Abstract
The biosynthesis of plumbagin is known to occur via the acetate polymalonate pathway; however there are several intermediary steps that remain unidentified that leads to its synthesis. The study identifies enzyme naphthoate synthase to catalyze the cyclization of O-malonyl benzoyl CoA to form an intermediate that is acted upon by thioesterase before the reaction proceeds to form plumbagin. Two possible structures were predicted for this intermediate using quantum mechanics studies. A total of 60 ns molecular dynamics simulations revealed the most probable intermediate structure of the predicted two.
1. Introduction
Plumbagin the quinoid constituent extracted from the root Plumbago zeylanica L [1] has important roles as a metabolite, an immunological adjuvant, an anticoagulant, an antineoplastic agent along with antimicrobial, anticancer properties and anti-proliferative activities in various tumor cell lines and animal tumor models [2]. A member of phenols and a hydroxy-1,4-naphthoquinone, plumbagin is named after the plant genus Plumbago, from the family Plumbaginaceae. Though the applications of plumbagin are well studied, the biosynthetic pathway of the same is not well established. It is known to be synthesized via the acetate polymalonate also known as the polyketide synthase pathway (PKS) where a polyketide reductase removes the oxygen atom of the third acetate unit before the initial cyclization. A proposed biosynthetic route for plumbagin synthesis involves conversion of alpha ketoglutarate to acetyl CoA, which after six intermediary reactions gives 4-[2-hydroxy-6-(2-oxoprpyl)phenyl]-2,4-dioxobutanoate. The same is reported to be the intermediate, which combines malonyl CoA and acetyl CoA catalyzed by the enzyme polyketide reductase. In either case this compound through a series of unknown reaction is seen to produce plumbagin. Another compound juglone identified to be a structural analog of plumbagin is formed by the shikimate pathway [3]. The structural similarity seen in plumbagin and the analog juglone made by the juglone biosynthesis pathway is the naphthoquinone scaffold the central chemical structure of many natural compounds such as menadione, juglone, 7-methyl juglone, droserone, etc. (Figure S1). The juglone biosynthesis pathway (JBP) and plumbagin biosynthesis pathway (PBP) are connected in this study to predict one of the unknown intermediate reaction steps in the PBP. The interaction of the O-malonyl benzoyl CoA (OMB CoA) a known intermediate of the PBP with enzyme naphthoate synthase (NS) is compared with the interaction of O-succinyl benzoyl CoA (OSB CoA), an analog of OMB CoA with NS in JBP. Quantum mechanics studies predicted two possible structures for this unknown CoA intermediate (IS 1 and IS 2) that follows this step in PBP. Docking and molecular dynamics simulations of the two structures with thioesterase revealed the most stable structure of the two.
2. Results
The reported enzymes in the JBP and PBP revealed the existence of O-succinyl benzoate and O-malonyl benzoate respectively. The network of reactions to derive the intermediary enzymes and intermediates that lead to synthesis of plumbagin is shown in Figure S2. The crucial step for the formation of these naphthoquinones is the cyclization of the intermediates to make the common scaffold. The rate limiting step in JBP is the cyclization of OSB-CoA to form DHNA-CoA by NS and then the removal of CoA, catalyzed by TE [4]. As we see the intermediate OMB in PBP, a compound similar to OSB in JBP, we proposed that OMB CoA was formed by CoA ligase in PBP, followed by the action of NS to do the cyclization. The removal of CoA is done by TE to form the naphthoquinone scaffold of plumbagin in PBP. We related the intermediary reactions in JBP to derive the same for PBP (Figure S2). This is validated by MD simulations of the TE-OMB complex.
2.1. Hypothetical Enzyme Reaction for Biosynthesis of Plumbagin
A hypothetical enzymatic reaction for the synthesis of plumbagin was derived based on the connection between JBP and acetate polymalonate pathway. Figure S3 indicates the formation of plumbagin from OMB CoA in two reaction steps. Comparing the two pathways we have proposed two possible structures IS 1 and IS 2, which can be synthesized by cyclization of OMB-CoA. These structures derived from QM studies were used for interaction studies with TE.
2.2. Enzyme Substrate Interaction of NS–OMB CoA
The NS sequence XP_010682844 of Beta vulgaris from the order Caryophyllales was modeled with the Swiss model, using the crystal structure 3T88 as a template. The ligand OMB CoA was modeled within this structure and energy minimized using quantum mechanics calculations using GAMESS. This structure was further used for docking and ideal conformations based on interactions and binding energy was chosen for simulation studies.
