A Kinetic Study on the Efficient Formation of High-Valent Mn(TPPS)-oxo Complexes by Various Oxidants


Abstract
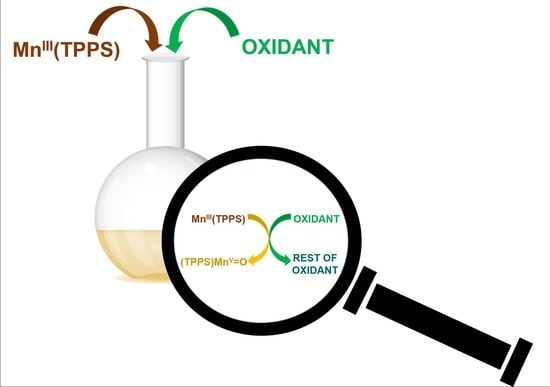
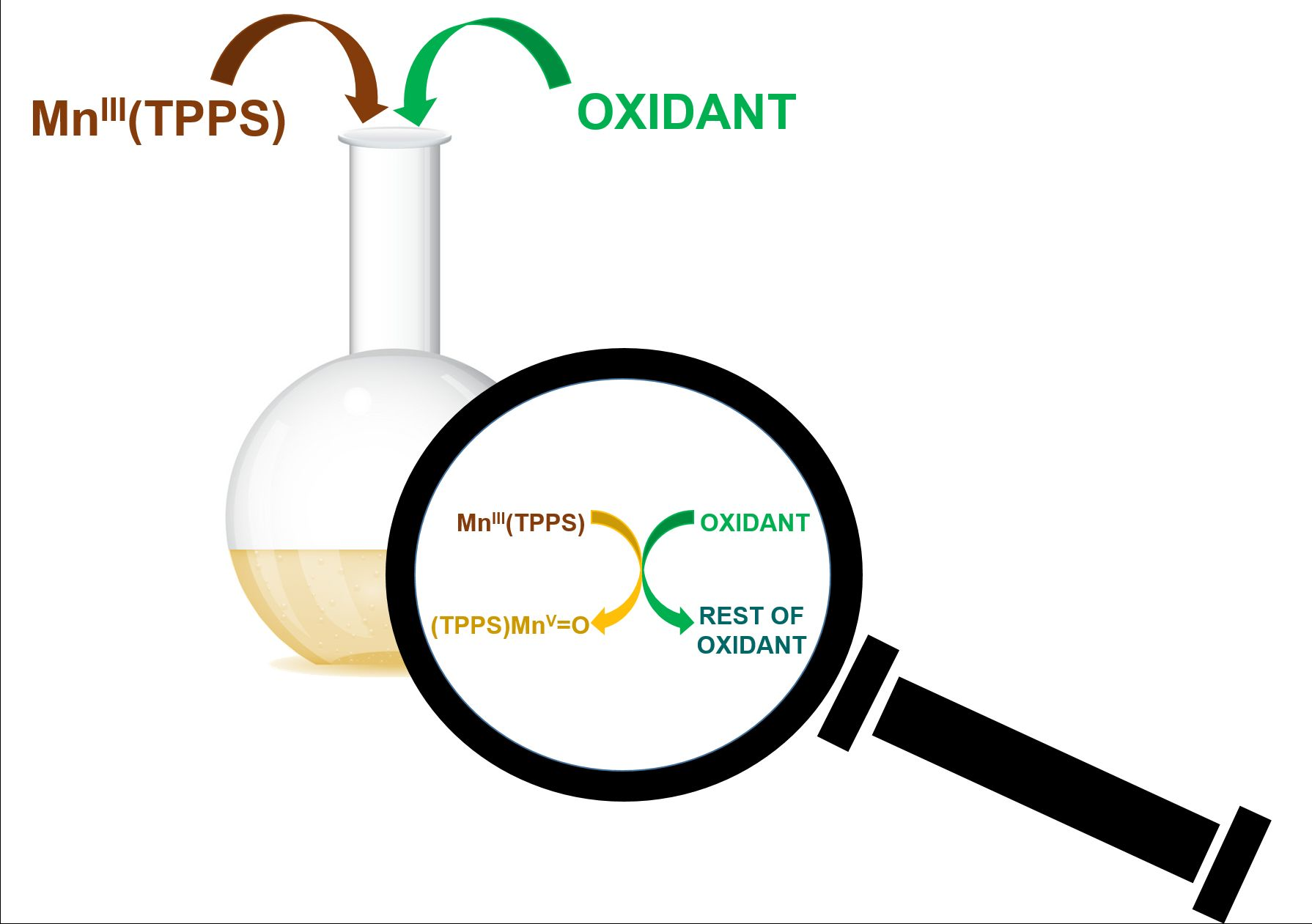
1. Introduction
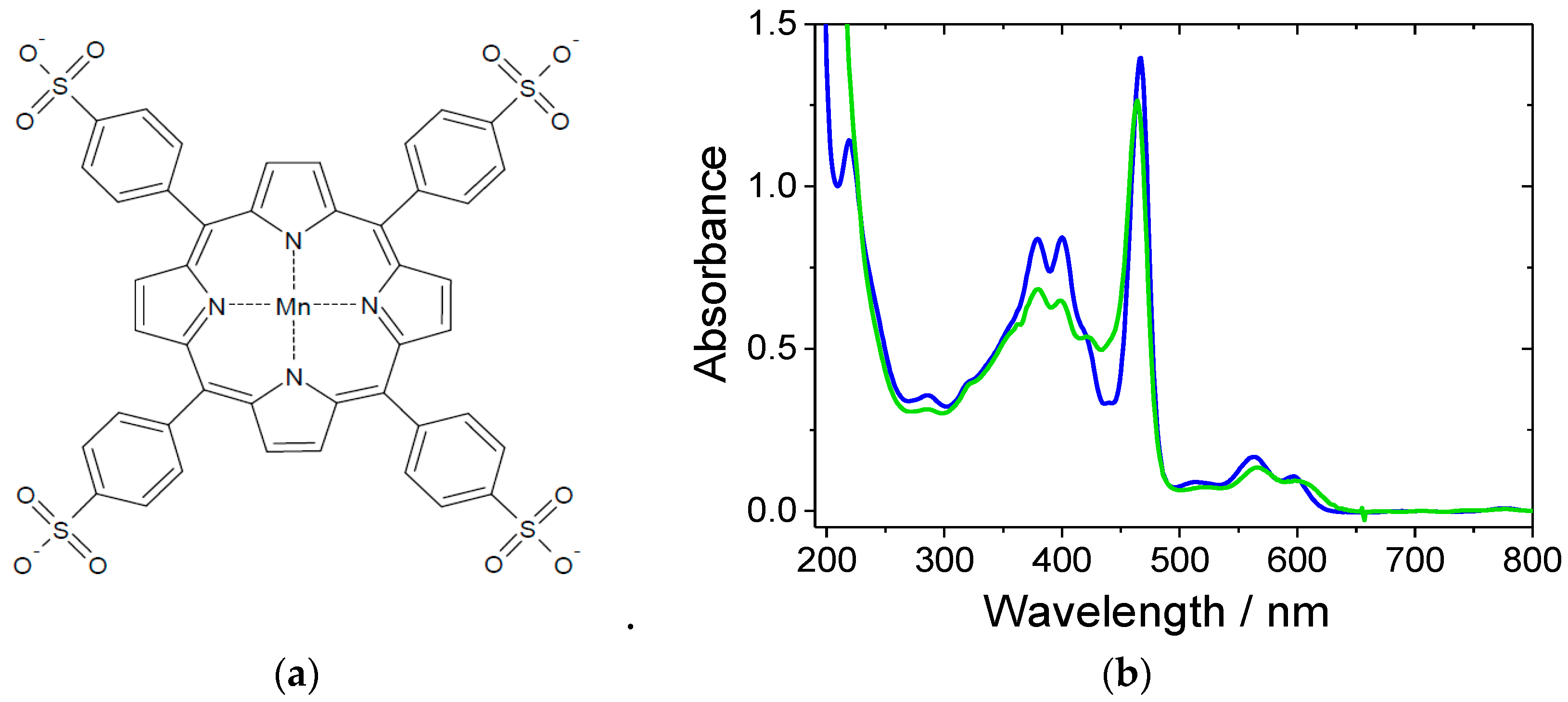
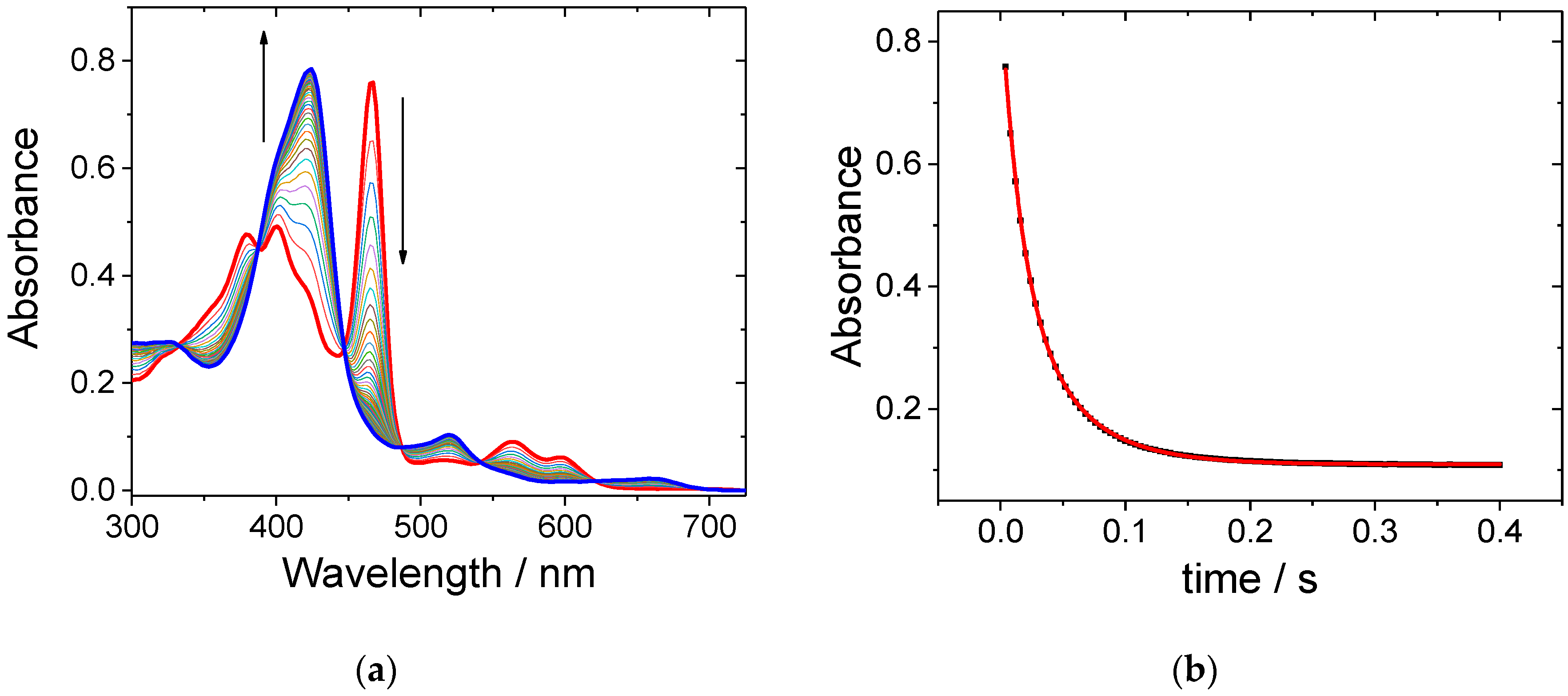
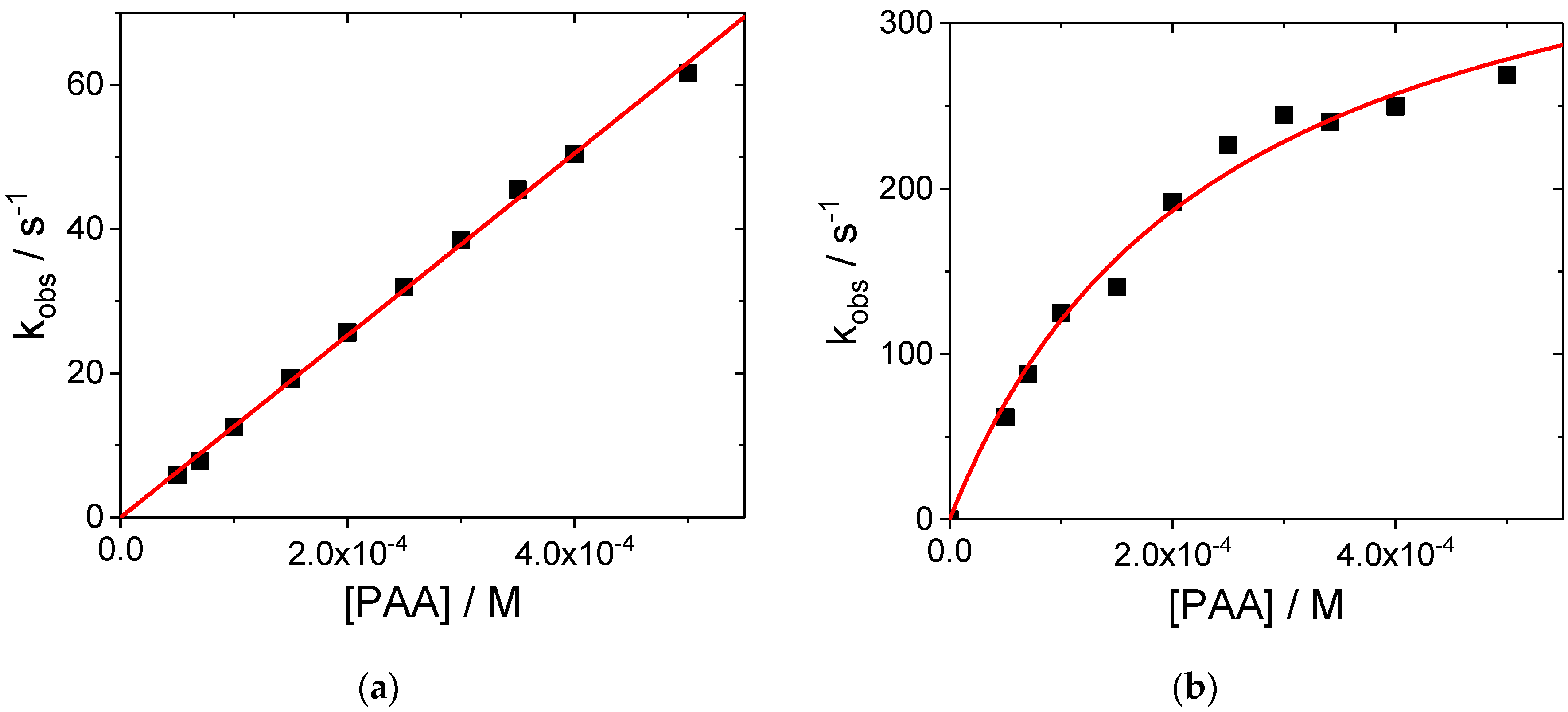
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pearce, C.I.; Lloyd, J.R.; Guthrie, J.T. The removal of colour from textile wastewater using whole bacterial cells: A review. Dyes Pigment. 2003, 58, 179–196. [Google Scholar] [CrossRef]
- Asghar, A.; Abdul Raman, A.A.; Wan Daud, W.M.A. Advanced oxidation processes for in-situ production of hydrogen peroxide/hydroxyl radical for textile wastewater treatment: A review. J. Clean. Prod. 2015, 87, 826–838. [Google Scholar] [CrossRef]
- Rajkumar, D.; Kim, J.G. Oxidation of various reactive dyes with in situ electro-generated active chlorine for textile dyeing industry wastewater treatment. J. Hazard. Mater. 2006, 136, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Ghodbane, H.; Hamdaoui, O. Intensification of sonochemical decolorization of anthraquinonic dye Acid Blue 25 using carbon tetrachloride. Ultrason. Sonochem. 2009, 16, 455–461. [Google Scholar] [CrossRef]
- Lin, S.H.; Chen, M.L. Treatment of textile wastewater by chemical methods for reuse. Water Res. 1997, 31, 868–876. [Google Scholar] [CrossRef]
- Can, O.T.; Kobya, M.; Demirbas, E.; Bayramoglu, M. Treatment of the textile wastewater by combined electrocoagulation. Chemosphere 2006, 62, 181–187. [Google Scholar] [CrossRef]
- Chen, G. Electrochemical technologies in wastewater treatment. Sep. Purif. Technol. 2004, 38, 11–41. [Google Scholar] [CrossRef]
- Chang, W.; Hong, S.; Park, J. Effect of zeolite media for the treatment of textile wastewater in a biological aerated filter. Process Biochem. 2002, 37, 693–698. [Google Scholar] [CrossRef]
- Ellouze, E.; Tahri, N.; Amar, R. Enhancement of textile wastewater treatment process using Nanofiltration. Desalination 2012, 286, 16–23. [Google Scholar] [CrossRef]
