Synthesis of Ce1−xPdxO2−δ Solid Solution in Molten Nitrate

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
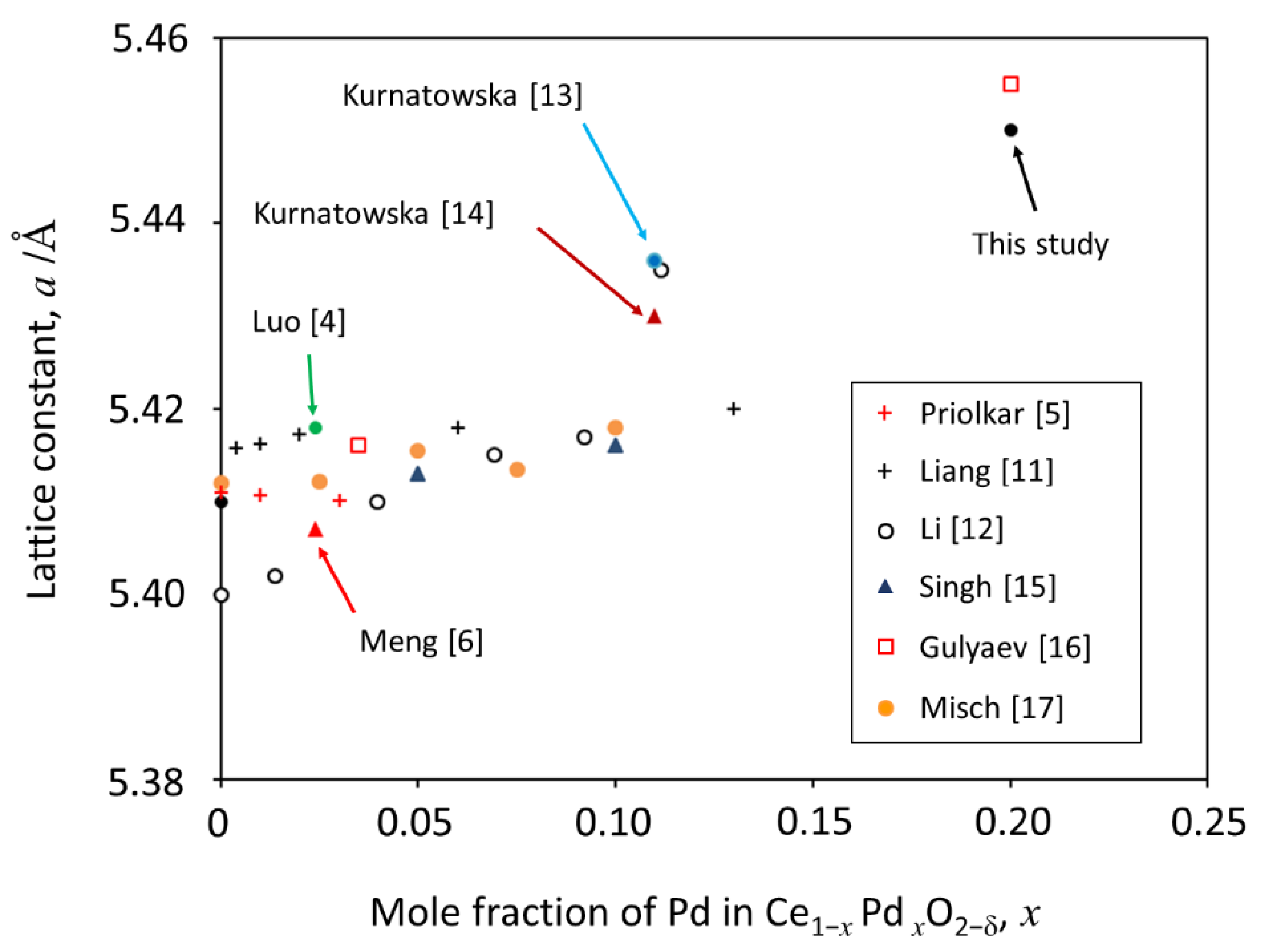
2.1. Crystal Structures of PdO−CeO2 Powders
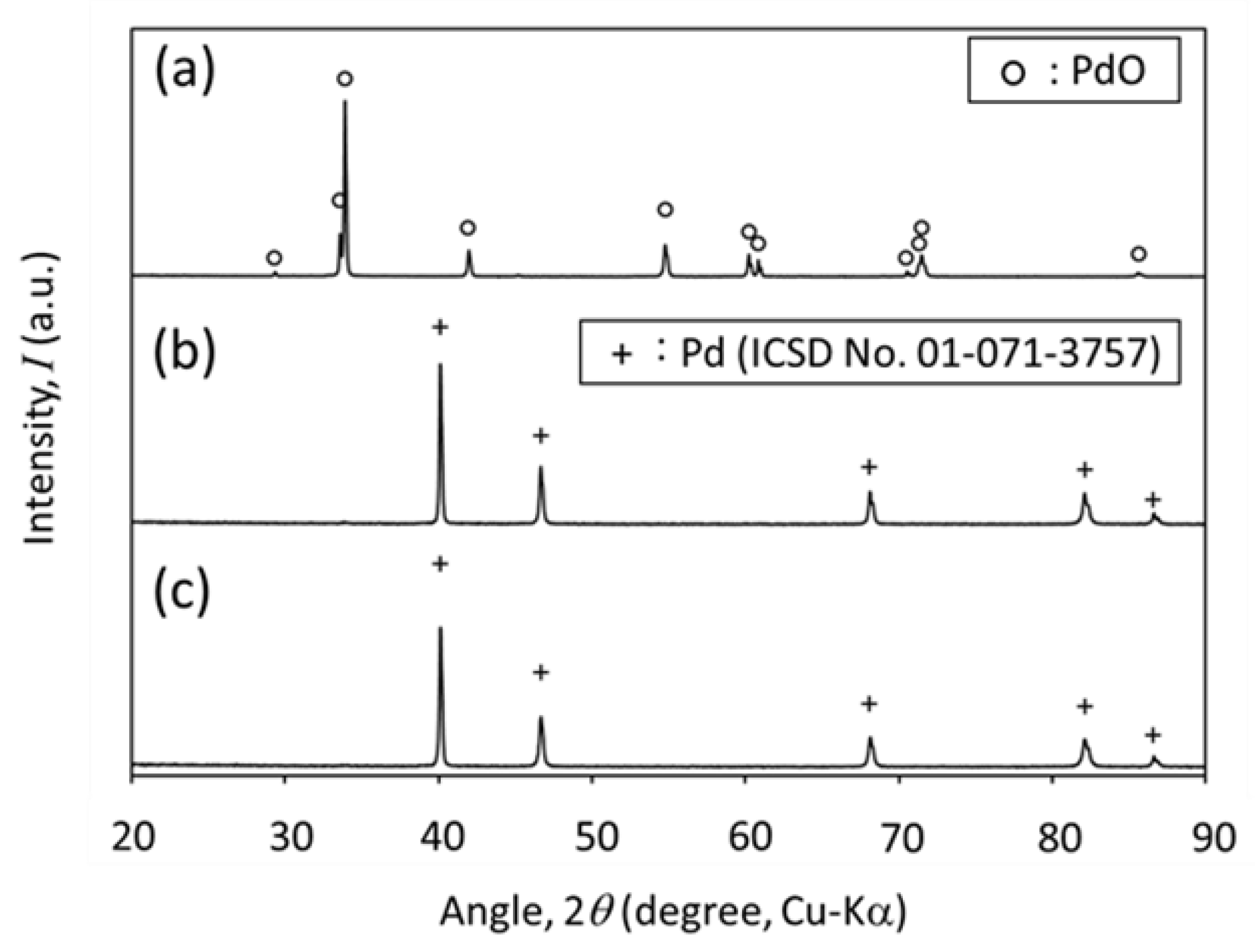
2.2. Thermal Decomposition of PdO−CeO2
2.3. Microscopic Observation
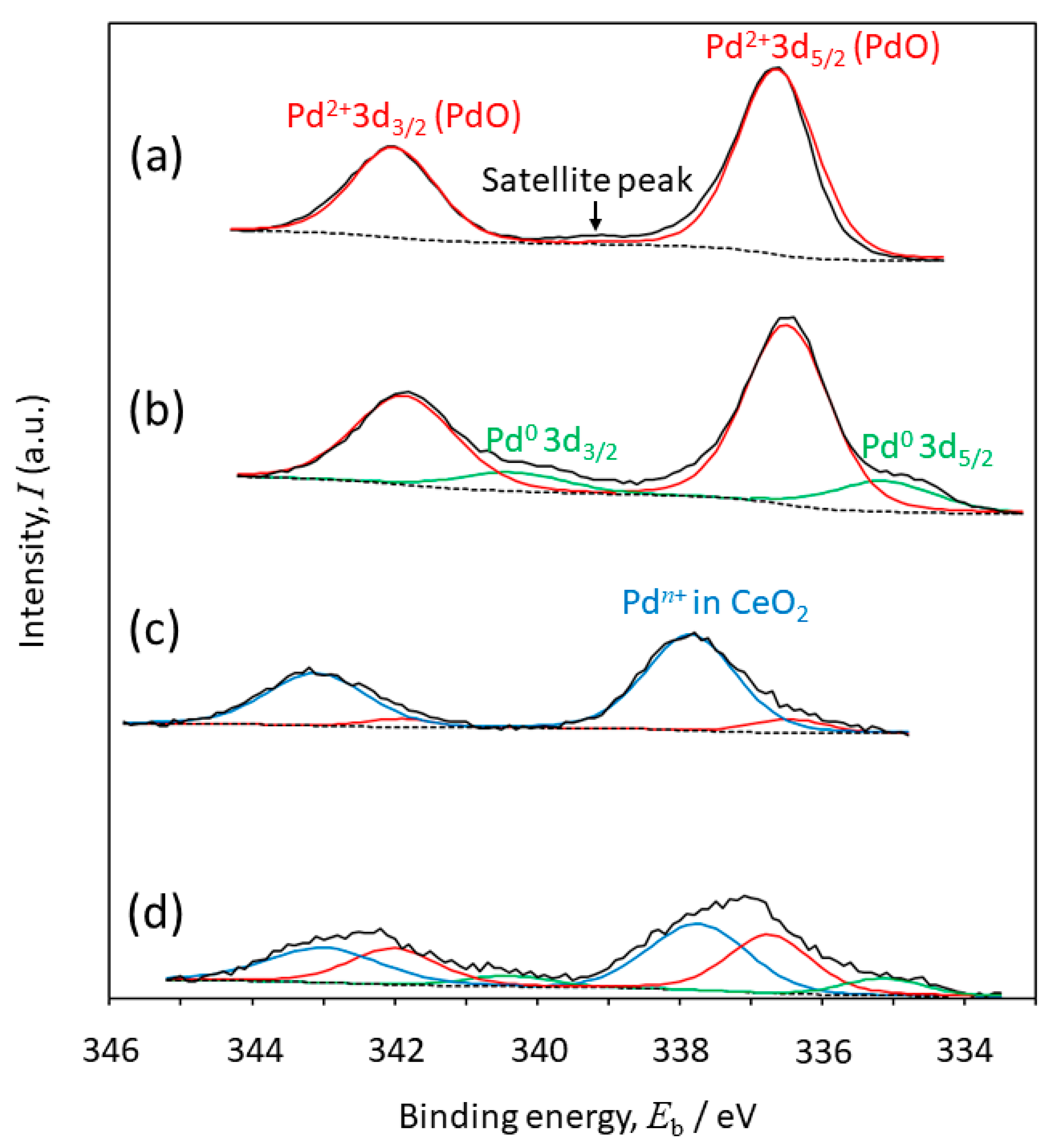
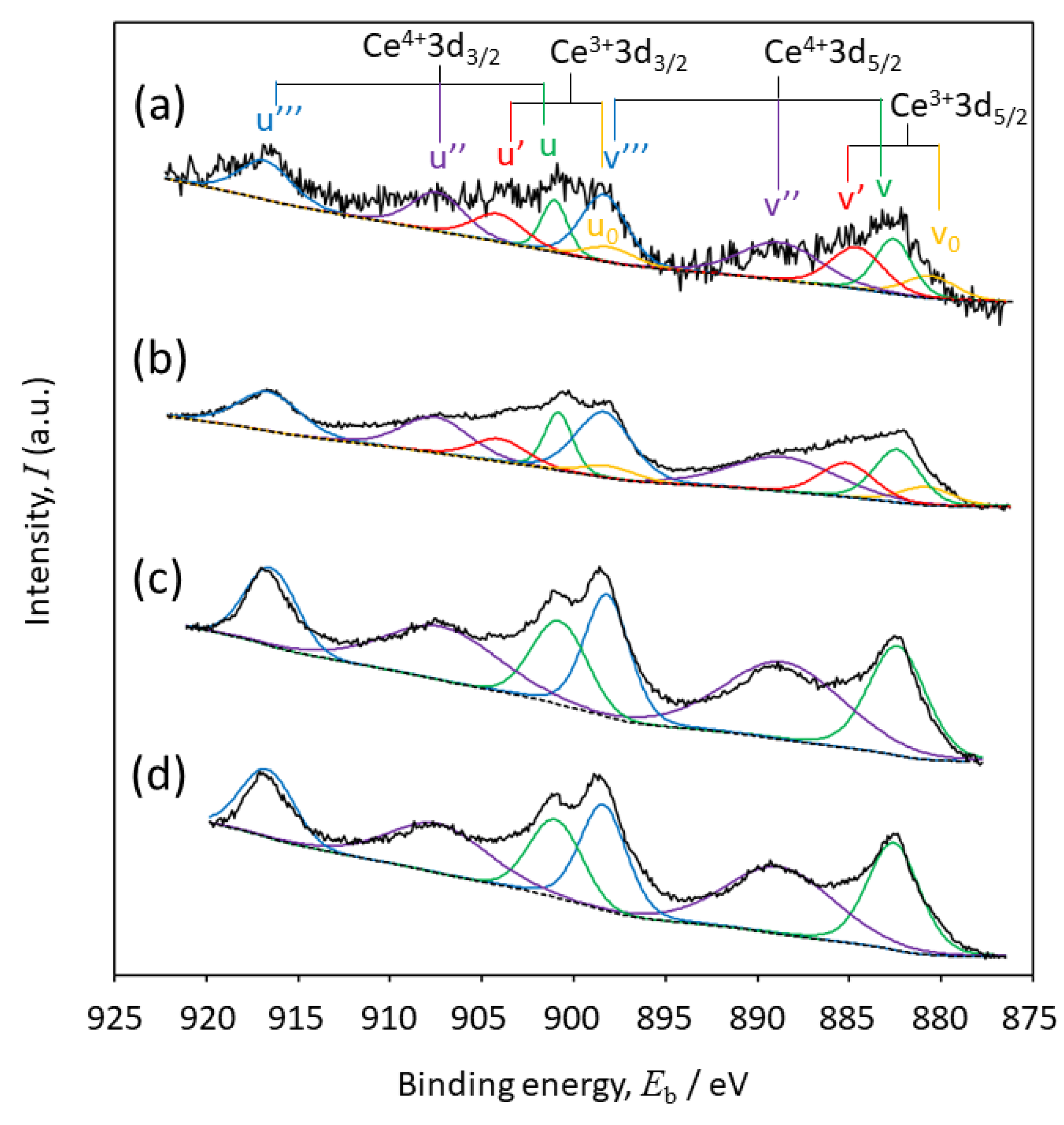
2.4. Oxidation States
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hegde, M.S.; Bera, P. Noble metal ion substituted CeO2 catalysts: Electronic interaction between noble metal ions and CeO2 lattice. Catal. Today 2015, 253, 40–50. [Google Scholar] [CrossRef]
- Feio, L.S.F.; Hori, C.E.; Mattos, L.V.; Zanchet, D.; Noronha, F.B.; Bueno, J.M.C. Partial oxidation and autothermal reforming of methane on Pd/CeO2–Al2O3 catalysts. Appl. Catal. A 2008, 348, 183–192. [Google Scholar] [CrossRef]
- Ryou, Y.; Lee, J.; Lee, H.; Kim, C.H.; Kim, D.H. Effect of sulfur aging and regeneration on low temperature NO adsorption over hydrothermally treated Pd/CeO2 and Pd/Ce0.58Zr0.42O2 catalysts. Catal. Today 2017, 297, 53–59. [Google Scholar] [CrossRef]
- Luo, Y.; Xiao, Y.; Cai, G.; Zheng, Y.; Wei, K. Complete methanol oxidation in carbon monoxide streams over Pd/CeO2 catalysts: Correlation between activity and properties. Appl. Catal. B 2013, 136–137, 317–324. [Google Scholar] [CrossRef]
