In Situ Raman Spectroscopy as a Tool for Discerning Subtle Structural Differences between Commercial (Ce,Zr)O2-Based OSC Materials of Identical Composition


 and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Temperature Evolution, Redox Cycling, and Structure Reinstatement
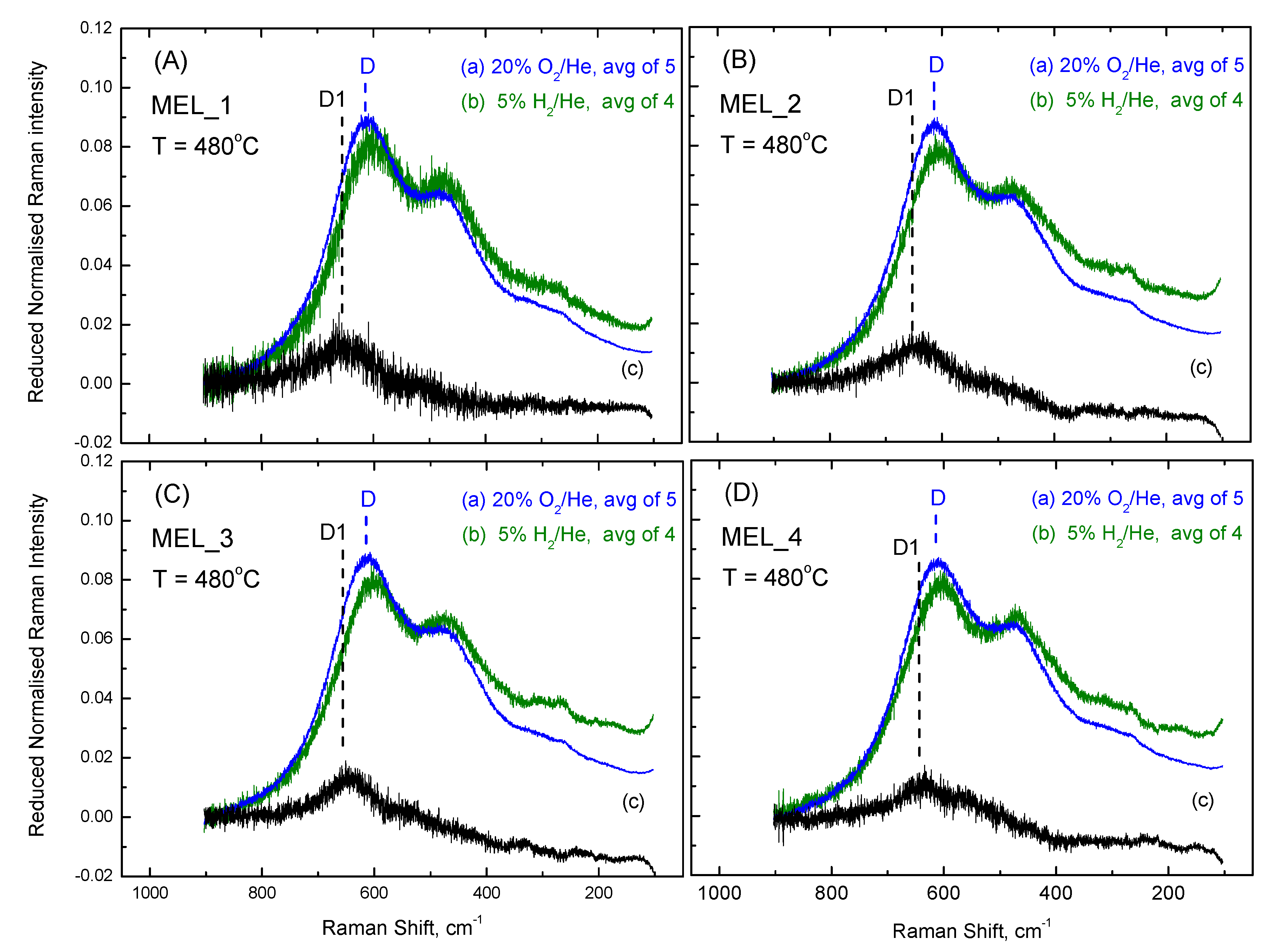
2.2. Assessing Anionic Sublattice Structures of CexZr1−xO2−δ-Based Materials as “Identical”
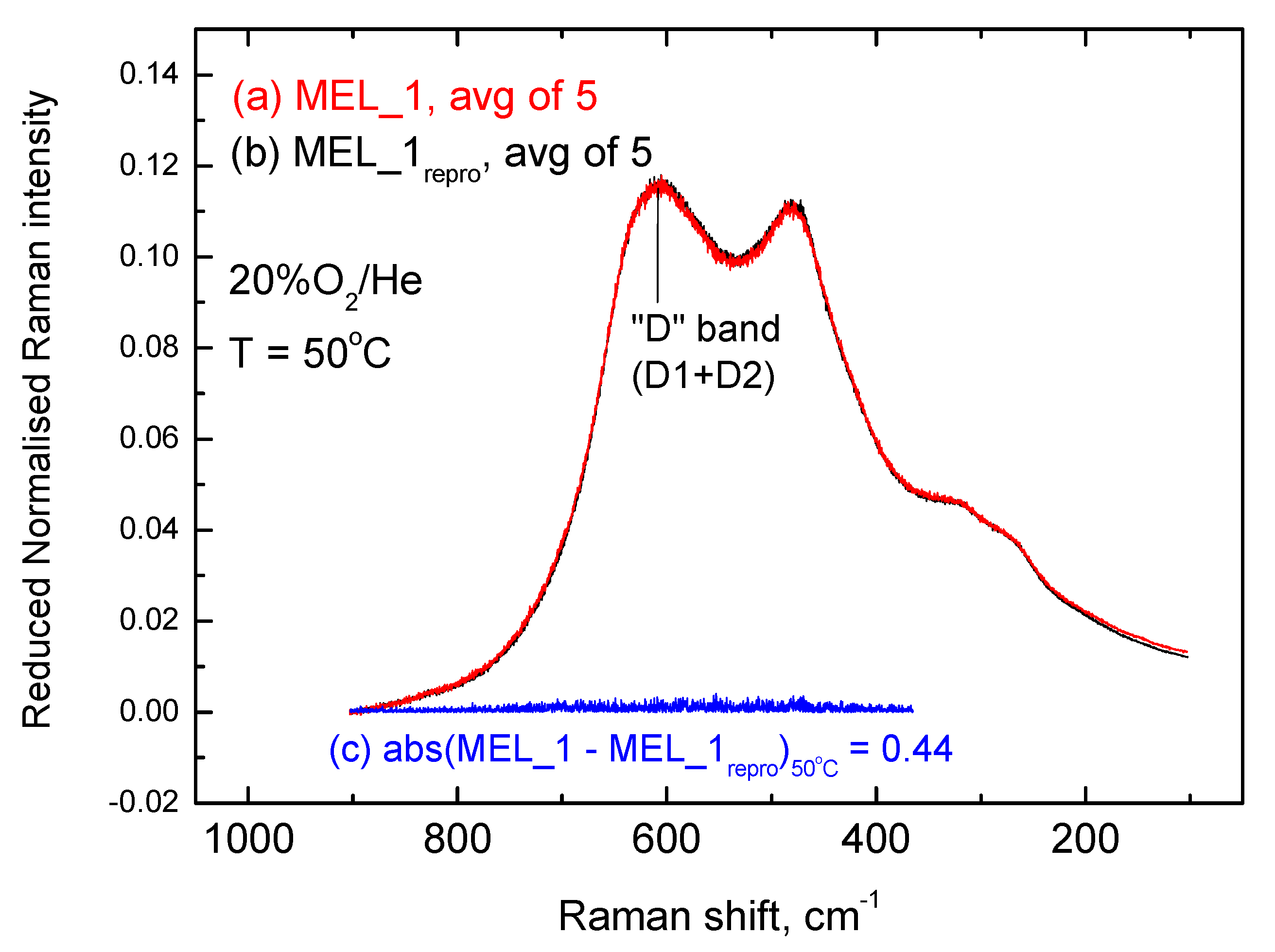
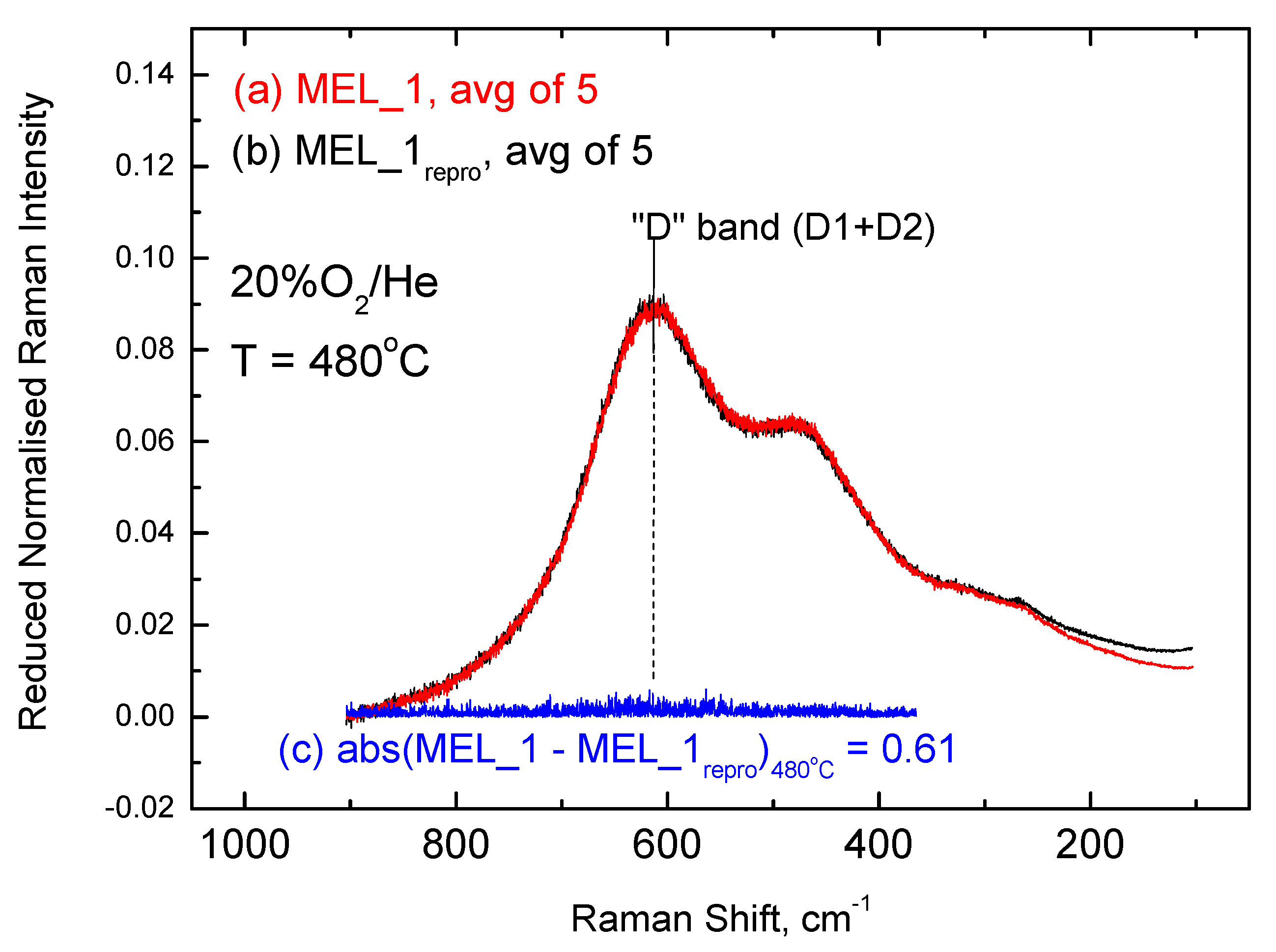
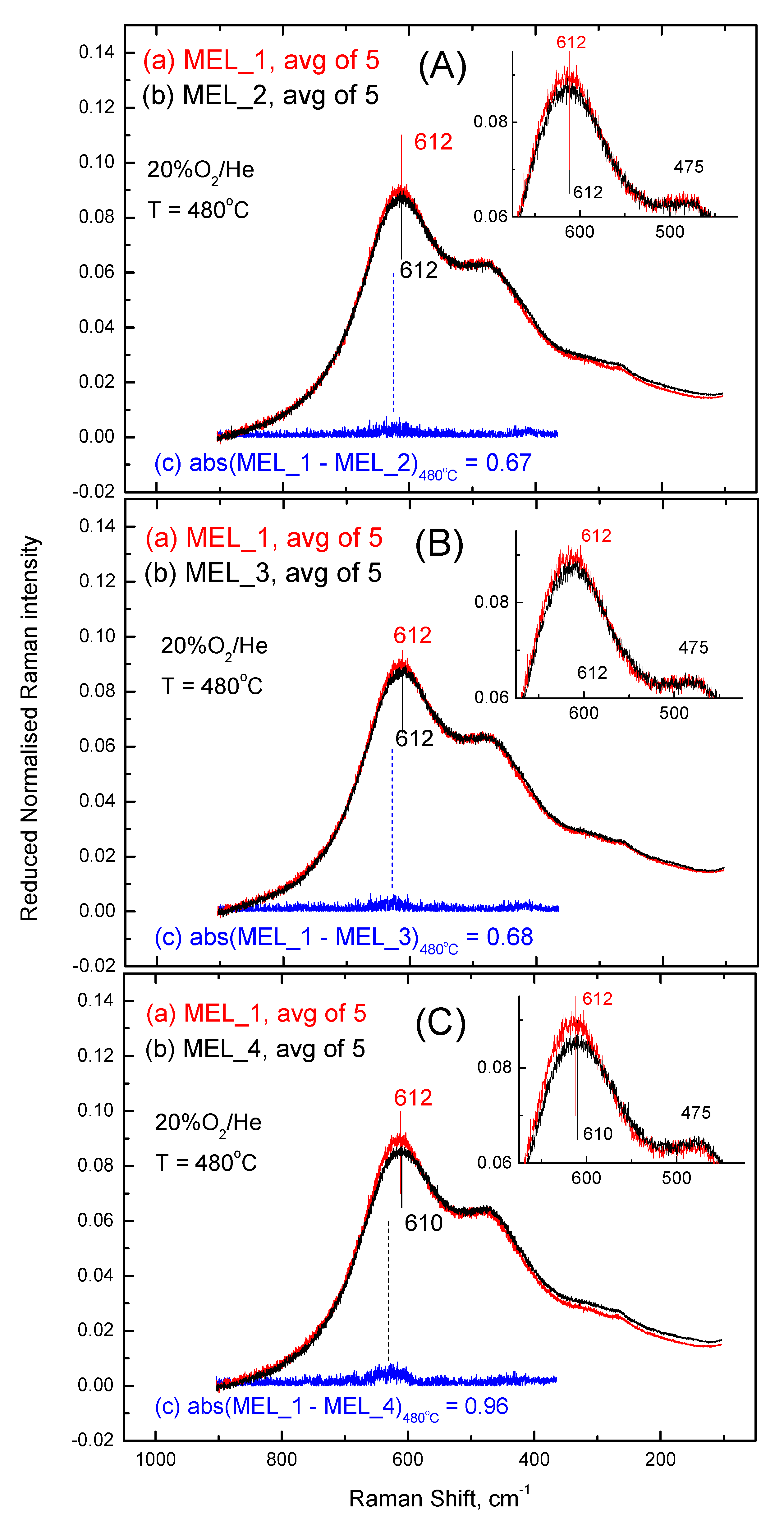
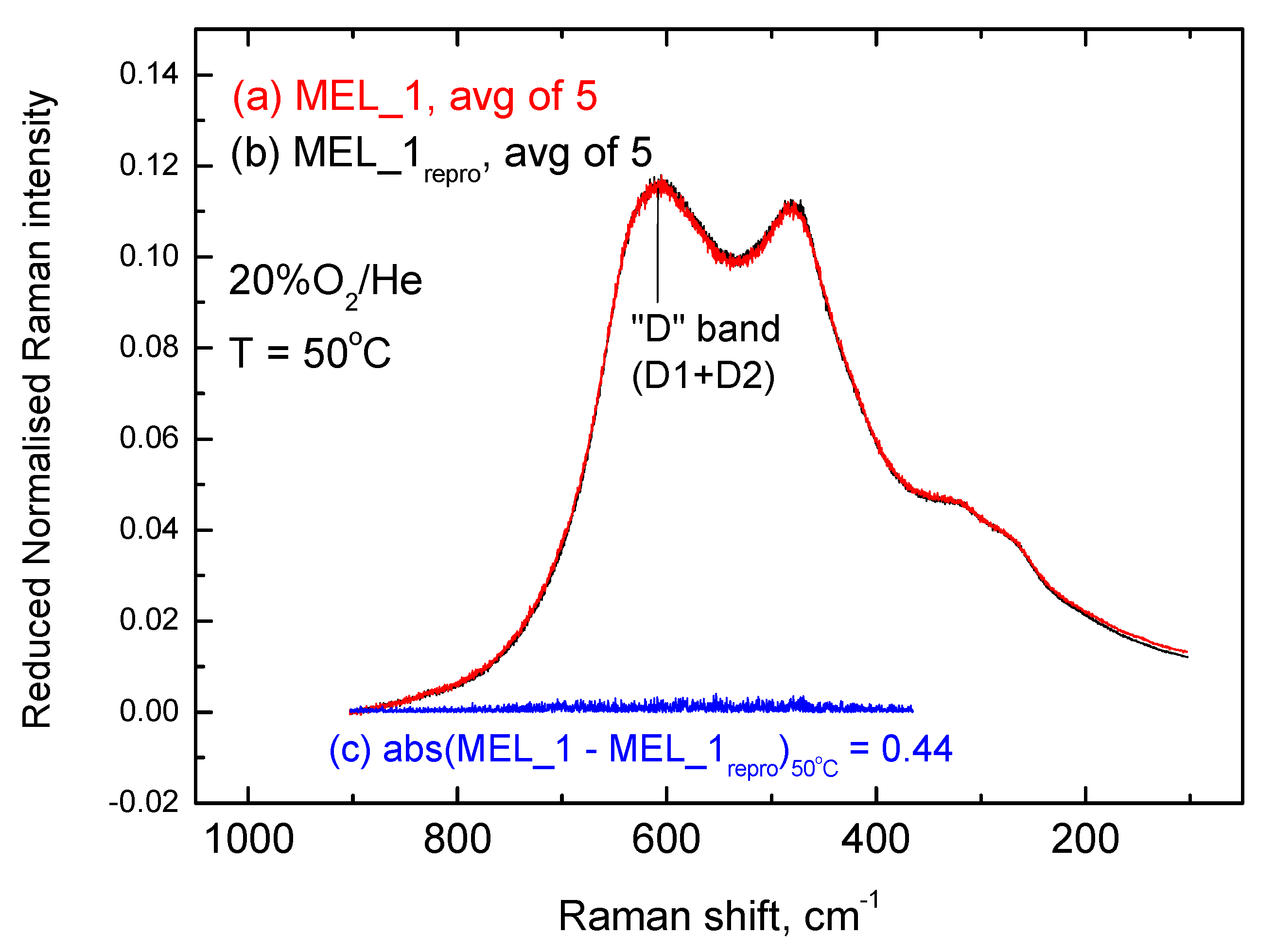
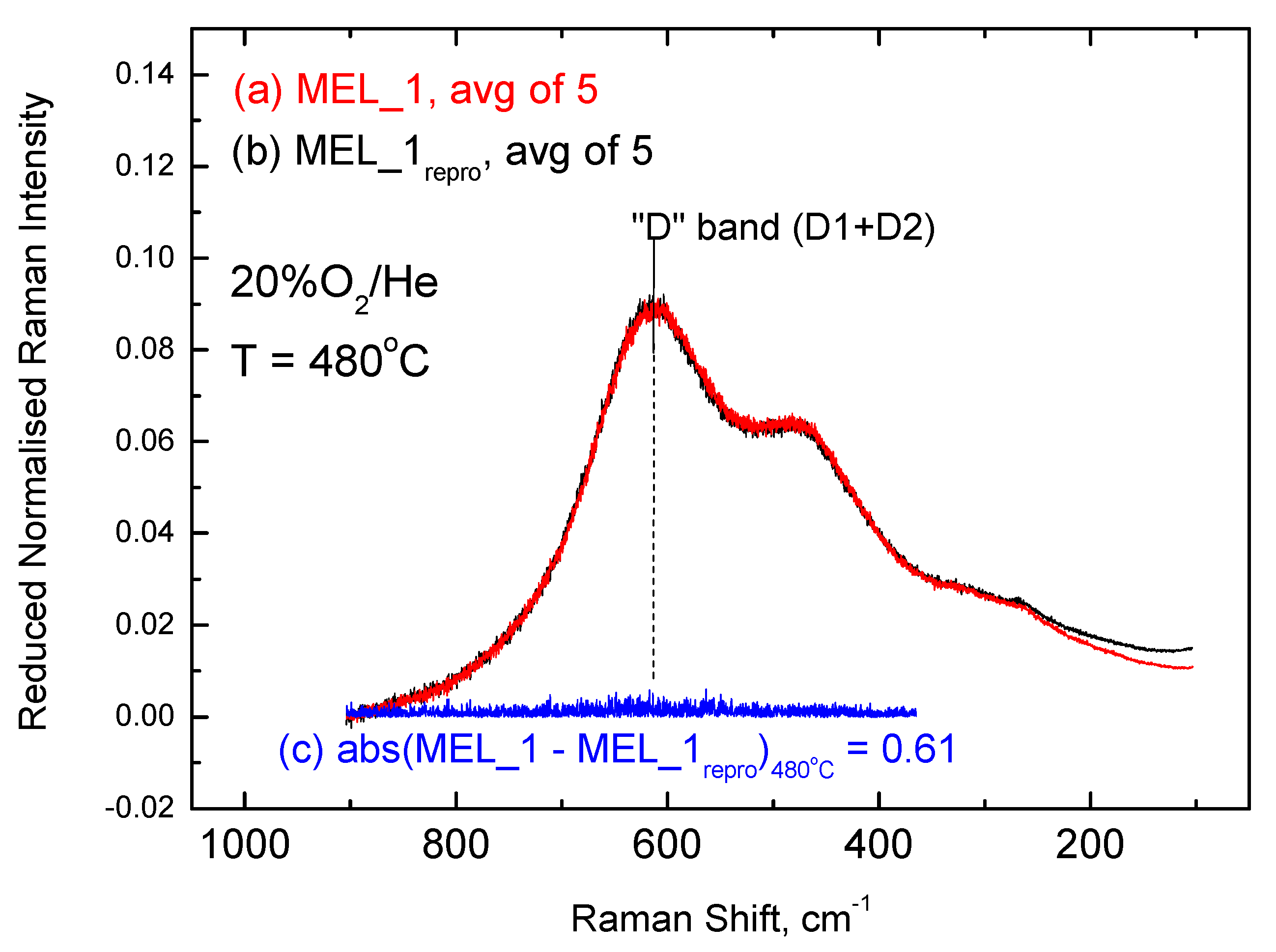
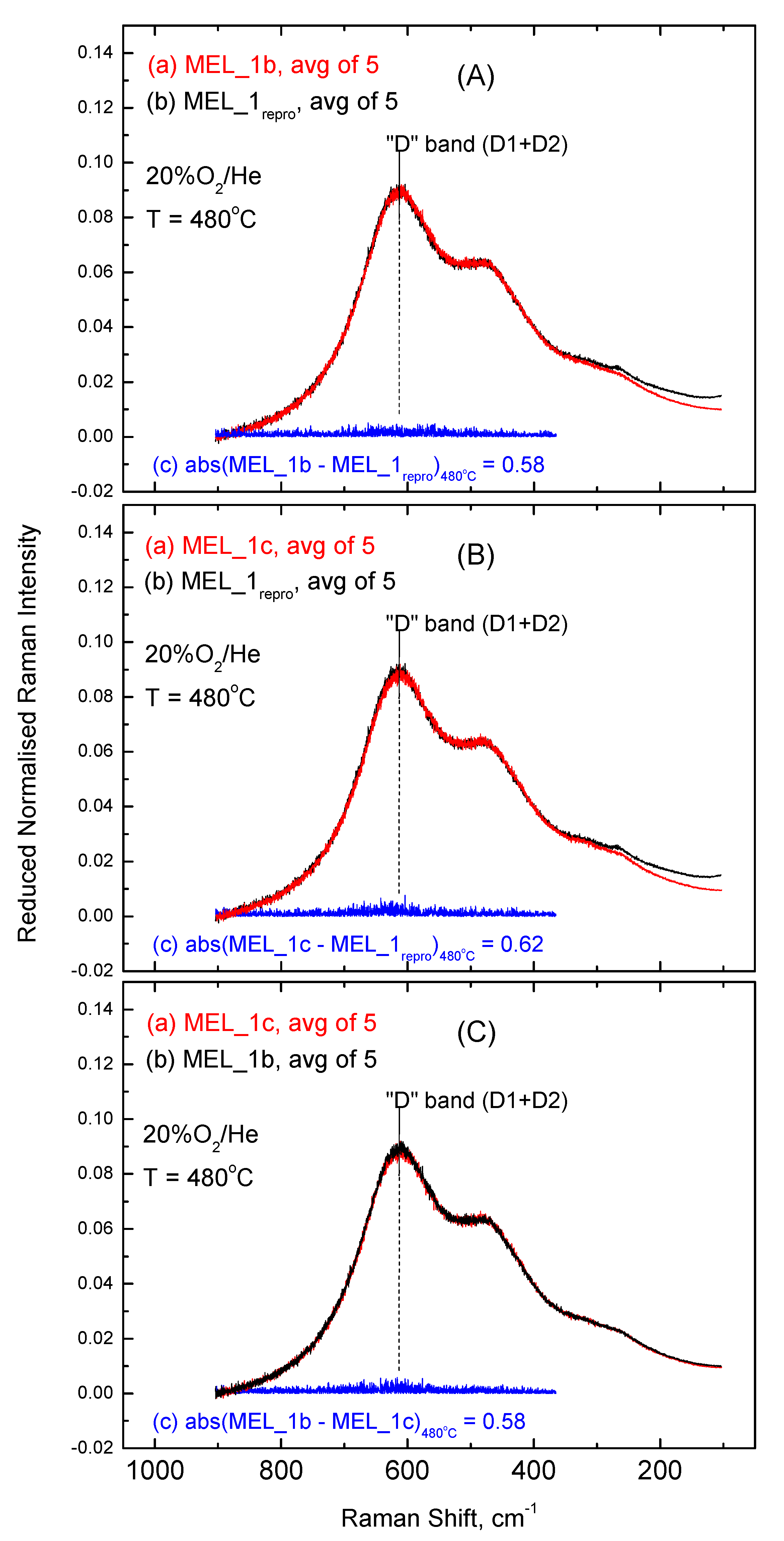
2.3. Inter-Batch Comparisons for Sample MEL_1 at 480 °C under 20% O2/He Gas Atmosphere
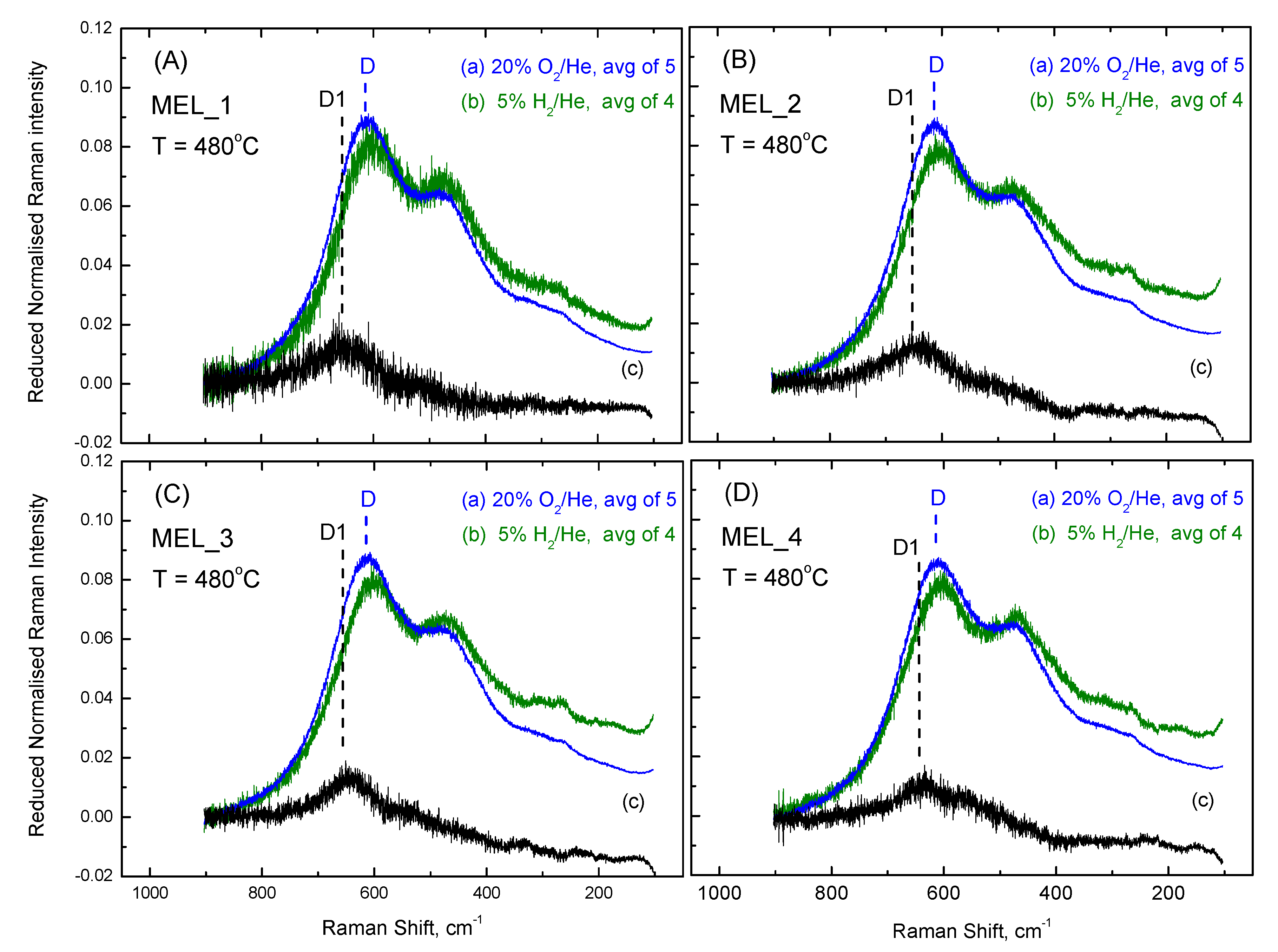
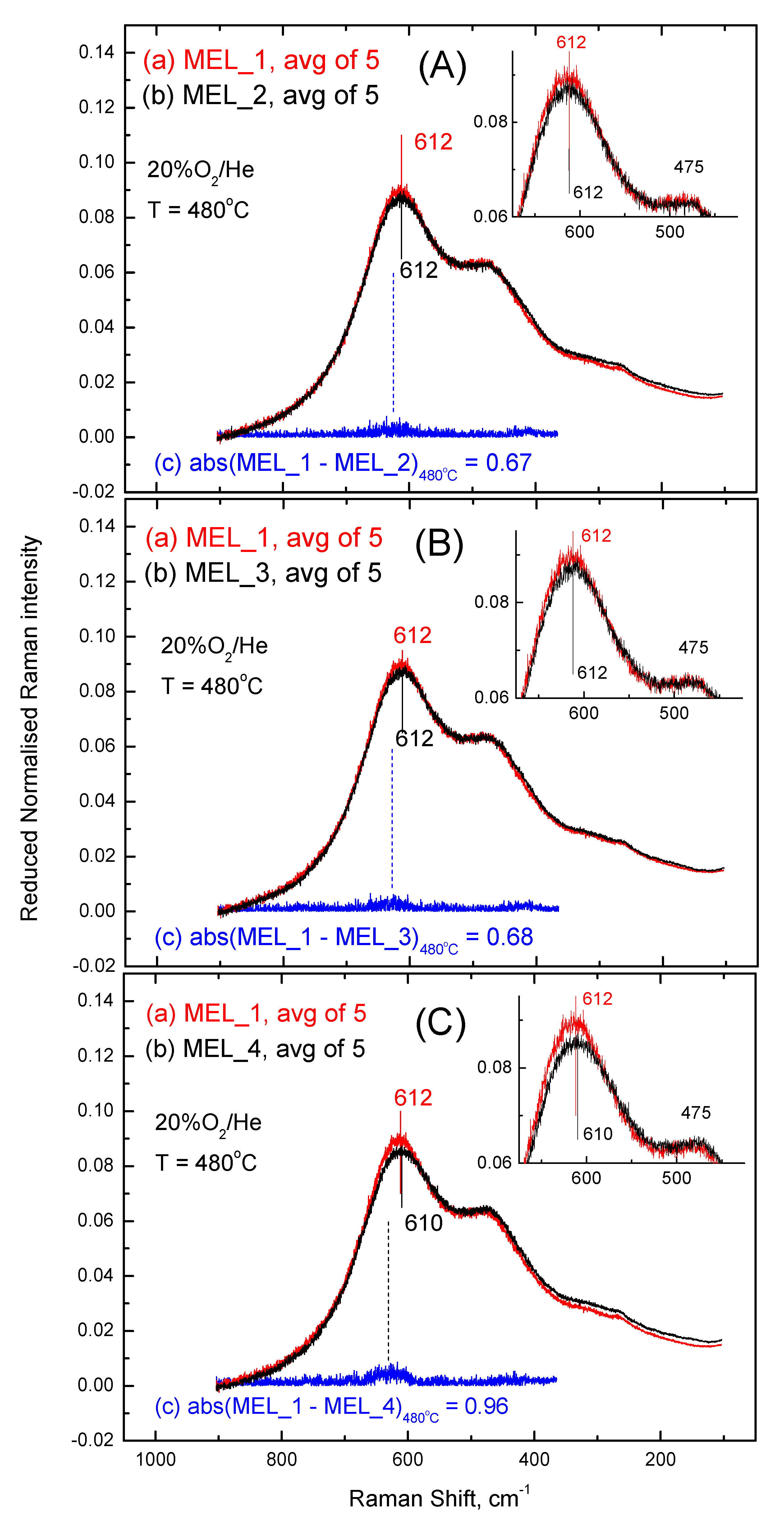
2.4. Comparisons of Anionic Sublattice Structures amongst CexZr1−xO2−δ-Based Materials under O2 Gas Atmosphere at 480 °C and 50 °C
2.5. Differences in Raman Spectra and Structural Implications
3. Experimental Methods and Protocols
3.1. Preparation of Materials
3.2. Protocol for In Situ Raman Analysis
3.2.1. General Remarks
3.2.2. Recording Sequence and Procedures
3.2.3. Data Handling
3.2.4. Protocol for Sample Comparisons
