The Effect of CO Partial Pressure on Important Kinetic Parameters of Methanation Reaction on Co-Based FTS Catalyst Studied by SSITKA-MS and Operando DRIFTS-MS Techniques

 ,
,  ,
, 
Abstract
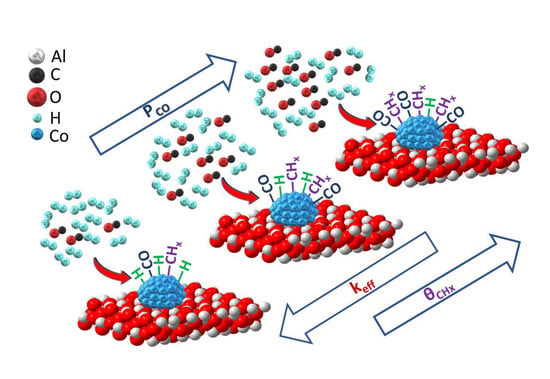
1. Introduction
- (i)
- The amount (NCO, μmol g−1) and surface coverage (θCO) of reversibly adsorbed CO-s;
- (ii)
- the amount (NCHx, μmol g−1) and surface coverage (θCHx) of active reaction CHx-s (Cα) intermediates;
- (iii)
- the mean residence time of CO-s (τCO, s) and active CHx-s (τCHx, s) intermediates;
- (iv)
- the turnover frequency leading to CH4, TOFchem, or TOFITK (s−1), respectively, estimated based on all Co surface metal atoms or on the active reaction intermediates (μmol g−1);
- (v)
- the amount (μmol g−1) of inactive carbonaceous species (Cβ) formed during FTS (230 °C) readily hydrogenated at 230 °C in 50% H2/Ar;
- (vi)
- the amount (μmol g−1) of the refractory carbonaceous species (Cγ) formed during FTS but hydrogenated at higher temperatures (230–550 °C);
- (vii)
- the relative reactivity (keff) of the various forms of linear-type adsorbed CO-s species formed over the Co surface during methanation after using operando transient DRIFTS-MS coupled with kinetic modelling.
2. Results
2.1. Co/γ-Al2O3 Catalyst Characterization
2.2. SSITKA-MS after 5 h in FTS
2.3. Transient Isothermal and Temperature-Programmed Hydrogenation (TIH/TPH)
2.4. Operando DRIFTS—Transient Isothermal Hydrogenation (TIH) of CO
3. Discussion
3.1. Influence of CO Partial Pressure on Intrinsic Kinetic Parameters of Methanation Reaction
3.2. The Influence of H2 Partial Pressure on the Reactivity of CO-s in the Methanation Reaction
3.3. Effect of CO Partial Pressure on the Formation of Inactive Cβ and Cγ Carbonaceous Species
4. Materials and Methods
4.1. Co/γ-Al2O3 Catalyst—Synthesis and Characterization
4.2. SSITKA-MS Following 5 h in FTS
4.3. Transient Isothermal (TIH) and Temperature-Programmed Hydrogenation (TPH) Experiments
4.4. Operando DRIFTS—Mass Spectrometry—Transient Isothermal Hydrogenation (TIH) of CO
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jahangiri, H.; Bennett, J.; Mahjoubi, P.; Wilson, K.; Gu, S. A review of advanced catalyst development for Fischer–Tropsch synthesis of hydrocarbons from biomass derived syn-gas. Catal. Sci. Technol. 2014, 4, 2210–2229. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the Development of Novel Cobalt Fischer−Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lin, T.; Dai, Y.; An, Y.; Yu, F.; Zhong, L.; Li, S.; Sun, Y. Recent advances in the investigation of nanoeffects of Fischer-Tropsch catalysts. Catal. Today 2018, 311, 8–22. [Google Scholar] [CrossRef]
- Krylova, A.Y. Products of the Fischer-Tropsch synthesis (A Review). Solid Fuel Chem. 2014, 48, 22–35. [Google Scholar] [CrossRef]
- Yang, J.; Ma, W.; Chen, D.; Holmen, A.; Davis, B.H. Fischer–Tropsch synthesis: A review of the effect of CO conversion on methane selectivity. Appl. Catal. A Gen. 2014, 470, 250–260. [Google Scholar] [CrossRef]