- Priolkar, K.R.; Bera, P.; Sarode, P.R.; Hegde, M.S.; Emura, S.; Kumashiro, R.; Lalla, N.P. Formation of Ce1−xPdxO2−δ solid solution in combustion-synthesized Pd/CeO2 catalyst: XRD, XPS, and EXAFS investigation. Chem. Mater. 2002, 14, 2120–2128. [Google Scholar] [CrossRef]
- Meng, L.; Lin, J.; Pu, Z.; Luo, L.; Jia, A.; Huang, W.; Luo, M.; Lu, J. Identification of active sites for CO and CH4 oxidation over PdO/Ce1−xPdxO2−δ catalysts. Appl. Catal. B 2012, 119–120, 117–122. [Google Scholar] [CrossRef]
- Bera, P.; Patil, K.C.; Jayaram, V.; Subbanna, G.N.; Hegde, M.S. Ionic dispersion of Pt and Pd on CeO2 by combustion method: Effect of metal-ceria interaction on catalytic activities for NO reduction and CO and hydrocarbon oxidation. J. Catal. 2000, 196, 293–301. [Google Scholar] [CrossRef]
- Sánchez-Rodríguez, D.; Yamaguchi, S.; Ihara, D.; Yamaura, H.; Yahiro, H. Self-propagating high-temperature synthesis of highly dispersed noble metals on ceria powder: Application to Pd/CeO2 catalyst. Ceram. Int. 2017, 43, 14533–14536. [Google Scholar] [CrossRef]
- Khader, M.M.; Al-Marri, M.J.; Ali, S.; Abdelmoneim, A.G. Active and stable methane oxidation nano-catalyst with highly-ionized palladium species prepared by solution combustion synthesis. Catalysts 2018, 8, 66. [Google Scholar] [CrossRef]
