Bioinspired Design and Computational Prediction of SCS Nickel Pincer Complexes for Hydrogenation of Carbon Dioxide


Abstract
:
1. Introduction
2. Results and Discussion
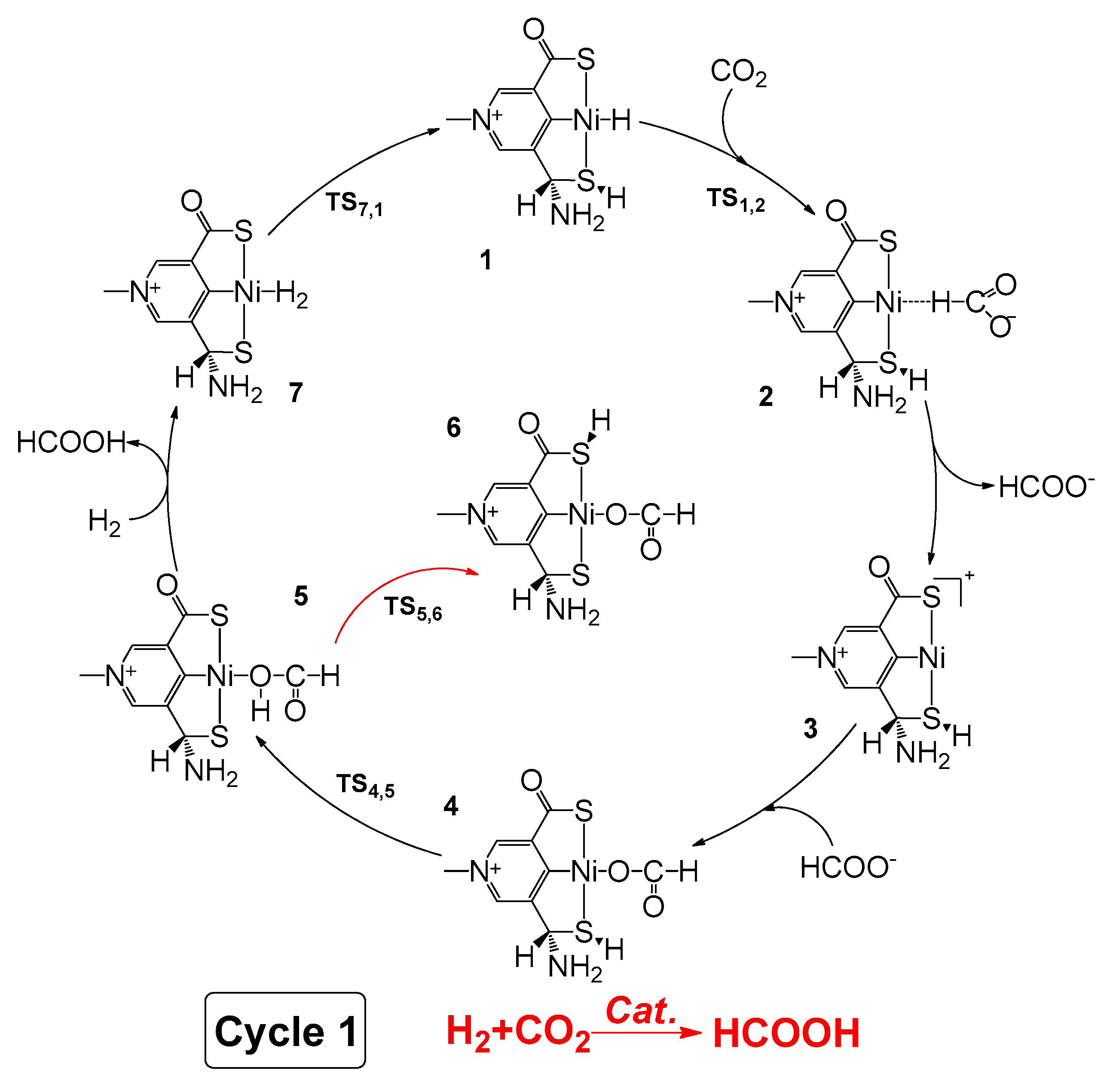
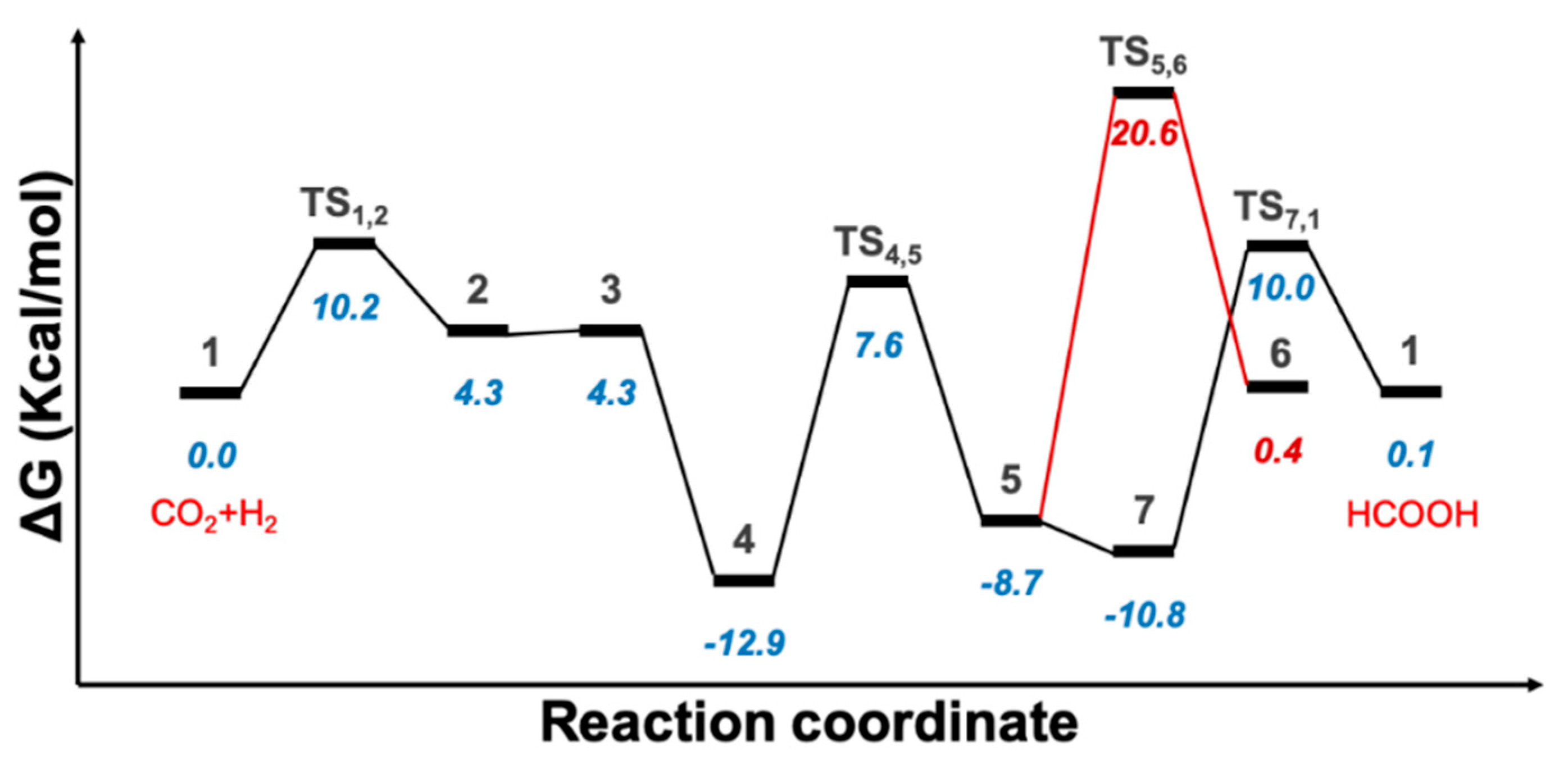
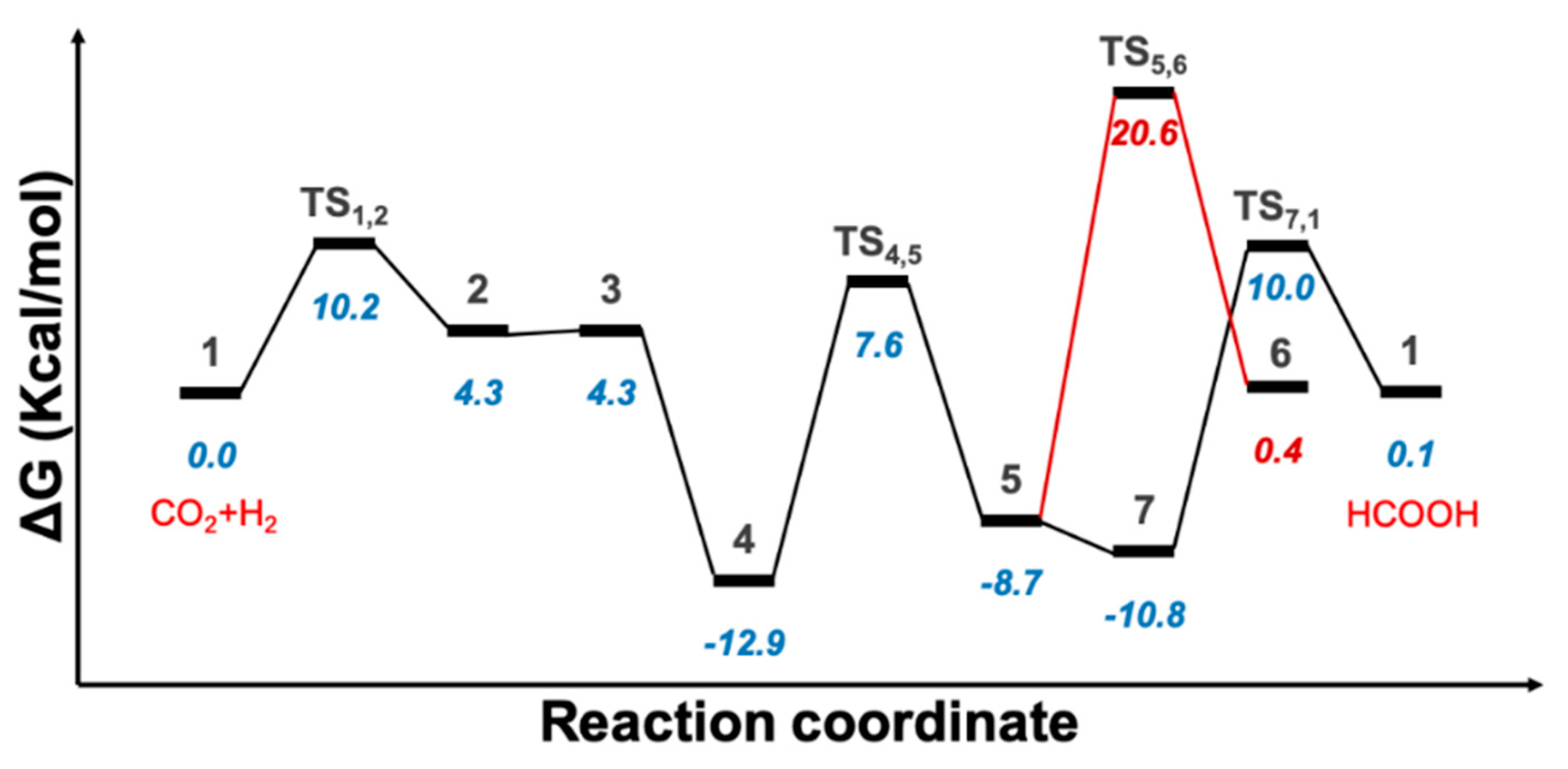
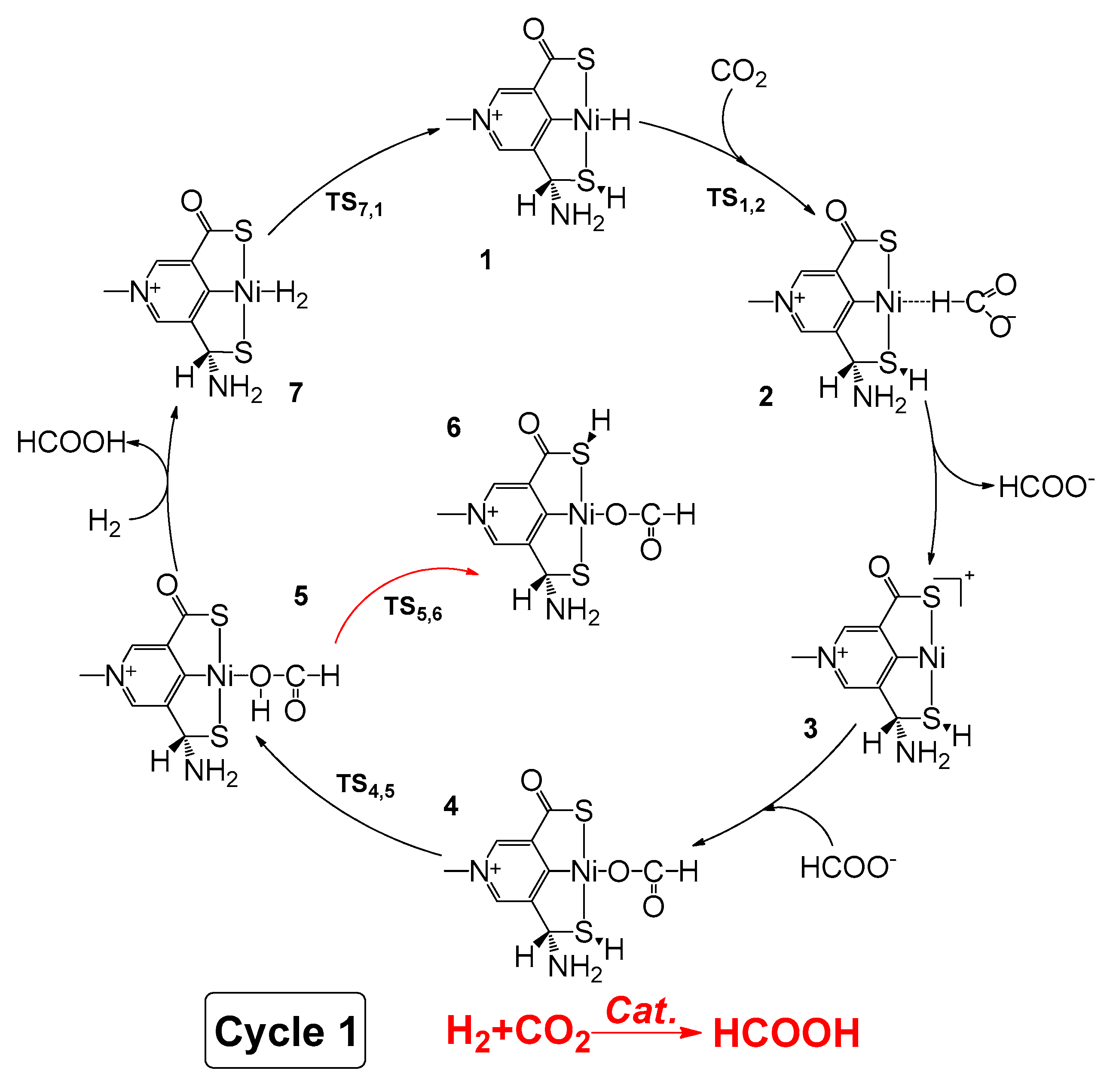
2.1. Predicted Catalytic Cycles for the Hydrogenation of CO2 to Formic Acid
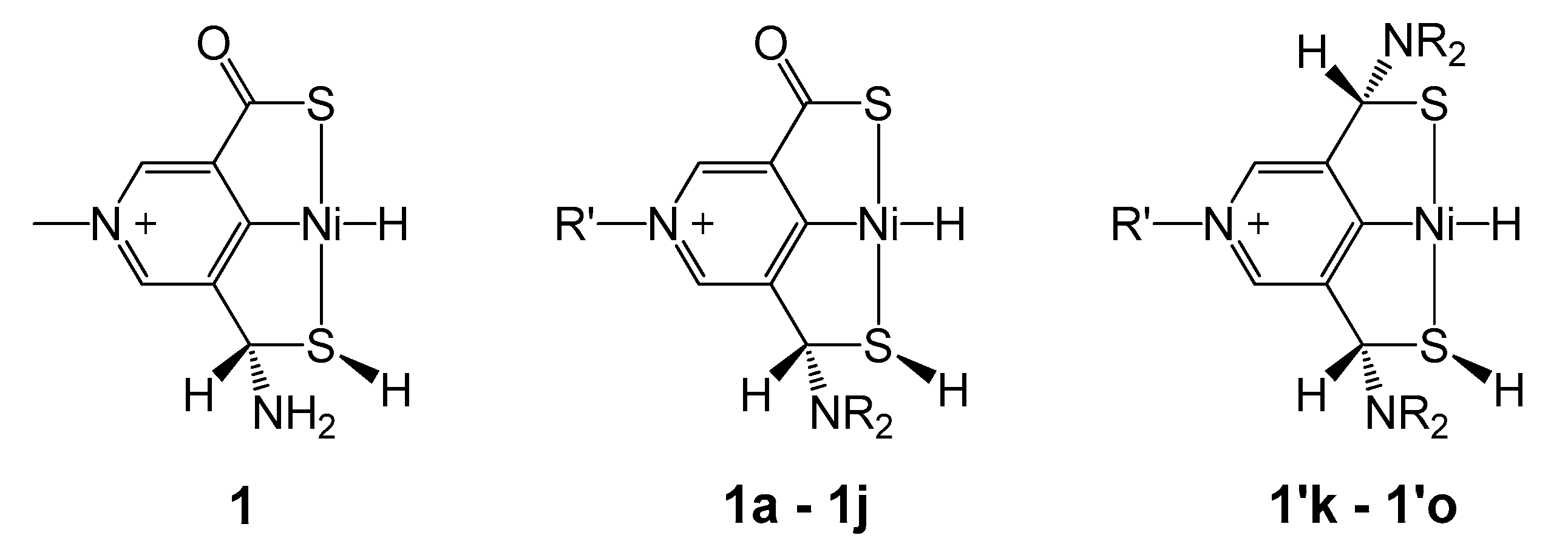
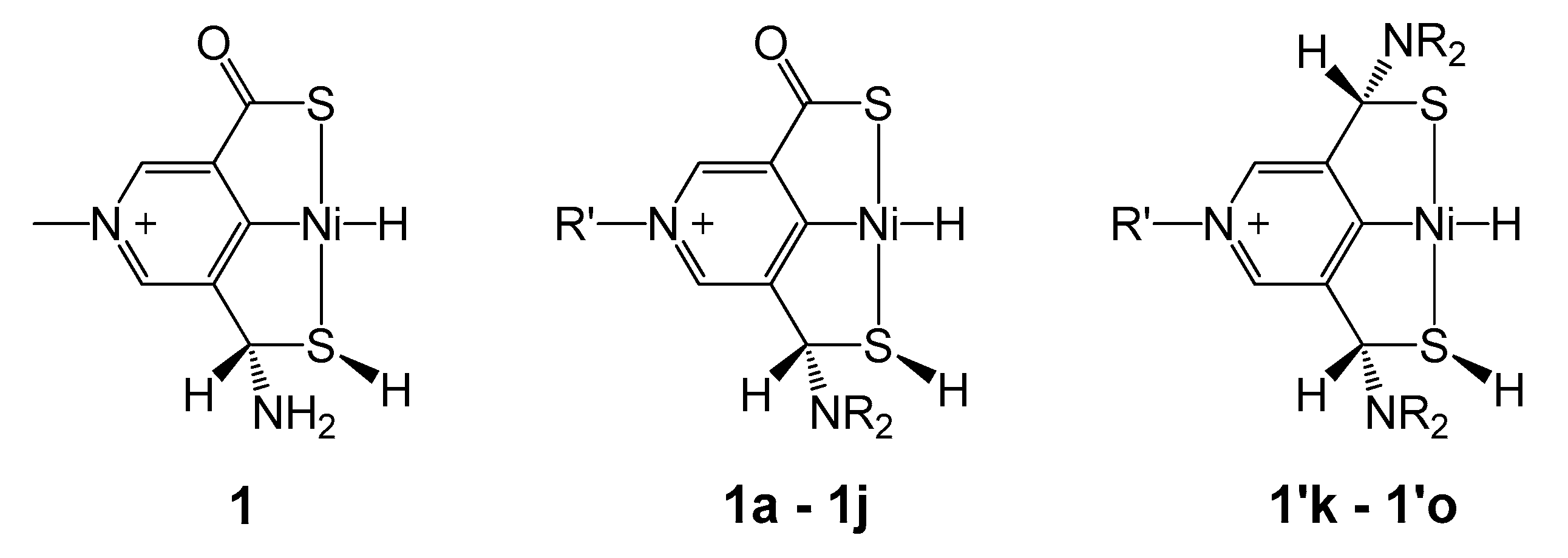
2.2. Influence of Substituents in the SCS Ligand
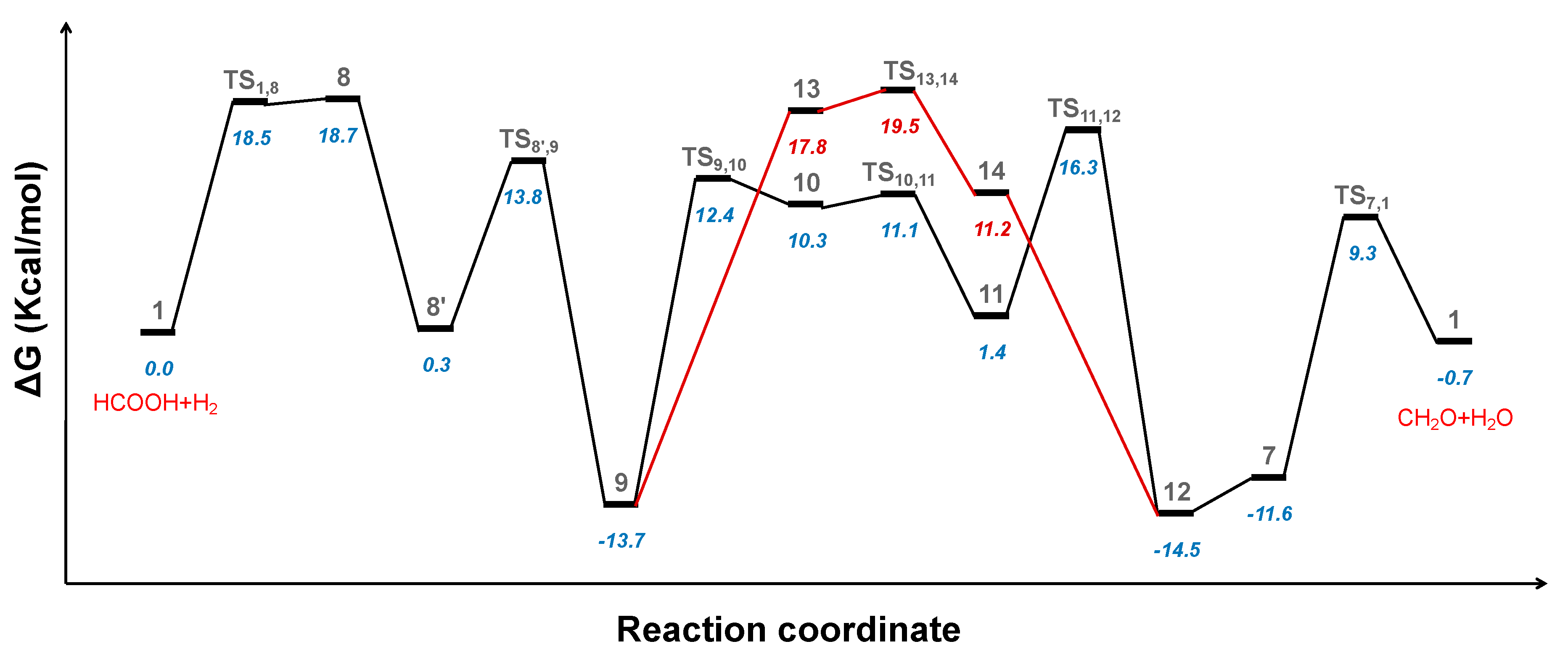
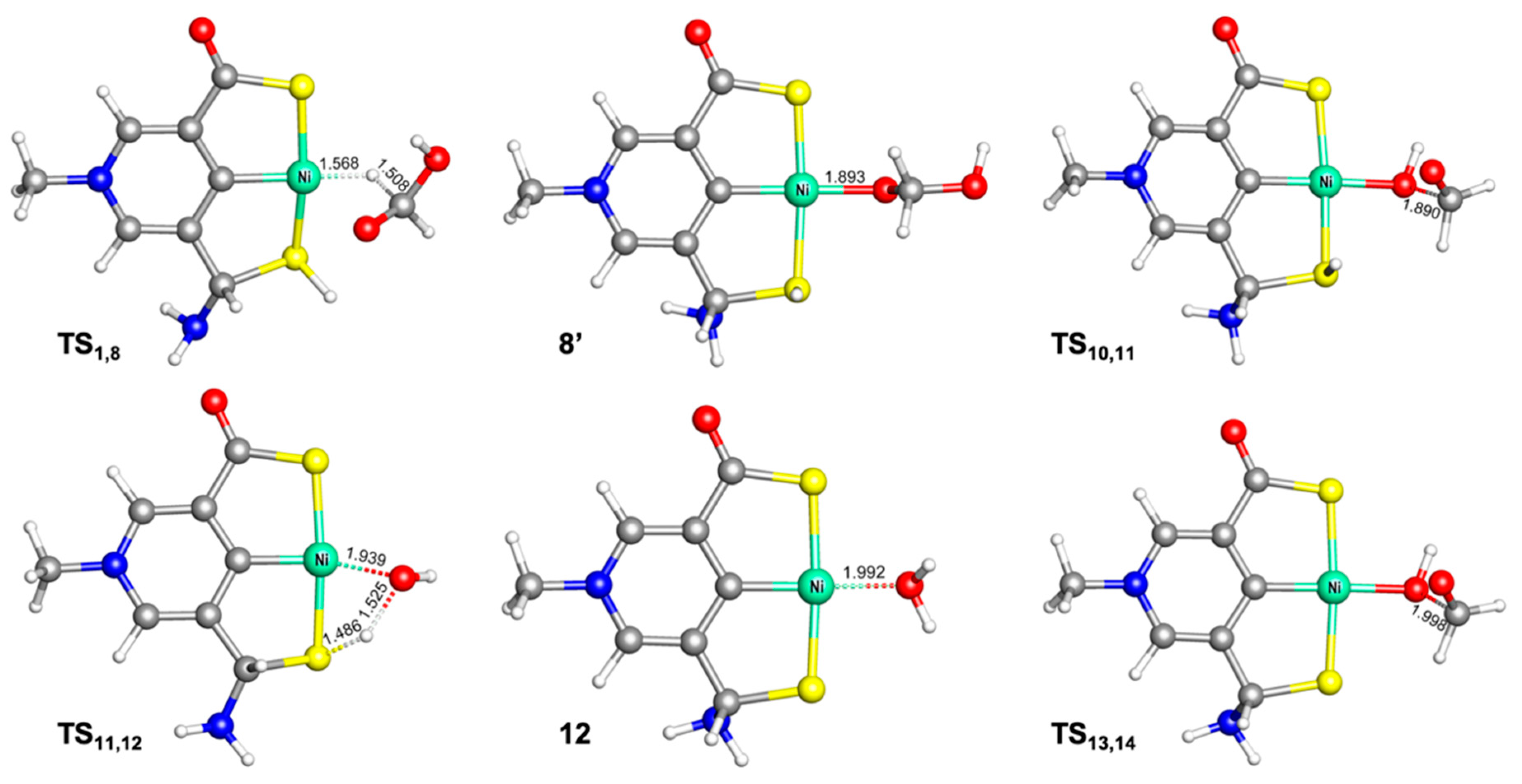
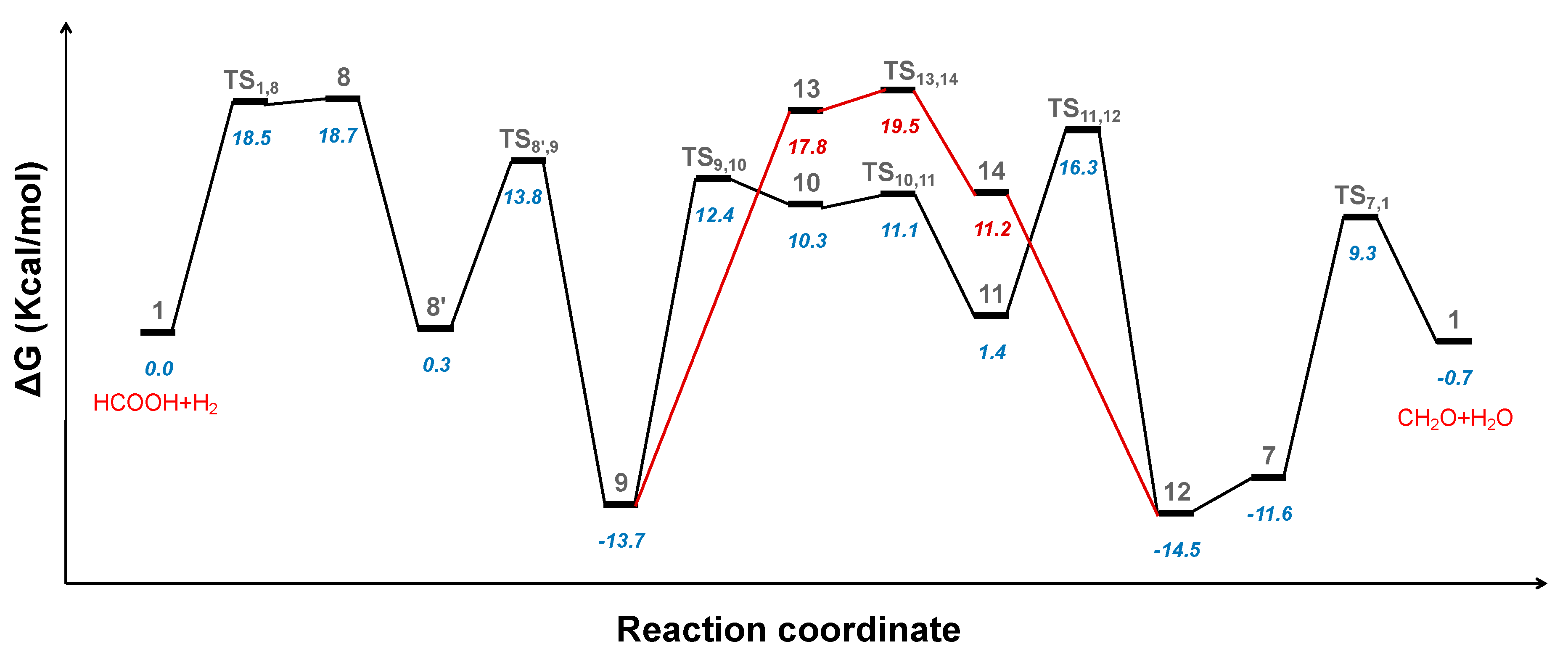
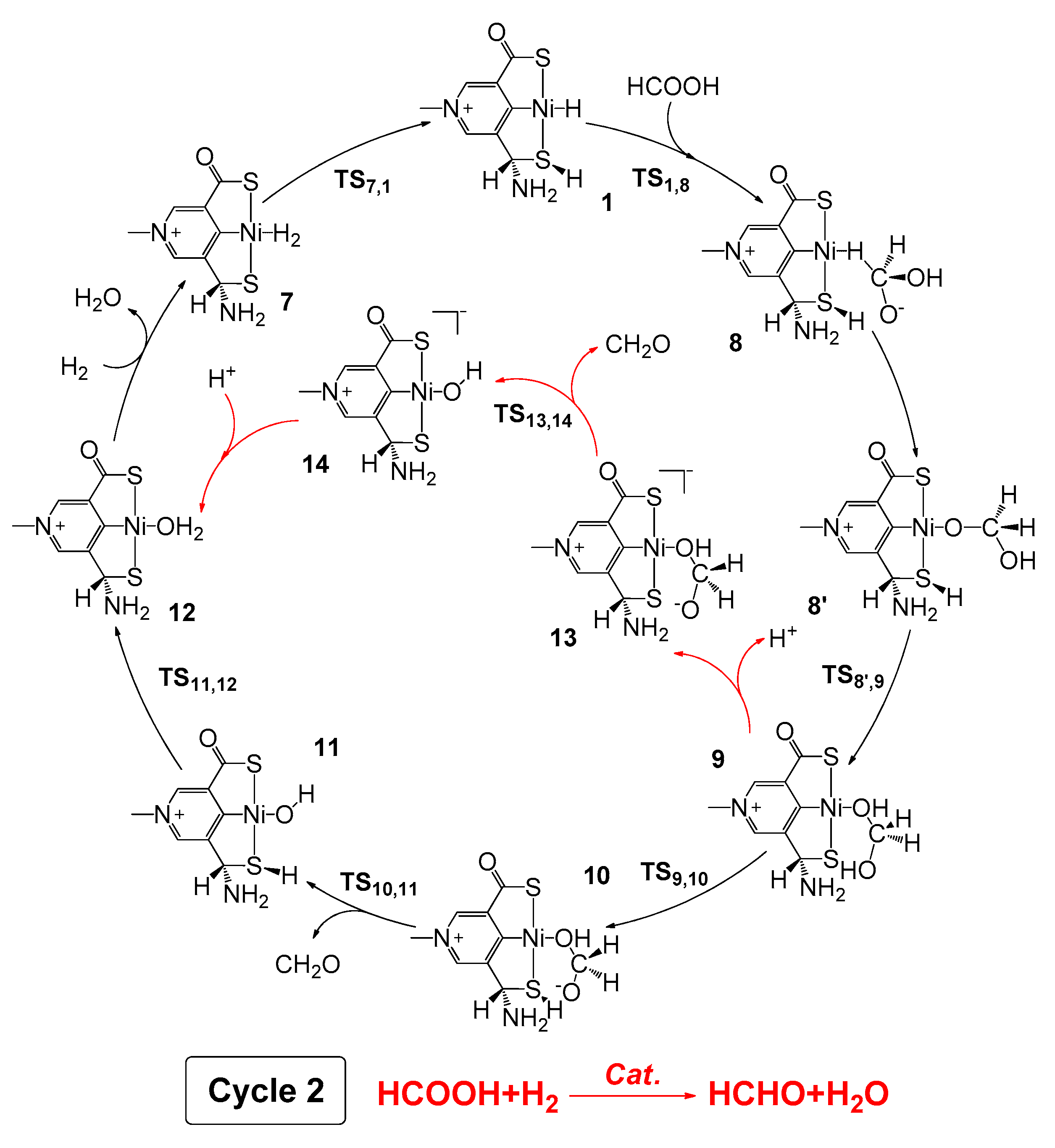
2.3. Hydrogenation of Formic Acid to Formaldehyde and Water
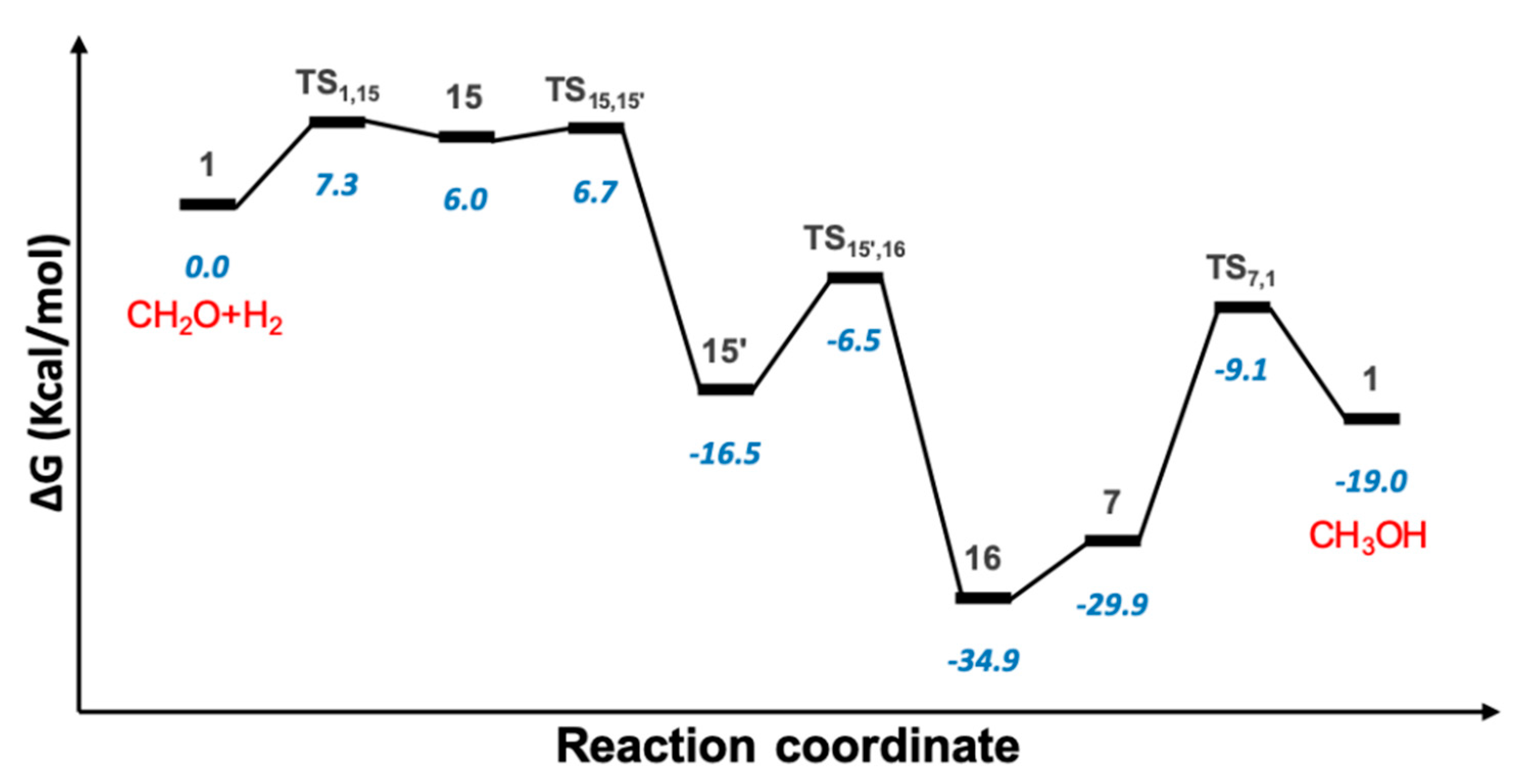
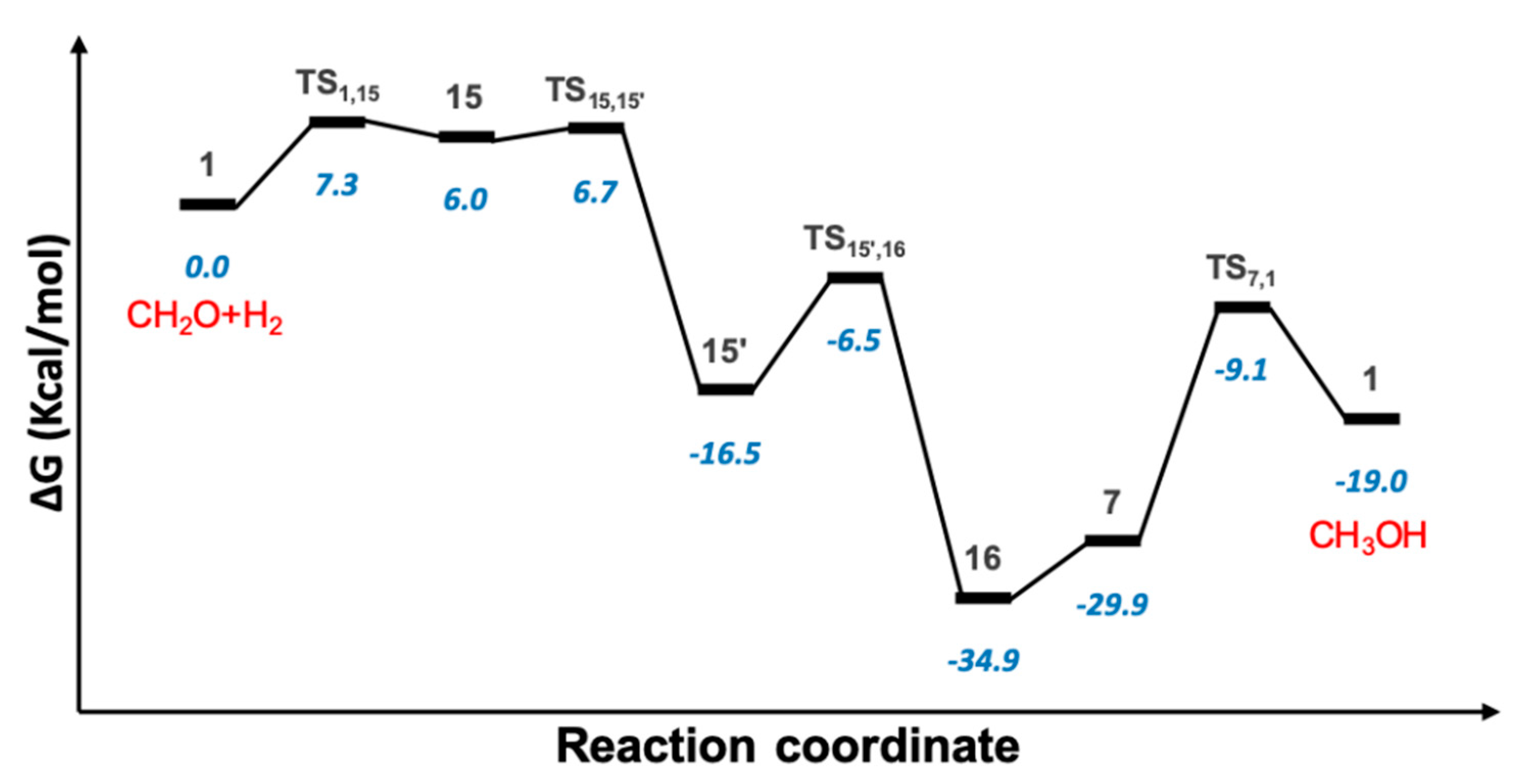
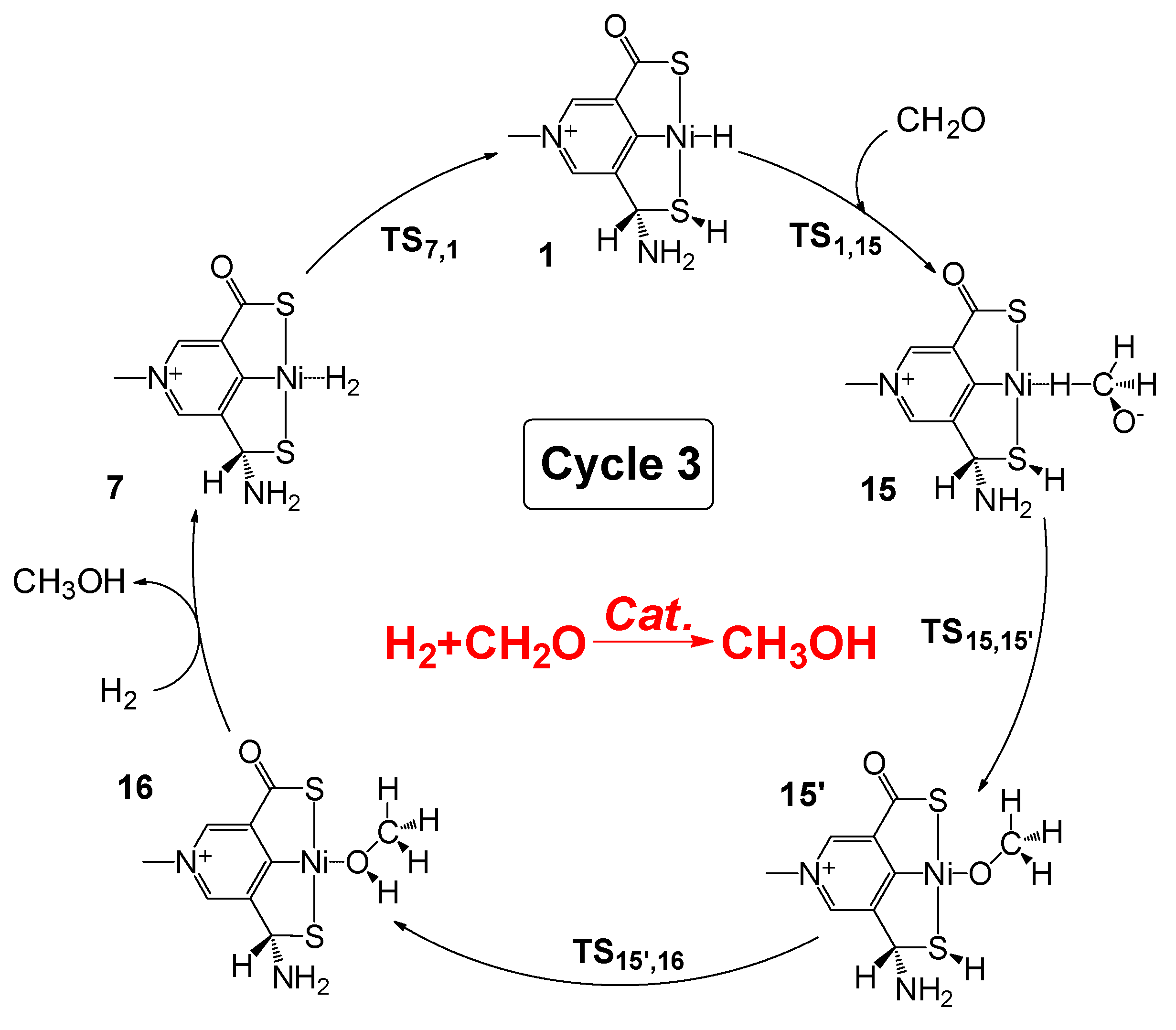
2.4. Hydrogenation of Formaldehyde to Methanol
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sakakura, T.; Choi, J.C.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W.A.; Kuhn, F.E. Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge? Angew. Chem. Int. Ed. Engl. 2011, 50, 8510–8537. [Google Scholar] [CrossRef] [PubMed]
- Eby, M.; Zickfeld, K.; Montenegro, A.; Archer, D.; Meissner, K.J.; Weaver, A.J. Lifetime of Anthropogenic Climate Change: Millennial Time Scales of Potential CO2 and Surface Temperature Perturbations. J. Clim. 2009, 22, 2501–2511. [Google Scholar] [CrossRef]