- Todic, B.; Ma, W.; Jacobs, G.; Davis, B.H.; Bukur, D.B. Effect of process conditions on the product distribution of Fischer-Tropsch synthesis over a Re-promoted cobalt-alumina catalyst using a stirred tank slurry reactor. J. Catal. 2014, 311, 325–338. [Google Scholar] [CrossRef]
- Den Breejen, J.P.; Sietsma, J.R.A.; Friedrich, H.; Bitter, J.H.; de Jong, K.P. Design of supported cobalt catalysts with maximum activity for the Fischer-Tropsch synthesis. J. Catal. 2010, 270, 146–152. [Google Scholar] [CrossRef]
- Den Breejen, J.P.; Radstake, P.B.; Bezemer, G.L.; Bitter, J.H.; Frøseth, V.; Holmen, A.; de Jong, K.P. On the Origin of the Cobalt Particle Size Effects in Fischer−Tropsch Catalysis. J. Am. Chem. Soc. 2009, 131, 7197–7203. [Google Scholar] [CrossRef]
- Iqbal, S.; Davies, T.E.; Hayward, J.S.; Morgan, D.J.; Karim, K.; Bartley, J.K.; Taylor, S.H.; Hutchings, G.J. Fischer Tropsch Synthesis using promoted cobalt-based catalysts. Catal. Today 2016, 272, 74–79. [Google Scholar] [CrossRef]
- Eschemann, T.O.; Oenema, J.; de Jong, K.P. Effects of noble metal promotion for Co/TiO2 Fischer-Tropsch catalysts. Catal. Today 2016, 261, 60–66. [Google Scholar] [CrossRef]
- Carvalho, A.; Ordomsky, V.V.; Luo, Y.; Marinova, M.; Muniz, A.R.; Marcilio, N.R.; Khodakov, A.Y. Elucidation of deactivation phenomena in cobalt catalyst for Fischer-Tropsch synthesis using SSITKA. J. Catal. 2016, 344, 669–679. [Google Scholar] [CrossRef]
- Chen, W.; Kimpel, T.F.; Song, Y.; Chiang, F.K.; Zijlstra, B.; Pestman, R.; Wang, P.; Hensen, E.J.M. Influence of Carbon Deposits on the Cobalt-Catalyzed Fischer-Tropsch Reaction: Evidence of a Two-Site Reaction Model. ACS Catal. 2018, 8, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Efstathiou, A.M.; Gleaves, J.T.; Yablonsky, G.S. Transient Techniques: Temporal Analysis of Products and Steady State Isotopic Transient Kinetic Analysis. In Characterization of Solid Materials and Heterogeneous Catalysts: From Structure to Surface Reactivity; Wiley—VCH: Weinheim, Germany, 2012; Volume 1&2, pp. 1013–1073. [Google Scholar]
- Pansare, S.; Sirijaruphan, A.; Goodwin, J.G. Investigation of Reaction at the Site Level Using SSITKA. In Isotopes in Heterogeneous Catalysis, Catalytic Science Series, v. 4; Imperial College Press: London, UK, 2006; pp. 183–211. [Google Scholar]
- Frøseth, V.; Storsæter, S.; Borg, O.; Blekkan, E.A.; Rønning, M.; Holmen, A. Steady state isotopic transient kinetic analysis (SSITKA) of CO hydrogenation on different Co catalysts. Appl. Catal. A Gen. 2005, 289, 10–15. [Google Scholar] [CrossRef]
- Yang, J.; Chen, D.; Holmen, A. Understanding the kinetics and Re promotion of carbon nanotube supported cobalt catalysts by SSITKA. Catal. Today 2012, 186, 99–108. [Google Scholar] [CrossRef]