- Jiang, J.; Lloyd, B. Progress in the development and use of ferrate(VI) salt as an oxidant and coagulant for water and wastewater treatment. Water Res. 2002, 36, 1397–1408. [Google Scholar] [CrossRef]
- Bulc, T.G.; Ojstršek, A. The use of constructed wetland for dye-rich textile wastewater treatment. J. Hazard. Mater. 2008, 155, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Peng, C.F. Treatment of textile wastewater by electrochemical method. Water Res. 1994, 28, 277–282. [Google Scholar] [CrossRef]
- Perkowski, J.; Kos, L.; Ledakowicz, S. Application of Ozone in Textile Wastewater Treatment. Ozone Sci. Eng. 1996, 18, 73–85. [Google Scholar] [CrossRef]
- Hage, R.; Lienke, A. Applications of Transition-Metal Catalysts to Textile and Wood-Pulp Bleaching. Angew. Chem. Int. Ed. 2006, 45, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Bäckvall, J.E. Modern Oxidation Methods; WILEY-VCH: Weinheim, Germany, 2010; pp. 371–381. [Google Scholar]
- Chatterjee, D.; Ember, E.; Pal, U.; Ghosh, S.; van Eldik, R. Remarkably high catalytic activity of the RuIII(edta)/H2O2 system towards degradation of the azo-dye Orange II. Dalton Trans. 2011, 40, 10473–10480. [Google Scholar] [CrossRef]
- Rothbart, S.; van Eldik, R. Manganese Compounds as Versatile Catalysts for the Oxidative Degradation of Organic Dyes. Adv. Inorg. Chem. 2013, 65, 165–215. [Google Scholar] [CrossRef]
- Gilbert, B.C.; Lindsay Smith, J.R.; Newton, M.S.; Oakes, J.; Prats, R.P.i. Azo dye oxidation with hydrogen peroxide catalysed by manganese 1,4,7-triazacyclononane complexes in aqueous solutions. Org. Biomol. Chem. 2003, 1, 1568–1577. [Google Scholar] [CrossRef]
- Zaharia, C.; Suteu, D.; Muresan, A.; Muresan, R.; Popescu, A. Textile wastewater treatment by homogeneous oxidation with hydrogen peroxide. Environ. Eng. Manag. J. 2009, 8, 1359–1369. [Google Scholar] [CrossRef]
