 °C S/E = 9 %C2H5OH = 10% | 2.6 | 100% after 22 h | 1.5 | |
| Pt@HBZ (HB zeolite) | T = 350 °C S/E = 4 %C2H5OH = 4% | 3.4 | 100% after 15 h | 0.23 | [134] |
| Pt-B (B zeolite) | T = 350 °C S/E = 4 %C2H5OH = 4% | 3.4 | 60% after 15 h | 0.46 | |
| 2.5Co/hydroxyapatite | T = 500 °C S/E = 6 %C2H5OH = 4.1% | 2.2 | 60% after 5 h | 17 | [135] |
| 5Co/hydroxyapatite | T = 500 °C S/E = 6 %C2H5OH = 4.1% | 2.2 | 40% after 5 h | 19 | |
| 7.5Co/hydroxyapatite | T = 500 °C S/E = 6 %C2H5OH = 4.1% | 2.2 | 30% after 5 h | 20 | |
| 20Ni/Attapulginte | T = 700 °C S/E = 3 %C2H5OH = 12.6% | 5.1 | 75% after 50 h | 0.25 | [136] |
| 20Ni/5Mg-Attapulgite | T = 700 °C S/E = 3 %C2H5OH = 12.6% | 5.1 | 85% after 50 h | 0.23 | |
| 20Ni/10Mg-Attapulgite | T = 700 °C S/E = 3 %C2H5OH = 12.6% | 5.1 | 98% after 50 h | 0.047 | |
| 20Ni/20Mg-Attapulgite | T = 700 °C S/E = 3 %C2H5OH = 12.6% | 5.1 | 87% after 50 h | 0.097 | |
| 10Ni/20Pr-CeO2 | T = 600 °C S/E = 5 %C2H5OH = 15.7% | 18.2 | 100% after 120 h | 0.0016 | [137] |
| Pd0.01Zn0.291Mg0.7Al2O4 | T = 450 °C S/E = 3 - | 3.1 | 100% after 30 h | 0.00017 | [138] |
| La0.7Ce0.3Ni0.7Fe0.3O3 | T = 500 °C S/E = 4 - | 1.2 | 98% after 50 h | 0.07 | [139] |
| 2.5Pt-1Cu@SiO2 | T = 400 °C S/E = 4 - | 2.9 | 100% after 30 h | 0.33 | [140] |
| 2.5Pt@SiO2 | T = 400 °C S/E = 4 - | 2.9 | 70% after 30 h | 0.60 | |
| 2.5Pt-1Cu/SiO2 | T = 400 °C S/E = 4 - | 2.9 | 80% after 30 h | 0.79 | |
| 10Ni/17CeO2ZrO25La2O3 | T = 500 °C S/E = 3 %C2H5OH = 5% | 10.1 | 86% after 4 h | 12 | [141] |
| 10Ni/ZrO25La2O3 | T = 500 °C S/E = 3 %C2H5OH = 5% | 10.1 | 57% after 4 h | 37 | |
| 1Rh/17CeO2ZrO25La2O3 | T = 500 °C S/E = 3 %C2H5OH = 5% | 10.1 | 92% after 4 h | 0.58 | |
| 1Rh/ZrO25La2O3 | T = 500 °C S/E = 3 %C2H5OH = 5% | 10.1 | 68% after 4 h | 5.2 | |
| Oxidative steam reforming of ethanol | |||||
| 1Pt3Ni/CeO2-SiO2 Cerium precursor: nitrate | T = 500 °C S/E = 4 O2/E = 0.5 %C2H5OH = 10% | 12.3 | 59% after 100 h | 0.0030 | [37] |
| 1Pt3Ni/CeO2-SiO2 Cerium precursor: ammonium nitrate | T = 500 °C S/E = 4 O2/E = 0.5 %C2H5OH = 10% | 12.3 | 60% after 100 h | 0.0029 | |
| 1Pt3Ni/CeO2-SiO2 Cerium precursor: acetyl acetonate | T = 500 °C S/E = 4 O2/E = 0.5 %C2H5OH = 10% | 12.3 | 73% after 100 h | 0.0014 | |
| 30Ni/CeO2-ZrO2 | T = 600 °C S/E = 9 O2/E = 0.35 | 5.1 | 95% after 36 h | 0.92 | [43] |