- Chen, W.; Filot, I.A.W.; Pestman, R.; Hensen, E.J.M. Mechanism of Cobalt-Catalyzed CO Hydrogenation: 2. Fischer–Tropsch Synthesis. ACS Catal. 2017, 7, 8061–8071. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Pestman, R.; Zijlstra, B.; Filot, I.A.W.; Hensen, E.J.M. Mechanism of Cobalt-Catalyzed CO Hydrogenation: 1. Methanation. ACS Catal. 2017, 7, 8050–8060. [Google Scholar] [CrossRef]
- Vasiliades, M.A.; Kalamaras, C.M.; Govender, N.S.; Govender, A.; Efstathiou, A.M. The effect of preparation route of commercial Co/γ-Al2O3 catalyst on important Fischer-Tropsch kinetic parameters studied by SSITKA and CO-DRIFTS transient hydrogenation techniques. J. Catal. 2019, 379, 60–77. [Google Scholar] [CrossRef]
- Frennet, A.; de Bocarmé, T.V.; Bastin, J.-M.; Kruse, N. Mechanism and Kinetics of the Catalytic CO−H2 Reaction: An Approach by Chemical Transients and Surface Relaxation Spectroscopy. J. Phys. Chem. B 2005, 109, 2350–2359. [Google Scholar] [CrossRef]
- Petallidou, K.C.; Vasiliades, M.A.; Efstathiou, A.M. Deactivation of Co/γ-Al2O3 in CO methanation studied by transient isotopic experiments: The effect of Co particle size. J. Catal. 2020. accepted for publication. [Google Scholar]
- Lorito, D.; Paredes-Nunez, A.; Mirodatos, C.; Schuurman, Y.; Meunier, F.C. Determination of formate decomposition rates and relation to product formation during CO hydrogenation over supported cobalt. Catal. Today 2016, 259, 192–196. [Google Scholar] [CrossRef]
- Paredes-Nunez, A.; Lorito, D.; Guilhaume, N.; Mirodatos, C.; Schuurman, Y.; Meunier, F.C. Nature and reactivity of the surface species observed over a supported cobalt catalyst under CO/H2 mixtures. Catal. Today 2015, 242, 178–183. [Google Scholar] [CrossRef]
- Efstathiou, A.M. Elucidation of Mechanistic and Kinetic Aspects of Water-Gas Shift Reaction on Supported Pt and Au Catalysts via Transient Isotopic Techniques. Catalysis 2016, 28, 175–236. [Google Scholar]
- Efstathiou, A.M.; Bennett, C.O. The CO/H2 reaction on Rh/Al2O3: II. Kinetic study by transient isotopic methods. J. Catal. 1989, 120, 137–156. [Google Scholar] [CrossRef]
- Kalamaras, C.M.; Americanou, S.; Efstathiou, A.M. “Redox” vs “associative formate with –OH group regeneration” WGS reaction mechanism on Pt/CeO2: Effect of platinum particle size. J. Catal. 2011, 279, 287–300. [Google Scholar] [CrossRef]
- Kalamaras, C.M.; Gonzalez, I.D.; Navarro, R.M.; Fierro, J.L.G.; Efstathiou, A.M. Effects of Reaction Temperature and Support Composition on the Mechanism of Water–Gas Shift Reaction over Supported-Pt Catalysts. J. Phys. Chem. C 2011, 115, 11595–11610. [Google Scholar] [CrossRef]
- Vasiliades, M.A.; Djinović, P.; Davlyatova, L.F.; Pintar, A.; Efstathiou, A.M. Origin and reactivity of active and inactive carbon formed during DRM over Ni/Ce0.38Zr0.62O2-δ studied by transient isotopic techniques. Catal. Today 2018, 299, 201–211. [Google Scholar] [CrossRef]