- Aresta, M. Carbon Dioxide as a Chemical Feedstock; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- He, M.; Sun, Y.; Han, B. Green Carbon Science: Scientific Basis for Integrating Carbon Resource Processing, Utilization, and Recycling. Angew. Chem. Int. Ed. 2013, 52, 9620–9633. [Google Scholar] [CrossRef]
- Jessop, P.G.; Ikariya, T.; Noyori, R. Homogeneous Hydrogenation of Carbon Dioxide. Chem. Rev. 1995, 95, 259–272. [Google Scholar] [CrossRef]
- Olah, G.A. Beyond Oil and Gas: The Methanol Economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef]
- Olah, G.A. The Methanol Economy. Chem. Eng. News 2003, 81, 5. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Surya Prakash, G.K. Chemical Recycling of Carbon Dioxide to Methanol and Dimethyl Ether: From Greenhouse Gas to Renewable, Environmentally Carbon Neutral Fuels and Synthetic Hydrocarbons. J. Org. Chem. 2009, 74, 487–498. [Google Scholar] [CrossRef]
- Nielsen, M.; Alberico, E.; Baumann, W.; Drexler, H.J.; Junge, H.; Gladiali, S.; Beller, M. Low-Temperature Aqueous-Phase Methanol Dehydrogenation to Hydrogen and Carbon Dioxide. Nature 2013, 495, 85–89. [Google Scholar] [CrossRef]
- Natte, K.; Neumann, H.; Beller, M.; Jagadeesh, R.V. Transition-Metal-Catalyzed Utilization of Methanol as a C1 Source in Organic Synthesis. Angew. Chem. Int. Ed. 2017, 56, 6384–6394. [Google Scholar] [CrossRef]
- Rodríguez-Lugo, R.E.; Trincado, M.; Vogt, M.; Tewes, F.; Santiso-Quinones, G.; Grützmacher, H. A Homogeneous Transition Metal Complex for Clean Hydrogen Production from Methanol-Water Mixtures. Nat. Chem. 2013, 5, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Onishi, N.; Laurenczy, G.; Beller, M.; Himeda, Y. Recent Progress for Reversible Homogeneous Catalytic Hydrogen Storage in Formic Acid and in Methanol. Coordin. Chem. Rev. 2018, 373, 317–332. [Google Scholar] [CrossRef]