- Slavinskaya, E.M.; Gulyaev, R.V.; Zadesenets, A.V.; Stonkus, O.A.; Zaikovskii, V.I.; Shubin, Y.V.; Korenev, S.V.; Boronin, A.I. Low-temperature CO oxidation by Pd/CeO2 catalysts synthesized using the coprecipitation method. Appl. Catal. B 2015, 166–167, 91–103. [Google Scholar] [CrossRef]
- Liang, H.; Raitano, J.M.; He, G.; Akey, A.J.; Herman, I.P.; Zhang, L.; Chan, S. Aqueous co-precipitation of Pd-doped cerium oxide nanoparticles: Chemistry, structure, and particle growth. J. Mater. Sci. 2012, 47, 299–307. [Google Scholar] [CrossRef]
- Li, G.; Li, L.; Jiang, D. Facile synthesis of highly active mesoporous PdCeOx solid solution for low-temperature CO oxidation. J. Phys. Chem. C 2015, 119, 12502–12507. [Google Scholar] [CrossRef]
- Kurnatowska, M.; Kepinski, L.; Mista, W. Structure evolution of nanocrystalline Ce1−xPdxO2−y mixed oxide in oxidizing and reducing atmosphere: Reduction-induced activity in low-temperature CO oxidation. Appl. Catal. B 2012, 117–118, 135–147. [Google Scholar] [CrossRef]
- Kurnatowska, M.; Schuster, M.E.; Mista, W.; Kepinski, L. Self-regenerative property of nanocrystalline Ce0.89M0.11O2−y (M=Pd, Rh) mixed oxides. ChemCatChem 2014, 6, 3125–3131. [Google Scholar] [CrossRef]
- Singh, P.; Hegde, M.S. Sonochemical synthesis of thermally stable hierarchical Ce1−xMxO2−δ (M=Pt or Pd, 0 ≤ x ≤ 0.10) nanocrystallites: Redox properties and methanol electro-oxidation activity. Cryst. Growth Des. 2010, 10, 2995–3004. [Google Scholar] [CrossRef]