- Vasiliades, M.A.; Djinović, P.; Pintar, A.; Kovač, J.; Efstathiou, A.M. The effect of CeO2-ZrO2 structural differences on the origin and reactivity of carbon formed during methane dry reforming over NiCo/CeO2-ZrO2 catalysts studied by transient techniques. Catal. Sci. Technol. 2017, 7, 5422–5434. [Google Scholar] [CrossRef]
- Vasiliades, M.A.; Makri, M.M.; Djinović, P.; Erjavec, B.; Pintar, A.; Efstathiou, A.M. Dry reforming of methane over 5 wt% Ni/Ce1-xPrxO2-δ catalysts: Performance and characterization of active and inactive carbon by transient isotopic techniques. Appl. Catal. B Environ. 2016, 197, 168–183. [Google Scholar] [CrossRef]
- Zhuo, M.; Tan, K.F.; Borgna, A.; Saeys, M. Density Functional Theory Study of the CO Insertion Mechanism for Fischer−Tropsch Synthesis over Co Catalysts. J. Phys. Chem. C 2009, 113, 8357–8365. [Google Scholar] [CrossRef]
- Yu, M.; Liu, L.; Jia, L.; Li, D.; Wang, Q.; Hou, B. Equilibrium morphology evolution of FCC cobalt nanoparticle under CO and hydrogen environments. Appl. Surf. Sci. 2020, 504, 144469. [Google Scholar] [CrossRef]
- Keyvanloo, K.; Fisher, M.J.; Hecker, W.C.; Lancee, R.J.; Jacobs, G.; Bartholomew, C.H. Kinetics of deactivation by carbon of a cobalt Fischer–Tropsch catalyst: Effects of CO and H2 partial pressures. J. Catal. 2015, 327, 33–47. [Google Scholar] [CrossRef]
- Zijlstra, B.; Broos, R.J.P.; Chen, W.; Oosterbeek, H.; Filot, I.A.W.; Hensen, E.J.M. Coverage Effects in CO Dissociation on Metallic Cobalt Nanoparticles. ACS Catal. 2019, 9, 7365–7372. [Google Scholar] [CrossRef]
- Chen, P.-P.; Liu, J.-X.; Li, W.-X. Carbon Monoxide Activation on Cobalt Carbide for Fischer–Tropsch Synthesis from First-Principles Theory. ACS Catal. 2019, 9, 8093–8103. [Google Scholar] [CrossRef]
- Petersen, M.A.; van den Berg, J.-A.; Ciobîcă, I.M.; van Helden, P. Revisiting CO Activation on Co Catalysts: Impact of Step and Kink Sites from DFT. ACS Catal. 2017, 7, 1984–1992. [Google Scholar] [CrossRef]
- Van Helden, P.; van den Berg, J.-A.; Weststrate, C.J. Hydrogen Adsorption on Co Surfaces: A Density Functional Theory and Temperature Programmed Desorption Study. ACS Catal. 2012, 2, 1097–1107. [Google Scholar] [CrossRef]
- Ledesma, C.; Yang, J.; Blekkan, E.A.; Holmen, A.; Chen, D. Carbon Number Dependence of Reaction Mechanism and Kinetics in CO Hydrogenation on a Co-Based Catalyst. ACS Catal. 2016, 6, 6674–6686. [Google Scholar] [CrossRef]
- Ledesma, C.; Yang, J.; Chen, D.; Holmen, A. Recent Approaches in Mechanistic and Kinetic Studies of Catalytic Reactions Using SSITKA Technique. ACS Catal. 2014, 4, 4527–4547. [Google Scholar] [CrossRef]
- Otarod, M.; Happel, J.; Walter, E. Transient isotopic tracing of methanation kinetics with parallel paths. Appl. Catal. A Gen. 1997, 160, 3–11. [Google Scholar] [CrossRef]
- Yang, J.; Qi, Y.; Zhu, J.; Zhu, Y.A.; Chen, D.; Holmen, A. Reaction mechanism of CO activation and methane formation on Co Fischer-Tropsch catalyst: A combined DFT, transient, and steady-state kinetic modeling. J. Catal. 2013, 308, 37–49. [Google Scholar] [CrossRef]
