Mechanism and Chemoselectivity of Mn-Catalyzed Intramolecular Nitrene Transfer Reaction: C–H Amination vs. C=C Aziridination

Abstract
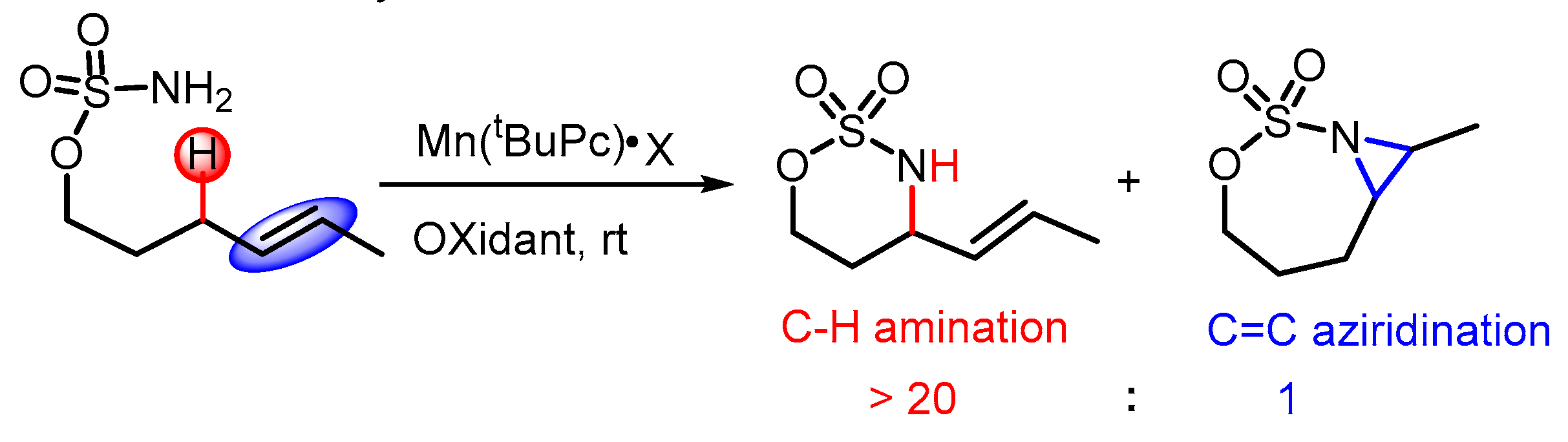
1. Introduction
2. Results and Discussion
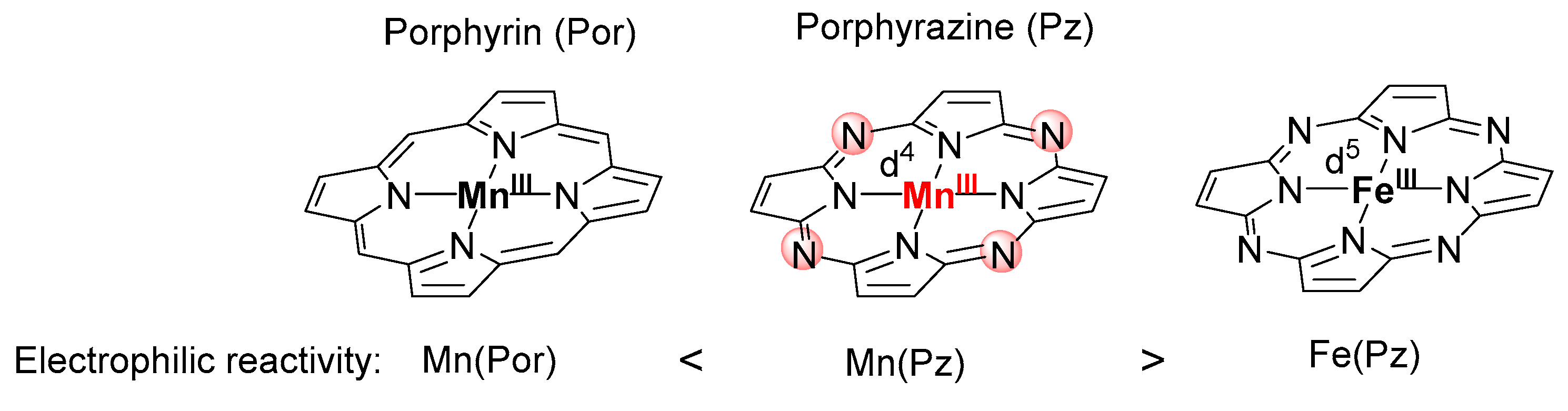
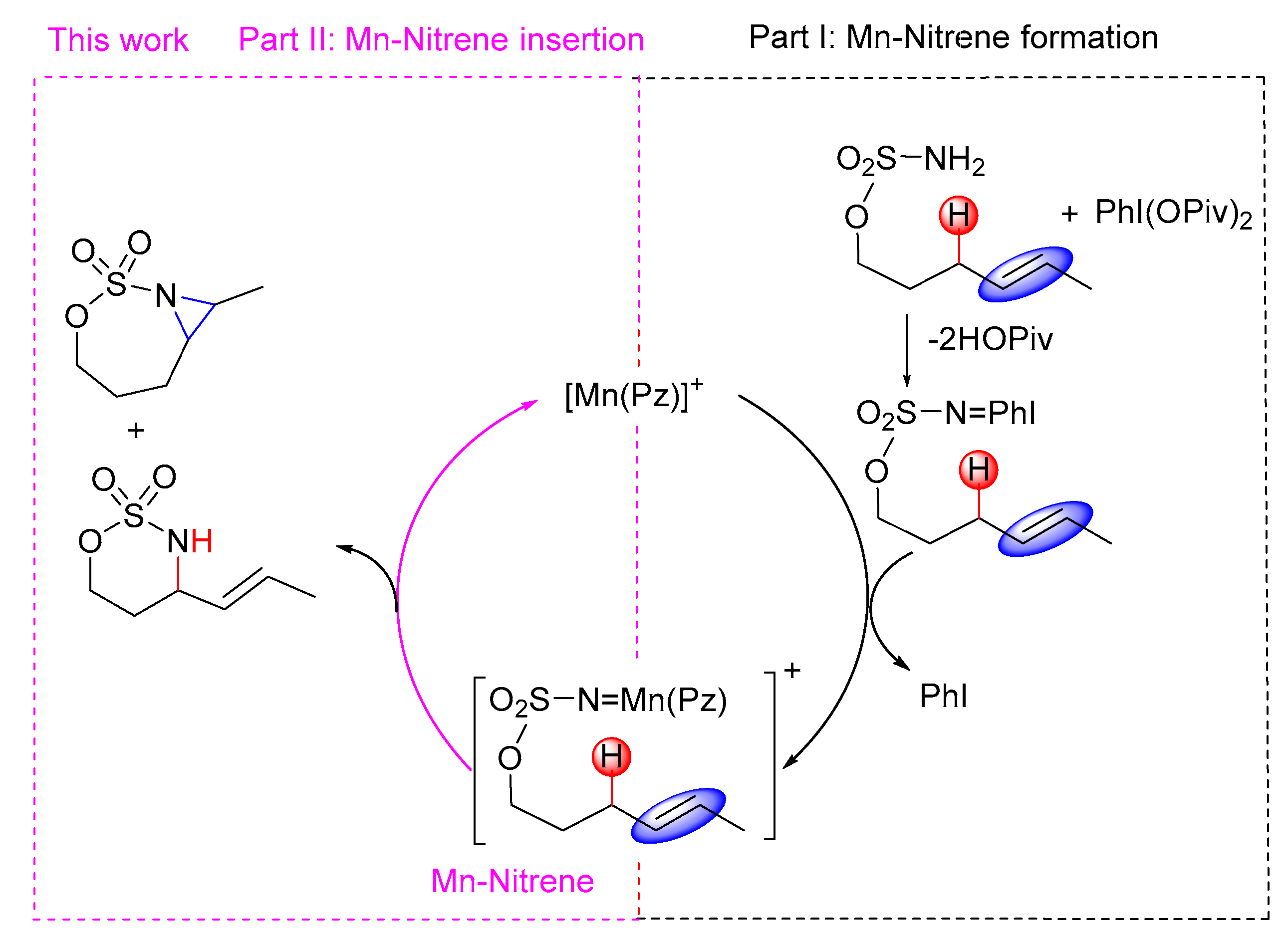
2.1. Electronic Structure and Reactivity of Mn-Nitrene
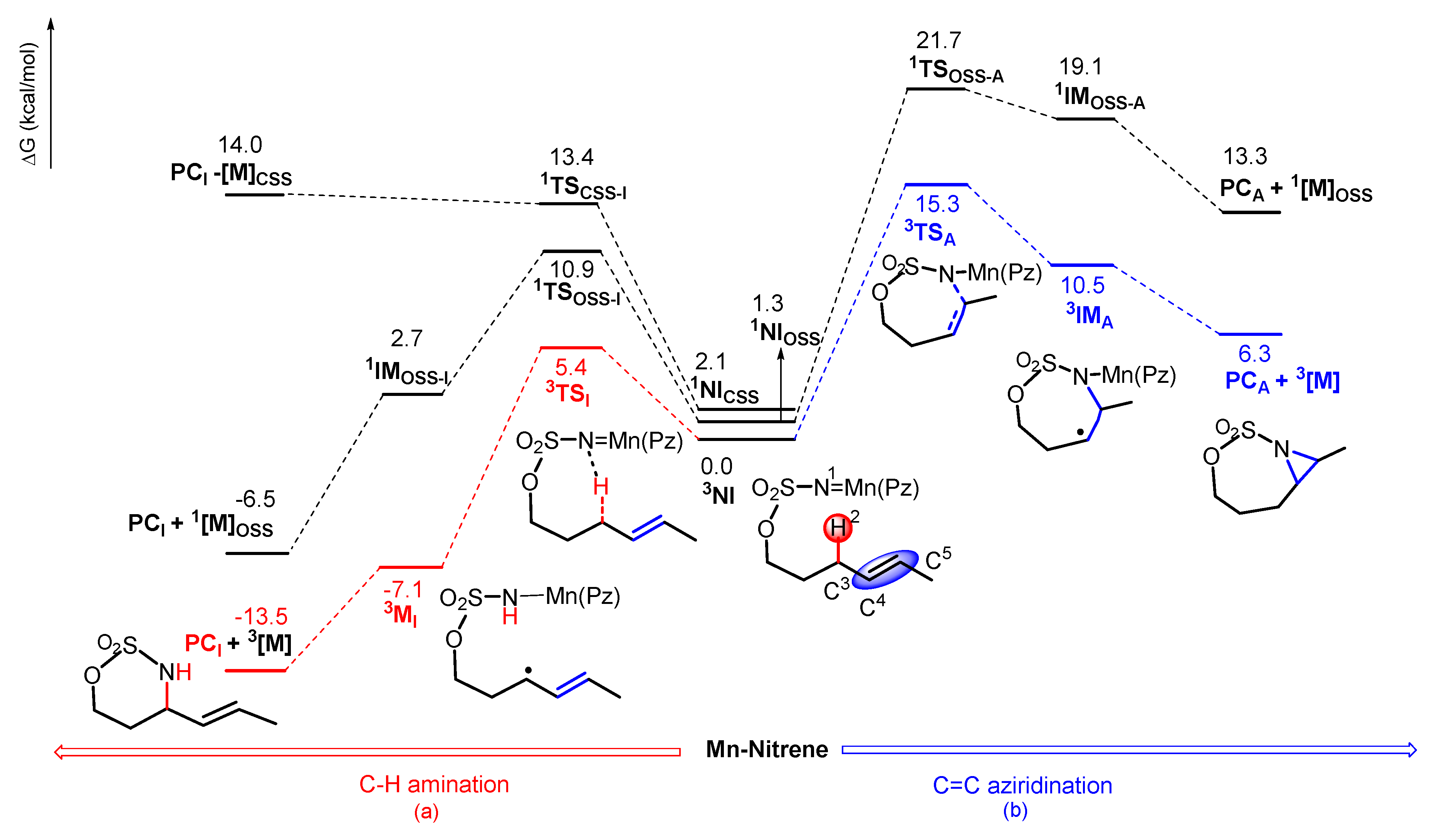
2.2. Mechanistic Investigation on C–H Amination and C=C Aziridination
2.2.1. C–H Amination Mechanism
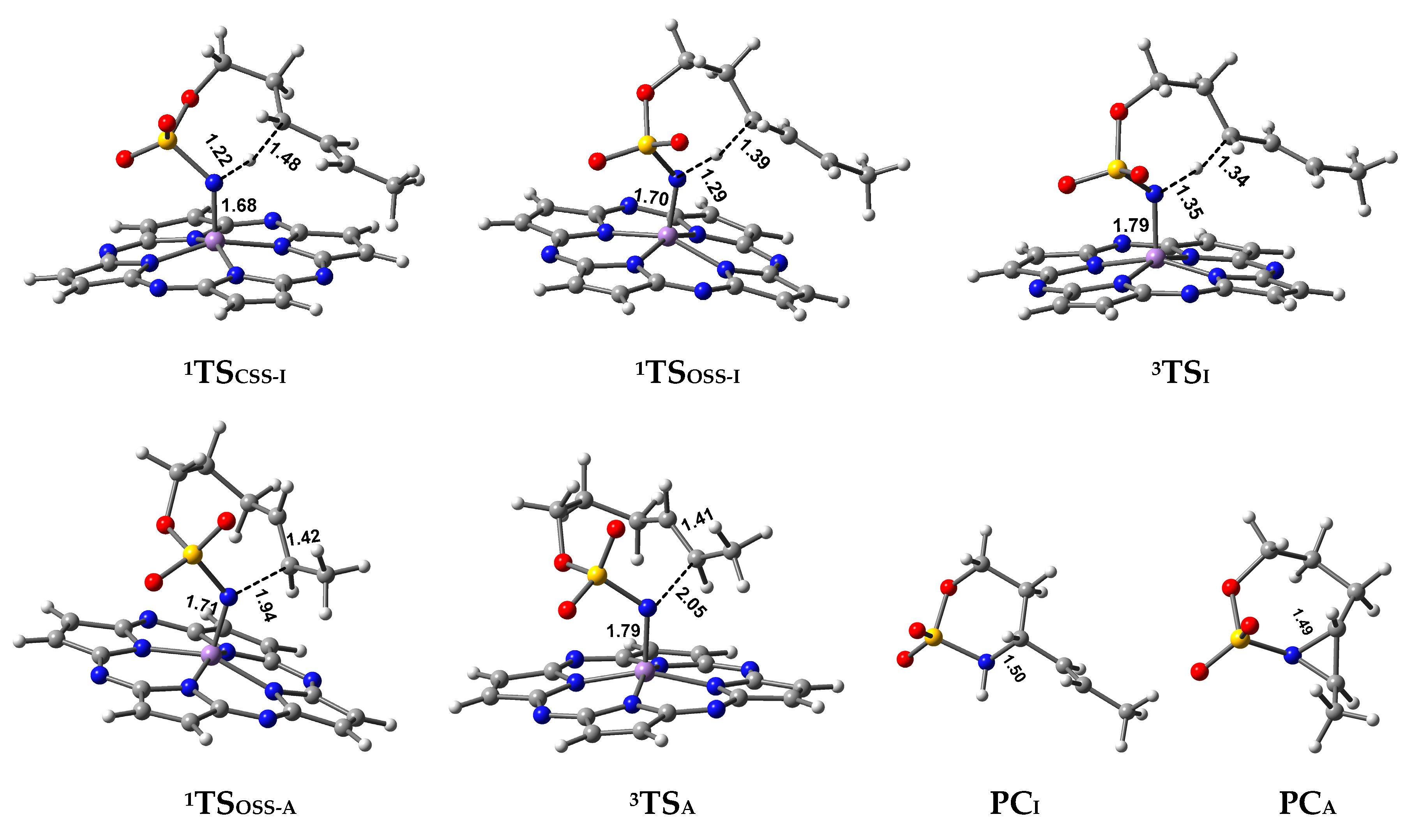
Closed-Shell Singlet (CSS) Pathway of Amination
Triplet and OSS Pathway of Amination
Preference of Singlet–Triplet Pathway for C–H Amination
2.2.2. C=C Aziridination Mechanism
Triplet and OSS Pathways of Aziridination
2.3. Chemoselectivity of C–H Amination vs. C=C Aziridination
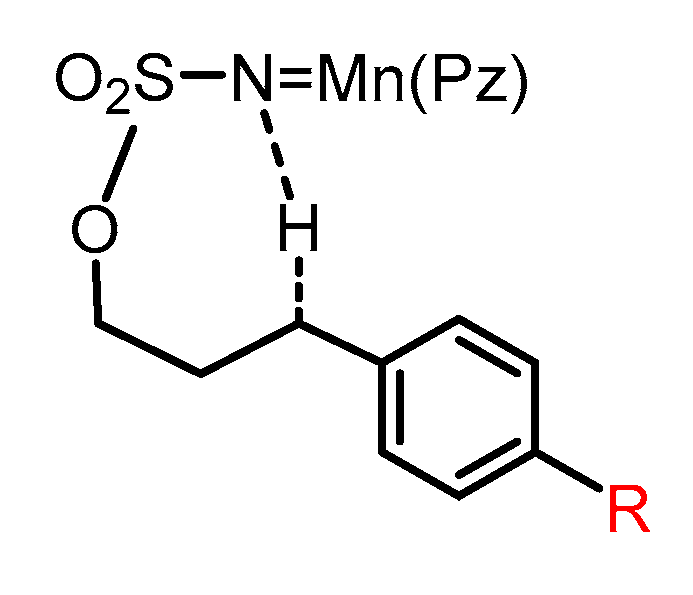
2.4. Substitution Effects on the Aminations
3. Computational Details
4. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| vs. | versus |
| Mn | Manganese |
| NT | nitrene transfer |
| I | Insertion |
| A | Aziridination |
| Pc | phthalocyanine |
| tBuPc | tert-butylphthalocyanine |
| Pz | Porphyrazine |
| CSS | closed-shell singlet state |
| OSS | open-shell singlet state |
| Por | Porphyrin |
| NBO | Natural Bond Orbital |
| ECPs | effective core potentials |
| BS | basis sets |
| IRC | Intrinsic reaction coordinate |
| SMD | solvation model based on density |
References
- Davies, H.M.L.; Morton, D. Collective approach to advancing C-H functionalization. ACS Cent. Sci. 2017, 3, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Darses, B.; Rodrigues, R.; Neuville, L.; Mazurais, M.; Dauban, P. Transition metal-catalyzed iodine(III)-mediated nitrene transfer reactions: Efficient tools for challenging syntheses. Chem. Commun. 2017, 53, 493–508. [Google Scholar] [CrossRef]