- Goeppert, A.; Czaun, M.; Jones, J.P.; Prakash, G.K.S.; Olah, G.A. Recycling of Carbon Dioxide to Methanol and Derived Products-Closing the Loop. Chem. Soc. Rev. 2014, 43, 7995–8048. [Google Scholar] [CrossRef] [PubMed]
- Bernskoetter, W.H.; Hazari, N. Reversible Hydrogenation of Carbon Dioxide to Formic Acid and Methanol: Lewis Acid Enhancement of Base Metal Catalysts. Acc. Chem. Res. 2017, 50, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.P.C.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Carbon Dioxide-to-Methanol Single-Pot Conversion using a C-scorpionate Iron (II) Catalyst. Green Chem. 2017, 19, 4811–4815. [Google Scholar] [CrossRef]
- Lane, E.M.; Zhang, Y.; Hazari, N.; Bernskoetter, W.H. Sequential Hydrogenation of CO2 to Methanol Using a Pincer Iron Catalyst. Organometallics 2019, 38, 15, 3084–3091. [Google Scholar] [CrossRef]
- Schneidewind, J.; Adam, R.; Baumann, W.; Jackstell, R.; Beller, M. Low-Temperature Hydrogenation of Carbon Dioxide to Methanol with a Homogeneous Cobalt Catalyst. Angew. Chem. Int. Ed. 2017, 56, 1890–1893. [Google Scholar] [CrossRef]
- Kar, S.; Goppert, A.; Kothandaraman, J.; Prakash, G.K.S. Manganese-Catalyzed Sequential Hydrogenation of CO2 to Methanol via Formamide. ACS Catal. 2017, 7, 9, 6347–6351. [Google Scholar] [CrossRef]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef]