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Trovarelli, A.; de Leitenberg, C.; Boaro, M.; Dolcetti, G. The Utilization of ceria in industrial catalysis. Catal. Today 1999, 50, 353–367. [Google Scholar] [CrossRef]
- Efstathiou, A.M.; Christou, S.Y. Investigation of the oxygen storage and release kinetics of model and commercial three-way catalytic materials by transient techniques. In Catalysis by Ceria and Related Materials, 2nd ed.; Trovarelli, A., Fornasiero, P., Eds.; Imperial College Press: London, UK, 2013; pp. 139–221. [Google Scholar]
- Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Active non-metallic Au and Pt species on ceria-based water-gas shift catalysts. Science 2003, 301, 935–938. [Google Scholar] [CrossRef]
- Steele, B.C.H.; Heizel, A. Materials for fuel-cell technologies. Nature 2001, 414, 345–352. [Google Scholar] [CrossRef]
- Heck, R.M.; Farrauto, R.J. Catalytic Air Pollution Control: Commercial Technology; John Wiley & Sons: New York, NY, USA, 1995. [Google Scholar]
- Di Monte, R.; Kaspar, J. Heterogeneous environmental catalysis–a gentle art: CeO2-ZrO2 mixed oxides as a case history. Catal. Today 2005, 100, 27–35. [Google Scholar] [CrossRef]