- Park, Y.; Kim, Y.; Chang, S. Transition metal-catalyzed C-H amination: Scope, mechanism, and applications. Chem. Rev. 2017, 117, 9247–9301. [Google Scholar] [CrossRef]
- Clark, J.R.; Feng, K.; Sookezian, A.; White, M.C. Manganese-catalysed benzylic C(sp3)–H amination for late-stage functionalization. Nat. Chem. 2018, 10, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Degennaro, L.; Trinchera, P.; Luisi, R. Recent advances in the stereoselective synthesis of aziridines. Chem. Rev. 2014, 114, 7881–7929. [Google Scholar] [CrossRef]
- Yang, Y.; Cho, I.; Qi, X.; Liu, P.; Arnold, F.H. An enzymatic platform for the asymmetric amination of primary, secondary and tertiary C(sp3)-H bonds. Nat. Chem. 2019, 11, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Zhang, R.K.; Buller, A.R.; Chen, S.B.; Arnold, F.H. Enantioselective, intermolecular benzylic C-H amination catalysed by an engineered iron-haem enzyme. Nat. Chem. 2017, 9, 629–634. [Google Scholar] [CrossRef]
- Roizen, J.L.; Harvey, M.E.; Du Bois, J. Metal-catalyzed nitrogen-atom transfer methods for the oxidation of aliphatic C-H bonds. Acc. Chem. Res. 2012, 45, 911–922. [Google Scholar] [CrossRef]
- Chiappini, N.D.; Mack, J.B.C.; Du Bois, J. Intermolecular sp3 C-H amination of complex molecules. Angew. Chem. Int. Ed. Engl. 2018, 18, 4956–4959. [Google Scholar] [CrossRef]
- Huang, M.; Yang, T.; Paretsky, J.D.; Berry, J.F.; Schomaker, J.M. Inverting steric effects: Using “attractive” noncovalent interactions to direct silver-catalyzed nitrene transfer. J. Am. Chem. Soc. 2017, 139, 17376–17386. [Google Scholar] [CrossRef]
- Goswami, M.; Lyaskovskyy, V.; Domingos, S.R.; Buma, W.J.; Woutersen, S.; Troeppner, O.; Ivanović-Burmazović, I.; Lu, H.; Cui, X.; Zhang, X.P.; et al. Characterization of porphyrin-Co(III)-‘nitrene radical’ species relevant in catalytic nitrene transfer reactions. J. Am. Chem. Soc. 2015, 137, 5468–5479. [Google Scholar] [CrossRef] [PubMed]
