Combining Exsolution and Infiltration for Redox, Low Temperature CH4 Conversion to Syngas



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
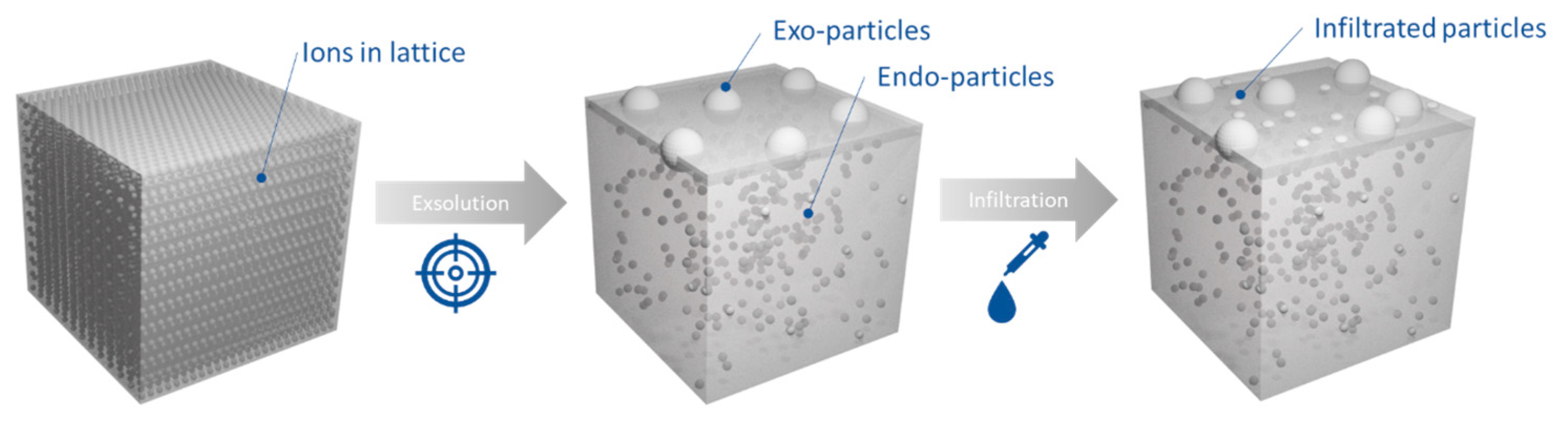
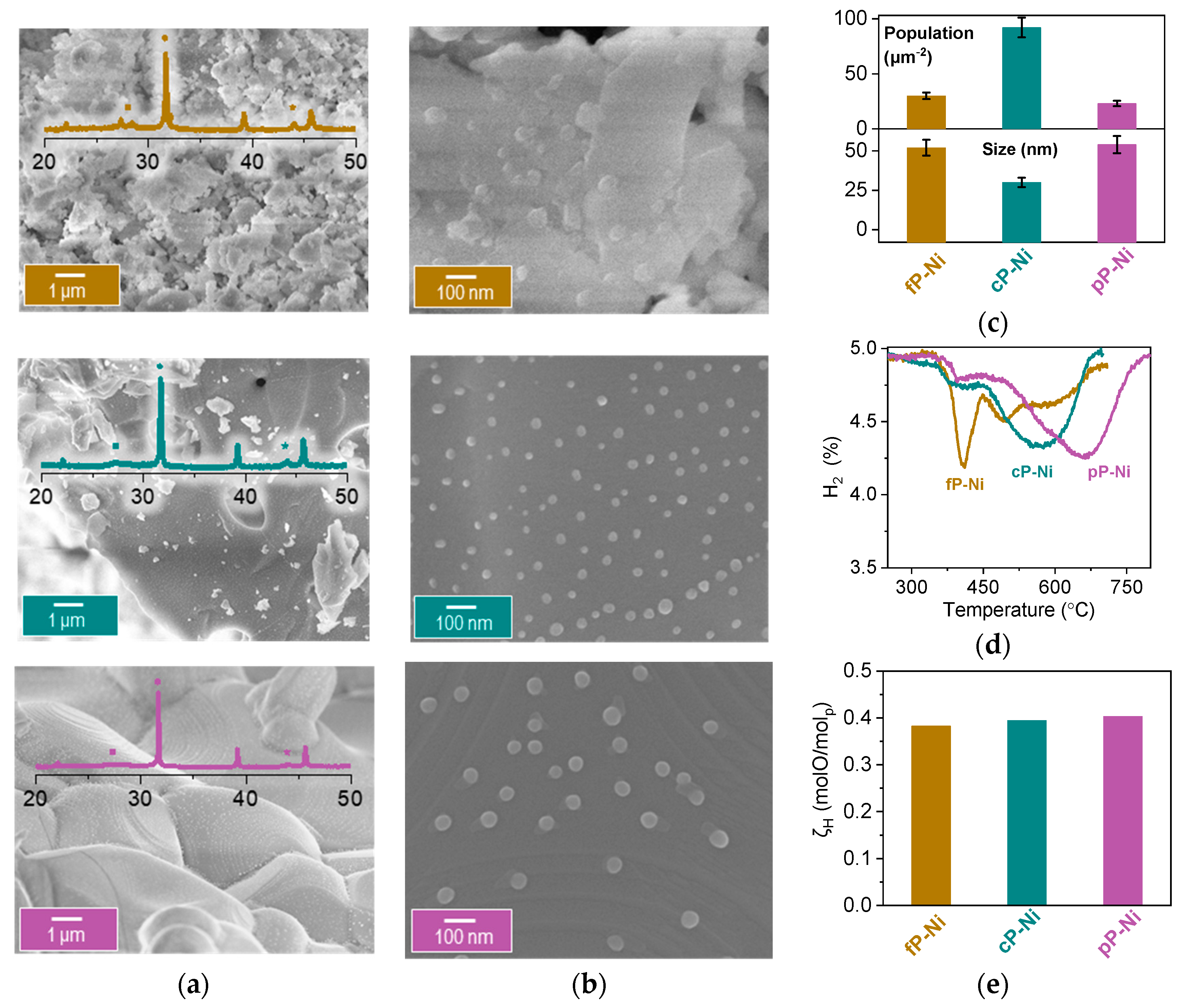
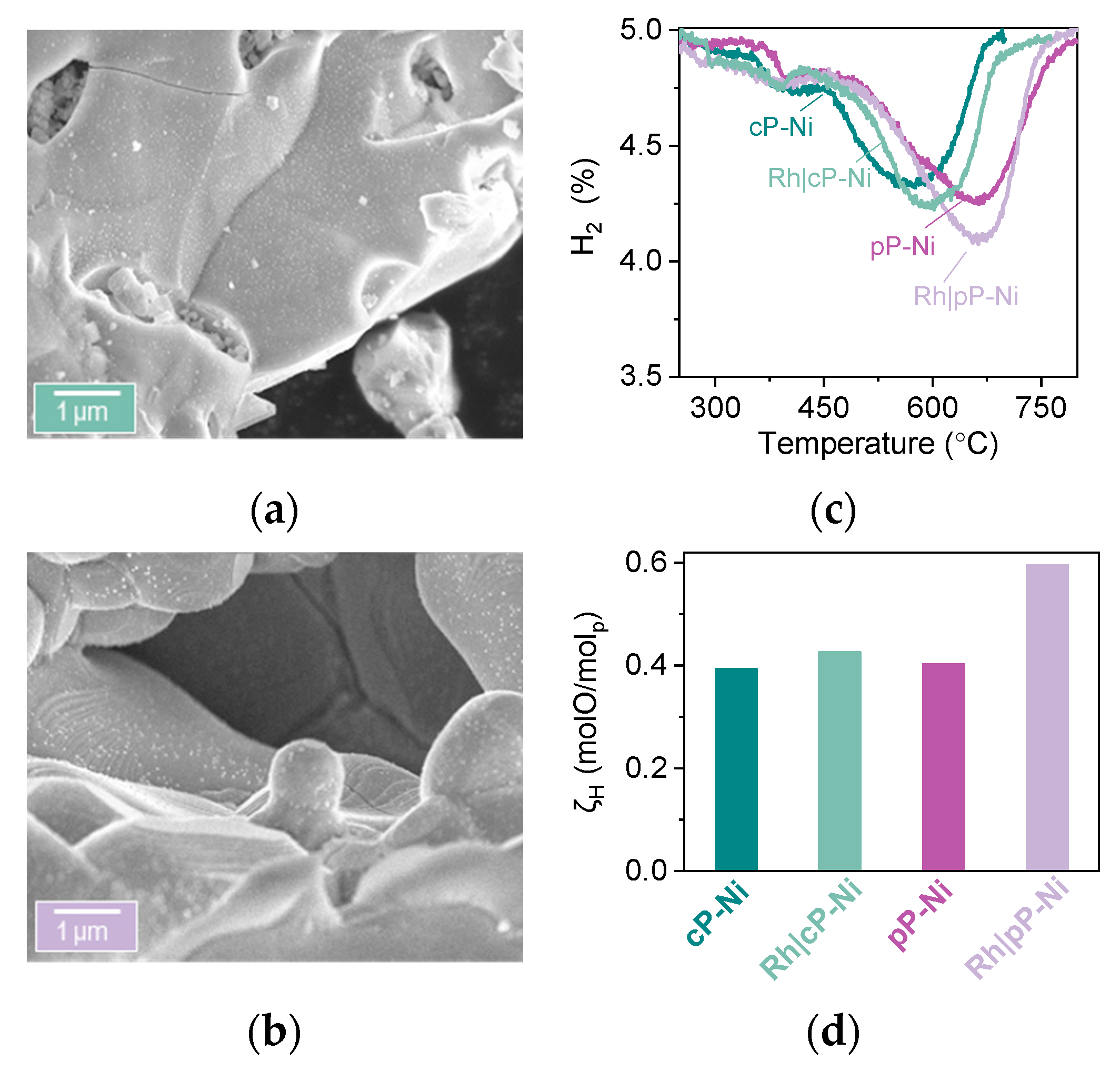
2.1. Microstructural Design of Perovskite System for Redox Methane Conversion
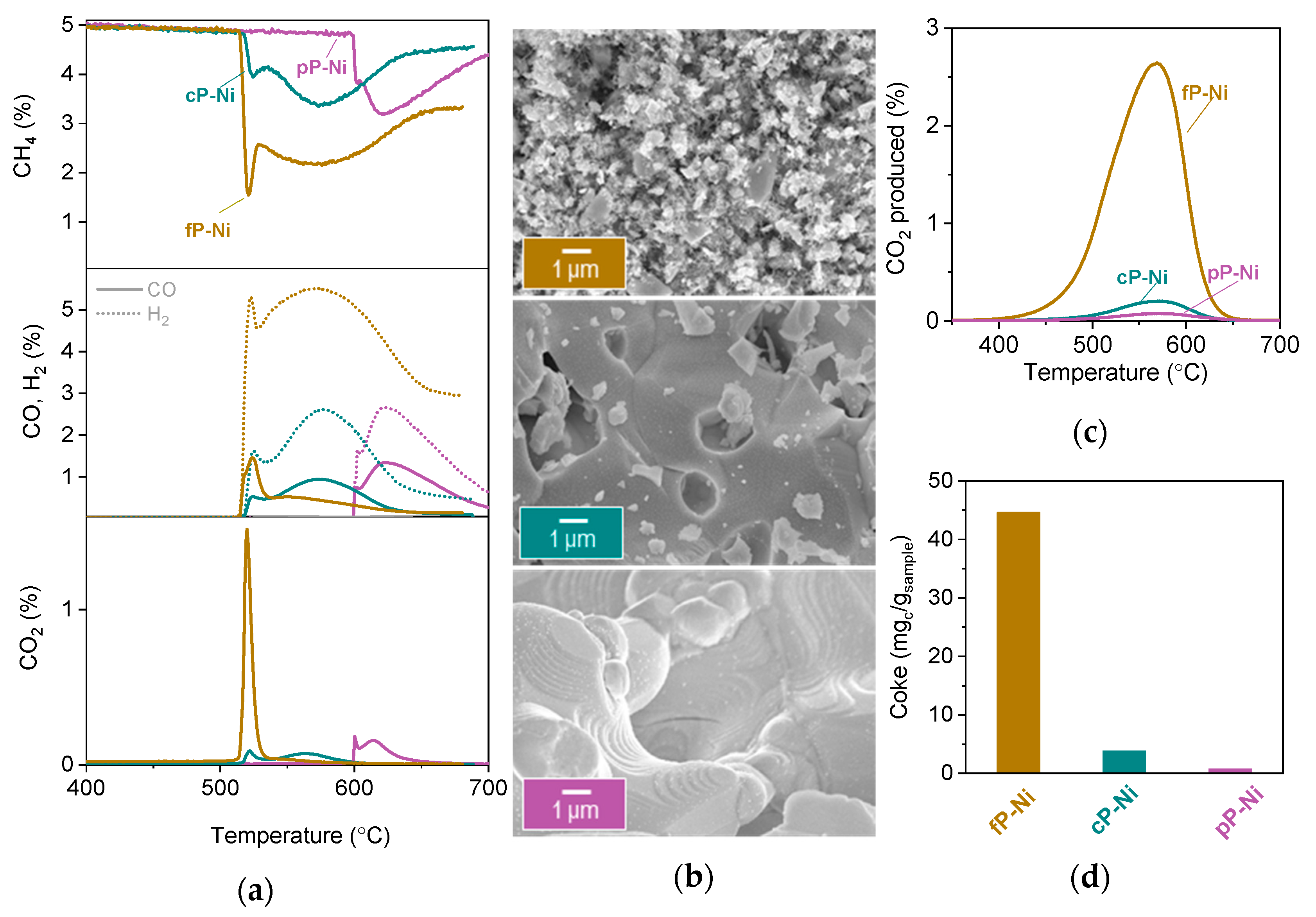
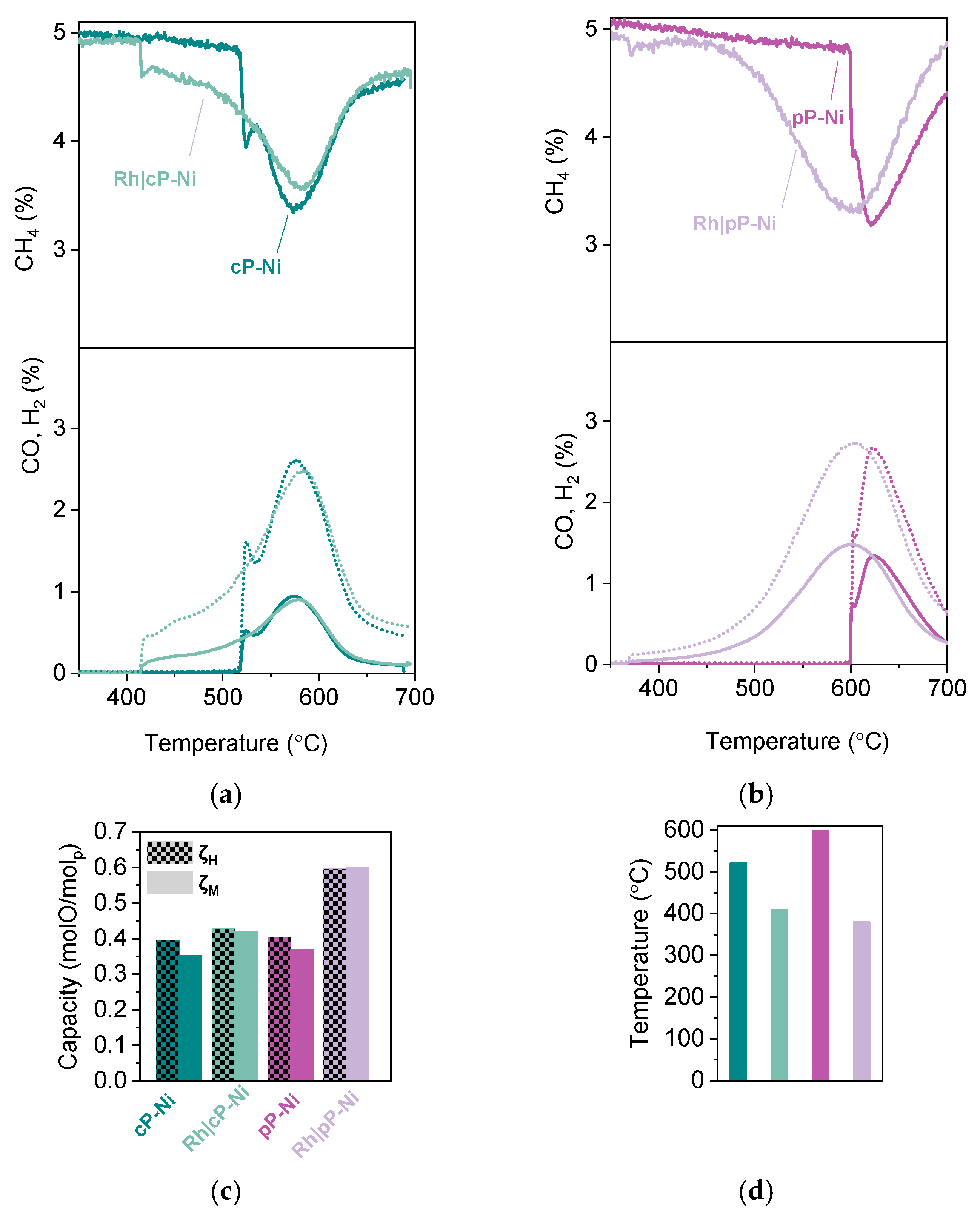
2.2. Probing the Effect of Microstructure on the Reactivity of the Exo/endo Particle System
2.3. Functionalization of the Surface Aiming to Access Low Temperature Activity
3. Materials and Methods
3.1. Sample Preparation
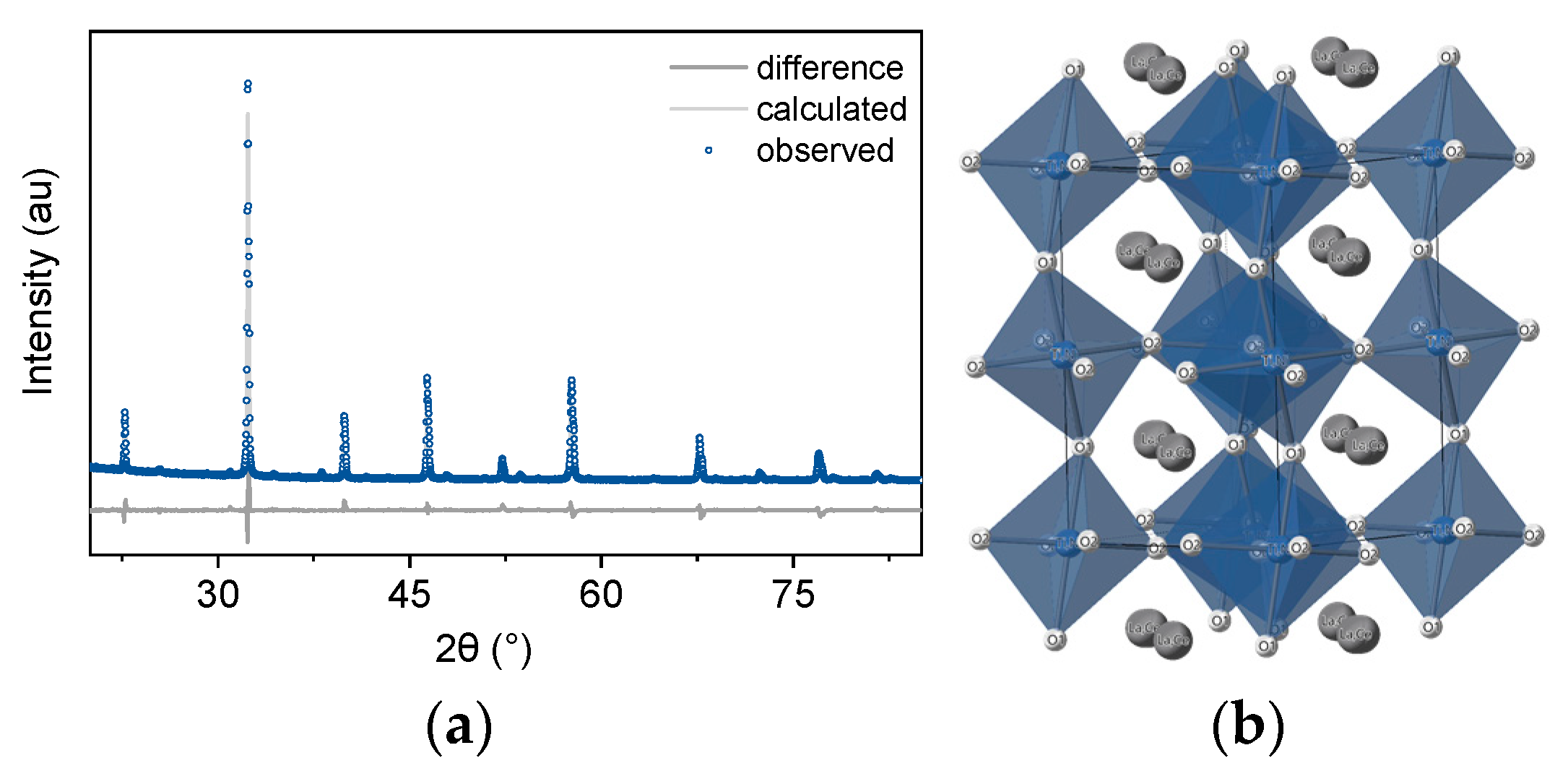
3.2. X-ray Diffraction
3.3. Electron Microscopy
Image Analysis
3.4. Reactor Set-up
3.4.1. Temperature Programmed Reduction
3.4.2. Temperature Programmed Oxidation
3.5. Calculations
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Data Availability
References
- Yang, J.; Cai, N.; Li, Z. Hydrogen Production from the Steam−Iron Process with Direct Reduction of Iron Oxide by Chemical Looping Combustion of Coal Char. Energy Fuels 2008, 22, 2570–2579. [Google Scholar] [CrossRef]