- Martinez-Arias, A.; Fernandez-Garcia, M.; Belver, C.; Conesa, J.C.; Soria, J. EPR study on oxygen handling properties of ceria, zirconia, and Zr-Ce(1:1) mixed oxide samples. Catal. Lett. 2000, 65, 197–204. [Google Scholar] [CrossRef]
- Fornasiero, P.; Balducci, G.; Di Monte, R.; Kaspar, J.; Sergo, V.; Gubitosa, G.; Ferrero, A.; Grazianni, M. Modification of the redox behavior of CeO2 induced by structural doping with ZrO2. J. Catal. 1996, 164, 173–183. [Google Scholar] [CrossRef]
- Fernandes-Garcia, M.; Martinez- Arias, A.; Hungria, A.B.; Iglesias-Juez, A.; Conesa, J.C.; Otero, C.; Soria, J. Thermal behavior of (Ce,Zr)Ox/Al2O3 complex oxides prepared by a microemulsion method. Phys. Chem. Chem. Phys. 2002, 4, 2473–2481. [Google Scholar] [CrossRef]
- Andriopoulou, C.; Trimpalis, A.; Petallidou, K.C.; Sgoura, A.; Efstathiou, A.M.; Boghosian, S. Structural and redox properties of Ce1-XZrxO2-δ and Ce0.8Zr0.15RE0.05O2-δ (RE: La, Nd, Pr, Y) solids studied by high temperature in situ Raman spectroscopy. J. Phys. Chem. C 2017, 121, 7931–7943. [Google Scholar] [CrossRef]
- Yashima, M.; Arashi, H.; Kakihana, M.; Yoshimura, M. Raman scattering study of cubic-tetragonal phase transition in Zr1-xCexO2 solid solution. J. Am. Ceram. Soc. 1994, 77, 1067–1071. [Google Scholar] [CrossRef]