- Harvey, M.E.; Musaev, D.G.; Du Bois, J. A diruthenium catalyst for selective, intramolecular allylic C-H amination: Reaction development and mechanistic insight gained through experiment and theory. J. Am. Chem. Soc. 2011, 133, 17207–17216. [Google Scholar] [CrossRef]
- Paradine, S.M.; White, M.C. Iron-catalyzed intramolecular allylic C-H amination. J. Am. Chem. Soc. 2012, 134, 2036–2039. [Google Scholar] [CrossRef] [PubMed]
- Weatherly, C.; Alderson, J.M.; Berry, J.F.; Hein, J.E.; Schomaker, J.M. Catalyst-controlled nitrene transfer by tuning metal:ligand ratios: Insight into the mechanisms of chemoselectivity. Organometallics 2017, 36, 1649–1661. [Google Scholar] [CrossRef]
- Li, J.; Cisar, J.S.; Zhou, C.-Y.; Vera, B.; Vera, W.H.; Rodríguez, A.D.; Cravatt, B.F.; Romo, D. Simultaneous structure–activity studies and arming of natural products by C-H amination reveal cellular targets of eupalmerin acetate. Nat. Chem. 2013, 5, 510–517. [Google Scholar] [CrossRef]
- Dolan, N.S.; Scamp, R.J.; Yang, T.; Berry, J.F.; Schomaker, J.M. Catalyst-controlled and tunable, chemoselective silver-catalyzed intermolecular nitrene transfer: Experimental and computational studies. J. Am. Chem. Soc. 2016, 138, 14658–14667. [Google Scholar] [CrossRef]
- Zalatan, D.N.; Du Bois, J. A chiral rhodium carboxamidate catalyst for enantioselective C-H amination. J. Am. Chem. Soc. 2008, 130, 9220–9221. [Google Scholar] [CrossRef]
- Lu, H.; Jiang, H.; Hu, Y.; Wojtas, L.; Zhang, X.P. Chemoselective intramolecular allylic C-H aminationversus C=C aziridination through Co(II)-based metalloradical catalysis. Chem. Sci. 2011, 2, 2361–2366. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, H.; Zhao, C. Mechanistic investigation of dirhodium-catalyzed intramolecular allylic C-H amination versus alkene aziridination. J. Org. Chem. 2014, 79, 9799–9811. [Google Scholar] [CrossRef]