- Van Berge, P.J.; van de Loosdrecht, J.; Visagie, J.L. Process for Producing Hydrocarbons with Cobalt Catalysts. Europe Patent EP1299503B1, 15 August 2007. [Google Scholar]
- Van de Loosdrecht, J.; Barradas, S.; Caricato, E.A.; Ngwenya, N.G.; Nkwanyana, P.S.; Rawat, M.A.S.; Sigwebela, B.H.; van Berge, P.J.; Visagie, J.L. Calcination of Co-Based Fischer–Tropsch Synthesis Catalysts. Top. Catal. 2003, 26, 121–127. [Google Scholar] [CrossRef]
- Borg, Ø.; Dietzel, P.D.C.; Spjelkavik, A.I.; Tveten, E.Z.; Walmsley, J.C.; Diplas, S.; Eri, S.; Holmen, A.; Rytter, E. Fischer–Tropsch synthesis: Cobalt particle size and support effects on intrinsic activity and product distribution. J. Catal. 2008, 259, 161–164. [Google Scholar] [CrossRef]
- Carvalho, A.; Ordomsky, V.V.; Marcilio, N.R.; Khodakov, A.Y. Number and intrinsic activity of cobalt surface sites in platinum promoted zeolite catalysts for carbon monoxide hydrogenation. Catal. Sci. Technol. 2020, 10, 2137–2144. [Google Scholar] [CrossRef]
- Weststrate, C.J.; Niemantsverdriet, J.W. Understanding FTS selectivity: The crucial role of surface hydrogen. Faraday Discuss. 2017, 197, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Jacobs, G.; Das, T.K.; Masuku, C.M.; Kang, J.; Ramana, V.; Pendyala, R.; Davis, B.H.; Klettlinger, J.L.S.; Yen, C.H. Fischer-Tropsch Synthesis: Kinetics and Water Effect on Methane Formation over 25%Co/γ-Al2O3 Catalyst. Ind. Eng. Chem. Res. 2014, 53, 2157–2166. [Google Scholar] [CrossRef]
- Bianchi, D.; Gass, J.L. Hydrogenation of carbonaceous adsorbed species on an iron/alumina catalyst, I. Experiments in isothermal conditions and exploitation by a kinetic model. J. Catal. 1990, 123, 298–309. [Google Scholar] [CrossRef]
- Efstathiou, A.M.; Chafik, T.; Bianchi, D.; Bennett, C.O. CO chemisorption and hydrogenation of surface carbon species formed after CO/He reaction on Rh/MgO: A transient kinetic study using FTIR and mass spectroscopy. J. Catal. 1994, 147, 24–37. [Google Scholar] [CrossRef]
- Couble, J.; Bianchi, D. Experimental Microkinetic Approach of the Surface Reconstruction of Cobalt Particles in Relationship with the CO/H2 Reaction on a Reduced 10% Co/Al2O3 Catalyst. J. Phys. Chem. C 2013, 117, 14544–14557. [Google Scholar] [CrossRef]
- Paredes-Nunez, A.; Lorito, D.; Schuurman, Y.; Guilhaume, N.; Meunier, F.C. Origins of the poisoning effect of chlorine on the CO hydrogenation activity of alumina-supported cobalt monitored by operando FT-IR spectroscopy. J. Catal. 2015, 329, 229–236. [Google Scholar] [CrossRef]
- Weststrate, C.J.; van de Loosdrecht, J.; Niemantsverdriet, J.W. Spectroscopic insights into cobalt-catalyzed Fischer-Tropsch synthesis: A review of the carbon monoxide interaction with single crystalline surfaces of cobalt. J. Catal. 2016, 342, 1–16. [Google Scholar] [CrossRef]
- Schweicher, J.; Bundhoo, A.; Frennet, A.; Kruse, N.; Daly, H.; Meunier, F.C. DRIFTS/MS studies during chemical transients and SSITKA of the CO/H2 reaction over Co-MgO catalysts. J. Phys. Chem. C 2010, 114, 2248–2255. [Google Scholar] [CrossRef]