- Yashima, M.; Sasaki, S.; Yamaguchi, Y.; Kakihana, M.; Yoshimura, M.; Mori, T. Internal distortion in ZrO2-CeO2 solid solutions: Neutron and high-resolution synchrotron X-ray diffraction study. Appl. Phys. Lett. 1998, 72, 182–184. [Google Scholar] [CrossRef]
- Mamontov, E.; Brezny, R.; Koranne, M.; Egami, T. Nanoscale heterogeneities and oxygen storage capacity of Ce0.5Zr0.5O2. J. Phys. Chem. B 2003, 107, 13007–13014. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, C.-H.; Hanson, J.C.; Robinson, R.D.; Herman, I.P.; Chan, S.W. Phases in ceria-zirconia binary oxide (1-x)CeO2-xZrO2 nanoparticles: The effect of particle size. J. Am. Ceram. Soc. 2006, 89, 1028–1036. [Google Scholar] [CrossRef]
- Kaspar, J.; Fornasiero, P.; Graziani, M. Use of CeO2-based oxides in the three-way catalysis. Catal. Today 1999, 50, 285–298. [Google Scholar] [CrossRef]
- Si, R.; Zhang, Y.-W.; Li, S.-J.; Lin, B.-X.; Yan, C.-H. Urea-based hydrothermally derived homogeneous nanostructured Ce1-xZrxO2 (x=0–0.8) solid solutions: A strong correlation between oxygen storage capacity and lattice strain. J. Phys. Chem. B 2004, 108, 12481–12488. [Google Scholar] [CrossRef]