- Rodrigues, R.; Lazib, Y.; Maury, J.; Neuville, L.; Leboeuf, D.; Dauban, P.; Darses, B. Approach to pactamycin analogues using rhodium(II)-catalyzed alkene aziridination and C(sp3)–H amination reactions. Org. Chem. Front. 2018, 5, 948–953. [Google Scholar] [CrossRef]
- Paudyal, M.P.; Adebesin, A.M.; Burt, S.R.; Ess, D.H.; Ma, Z.; Kürti, L.; Falck, J.R. Dirhodium-catalyzed C-H amination using hydroxylamines. Science 2016, 353, 1144–1147. [Google Scholar] [CrossRef]
- Varela-Álvarez, A.; Yang, T.; Jennings, H.; Kornecki, K.P.; Macmillan, S.N.; Lancaster, K.M.; Mack, J.B.C.; Du Bois, J.; Berry, J.F.; Musaev, D.G. Rh2(II,III) catalysts with chelating carboxylate and carboxamidate supports: Electronic structure and nitrene transfer reactivity. J. Am. Chem. Soc. 2016, 138, 2327–2341. [Google Scholar] [CrossRef]
- Alderson, J.M.; Corbin, J.R.; Schomaker, J.M. Tunable, chemo- and site-selective nitrene transfer reactions through the rational design of silver(I) catalysts. Acc. Chem. Res. 2017, 50, 2147–2158. [Google Scholar] [CrossRef] [PubMed]
- Scamp, R.J.; Jirak, J.G.; Dolan, N.S.; Guzei, I.A.; Schomaker, J.M. A general catalyst for site-selective C(sp3)-H bond amination of activated secondary over tertiary alkyl C(sp3)-H bonds. Org. Lett. 2016, 18, 3014–3017. [Google Scholar] [CrossRef] [PubMed]
- Ju, M.; Weatherly, C.D.; Guzei, I.A.; Schomaker, J.M. Chemo- and enantioselective intramolecular silver-catalyzed aziridinations. Angew. Chem. 2017, 129, 10076–10080. [Google Scholar] [CrossRef]
- Manca, G.; Gallo, E.; Intrieri, D.; Mealli, C. DFT mechanistic proposal of the ruthenium porphyrin-catalyzed allylic amination by organic azides. ACS Catal. 2014, 4, 823–832. [Google Scholar] [CrossRef]