- Gulyaev, R.V.; Slavinskaya, E.M.; Novopashin, S.A.; Smovzh, D.V.; Zaikovskii, A.V.; Osadchii, D.Y.; Bulavchenko, O.A.; Korenev, S.V.; Boronin, A.I. Highly active PdCeOx composite catalysts for low-temperature CO oxidation, prepared by plasma-arc synthesis. Appl. Catal. B 2014, 147, 132–143. [Google Scholar] [CrossRef]
- Misch, L.M.; Kurzman, J.A.; Derk, A.R.; Kim, Y.; Seshadri, R.; Metiu, H.; McFarland, E.W.; Stucky, G.D. C−H bond activation by Pd-substituted CeO2: Substituted ions versus reduced species. Chem. Mater. 2011, 23, 5432–5439. [Google Scholar] [CrossRef]
- Sasaki, H.; Matsushita, H.; Sakamoto, K.; Sakamoto, T.; Maeda, M. Formation of Pt3O4 particles on PtO2−CeO2 solid solution. J. Phys. Chem. Solids 2019, 135, 109097. [Google Scholar] [CrossRef]
- Voorhees, V.; Adams, R. The use of the oxides of platinum for the catalytic reduction of organic compounds. I. J. Am. Chem. Soc. 1922, 44, 1397–1405. [Google Scholar] [CrossRef]
- Adams, R.; Voorhees, V.; Shriner, R.L. Platinum catalyst for reductions. Org. Synth. 1928, 8, 92. [Google Scholar]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chaleogenides. Acta Cryst. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Deshpande, S.; Patil, S.; Kuchibhatla, S.V.; Seal, S. Size dependency variation in lattice parameter and valency states in nanocrystalline cerium oxide. Appl. Phys. Lett. 2005, 87, 133113. [Google Scholar] [CrossRef]