- Cheng, Q.; Tian, Y.; Lyu, S.; Zhao, N.; Ma, K.; Ding, T.; Jiang, Z.; Wang, L.; Zhang, J.; Zheng, L.; et al. Confined small-sized cobalt catalysts stimulate carbon-chain growth reversely by modifying ASF law of Fischer-Tropsch synthesis. Nat. Commun. 2018, 9, 3250. [Google Scholar] [CrossRef] [PubMed]
- Hibbitts, D.; Dybeck, E.; Lawlor, T.; Neurock, M.; Iglesia, E. Preferential activation of CO near hydrocarbon chains during Fischer–Tropsch synthesis on Ru. J. Catal. 2016, 337, 91–101. [Google Scholar] [CrossRef]
- Ojeda, M.; Li, A.; Nabar, R.; Nilekar, A.U.; Mavrikakis, M.; Iglesia, E. Kinetically Relevant Steps and H2/D2 Isotope Effects in Fischer−Tropsch Synthesis on Fe and Co Catalysts. J. Phys. Chem. C 2010, 114, 19761–19770. [Google Scholar] [CrossRef]
- Ojeda, M.; Nabar, R.; Nilekar, A.U.; Ishikawa, A.; Mavrikakis, M.; Iglesia, E. CO activation pathways and the mechanism of Fischer–Tropsch synthesis. J. Catal. 2010, 272, 287–297. [Google Scholar] [CrossRef]
- Vada, S.; Chen, B.; Goodwin, J.G., Jr. Isotopic transient study of La promotion of Co/Al2O3 on CO hydrogenation. J. Catal. 1995, 153, 224–231. [Google Scholar] [CrossRef]
- Yang, J.; Frøseth, V.; Chen, D.; Holmen, A. Particle size effect for cobalt Fischer-Tropsch catalysts based on in situ CO chemisorption. Surf. Sci. 2016, 648, 67–73. [Google Scholar] [CrossRef]
- Peña, D.; Griboval-Constant, A.; Lancelot, C.; Quijada, M.; Visez, N.; Stéphan, O.; Lecocq, V.; Diehl, F.; Khodakov, A.Y. Molecular structure and localization of carbon species in alumina supported cobalt Fischer-Tropsch catalysts in a slurry reactor. Catal. Today 2014, 228, 65–76. [Google Scholar] [CrossRef]
- De Pontes, M.; Yokomizo, G.H.; Bell, A.T. A novel method for analyzing transient response data obtained in isotopic tracer studies of CO hydrogenation. J. Catal. 1987, 104, 147–155. [Google Scholar] [CrossRef]
- Van Helden, P.; van den Berg, J.A.; Ciobîcǎ, I.M. Hydrogen-assisted CO dissociation on the Co(211) stepped surface. Catal. Sci. Technol. 2012, 2, 491–494. [Google Scholar] [CrossRef]
- Moodley, D.J.; van de Loosdrecht, J.; Saib, A.M.; Niemantsverdriet, J.W. Advances in Fischer-Tropsch Synthesis, Catalysts and Catalysis; Davis, B.H., Occelli, M.L., Speight, J.G., Eds.; CRC Press: Boca Raton, FL, USA, 2010; Chapter 4; pp. 49–81. [Google Scholar]
- Sadeqzadeh, M.; Chambrey, S.; Piché, S.; Fongarland, P.; Luck, F.; Curulla-Ferré, D.; Schweich, D.; Bousquet, J.; Khodakov, A.Y. Deactivation of a Co/Al2O3 Fischer–Tropsch catalyst by water-induced sintering in slurry reactor: Modeling and experimental investigations. Catal. Today 2013, 215, 52–59. [Google Scholar] [CrossRef]
- Peña, D.; Griboval-Constant, A.; Lecocq, V.; Diehl, F.; Khodakov, A.Y. Influence of operating conditions in a continuously stirred tank reactor on the formation of carbon species on alumina supported cobalt Fischer-Tropsch catalysts. Catal. Today 2013, 215, 43–51. [Google Scholar] [CrossRef]