| 1Rh-30Ni/CeO2-ZrO2 | T = 600 °C S/E = 9 O2/E = 0.35 | 5.1 | 85% after 36 h | 0.45 | |
| NiCo-MgAl (Ni+Co = 20 wt.%) Conventional synthesis | T = 550 °C S/E = 3 O2/E = 0.5 %C2H5OH = 12.8% | 91.6 | 100% after 100 h | 7.7 | [58] |
| NiCo-MgAl (Ni+Co = 20 wt.%) Microwave-assisted co-precipitation | T = 550 °C S/E = 3 O2/E = 0.5 %C2H5OH = 12.8% | 91.6 | 100% after 100 h | 7 | |
| NiCo-MgAl (Ni+Co = 20 wt.%) Sonication-assisted co-precipitation | T = 550 °C S/E = 3 O2/E = 0.5 %C2H5OH = 12.8% | 91.6 | 100% after 100 h | 5.4 | |
| 1Pt-3Ni/CeO2-SiO2 CeO2/SiO2 ratio = 25 | T = 500 °C S/E = 4 O2/E = 0.5 %C2H5OH = 10% | 4.1 | 100% after 100 h | 0.0076 | [60] |
| 1Pt-3Ni/CeO2-SiO2 CeO2/SiO2 ratio = 30 | T = 500 °C S/E = 4 O2/E = 0.5 %C2H5OH = 10% | 4.1 | 100% after 135 h | 0.0012 | |
| 1Pt-3Ni/CeO2-SiO2 CeO2/SiO2 ratio = 40 | T = 500 °C S/E = 4 O2/E = 0.5 %C2H5OH = 10% | 4.1 | 100% after 120 h | 0.0065 | |
| 15Ni/MgAl2O4 | T = 500 °C S/E = 3 O2/E = 0.5 %C2H5OH = 2.5% | 9.2 | 80% after 28 h | 22 | [94] |
| 4Co11Ni/MgAl2O4 | T = 500 °C S/E = 3 O2/E = 0.5 %C2H5OH = 2.5% | 9.2 | 70% after 28 h | 21 | |
| 7.5Co7.5Ni/MgAl2O4 | T = 500 °C S/E = 3 O2/E = 0.5 %C2H5OH = 2.5% | 9.2 | 70% after 28 h | 7.1 | |
| 11Co4Ni/MgAl2O4 | T = 500 °C S/E = 3 O2/E = 0.5 %C2H5OH = 2.5% | 9.2 | 60% after 28 h | 6.7 | |
| 15Co/MgAl2O4 | T = 500 °C S/E = 3 O2/E = 0.5 %C2H5OH = 2.5% | 9.2 | 60% after 28 h | 1.9 | |
| 4Co4Ni/MgAl2O4 | T = 500 °C S/E = 3 O2/E = 0.5 %C2H5OH = 2.5% | 9.2 | 60% after 28 h | 0.11 | |
| La0.6Sr0.4CoO3-δ | T = 600 °C S/E = 3 O2/E = 0.5 %C2H5OH = 4.4% | 3.6 | 96% after 5 h | 2.1 | [142] |
| Mg2AlNi3HzOy | T = 260 °C S/E = 3 O2/E = 1.6 %C2H5OH = 14.4% | 81.9 | 100% after 75 h | 0.31 | [143] |
| NiCo-MgAl (Ni+Co = 20 wt.%) Conventional synthesis | T = 550 °C S/E = 3 O2/E = 0.5 %C2H5OH = 12.8% | 47.3 | 100% after 100 h | 0.072 | [144] |
| NiCo-5PrMgAl | T = 550 °C S/E = 3 O2/E = 0.5 %C2H5OH = 12.8% | 47.3 | 100% after 100 h | 0.044 | |
| NiCo-5CeMgAl | T = 550 °C S/E = 3 O2/E = 0.5 %C2H5OH = 12.8% | 47.3 | 100% after 100 h | 0.049 | |
| La2Ce1.8Ru0.2O7/La2Zr2O7 | T = 400 °C S/E = 3 O2/E = 0.6 %C2H5OH = 14.6% | 28.1 | 100% after 100 h | 0.0013 | [145] |
| MgxLa2-xCe1.8Ru0.2O7/La2Zr2O7-δ | T = 400 °C S/E = 3 O2/E = 0.6 %C2H5OH = 14.6% | 28.1 | 100% after 100 h | 0.00021 | |
| CaxLa2-xCe1.8Ru0.2O7/La2Zr2O7-δ | T = 400 °C S/E = 3 O2/E = 0.6 %C2H5OH = 14.6% | 28.1 | 100% after 100 h | 0.0011 | |