- Si, R.; Zhang, Y.-W.; Wang, L.-M.; Li, S.-J.; Lin, B.-X.; Chu, W.-S.; Wu, Z.-Y.; Yan, C.-H. Enhanced thermal stability and oxygen storage capacity for CexZr1-xO2 (x=0.4–0.6) solid solutions by hydrothermally homogeneous doping of trivalent rare earths. J. Phys. Chem. B 2007, 111, 787–794. [Google Scholar]
- Ilieva, L.; Petrova, P.; Pantaleo, G.; Zanella, R.; Liotta, L.F.; Georgiev, V.; Boghosian, S.; Kaszkur, Z.; Venezia, A.M.; Tabakova, T. Gold catalysts supported on Y-modified ceria for CO-free hydrogen production via PROX. Appl. Catal. B Environ. 2016, 188, 154–168. [Google Scholar] [CrossRef]
- Lykaki, M.; Pachatouridou, E.; Carabineiro, S.A.C.; Iliopoulou, E.; Andriopoulou, C.; Kallithrakas-Kontos, N.; Boghosian, S.; Konsolakis, M. Ceria nanoparticles shape effects on the structural defects and surface chemistry: Implications in CO oxidation by CuO/Ceria Oxides. Appl. Catal. B Environ. 2018, 230, 18–28. [Google Scholar] [CrossRef]
- Vasiliades, M.A.; Harris, D.; Stephenson, H.; Boghosian, S.; Efstathiou, A.M. A novel analysis of transient isothermal 18O isotopic Exchange on commercial CexZr1-xO2-based materials. Top. Catal. 2019, 62, 219–226. [Google Scholar] [CrossRef]
- Takao, Y.; Norman, C.; Edwards, G. Process for Preparing Zirconium-Cerium-Based Mixed Oxides. U.S. Patent 7,431,910, 7 October 2008. [Google Scholar]