- Qin, J.; Zhou, Z.; Cui, T.; Hemming, M.; Meggers, E. Enantioselective intramolecular C-H amination of aliphatic azides by dual ruthenium and phosphine catalysis. Chem. Sci. 2019, 10, 3202–3207. [Google Scholar] [CrossRef]
- Kohler, D.G.; Gockel, S.N.; Kennemur, J.L.; Waller, P.J.; Hull, K.L. Palladium-catalysed anti-Markovnikov selective oxidative amination. Nat. Chem. 2018, 10, 333–340. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, X.-J.; Cao, P.; Liu, J.-K.; Wu, B. Polycyclic azetidines and pyrrolidines via palladium-catalyzed intramolecular amination of unactivated C(sp3)-H bonds. Org. Lett. 2017, 19, 4880–4883. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, E.T.; Betley, T.A. Complex N-heterocycle synthesis via iron-catalyzed, direct C-H bond amination. Science 2013, 340, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Wilding, M.J.T.; Iovan, D.A.; Betley, T.A. High-spin iron imido complexes competent for C-H bond amination. J. Am. Chem. Soc. 2017, 139, 12043–12049. [Google Scholar] [CrossRef] [PubMed]
- Bagh, B.; Broere, D.L.J.; Sinha, V.; Kuijpers, P.F.; van Leest, N.P.; de Bruin, B.; Demeshko, S.; Siegler, M.A.; van der Vlugt, J.I. Catalytic synthesis of n-heterocycles via direct C(sp3)-H amination using an air-stable iron(III) species with a redox-active ligand. J. Am. Chem. Soc. 2017, 139, 5117–5124. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, V.; Kalra, A.; Das, P.; Paraskevopoulou, P.; Gorla, S.; Ai, L.; Wang, Q.; Mohapatra, S.; Choudhury, A.; Sun, Z.; et al. Comparative Nitrene-transfer chemistry to olefinic substrates mediated by a library of anionic Mn(II) triphenylamido-amine reagents and M(II) congeners (M = Fe, Co, Ni) favoring aromatic over aliphatic alkenes. ACS Catal. 2018, 8, 9183–9206. [Google Scholar] [CrossRef]