- Pagano, M.; Ciannarella, R.; Locaputo, V.; Mascolo, G.; Volpe, A. Oxidation of azo and anthraquinonic dyes by peroxymonosulphate activated by UV light. J. Environ. Sci. Health A 2018, 53, 393–404. [Google Scholar] [CrossRef]
- Lin, K.A.; Lin, T. Degradation of Acid Azo Dyes Using Oxone Activated by Cobalt Titanate Perovskite. Water Air Soil Pollut. 2017, 229, 10. [Google Scholar] [CrossRef]
- Yuan, Z.; Ni, Y.; Van Heiningen, A.R.P. Kinetics of peracetic acid decomposition: Part I: Spontaneous decomposition at typical pulp bleaching conditions. Can. J. Chem. Eng. 1997, 75, 37–41. [Google Scholar] [CrossRef]
- Emmons, W.D. The Oxidation of Amines with Peracetic Acid. J. Am. Chem. Soc. 1957, 79, 5528–5530. [Google Scholar] [CrossRef]
- Appels, L.; Assche, A.V.; Willems, K.; Degrève, J.; Van Impe, J.; Dewil, R. Peracetic acid oxidation as an alternative pre-treatment for the anaerobic digestion of waste activated sludge. Bioresour.Technol. 2011, 102, 4124–4130. [Google Scholar] [CrossRef] [PubMed]
- Kitis, M. Disinfection of wastewater with peracetic acid: A review. Environ. Int. 2004, 30, 47–55. [Google Scholar] [CrossRef]
- Wagner, M.; Brumelis, D.; Gehr, R. Disinfection of Wastewater by Hydrogen Peroxide or Peracetic Acid: Development of Procedures for Measurement of Residual Disinfectant and Application to a Physicochemically Treated Municipal Effluent. Water Environ. Res. 2002, 74, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Suess, H.U. Pulp Bleaching Today; De Gruyter: Berlin, Germany, 2010. [Google Scholar] [CrossRef]
- Zhao, R.; Tang, Y.; Wei, S.J.; Xu, X.; Shi, X.; Zhang, G. Electrosynthesis of sodium hypochlorite in room temperature ionic liquids and in situ electrochemical epoxidation of olefins. React. Kinet. Mech. Cat. 2012, 106, 37–47. [Google Scholar] [CrossRef]
- Page, P.C.B.; Parker, P.; Buckley, B.R.; Rassias, G.A.; Bethell, D. Organocatalysis of asymmetric epoxidation by iminium salts using sodium hypochlorite as the stoichiometric oxidant. Tetrahedron 2009, 65, 2910–2915. [Google Scholar] [CrossRef]