- Metcalfe, I.S.; Ray, B.; Dejoie, C.; Hu, W.; de Leeuwe, C.; Dueso, C.; García-García, F.R.; Mak, C.-M.; Papaioannou, E.I.; Thompson, C.R.; et al. Overcoming chemical equilibrium limitations using a thermodynamically reversible chemical reactor. Nat. Chem. 2019, 11, 638. [Google Scholar] [CrossRef] [PubMed]
- Adiya, Z.I.S.; Dupont, V.; Mahmud, T. Steam reforming of shale gas in a packed bed reactor with and without chemical looping using nickel based oxygen carrier. Int. J. Hydrogen Energy 2018, 43, 6904–6917. [Google Scholar] [CrossRef]
- Pujara, M.; Sheth, M.; Rachchh, N.; Bhoraniya, R.; Harichandan, A.B. Chemical Looping Reforming (CLR) System for H2 Production—A Review. In Proceedings of the Renewable Energy and Climate Change; Deb, D., Dixit, A., Chandra, L., Eds.; Springer: Singapore, 2020; pp. 267–276. [Google Scholar]
- Ipsakis, D.; Heracleous, E.; Silvester, L.; Bukur, D.B.; Lemonidou, A.A. Reaction-based kinetic model for the reduction of supported NiO oxygen transfer materials by CH4. Catal. Today 2019. [Google Scholar] [CrossRef]
- Zeng, L.; Cheng, Z.; Fan, J.A.; Fan, L.-S.; Gong, J. Metal oxide redox chemistry for chemical looping processes. Nat. Rev. Chem. 2018, 2, 349–364. [Google Scholar] [CrossRef]