- Hailstone, R.K.; DiFrancesco, A.G.; Leong, J.G.; Allston, T.D.; Reed, K.J. A study of lattice expansion in CeO2 nanoparticles by transmission electron microscopy. J. Phys. Chem. C 2009, 113, 15155–15159. [Google Scholar] [CrossRef]
- Gulyaev, R.V.; Kardash, T.Y.; Malykhin, S.E.; Stonkus, O.A.; Ivanova, A.S.; Boronin, A.I. The local structure of PdxCe1−xO2−x−δ solid solutions. Phys. Chem. Chem. Phys. 2014, 16, 13523–13539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gromek, J.; Fernando, G.W.; Boorse, S.; Marcus, H.L. PdO/Pd system equilibrium phase diagram under a gas mixture of oxygen and nitrogen. J. Phase Equilibria 2002, 23, 246–248. [Google Scholar] [CrossRef]
- Hinokuma, S.; Fujii, H.; Okamoto, M.; Ikeue, K.; Machida, M. Metallic Pd nanoparticles formed by Pd-O-Ce interaction: A reason for sintering-induced activation for CO oxidation. Chem. Mater. 2010, 22, 6183–6190. [Google Scholar] [CrossRef]
- Pillo, T.; Zimmermann, R.; Steiner, P.; Hüfner, S. The electronic structure of PdO found by photoemission (UPS and XPS) and inverse photoemission (BIS). J. Phys. Condens. Matter 1997, 9, 3987–3999. [Google Scholar] [CrossRef]