- Kirihara, M.; Okada, T.; Sugiyama, Y.; Akiyoshi, M.; Matsunaga, T.; Kimura, Y. Sodium Hypochlorite Pentahydrate Crystals (NaOCl·5H2O): A Convenient and Environmentally Benign Oxidant for Organic Synthesis. Org. Process Res. Dev. 2017, 21, 1925–1937. [Google Scholar] [CrossRef]
- Zhang, W.; Jacobsen, E.N. Asymmetric olefin epoxidation with sodium hypochlorite catalyzed by easily prepared chiral manganese(III) salen complexes. J. Org. Chem. 1991, 56, 2296–2298. [Google Scholar] [CrossRef]
- Gonsalvi, L.; Arends, I.W.C.E.; Moilanen, P.; Sheldon, R.A. The Effect of pH Control on the Selective Ruthenium-Catalyzed Oxidation of Ethers and Alcohols with Sodium Hypochlorite. Adv. Synth. Catal. 2003, 345, 1321–1328. [Google Scholar] [CrossRef]
- Chellamani, A.; Harikengaram, S. Mechanism of oxidation of aryl methyl sulfoxides with sodium hypochlorite catalyzed by (salen)MnIII complexes. J. Mol. Catal. A Chem. 2006, 247, 260–267. [Google Scholar] [CrossRef]
- Chellamani, A.; Harikengaram, S. Mechanism of (Salen)manganese(III)-Catalyzed Oxidation of Aryl Phenyl Sulfides with Sodium Hypochlorite. Helv. Chim. Acta 2011, 94, 453–463. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, Q.; Ma, W.; Zhao, J. Enantioselective oxidation of racemic secondary alcohols catalyzed by chiral Mn(III)-salen complex with sodium hypochlorite as oxidant. Catal. Commun. 2014, 45, 114–117. [Google Scholar] [CrossRef]
- Patil, R.D.; Sasson, Y. Naphthalenes Oxidation by Aqueous Sodium Hypochlorite Catalyzed by Ruthenium Salts Under Phase-Transfer Catalytic Conditions. Catal. Lett. 2016, 146, 991–997. [Google Scholar] [CrossRef]
- Mirkhani, V.; Tangestaninejad, S.; Moghadam, M.; Yadollahi, B. Efficient and selective epoxidation of alkenes by supported manganese porphyrin under ultrasonic irradiation. J. Chem. Res. 2000, 2000, 515–517. [Google Scholar] [CrossRef]