- Hu, J.; Galvita, V.; Poelman, H.; Marin, G. Advanced Chemical Looping Materials for CO2 Utilization: A Review. Materials 2018, 11, 1187. [Google Scholar] [CrossRef]
- Buelens, L.C.; Galvita, V.V.; Poelman, H.; Detavernier, C.; Marin, G.B. Super-dry reforming of methane intensifies CO2 utilization via Le Chateliers principle. Science 2016, 354, 449–452. [Google Scholar] [CrossRef]
- Neagu, D.; Papaioannou, E.I.; Ramli, W.K.W.; Miller, D.N.; Murdoch, B.J.; Ménard, H.; Umar, A.; Barlow, A.J.; Cumpson, P.J.; Irvine, J.T.S.; et al. Demonstration of chemistry at a point through restructuring and catalytic activation at anchored nanoparticles. Nat. Commun. 2017, 8, 1855. [Google Scholar] [CrossRef]
- Neagu, D.; Oh, T.-S.; Miller, D.N.; Ménard, H.; Bukhari, S.M.; Gamble, S.R.; Gorte, R.J.; Vohs, J.M.; Irvine, J.T.S. Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution. Nat. Commun. 2015, 6, 8120. [Google Scholar] [CrossRef]
- Neagu, D.; Kyriakou, V.; Roiban, I.-L.; Aouine, M.; Tang, C.; Caravaca, A.; Kousi, K.; Schreur-Piet, I.; Metcalfe, I.S.; Vernoux, P.; et al. In Situ Observation of Nanoparticle Exsolution from Perovskite Oxides: From Atomic Scale Mechanistic Insight to Nanostructure Tailoring. ACS Nano 2019, 13, 12996–13005. [Google Scholar] [CrossRef]