- Brun, M.; Berthet, A.; Bertolini, J.C. XPS, AES and Auger parameter of Pd and PdO. J. Electron Spectrosc. Relat. Phenom. 1999, 104, 55–60. [Google Scholar] [CrossRef]
- Slavinskaya, E.M.; Boronin, A.I.; Danilova, I.G.; Amosov, Y.I.; Ivanova, A.S.; Kuznetsov, P.A.; Polukhina, I.A.; Gulyaev, R.V.; Stadnichenko, A.I.; Koshcheev, S.V.; et al. Synthesis and physicochemical characterization of palladium-cerium oxide catalysts for the low-temperature oxidation of carbon monoxide. Kinet. Catal. 2009, 50, 819–823. [Google Scholar] [CrossRef]
- Gulyaev, R.V.; Stadnichenko, A.I.; Slavinskaya, E.M.; Ivanova, A.S.; Koscheev, S.V.; Boronin, A.I. In situ preparation and investigation of Pd/CeO2 catalysts for the low-temperature oxidation of CO. Appl. Catal. A 2012, 439–440, 41–50. [Google Scholar] [CrossRef]
- Kim, K.S.; Gossmann, A.F.; Winograd, N. X-ray photoelectron spectroscopic studies of palladium oxides and the palladium-oxygen electrode. Anal. Chem. 1974, 46, 197–200. [Google Scholar] [CrossRef]
- Paparazzo, E. Use and mis-use of X-ray photoemission spectroscopy Ce3d spectra of Ce2O3 and CeO2. J. Phys. Condens. Matter 2018, 30, 343003. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Liu, J.; Filot, I.A.W.; Zhang, L.; Hensen, E.J.M. Highly active and stable CH4 oxidation by substitution of Ce4+ by two Pd2+ ions in CeO2(111). ACS Catal. 2018, 8, 6552–6559. [Google Scholar] [CrossRef] [PubMed]