- Tangestaninejad, S.; Moghadam, M.; Mirkhani, V.; Mohammadpoor-Baltork, I.; Hoseini, N. Efficient and Selective Hydrocarbon Oxidation with Sodium Periodate Catalyzed by Supported Manganese(III) Porphyrin. J. Iran. Chem. Soc. 2010, 7, 663–672. [Google Scholar] [CrossRef]
- Zakavi, S.; Ebadi, S.; Javanmard, M. Nanosized cationic and anionic manganese porphyrins as mesoporous catalysts for the oxidation of olefins: Nano versus bulk aggregates. Appl. Organomet. Chem. 2018, 32, e4175. [Google Scholar] [CrossRef]
- Song, R.; Sorokin, A.; Bernadou, J.; Meunier, B. Metalloporphyrin-Catalyzed Oxidation of 2-Methylnaphthalene to Vitamin K3 and 6-Methyl-1,4-naphthoquinone by Potassium Monopersulfate in Aqueous Solution. J. Org. Chem. 1997, 62, 673–678. [Google Scholar] [CrossRef]
- Procner, M.; Orzeł, Ł.; Stochel, G.; van Eldik, R. Spectroscopic and kinetic evidence for redox cycling, catalase and degradation activities of MnIII(TPPS) in a basic aqueous peroxide medium. Chem. Commun. 2016, 52, 5297–5300. [Google Scholar] [CrossRef]
- Procner, M.; Orzeł, Ł.; Stochel, G.; van Eldik, R. Catalytic Degradation of Orange II by MnIII(TPPS) in Basic Hydrogen Peroxide Medium: A Detailed Kinetic Analysis. Eur. J. Inorg. Chem. 2018, 2018, 3462–3471. [Google Scholar] [CrossRef]
- Arasasingham, R.D.; He, G.X.; Bruice, T.C. Mechanism of manganese porphyrin-catalyzed oxidation of alkenes. Role of manganese(IV)-oxo species. J. Am. Chem. Soc. 1993, 115, 7985–7991. [Google Scholar] [CrossRef]
- Nam, W.; Kim, I.; Lim, M.H.; Choi, H.J.; Lee, J.S.; Jang, H.G. Isolation of an oxomanganese(v) porphyrin intermediate in the reaction of a manganese(III) porphyrin complex and H2O2 in aqueous solution. Chem.-Eur. J. 2002, 8, 2067–2071. [Google Scholar] [CrossRef]
- Lahaye, T.; Groves, J.T. Modeling the haloperoxidases: Reversible oxygen atom transfer between bromide ion and an oxo-Mn(V) porphyrin. J. Inorg. Biochem. 2007, 101, 1786–1797. [Google Scholar] [CrossRef] [PubMed]
- Ember, E.; Rothbart, S.; Puchta, R.; van Eldik, R. Metal ion-catalyzed oxidative degradation of Orange II by H 2O2. High catalytic activity of simple manganese salts. New J. Chem. 2009, 33, 34–49. [Google Scholar] [CrossRef]