- Papaioannou, E.I.; Neagu, D.; Ramli, W.K.W.; Irvine, J.T.S.; Metcalfe, I.S. Sulfur-Tolerant, Exsolved Fe–Ni Alloy Nanoparticles for CO Oxidation. Top. Catal. 2018. [Google Scholar] [CrossRef]
- Komarala, E.P.; Komissarov, I.; Rosen, B.A. Effect of Fe and Mn Substitution in LaNiO3 on Exsolution, Activity, and Stability for Methane Dry Reforming. Catalysts 2020, 10, 27. [Google Scholar] [CrossRef]
- Otto, S.-K.; Kousi, K.; Neagu, D.; Bekris, L.; Janek, J.; Metcalfe, I.S. Exsolved Nickel Nanoparticles Acting as Oxygen Storage Reservoirs and Active Sites for Redox CH4 Conversion. ACS Appl. Energy Mater. 2019, 2, 7288–7298. [Google Scholar] [CrossRef]
- Hosseini, D.; Donat, F.; Abdala, P.M.; Kim, S.M.; Kierzkowska, A.M.; Müller, C.R. Reversible Exsolution of Dopant Improves the Performance of Ca2Fe2O5 for Chemical Looping Hydrogen Production. ACS Appl. Mater. Interfaces 2019. [Google Scholar] [CrossRef]
- Zeng, D.; Qiu, Y.; Peng, S.; Chen, C.; Zeng, J.; Zhang, S.; Xiao, R. Enhanced hydrogen production performance through controllable redox exsolution within CoFeAlOx spinel oxygen carrier materials. J. Mater. Chem. A 2018, 6, 11306–11316. [Google Scholar] [CrossRef]