- Lang, K.; Torker, S.; Wojtas, L.; Peter Zhang, X. Asymmetric induction and enantiodivergence in catalytic radical C-H amination via Enantiodifferentiative H-Atom abstraction and stereoretentive radical substitution. J. Am. Chem. Soc. 2019, 141, 12388–12396. [Google Scholar] [CrossRef]
- Meng, D.; Tang, Y.; Wei, J.; Shi, X.; Yang, M. Copper-catalyzed remote (δ) C(sp3)-H bond amination: A practical strategy to construct pyrrolidine derivatives. Chem. Commun. 2017, 53, 5744–5747. [Google Scholar] [CrossRef]
- Bagchi, V.; Paraskevopoulou, P.; Das, P.; Chi, L.; Wang, Q.; Choudhury, A.; Mathieson, J.S.; Cronin, L.; Pardue, D.B.; Cundari, T.R.; et al. A versatile tripodal Cu(I) reagent for C-N bond construction via nitrene-transfer chemistry: Catalytic perspectives and mechanistic insights on C-H aminations/amidinations and olefin aziridinations. J. Am. Chem. Soc. 2014, 136, 11362–11381. [Google Scholar] [CrossRef]
- Fiori, K.W.; Espino, C.G.; Brodsky, B.H.; Du Bois, J. A mechanistic analysis of the Rh-catalyzed intramolecular C-H amination reaction. Tetrahedron 2009, 65, 3042–3051. [Google Scholar] [CrossRef]
- Paradine, S.M.; Griffin, J.R.; Zhao, J.; Petronico, A.L.; Miller, S.M.; White, M.C. A Manganese catalyst for highly reactive yet chemoselective intramolecular C(sp3)-H aminations. Nat. Chem. 2015, 7, 987–994. [Google Scholar] [CrossRef]
- Zhang, X.; Chung, L.W.; Wu, Y.-D. New mechanistic insights on the selectivity of transition-metal-catalyzed organic reactions: The role of computational chemistry. Acc. Chem. Res. 2016, 49, 1302–1310. [Google Scholar] [CrossRef]
- Davies, D.L.; Macgregor, S.A.; McMullin, C.L. Computational studies of carboxylate-assisted C-H activation and functionalization at group 8-10 transition metal centers. Chem. Rev. 2017, 117, 8649–8709. [Google Scholar] [CrossRef]