- Rothbart, S.; Ember, E.; van Eldik, R. Comparative study of the catalytic activity of [MnII(bpy)2Cl2] and [Mn2III/IV(μ-O)2(bpy)4](ClO4)3 in the H2O2 induced oxidation of organic dyes in carbonate buffered aqueous solution. Dalton Trans. 2010, 39, 3264–3272. [Google Scholar] [CrossRef]
- Richardson, D.E.; Yao, H.; Frank, K.M.; Bennett, D.A. Equilibria, Kinetics, and Mechanism in the Bicarbonate Activation of Hydrogen Peroxide: Oxidation of Sulfides by Peroxymonocarbonate. J. Am. Chem. Soc. 2000, 122, 1729–1739. [Google Scholar] [CrossRef]
- Bakhmutova-Albert, E.V.; Yao, H.; Denevan, D.E.; Richardson, D.E. Kinetics and mechanism of peroxymonocarbonate formation. Inorg. Chem. 2010, 49, 11287–11296. [Google Scholar] [CrossRef]
- Swern, D. Organic Peroxides; Wiley: New York, NY, USA, 1970; Volume I. [Google Scholar] [CrossRef]
- Rothbart, S.; Ember, E.; van Eldik, R. Mechanistic studies on the oxidative degradation of Orange II by peracetic acid catalyzed by simple manganese(ii) salts. Tuning the lifetime of the catalyst. New J. Chem. 2012, 36, 732–748. [Google Scholar] [CrossRef]
- Evans, D.F.; Upton, M.W. Studies on singlet oxygen in aqueous solution. Part 3. The decomposition of peroxy-acids. J. Chem. Soc. Dalton Trans. 1985, 1151–1153. [Google Scholar] [CrossRef]
- Koubek, E.; Haggett, M.L.; Battaglia, C.J.; Ibne-Rasa, K.M.; Pyun, H.Y.; Edwards, J.O. Kinetics and Mechanism of the Spontaneous Decompositions of Some Peroxoacids, Hydrogen Peroxide and t-Butyl Hydroperoxide. J. Am. Chem. Soc. 1963, 85, 2263–2268. [Google Scholar] [CrossRef]
- Ball, D.L.; Edwards, J.O. The Kinetics and Mechanism of the Decomposition of Caro’s Acid. I. J. Am. Chem. Soc. 1956, 78, 1125–1129. [Google Scholar] [CrossRef]
- Awad, M.I.; Harnoode, C.; Tokuda, K.; Ohsaka, T. Simultaneous Electroanalysis of Peroxyacetic Acid and Hydrogen Peroxide. Anal. Chem. 2001, 73, 1839–1843. [Google Scholar] [CrossRef]