- Lindenthal, L.; Rameshan, R.; Summerer, H.; Ruh, T.; Popovic, J.; Nenning, A.; Löffler, S.; Opitz, A.K.; Blaha, P.; Rameshan, C. Modifying the Surface Structure of Perovskite-Based Catalysts by Nanoparticle Exsolution. Catalysts 2020, 10, 268. [Google Scholar] [CrossRef]
- Kousi, K.; Neagu, D.; Bekris, L.; Papaioannou, E.I.; Metcalfe, I.S. Endogenous Nanoparticles Strain Perovskite Host Lattice Providing Oxygen Capacity and Driving Oxygen Exchange and CH4 Conversion to Syngas. Angew. Chem. Int. Ed. 2020, 59, 2510–2519. [Google Scholar] [CrossRef]
- Wan Abu Bakar, W.A.; Ali, R.; Mohammad, N.S. The effect of noble metals on catalytic methanation reaction over supported Mn/Ni oxide based catalysts. Arab. J. Chem. 2015, 8, 632–643. [Google Scholar] [CrossRef]
- Hou, Z.; Yashima, T. Small amounts of Rh-promoted Ni catalysts for methane reforming with CO2. Catal. Lett. 2003, 89, 193–197. [Google Scholar] [CrossRef]
- Zhu, X.; Imtiaz, Q.; Donat, F.; Müller, C.R.; Li, F. Chemical looping beyond combustion—A perspective. Energy Environ. Sci. 2020, 13, 772–804. [Google Scholar] [CrossRef]
- Tang, C.; Kousi, K.; Neagu, D.; Portolés, J.; Papaioannou, E.I.; Metcalfe, I.S. Towards efficient use of noble metals via exsolution exemplified for CO oxidation. Nanoscale 2019, 11, 16935–16944. [Google Scholar] [CrossRef] [PubMed]