| Ethanol dry reforming | |||||
| 1Rh/CeO2 | T = 700 °C CO2/E = 1 - | 18.5 | 88% after 65 h | 0.0089 | [63] |
| 1Rh/CeO2 | T = 700 °C CO2/E = 3 - | 18.5 | 100% after 65 h | 0.0033 | |
| 2Rh/CeO2 | T = 700 °C CO2/E = 1 - | 4.6 | 100% after 70 h | 0.035 | [82] |
| 15Cu/CeO2 | T = 700 °C CO2/E = 1 %C2H5OH = 30% | 6.2 | 75% after 90 h | 0.0061 | [98] |
| 15Cu/ZrO2 | T = 700 °C CO2/E = 1 %C2H5OH = 30% | 6.2 | 64% after 90 h | 0.0093 | |
| 15Cu/CeO2-ZrO2 Ce/Zr mol ratio = 1 | T = 700 °C CO2/E = 1 %C2H5OH = 30% | 6.2 | 100% after 90 h | 0.0045 | |
| 10Co/Al2O3 | T = 700 °C CO2/E = 1 %C2H5OH = 20% | 17.2 | 20% after 8 h | 8.9 | [123] |
| 2Ce-10Co/Al2O3 | T = 700 °C CO2/E = 1 %C2H5OH = 20% | 17.2 | 38% after 8 h | 6.6 | |
| 3Ce-10Co/Al2O3 | T = 700 °C CO2/E = 1 %C2H5OH = 20% | 17.2 | 50% after 8 h | 4.7 | |
| 4Ce-10Co/Al2O3 | T = 700 °C CO2/E = 1 %C2H5OH = 20% | 17.2 | 37% after 8 h | 5.1 | |
| 5Ce-10Co/Al2O3 | T = 700 °C CO2/E = 1 %C2H5OH = 20% | 17.2 | 34% after 8 h | 5.2 | |
| 10Co/Al2O3 | T = 700 °C CO2/E = 1 %C2H5OH = 20% | 17.2 | 50% after 72 h | 0.12 | [124] |
| 3La10Co/Al2O3 | T = 700 °C CO2/E = 1 %C2H5OH = 20% | 17.2 | 30% after 72 h | 0.078 | |
| 10Ni/SiO2-Al2O3 | T = 750 °C CO2/E = 1.4 - | 1.8 | 97% after 10 h | 2.8 | [146] |
| 10Ni/ Al2O3 calcined at 500 °C | T = 700 °C CO2/E = 3 - | 36.9 | 100% after 4 h | 5.5 | [147] |
| 10Ni/ Al2O3 calcined at 600 °C | T = 700 °C CO2/E = 3 - | 36.9 | 100% after 4 h | 6.7 | |
| 10Ni/ Al2O3 calcined at 700 °C | T = 700 °C CO2/E = 3 - | 36.9 | 100% after 4 h | 9.7 | |
| Catalyst a | Operative Conditions b | T (°C) | WHSV c (h−1) | X MeOH (%) | Carbon Formation Rate d (MULTIPLIED for 1000) | Ref. |
|---|---|---|---|---|---|---|
| Methanol Steam Reforming | ||||||
| 15%Cu-MCM-41 | H2O/CH3OH = 3/1 | 250 | 1.0 | ≈73 after 48 h | 0.015 | [158] |
| 20%Cu-MCM-41 | H2O/CH3OH = 3/1 | 250 | 1.0 | ≈60 after 48 h | 0.024 | [158] |
| Cu-ZnO-Al2O3-ZrO2-Ga2O3 Cu/Zn/Al/Zr/Ga = 14.9:30.9:3.9:10.8:1.9 mass ratio | H2O/CH3OH = 1/1 | 275 | 4.3 | ≈85 after 70 h | 0.010 | [159] |
| Cu-ZnO-Al2O3-ZrO2-Ga2O3 Cu/Zn/Al/Zr/Ga = 13.3:28.2:3.9:10.0:1.8 mass ratio | H2O/CH3OH = 1/1 | 275 | 4.3 | ≈70 after 70 h | 0.014 | [159] |
| Pd-MCM-41 | H2O/CH3OH = 3/1 | 300 | 1.0 | ≈32 after 40 h | 0.041 | [163] |