- Iovan, D.A.; Wilding, M.J.T.; Baek, Y.; Hennessy, E.T.; Betley, T.A. Diastereoselective C-H bond amination for disubstituted pyrrolidines. Angew. Chem. 2017, 129, 15805–15808. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, C.; Weng, Y.; Xu, H. Insight into the mechanism and site-selectivity of Rh2II,II(esp)2-catalyzed intermolecular C-H amination. Catal. Sci. Technol. 2016, 6, 5292–5303. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, K.; Lin, B.; Weng, Y. A comparative study of inter- and intramolecular C–H aminations: Mechanism and site selectivity. RSC Adv. 2017, 7, 34783–34794. [Google Scholar] [CrossRef]
- Wang, J. Theoretical Studies on the Mechanisms of C-N/C Formation and Cleavage Promoted by Transition Metal Complexes. Ph.D. Thesis, Sun Yat-sen University, Guangzhou, China, 2010. [Google Scholar]
- Wang, J.; Xu, H.; Gao, H.; Su, C.-Y.; Zhao, C.; Phillips, D.L. DFT study on the mechanism of amides to aldehydes using Cp2Zr(H)Cl. Organometallics 2010, 29, 42–51. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Jang, E.S.; McMullin, C.L.; Käß, M.; Meyer, K.; Cundari, T.R.; Warren, T.H. Copper(II) anilides in sp3 C-H amination. J. Am. Chem. Soc. 2014, 136, 10930–10940. [Google Scholar] [CrossRef] [PubMed]
- Wilding, M.J.T.; Iovan, D.A.; Wrobel, A.T.; Lukens, J.T.; MacMillan, S.N.; Lancaster, K.M.; Betley, T.A. Direct comparison of C-H bond amination efficacy through manipulation of nitrogen-valence centered redox: Imido versus iminyl. J. Am. Chem. Soc. 2017, 139, 14757–14766. [Google Scholar] [CrossRef] [PubMed]
- Lyaskovskyy, V.; Suarez, A.I.O.; Lu, H.; Jiang, H.; Zhang, X.P.; de Bruin, B. Mechanism of cobalt(II) porphyrin-catalyzed C-H amination with organic azides: Radical nature and H-atom abstraction ability of the key cobalt(III)-nitrene intermediates. J. Am. Chem. Soc. 2011, 133, 12264–12273. [Google Scholar] [CrossRef]