- Neagu, D.; Tsekouras, G.; Miller, D.N.; Ménard, H.; Irvine, J.T.S. In situ growth of nanoparticles through control of non-stoichiometry. Nat. Chem. 2013, 5, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Shafiefarhood, A.; Zhang, J.; Neal, L.M.; Li, F. Rh-promoted mixed oxides for “low-temperature” methane partial oxidation in the absence of gaseous oxidants. J. Mater. Chem. A 2017, 5, 11930–11939. [Google Scholar] [CrossRef]
- Palcheva, R.; Olsbye, U.; Palcut, M.; Rauwel, P.; Tyuliev, G.; Velinov, N.; Fjellvåg, H.H. Rh promoted La0.75Sr0.25(Fe0.8Co0.2)1−xGaxO3-δ perovskite catalysts: Characterization and catalytic performance for methane partial oxidation to synthesis gas. Appl. Surf. Sci. 2015, 357, 45–54. [Google Scholar] [CrossRef]
- Hou, Z.; Chen, P.; Fang, H.; Zheng, X.; Yashima, T. Production of synthesis gas via methane reforming with CO2 on noble metals and small amount of noble-(Rh-) promoted Ni catalysts. Int. J. Hydrogen Energy 2006, 31, 555–561. [Google Scholar] [CrossRef]
- Wu, J.C.S.; Chou, H.-C. Bimetallic Rh–Ni/BN catalyst for methane reforming with CO2. Chem. Eng. J. 2009, 148, 539–545. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Goula, G.; Hatzisymeon, M.; Betsi-Argyropoulou, I.; Botzolaki, G.; Kousi, K.; Kondarides, D.I.; Taylor, M.J.; Parlett, C.M.A.; Osatiashtiani, A.; et al. Effect of support oxygen storage capacity on the catalytic performance of Rh nanoparticles for CO2 reforming of methane. Appl. Catal. B Environ. 2019, 243, 490–501. [Google Scholar] [CrossRef]
- Kyriakou, V.; Neagu, D.; Zafeiropoulos, G.; Sharma, R.K.; Tang, C.; Kousi, K.; Metcalfe, I.S.; van de Sanden, M.C.M.; Tsampas, M.N. Symmetrical Exsolution of Rh Nanoparticles in Solid Oxide Cells for Efficient Syngas Production from Greenhouse Gases. ACS Catal. 2019. [Google Scholar] [CrossRef]
- Vayssilov, G.N.; Lykhach, Y.; Migani, A.; Staudt, T.; Petrova, G.P.; Tsud, N.; Skála, T.; Bruix, A.; Illas, F.; Prince, K.C.; et al. Support nanostructure boosts oxygen transfer to catalytically active platinum nanoparticles. Nat. Mater. 2011, 10, 310–315. [Google Scholar] [CrossRef]
- Varga, E.; Pusztai, P.; Óvári, L.; Oszkó, A.; Erdőhelyi, A.; Papp, C.; Steinrück, H.-P.; Kónya, Z.; Kiss, J. Probing the interaction of Rh, Co and bimetallic Rh–Co nanoparticles with the CeO2 support: Catalytic materials for alternative energy generation. Phys. Chem. Chem. Phys. 2015, 17, 27154–27166. [Google Scholar] [CrossRef]