| Zn-MCM-41 | H2O/CH3OH = 3/1 | 300 | 1.0 | ≈5 after 40 h | 0.15 | [163] |
| Ni-MCM-41 | H2O/CH3OH = 3/1 | 300 | 1.0 | ≈15 after 40 h | 0.16 | [163] |
| Cu-MCM-41 | H2O/CH3OH = 3/1 | 300 | 3.0 | ≈75 after 40 h | 0.049 | [163] |
| Cu/Al2O3 | H2O/CH3OH = 3/2 | 500 | 0.8 | ≈91 after 5 h | 10 | [165] |
| NixMgyO Impregnation | H2O/CH3OH = 1/1 | 600 | 65.7 | 51.4 after 20 h | 0.0079 | [168] |
| NixMgyO Hydrothermal method | H2O/CH3OH = 1/1 | 600 | 65.7 | 58.3 after 20 h | 0.0030 | [168] |
| NixMgyO Co-precipitation | H2O/CH3OH = 1/1 | 600 | 65.7 | 57.3 after 20 h | 0.085 | [168] |
| Cu/cubic-ZnTiO3 | N2/H2O/CH3OH = 1/2/1 | 250 | 1 | ≈63 after 42 h | 1.1 | [177] |
| Cu/hexagonal-ZnTiO3 | N2/H2O/CH3OH = 1/2/1 | 250 | 1 | ≈5 after 42 h | 6.7 | [177] |
| 10%Cu-10%Zn/MCM-41 | H2O/CH3OH = 2/1 | 300 | 1.62 | ≈75 after 60 h | 0.0011 | [183] |
| 10%Cu-10%Zn-2%Zr/MCM-41 Impregnated | H2O/CH3OH = 2/1 | 300 | 1.62 | ≈83 after 60 h | 0.00095 | [183] |
| 10%Cu-10%Zn-2%Zr/MCM-41 Sol–gel method | H2O/CH3OH = 2/1 | 300 | 1.62 | 90.2 after 60 h | 0.00079 | [183] |
| 10%Cu-10%Zn-2%Zr/MCM-41 MCM-41 pretreated with acetic acid | H2O/CH3OH = 2/1 | 300 | 1.62 | 92.8 after 60 h | 0.00063 | [183] |
| 10%Cu/SBA-15 | H2O/CH3OH = 2/1 | 300 | 43.7 | ≈64 after 60 h | 0.0031 | [198] |
| 10%Cu/5%ZnO/SBA-15 | H2O/CH3OH = 2/1 | 300 | 43.7 | ≈74 after 60 h | 0.0025 | [198] |
| 10%Cu/5%ZnO/2%CeO2/SBA-15 | H2O/CH3OH = 2/1 | 300 | 43.7 | ≈85 after 60 h | 0.0013 | [198] |
| 10%Cu/5%ZnO/2%ZrO2/SBA-15 | H2O/CH3OH = 2/1 | 300 | 43.7 | ≈84 after 60 h | 0.0031 | [198] |
| 10%Cu/5%ZnO/2%CeO2/2%ZrO2/SBA-15 | H2O/CH3OH = 2/1 | 300 | 43.7 | ≈86 after 60 h | 0.0031 | [198] |
| Oxidative Steam Reforming of Methanol | ||||||
| xAu/CeO2–Fe2O3 x = 3 wt.%, Ce/(Ce+Fe) = 0.25. | O2/H2O/CH3OH = 0.6/2/1. | 350 | 11.9 | 91 after 12 h | 0.098 | [209] |
| xAu/CeO2–Fe2O3 x = 3 wt.%, Ce/(Ce+Fe) = 0.25. | O2/H2O/CH3OH = 0.6/2/1. O2 pretreatment | 350 | 11.9 | 92 after 12 h | 0.0082 | [209] |
| 9 wt.%Cu/CeO2 | H2O/CH3OH = 2/1; O2 = 5 mL·min−1 | 350 | 0.9 | 65 after 2.8 h | 0.0042 | [212] |
| 9 wt.%Cu/CeZrO4 | H2O/CH3OH = 2/1; O2 = 5 mL·min−1 | 300 | 0.9 | 65 after 2.8 h | 0.0032 | [212] |
| Catalyst a | Operative Conditions b | T (°C) | WHSV c (h−1) | X Glycerol (%) | Carbon Formation Rate d (Multiplied for 1000) | Ref. |
|---|---|---|---|---|---|---|
| Glycerol steam reforming | ||||||