- Suarez, A.I.O.; Jiang, H.; Zhang, X.P.; de Bruin, B. The radical mechanism of cobalt(II) porphyrin-catalyzed olefin aziridination and the importance of cooperative H-bonding. Dalton Trans. 2011, 40, 5697–5705. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270. [Google Scholar] [CrossRef]
- Roy, L.E.; Hay, P.J.; Martin, R.L. Revised basis sets for the LANL effective core potentials. J. Chem. Theory Comput. 2008, 4, 1029–1031. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; revision D.01; Gaussian, Inc.: Wallingford, UK, 2013. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spieces | ΔG(CH3) | ΔG(H) | ΔG(F) |
|---|---|---|---|
| R-Bn-3NI | 0.0 | 0.0 | 0.0 |
| R-Bn-3TS | 7.2 | 8.7 | 9.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Zheng, K.; Li, T.; Zhan, X. Mechanism and Chemoselectivity of Mn-Catalyzed Intramolecular Nitrene Transfer Reaction: C–H Amination vs. C=C Aziridination. Catalysts 2020, 10, 292. https://doi.org/10.3390/catal10030292
Wang J, Zheng K, Li T, Zhan X. Mechanism and Chemoselectivity of Mn-Catalyzed Intramolecular Nitrene Transfer Reaction: C–H Amination vs. C=C Aziridination. Catalysts. 2020; 10(3):292. https://doi.org/10.3390/catal10030292
Chicago/Turabian StyleWang, Juping, Kangcheng Zheng, Ting Li, and Xiaojing Zhan. 2020. "Mechanism and Chemoselectivity of Mn-Catalyzed Intramolecular Nitrene Transfer Reaction: C–H Amination vs. C=C Aziridination" Catalysts 10, no. 3: 292. https://doi.org/10.3390/catal10030292
APA StyleWang, J., Zheng, K., Li, T., & Zhan, X. (2020). Mechanism and Chemoselectivity of Mn-Catalyzed Intramolecular Nitrene Transfer Reaction: C–H Amination vs. C=C Aziridination. Catalysts, 10(3), 292. https://doi.org/10.3390/catal10030292