- Kar, S.; Kothandaraman, J.; Goeppert, A.; Surya Prakash, G.K. Advances in Catalytic Homogeneous Hydrogenation of Carbon Dioxide to Methanol. J. CO2 Util. 2018, 23, 212–218. [Google Scholar] [CrossRef]
- Schmeier, T.J.; Dobereiner, G.E.; Crabtree, R.H.; Hazari, N. Secondary Coordination Sphere Interactions Facilitate the Insertion Step in an Iridium (III) CO2 Reduction Catalyst. J. Am. Chem. Soc. 2011, 133, 9274–9277. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Sasaki, Y.; Watanabe, T.; Saito, M. Homogeneous Hydrogenation of Carbon Dioxide to Methanol Catalyzed by Ruthenium Cluster Anions in the Presence of Halide Anions. Bull. Chem. Soc. Jpn. 1995, 68, 2837–2842. [Google Scholar] [CrossRef]
- Kar, S.; Sen, R.; Goeppert, A.; Prakash, G.K.S. Integrative CO2 Capture and Hydrogenation to Methanol with Reusable Catalyst and Amine: Toward a Carbon Neutral Methanol Economy. J. Am. Chem. Soc. 2018, 140, 1580–1583. [Google Scholar] [CrossRef] [PubMed]
- Huff, C.A.; Sanford, M.S. Cascade Catalysis for the Homogeneous Hydrogenation of CO2 to Methanol. J. Am. Chem. Soc. 2011, 133, 18122–18125. [Google Scholar] [CrossRef] [PubMed]
- Rezayee, N.M.; Huff, C.A.; Sanford, M.S. Tandem Amine and Ruthenium Catalyzed Hydrogenation of CO2 to Methanol. J. Am. Chem. Soc. 2015, 137, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.-I.; Sasaki, Y.; Kawai, M.; Watanabe, T.; Saito, M. Ruthenium Complex Catalysed Hydrogenation of Carbon Dioxide to Carbon Monoxide, Methanol and Methane. J. Chem. Soc. Chem. Commun. 1993, 629–631. [Google Scholar] [CrossRef]
- Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G.A.; Prakash, G.K.S. Conversion of CO2 from Air into Methanol Using a Polyamine and a Homogeneous Ruthenium Catalyst. J. Am. Chem. Soc. 2016, 138, 778–781. [Google Scholar] [CrossRef]
- Wesselbaum, S.; Moha, V.; Meuresch, M.; Brosinski, S.; Thenert, K.M.; Kothe, J.; vom Stein, T.; Englert, U.; Hölscher, M.; Klankermayer, J.; et al. Hydrogenation of Carbon Dioxide to Methanol using a Homogeneous Ruthenium−Triphos Catalyst: From Mechanistic Investigations to Multiphase Catalysis. Chem. Sci. 2015, 6, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Wesselbaum, S.; vom Stein, T.; Klankermayer, J.; Leitner, W. Hydrogenation of Carbon Dioxide to Methanol by Using a Homogeneous Ruthenium Phosphine Catalyst. Angew. Chem. Int. Ed. 2012, 51, 7499–7502. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.; Sen, R.; Kothandaraman, J.; Goeppert, A.; Chowdhury, R.; Munoz, S.B.; Haiges, R.; Prakash, G.K.S. Mechanistic Insights into Ruthenium-Pincer-Catalyzed Amine-Assisted Homogeneous Hydrogenation of CO2 to Methanol. J. Am. Chem. Soc. 2019, 141, 7, 3160–3170. [Google Scholar] [CrossRef]
- Tsurusaki, A.; Murata, K.; Onishi, N.; Sordakis, K.; Laurenczy, G.; Himeda, Y. Investigation of Hydrogenation of Formic Acid to Methanol using H2 or Formic Acid as a Hydrogen Source. ACS Catal. 2017, 7, 1123–1131. [Google Scholar] [CrossRef]
- Wang, W.; Qiu, B.; Yang, X. Computational Prediction of Pentadentate Iron and Cobalt Complexes as a Mimic of Mono-iron Hydrogenase for the Hydrogenation of Carbon Dioxide to Methanol. Dalton Trans. 2019, 48, 8034–8038. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Ge, H.; Yang, X. Hydrogenation of CO2 to Methanol Catalyzed by Cp*Co Complexes: Mechanistic Insights and Ligand Design. Inorg. Chem. 2019, 58, 5494–5502. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Chen, X.; Yang, X. Hydrogenation of carbon dioxide to methanol catalyzed by iron, cobalt, and manganese cyclopentadienone complexes: Mechanistic insights and computational design. Chem. Eur. J. 2017, 23, 8850–8856. [Google Scholar] [CrossRef]
- Chen, X.; Ge, H.; Yang, X. Newly designed manganese and cobalt complexes with pendant amines for the hydrogenation of CO2 to methanol: A DFT study. Catal. Sci. Technol. 2017, 7, 348–355. [Google Scholar] [CrossRef]
- Chen, X.; Yang, X. Bioinspired Design and Computational Prediction of Iron Complexes with Pendant Amines for the Production of Methanol from CO2 and H2. J. Phys. Chem. Lett. 2016, 7, 1035–1041. [Google Scholar] [CrossRef]
- Desguin, B.; Zhang, T.; Soumillion, P.; Hols, P.; Hu, J.; Hausinger, R.P. A Tethered Niacin-Derived Pincer Complex with a Nickel-Carbon Bond in Lactate Racemase. Science 2015, 349, 66–69. [Google Scholar] [CrossRef]
- Qiu, B.; Yang, X. A Bio-inspired Design and Computational Prediction of Scorpion-Like SCS Nickel Pincer Complexes for Lactate Racemization. Chem. Commun. 2017, 53, 11410–11413. [Google Scholar] [CrossRef]
- Qiu, B.; Wang, W.; Yang, X. Computational Prediction of Ammonia-Borane Dehydrocoupling and Transfer Hydrogenation of Ketones and Imines Catalyzed by SCS Nickel Pincer Complexes. Front. Chem. 2019, 7, 627. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Hall, M.B. Monoiron Hydrogenase Catalysis: Hydrogen Activation with the Formation of a Dihydrogen, Fe−Hδ−⋯Hδ+−O, Bond and Methenyl-H4MPT+ Triggered Hydride Transfer. J. Am. Chem. Soc. 2009, 131, 10901–10908. [Google Scholar] [CrossRef]
- Shi, R.; Wodrich, M.D.; Pan, H.; Tirani, F.F.; Hu, X. Functional Models of the Nickel Pincer Nucleotide Cofactor of Lactate Racemase. Angew. Chem. Int. Ed. 2019, 58, 16869–16872. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wodrich, M.D.; Scopelliti, R.; Corminboeuf, C.; Hu, X. Nickel Pincer Model of the Active Site of Lactate Racemase Involves Ligand Participation in Hydride Transfer. Proc. Natl. Acad. Sci. USA 2017, 114, 1242–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topol, I.A.; Tawa, G.J.; Burt, S.K.; Rashin, A.A. Calculation of Absolute and Relative Acidities of Substituted Imidazoles in Aqueous Solvent. J. Phys. Chem. A. 1997, 101, 10075–10081. [Google Scholar] [CrossRef]
- Wiedner, E.S.; Linehan, J.C. Making a Splash in Homogeneous CO2 Hydrogenation: Elucidating the Impact of Solvent on Catalytic Mechanisms. Chem. Eur. J. 2018, 24, 16964–16971. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Sundermann, A. Correlation Consistent Valence Basis Sets for Use with the Stuttgart-Dresden-Bonn Relativistic Effective Core Potentials: The Atoms Ga-Kr and In-Xe. J. Chem. Phys. 2001, 114, 3408–3420. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.; Webster, C.E.; Hall, M.B. JIMP2, version 0.091, A Free Program for Visualizing and Manipulating Molecules; Texas A&M University: College Station, TX, USA, 2006. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | R′ | 4 → TS1,2 | 4 → TS7,1 | |