- Ferencz, Z.; Erdőhelyi, A.; Baán, K.; Oszkó, A.; Óvári, L.; Kónya, Z.; Papp, C.; Steinrück, H.-P.; Kiss, J. Effects of Support and Rh Additive on Co-Based Catalysts in the Ethanol Steam Reforming Reaction. ACS Catal. 2014, 4, 1205–1218. [Google Scholar] [CrossRef]
- Zhu, X.; Li, K.; Neal, L.; Li, F. Perovskites as Geo-inspired Oxygen Storage Materials for Chemical Looping and Three-Way Catalysis: A Perspective. ACS Catal. 2018, 8, 8213–8236. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kousi, K.; Neagu, D.; Metcalfe, I.S. Combining Exsolution and Infiltration for Redox, Low Temperature CH4 Conversion to Syngas. Catalysts 2020, 10, 468. https://doi.org/10.3390/catal10050468
Kousi K, Neagu D, Metcalfe IS. Combining Exsolution and Infiltration for Redox, Low Temperature CH4 Conversion to Syngas. Catalysts. 2020; 10(5):468. https://doi.org/10.3390/catal10050468
Chicago/Turabian StyleKousi, Kalliopi, Dragos Neagu, and Ian S. Metcalfe. 2020. "Combining Exsolution and Infiltration for Redox, Low Temperature CH4 Conversion to Syngas" Catalysts 10, no. 5: 468. https://doi.org/10.3390/catal10050468
APA StyleKousi, K., Neagu, D., & Metcalfe, I. S. (2020). Combining Exsolution and Infiltration for Redox, Low Temperature CH4 Conversion to Syngas. Catalysts, 10(5), 468. https://doi.org/10.3390/catal10050468