- Costa, C.N.; Christou, S.Y.; Georgiou, G.; Efstathiou, A.M. Mathematical modeling of the oxygen storage capacity phenomenon studied by CO pulse transient experiments over Pd/CeO2 catalyst. J. Catal. 2003, 219, 259–272. [Google Scholar] [CrossRef]
- Rebmann, E.; Fongarland, P.; Lecocq, V.; Diehl, F.; Schuurman, Y. Kinetic modeling of transient Fischer-Tropsch experiments over Co/Al2O3 catalysts with different microstructures. Catal. Today 2016, 275, 20–26. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CO | NCO | θCO | NCHx | θCHx | yCH4, s.s (ppm) | TOFchem | TOFITK | τCHx (s) | keff | SCH4 (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| vol% | (μmol g−1) | (μmol g−1) | (× 103, s−1) | (× 103, s−1) | (× 102, s−1) | |||||
| 3 | 10.0 | 0.029 | 15.8 | 0.047 | 3870 | 9.7 | 127.7 | 4.8 | 20.8 | 60 |
| 5 | 10.8 | 0.032 | 28.8 | 0.085 | 2690 | 7.4 | 61.5 | 12.0 | 8.3 | 38.5 |
| 7 | 16.5 | 0.048 | 39.7 | 0.117 | 4380 | 7.3 | 44.2 | 16.0 | 6.3 | 35 |
| vol% CO | ΤΙH | ΤΙH | ΤΙH | ΤΙH | ΤPH | ΤPH |
|---|---|---|---|---|---|---|
| N (12CH4) or Cβ | θ (12CH4) | N (13CH4) | θ (13CH4) | N (12CH4) or Cγ | θ (12CH4) | |
| (μmol g−1) | (μmol g−1) | (μmol g−1) | ||||
| 3 | 34.3 | 0.10 | 27.6 | 0.08 | 128 | 0.38 |
| 5 | 27.6 | 0.08 | 40.2 | 0.12 | 228 | 0.67 |
| 7 | 30.4 | 0.09 | 56.3 | 0.17 | 252 | 0.74 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasiliades, M.A.; Kyprianou, K.K.; Govender, N.S.; Govender, A.; Crous, R.; Moodley, D.; Efstathiou, A.M. The Effect of CO Partial Pressure on Important Kinetic Parameters of Methanation Reaction on Co-Based FTS Catalyst Studied by SSITKA-MS and Operando DRIFTS-MS Techniques. Catalysts 2020, 10, 583. https://doi.org/10.3390/catal10050583
Vasiliades MA, Kyprianou KK, Govender NS, Govender A, Crous R, Moodley D, Efstathiou AM. The Effect of CO Partial Pressure on Important Kinetic Parameters of Methanation Reaction on Co-Based FTS Catalyst Studied by SSITKA-MS and Operando DRIFTS-MS Techniques. Catalysts. 2020; 10(5):583. https://doi.org/10.3390/catal10050583
Chicago/Turabian StyleVasiliades, Michalis A., Konstantina K. Kyprianou, Nilenindran S. Govender, Ashriti Govender, Renier Crous, Denzil Moodley, and Angelos M. Efstathiou. 2020. "The Effect of CO Partial Pressure on Important Kinetic Parameters of Methanation Reaction on Co-Based FTS Catalyst Studied by SSITKA-MS and Operando DRIFTS-MS Techniques" Catalysts 10, no. 5: 583. https://doi.org/10.3390/catal10050583
APA StyleVasiliades, M. A., Kyprianou, K. K., Govender, N. S., Govender, A., Crous, R., Moodley, D., & Efstathiou, A. M. (2020). The Effect of CO Partial Pressure on Important Kinetic Parameters of Methanation Reaction on Co-Based FTS Catalyst Studied by SSITKA-MS and Operando DRIFTS-MS Techniques. Catalysts, 10(5), 583. https://doi.org/10.3390/catal10050583