| 8Ni/Al2O3 8Ni/ 4La2O3-Al2O3 | mcat = 200 mg 31 v.v. % C3H8O3 and H2O (63% H2O, 7% C3H8O3 and 30% He) TOS = 4 h | 700 | GHSV = 50,000 mL g−1h−1 | ~75 ~80 After 4 h | 0.141 0.152 | [221] |
| Rh/alumina | 0.1 mL min−1 of aqueous glycerol, P = 4.5 bar mcat = 800 mg TOS = 13 h | 400 | 7.8 | ~92 After 13 h | 0.045 | [227] |
| 14.5Ni/SBA-15 14.5Ni-4Co/SBA-15 14.3Ni-3.6Cr/SBA-15 15Ni-4Cu/SBA-15 | S/C = 2 Water/glycerol = 6 mol/mol mcat = 300 mg TOS = 5 h | 600 | 7.7 | ~92 ~94 ~95 ~88 After 5 h | 27.879 5.092 3.009 51.157 | [231] |
| 8Ni/Al2O3 8Ni/CaO-MgO-Al2O3 | mcat = 200 mg 31 v.v. % C3H8O3 and H2O (63% H2O, 7% C3H8O3 and 30% He) TOS = 20 h | 600 | GHSV = 50,000 mL g−1h−1 | ~70 ~80 After 20 h | 0.057 0.048 | [241] |
| 10% CeO2 addition to NiAl2O4 spinel | mcat = 500 mg Water/glycerol = 9 (glycerol solution)/Ar = 1 TOS = 16 h | 630 | GHSV = 19600 cm3gcat−1h−1 | 90 After 16 h | 0.16 | [245] |
| 10Ni/Al2O3 10Ni/Al2O3/5CeO2 | mcat = 1 g 30 wt.% glycerol feed TOS = 16 h | 650 | 12 | - | 0.00067 0.000424 | [248] |
| 7Co/SBA-15 7Co-8.5Zr/SBA-15 7Co-8.5Ce/SBA-15 7Co-8.5La/SBA-15 | S/C = 2 Water/glycerol = 6 mol/mol mcat = 300 mg TOS = 5 h | 600 | 7.7 | ~75 >90 >90 >90 After 5 h | 5.092 5.555 5.555 4.629 | [249] |
| 12.5Ni-UGS | mcat = 500 mg S/C = 3 Water/glycerol = 9 (Water+glycerol)/Ar = 1:4 TOS = 48 h | 580 | GHSV = 20600 cm3gcat−1h−1 | 90 After 48 h | 0.17 | [251] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palma, V.; Ruocco, C.; Cortese, M.; Martino, M. Bioalcohol Reforming: An Overview of the Recent Advances for the Enhancement of Catalyst Stability. Catalysts 2020, 10, 665. https://doi.org/10.3390/catal10060665
Palma V, Ruocco C, Cortese M, Martino M. Bioalcohol Reforming: An Overview of the Recent Advances for the Enhancement of Catalyst Stability. Catalysts. 2020; 10(6):665. https://doi.org/10.3390/catal10060665
Chicago/Turabian StylePalma, Vincenzo, Concetta Ruocco, Marta Cortese, and Marco Martino. 2020. "Bioalcohol Reforming: An Overview of the Recent Advances for the Enhancement of Catalyst Stability" Catalysts 10, no. 6: 665. https://doi.org/10.3390/catal10060665
APA StylePalma, V., Ruocco, C., Cortese, M., & Martino, M. (2020). Bioalcohol Reforming: An Overview of the Recent Advances for the Enhancement of Catalyst Stability. Catalysts, 10(6), 665. https://doi.org/10.3390/catal10060665