|---|---|---|---|---|
| (kcal/mol) | ||||
| 1 | H | CH3 | 23.2 | 23.0 |
| 1a | F | CH3 | 23.9 | 23.7 |
| 1b | Cl | CH3 | 22.9 | 22.5 |
| 1c | Br | CH3 | 22.2 | 21.3 |
| 1d | CH3 | CH3 | 22.6 | 23.0 |
| 1e | H | H | 23.6 | 23.5 |
| 1f | H | COOH | 23.4 | 21.0 |
| 1g | H | NH2 | 22.9 | 23.3 |
| 1h | H | F | 23.3 | 22.5 |
| 1i | H | Cl | 23.3 | 20.9 |
| 1j | H | CN | 23.2 | 22.0 |
| 1′k | H | CH3 | 24.6 | 25.2 |
| 1′l | F | CH3 | 25.3 | 24.5 |
| 1′m | Cl | CH3 | 22.9 | 22.2 |
| 1′n | Br | CH3 | 22.5 | 20.8 |
| 1′o | CH3 | CH3 | 24.4 | 25.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Qiu, B.; Yang, X. Bioinspired Design and Computational Prediction of SCS Nickel Pincer Complexes for Hydrogenation of Carbon Dioxide. Catalysts 2020, 10, 319. https://doi.org/10.3390/catal10030319
Liu X, Qiu B, Yang X. Bioinspired Design and Computational Prediction of SCS Nickel Pincer Complexes for Hydrogenation of Carbon Dioxide. Catalysts. 2020; 10(3):319. https://doi.org/10.3390/catal10030319
Chicago/Turabian StyleLiu, Xiaoyun, Bing Qiu, and Xinzheng Yang. 2020. "Bioinspired Design and Computational Prediction of SCS Nickel Pincer Complexes for Hydrogenation of Carbon Dioxide" Catalysts 10, no. 3: 319. https://doi.org/10.3390/catal10030319
APA StyleLiu, X., Qiu, B., & Yang, X. (2020). Bioinspired Design and Computational Prediction of SCS Nickel Pincer Complexes for Hydrogenation of Carbon Dioxide. Catalysts, 10(3), 319. https://doi.org/10.3390/catal10030319