- Urano, H.; Fukuzaki, S. The Mode of Action of Sodium Hypochlorite in the Decolorization of Azo Dye Orange II in Aqueous Solution. Biocontrol Sci. 2011, 16, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C. The acid ionization constant of HOCl from 5 to 35. J. Phys. Chem. 1966, 70, 3798–3805. [Google Scholar] [CrossRef]
- Lister, M.W. Decomposition of sodium hypochlorite: The uncatalyzed reaction. Can. J. Chem. 1956, 34, 465–478. [Google Scholar] [CrossRef]
- Zheng, T.; Richardson, D.E. Homogeneous aqueous oxidation of organic molecules by Oxone® and catalysis by a water-soluble manganese porphyrin complex. Tetrahedron Lett. 1995, 36, 833–836. [Google Scholar] [CrossRef]
- Ghanbari, F.; Moradi, M. Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants: Review. Chem. Eng. J. 2017, 310, 41–62. [Google Scholar] [CrossRef]
- Durrant, M.C.; Davies, D.M.; Deary, M.E. Dioxaborirane: A highly reactive peroxide that is the likely intermediate in borate catalysed electrophilic reactions of hydrogen peroxide in alkaline aqueous solution. Org. Biomol. Chem. 2011, 9, 7249–7254. [Google Scholar] [CrossRef]
- Pizer, R.; Tihal, C. Peroxoborates. Interaction of boric acid and hydrogen peroxide in aqueous solution. Inorg. Chem. 1987, 26, 3639–3642. [Google Scholar] [CrossRef]
- Deary, M.E.; Durrant, M.C.; Davies, D.M. A kinetic and theoretical study of the borate catalysed reactions of hydrogen peroxide: The role of dioxaborirane as the catalytic intermediate for a wide range of substrates. Org. Biomol. Chem. 2013, 11, 309–317. [Google Scholar] [CrossRef]
- Davies, D.M.; Deary, M.E.; Quill, K.; Smith, R.A. Borate-Catalyzed Reactions of Hydrogen Peroxide: Kinetics and Mechanism of the Oxidation of Organic Sulfides by Peroxoborates. Chem. Eur. J. 2005, 11, 3552–3558. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxidant | Experimental Conditions | Second-Order Rate Constant M−1 s−1 |
|---|---|---|
| H2O2 | pH = 9.3, NaOH | 1.08 ± 0.04 |
| pH = 9.3, 0.5 M KNO3 + NaOH | (3.2 ± 0.2) × 102 | |