On comparing the OMB CoA docked results, with previously reported interactions of NS with OSB CoA [5], we saw similar interactions in both cases, which indicates that similar intermediates existed in JBP and PBP. The modeling and docking of NS with OMB CoA shows canonical interactions as in NS-OSB CoA (Figure S4). The model resulted in a hexamer with six chains, the dimer of which was chosen for simulation studies as it was seen that the ligand was bound between two chains (Figure 1a). The OMB CoA binding region includes Gly 225.B, Leu 110.A, Ile 135.A and makes contacts with Tyr 101.A, Tyr 262.B, Thr 258.B, Ser 165.A, Thr 159.A and Val 112.A (Figure 1b).
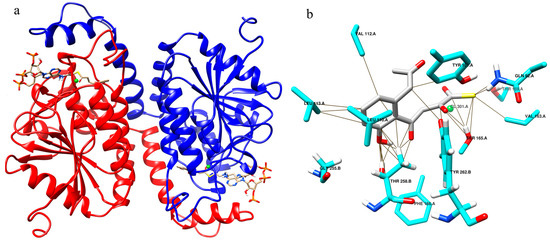
Figure 1.
Structure of naphthoate synthase with OMB CoA in the active site. (a) Dimer of naphthoate synthase with substrate OMB CoA. (b) Neighbors and residues making contacts with OMB in naphthoate synthase.
2.3. Transition and Intermediate Sates of the Intermediary Compound
The QM studies of OMB, the starter molecule in this study, show the existence of two transition states (TSs) and two intermediate states (ISs). In TS 1 the reactive positively charged carbon interacts with polar carbonyl carbon with a distance of 1.56 Å and ΔG of 62.7 Kcal/mol. This initiates the reaction and proceeds to show two intermediate states IS 1 and IS 2 that corresponds to structures b and c in Figure 2.
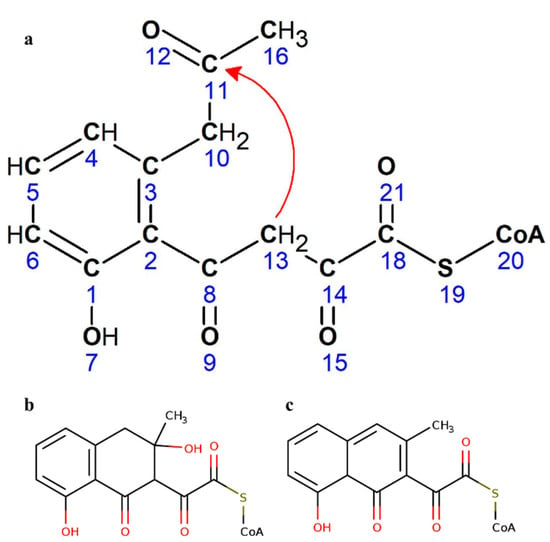
Figure 2.
Possible structures of predicted intermediate (a) Cyclization of OMB CoA. (b) Structure 1 possible for the predicted intermediate. (c) Structure 2 possible for the predicted intermediate.
The final state is the transition state TS 2 where a prominent bond formation with conformational changes was observed to give a final low energy state molecule (Video S1). The energy of the starting and final conformations is −502.01 kcal/mol and −690.26 kcal/mol respectively, which indicates a stable reaction for the production of the final molecule. This reaction barrier seems to be circumvented by NS quite efficiently. We proposed that OMB was cyclized by NS by the Claisen condensation mechanism where the thirteenth carbon attacks the eleventh carbon (Figure 2a), forming the ring with the hydroxyl and methyl group attached to form IS 1 (Figure 2b). However we observed that in IS 2 (Figure 2c), the hydroxyl group seemed removed possibly by a spontaneous hydrolysis. The IS 1 or IS 2 in the CoA bound form can act as the substrate for TE from which the CoA is removed by the esterase action. Quantum mechanics optimization of OMB in water was done using DFT. The plausible intermediates and the heat of formation of these intermediates are shown in Figure 3a. The movie of the QM optimized intermediate formation is shown in Video S2; TS 2 goes through considerable conformational changes to form the final product. Figure 3b,c shows the trajectory of the QM optimization, the formation of the naphthalene intermediate from OMB.