- Mamontov, E.; Egami, T.; Brezny, R.; Koranne, M.; Tyagi, S. Lattice defects and oxygen storage capacity of nanocrystalline ceria and ceria-zirconia. J. Phys. Chem. B 2000, 104, 11110–11116. [Google Scholar] [CrossRef]
- Wu, Z.; Li, M.; Howe, J.; Meyer, H.M.; Overbury, S.H. Probing defect sites on CeO2 nanocrystals with well-defined strface planes by Raman spectroscopy and O2 absorption. Langmuir 2010, 26, 16595–16606. [Google Scholar] [CrossRef]
- Taniguchi, T.; Watanabe, T.; Sugiyama, N.; Subramani, A.K.; Wagata, H.; Matsushita, N.; Yoshimura, M. Identifying defects in ceria-based nanocrystals by UV resonance Raman spectroscopy. J. Phys. Chem. C 2009, 113, 19789–19793. [Google Scholar] [CrossRef]
- Yashima, M.; Morimoto, K.; Ishizawa, N.; Yoshimura, M. Zirconia-ceria solid solution synthesis and the temperature-time-trasformation diagram for the 1:1 composition. J. Am. Ceram. Soc. 1993, 76, 1745–1750. [Google Scholar] [CrossRef]
- Martinez-Arias, A.; Fernandez-Garcia, M.; Ballesteros, V.; Salamanca, L.N.; Conesa, J.C.; Otero, C.; Soria, J. Characterization of high surface area Zr-Ce (1:1) mixed oxide prepared by the microemulsion method. Langmuir 1999, 15, 4796–4802. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Boghosian, S. On the configuration of MoOx sites on alumina, zirconia, titania and silica. Vibrational properties, molecular structure and vibrational isotope effects. Catal. Sci. Technol. 2013, 3, 1869–1888. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Panagiotou, G.D.; Stathi, P.; Kalampounias, A.G.; Bourikas, K.; Kordulis, C.; Deligiannakis, Y.; Boghosian, S.; Lycourghiotis, A. Molybdena Deposited on Titania by Equilibrium Deposition Filtration: Evolution of the Structural Configuration of Oxo-Molybdenum (VI) Sites with Temperature. Phys. Chem. Chem. Phys. 2016, 18, 23980–23989. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | ||||||
| abs(MEL_1–MEL_i) | 480 °C | 50 °C | ||||
| Integrated Absolute Differences | Integrated Absolute Difference (AD) | Net Absolute Difference (NAD) | Relative Net Absolute Difference (RNAD) | Integrated Absolute Difference (AD) | Net Absolute Difference (NAD) | Relative Net Absolute Difference (RNAD) |
| abs(MEL_2–MEL_1) | 0.67 | 0.07 (1) | 0.0030 (2) | 0.52 | 0.08 (3) | 0.0025 (4) |
| abs(MEL_3–MEL_1) | 0.68 | 0.08 | 0.0035 | 0.51 | 0.07 | 0.0022 |
| abs(MEL_4–MEL_1) | 0.96 | 0.36 | 0.0155 | 0.69 | 0.25 | 0.0078 |
| (B) | ||||||
| (a) | ||||||
| (b) | ||||||
| (c) | ||||||
| (d) | ||||||
- (1)
- (2)
- (3)
- (4)