- Shriner, R.L.; Adams, R. The Preparation of palladous oxide and its use as a catalyst in the reduction of organic compounds. VI. J. Am. Chem. Soc. 1924, 46, 1683–1693. [Google Scholar] [CrossRef]
- Naguib, H.M.; Kelly, R. Criteria for bombardment-induced structural changes in non-metallic solids. Radiat. Eff. 1975, 25, 1–12. [Google Scholar] [CrossRef]
- Holgado, J.P.; Alvarez, R.; Munuera, G. Study of CeO2 XPS spectra by factor analysis: Reduction of CeO2. Appl. Surf. Sci. 2000, 161, 301–315. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sasaki, H.; Sakamoto, K.; Mori, M.; Sakamoto, T. Synthesis of Ce1−xPdxO2−δ Solid Solution in Molten Nitrate. Catalysts 2020, 10, 640. https://doi.org/10.3390/catal10060640
Sasaki H, Sakamoto K, Mori M, Sakamoto T. Synthesis of Ce1−xPdxO2−δ Solid Solution in Molten Nitrate. Catalysts. 2020; 10(6):640. https://doi.org/10.3390/catal10060640
Chicago/Turabian StyleSasaki, Hideaki, Keisuke Sakamoto, Masami Mori, and Tatsuaki Sakamoto. 2020. "Synthesis of Ce1−xPdxO2−δ Solid Solution in Molten Nitrate" Catalysts 10, no. 6: 640. https://doi.org/10.3390/catal10060640
APA StyleSasaki, H., Sakamoto, K., Mori, M., & Sakamoto, T. (2020). Synthesis of Ce1−xPdxO2−δ Solid Solution in Molten Nitrate. Catalysts, 10(6), 640. https://doi.org/10.3390/catal10060640