| pH = 9.3, 0.5 M NaHCO3 + Na2CO3 | (7.2 ± 0.2) × 103 | |
| pH = 11, NaOH | (2.20 ± 0.06) × 103 | |
| pH = 11, 0.5 M KNO3 + NaOH | (4.1 ± 0.7) × 104 [a] | |
| pH = 11, 0.5 M NaHCO3 + Na2CO3 | (9.3 ± 0.7) × 104 | |
| PAA | pH = 9.3, 0.5 M NaHCO3 + Na2CO3 | (1.26 ± 0.02) × 105 |
| pH = 11, NaOH | (2.19 ± 0.06) × 104 | |
| pH = 11, 0.5 M KNO3 + NaOH | (9.3 ± 2.0) × 104 [a] | |
| pH = 11, 0.5 M NaHCO3 + Na2CO3 | (1.7 ± 0.4) × 106 [a] | |
| NaOCl | pH = 9.3, NaOH | (4.1 ± 0.2) × 102 |
| pH = 9.3, 0.5 M KNO3 + NaOH | (1.42 ± 0.07) × 103 | |
| pH = 9.3, 0.5 M NaHCO3 + Na2CO3 | (1.46 ± 0.04) × 103 | |
| pH = 11, NaOH | (9.6 ± 0.4) × 102 | |
| pH = 11, 0.5 M KNO3 + NaOH | (3.65 ± 0.06) × 103 | |
| pH = 11, 0.5 M NaHCO3 + Na2CO3 | (1.19 ± 0.03) × 104 | |
| peroxomonosulfate | pH = 11, 0.5 M KNO3 + NaOH | (4.4 ± 1.1) × 104 [a] |
| pH = 11, 0.5 M NaHCO3 + Na2CO3 | (1.17 ± 0.02) × 105 | |
| perborate | pH = 11, 0.5 M KNO3 + NaOH | (8.8 ± 0.2) × 102 |
| pH = 11, 0.5 M NaHCO3 + Na2CO3 | (3.7 ± 0.2) × 104 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Procner, M.; Orzel, Ł.; Stochel, G.; van Eldik, R. A Kinetic Study on the Efficient Formation of High-Valent Mn(TPPS)-oxo Complexes by Various Oxidants. Catalysts 2020, 10, 610. https://doi.org/10.3390/catal10060610
Procner M, Orzel Ł, Stochel G, van Eldik R. A Kinetic Study on the Efficient Formation of High-Valent Mn(TPPS)-oxo Complexes by Various Oxidants. Catalysts. 2020; 10(6):610. https://doi.org/10.3390/catal10060610
Chicago/Turabian StyleProcner, Magdalena, Łukasz Orzel, Grażyna Stochel, and Rudi van Eldik. 2020. "A Kinetic Study on the Efficient Formation of High-Valent Mn(TPPS)-oxo Complexes by Various Oxidants" Catalysts 10, no. 6: 610. https://doi.org/10.3390/catal10060610
APA StyleProcner, M., Orzel, Ł., Stochel, G., & van Eldik, R. (2020). A Kinetic Study on the Efficient Formation of High-Valent Mn(TPPS)-oxo Complexes by Various Oxidants. Catalysts, 10(6), 610. https://doi.org/10.3390/catal10060610