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andriopoulou, C.; Harris, D.; Stephenson, H.; Efstathiou, A.M.; Boghosian, S. In Situ Raman Spectroscopy as a Tool for Discerning Subtle Structural Differences between Commercial (Ce,Zr)O2-Based OSC Materials of Identical Composition. Catalysts 2020, 10, 462. https://doi.org/10.3390/catal10040462
Andriopoulou C, Harris D, Stephenson H, Efstathiou AM, Boghosian S. In Situ Raman Spectroscopy as a Tool for Discerning Subtle Structural Differences between Commercial (Ce,Zr)O2-Based OSC Materials of Identical Composition. Catalysts. 2020; 10(4):462. https://doi.org/10.3390/catal10040462
Chicago/Turabian StyleAndriopoulou, Chrysanthi, Deb Harris, Hazel Stephenson, Angelos M. Efstathiou, and Soghomon Boghosian. 2020. "In Situ Raman Spectroscopy as a Tool for Discerning Subtle Structural Differences between Commercial (Ce,Zr)O2-Based OSC Materials of Identical Composition" Catalysts 10, no. 4: 462. https://doi.org/10.3390/catal10040462
APA StyleAndriopoulou, C., Harris, D., Stephenson, H., Efstathiou, A. M., & Boghosian, S. (2020). In Situ Raman Spectroscopy as a Tool for Discerning Subtle Structural Differences between Commercial (Ce,Zr)O2-Based OSC Materials of Identical Composition. Catalysts, 10(4), 462. https://doi.org/10.3390/catal10040462