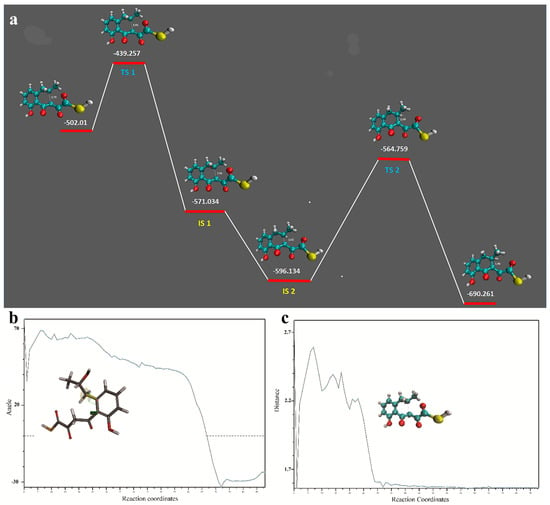
Figure 3.
Quantum mechanics studies of OMB conversion. (a) The transition and intermediate states of the OMB to naphthalene intermediate. Dotted lines represent distance between the reactive atoms. (b) The angle between the selected atoms shown in the insert. After bond formation it is evidently seen in figure (c) that there was considerable conformational changes that happened from TS2 to form the final product.
2.4. Enzyme Substrate Interaction Studies of Intermediate Structure 1 and 2 with Thioesterase
TE from Spinacia oleracea was modeled using the Swiss model with the template 1Q4S. The predicted intermediates were built in this model by induced fit method, energy minimized using quantum mechanics calculations and used for docking and simulation studies with IS 1 (Figure 4a) and with IS 2 (Figure 5a).
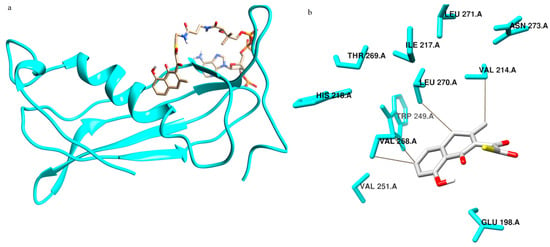
Figure 4.
Structure of thioesterase with IS 1 in the active site. (a) Thioesterase in the ribbon model with IS 1 in the stick model. (b) Residues making contacts with IS 1 in thioesterase.
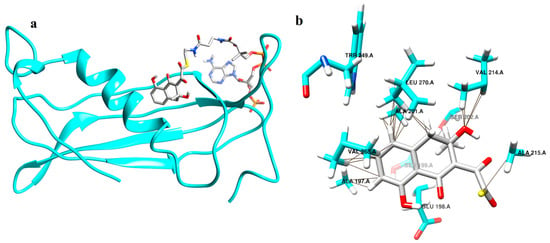
Figure 5.
Structure of thioesterase with IS 2 in the active site. (a) Thioesterase in the ribbon model with IS 2 in the stick model. (b) Residues making contacts with IS 2 in thioesterase.
Docking studies were conducted with the A chain of TE. In the case of IS 1 the binding site residues were Ala 194, Ala 197, Glu 198, Ala 201, Ser 202, Lys 212, Val 214, His 218, Trp 249, Val 251, Ser 266, Val 268, Leu 270 and Asn 273 of the A chain. It was seen that the amino acids in the binding site of IS 2 include Glu 198, Val 214, Ile 217, His 218, Trp 249, Val 251, Val 268, Thr 269, Leu 270 and Asn 273. Interaction studies on TE revealed that IS 1 made contact with residues Ala 197, Ala 201, Ser 202, Val 214, Ala 215, Val 268, Thr 269 and Leu 270 (Figure 4b) and IS 2 made contacts with Val 214 and Val 268 (Figure 5b).
2.5. Results of Molecular Dynamics Simulation
2.5.1. Free Energy of Binding of OMB with Naphthoate Synthase after Simulations Reveals Stable Interactions
Free energy of binding calculated for the NS–OMB simulation that was carried out for 20 ns. It was observed that at about 2 ns, there was a slight fluctuation observed in the binding energy, however from 3 to 20 ns across the simulation, interactions seemed to be stable (Video S2, Figure 6). The average binding energy was −190 ± 0.19 kJ/mol. Across 20 ns OMB seemed to stay very stable within the active site (Figure 6a). Hydroxyl group of the OMB head rearranged to form hydrogen bond with the backbone oxygen of Phe 166, which in turn stabilized the E–S complex. The per residue energy contribution reveal that residues Leu 113, Val 112 and Val 140 of the A chain were those that very much stabilized the reaction with mean energy approximately −4 kcal/mol (Figure 6b).
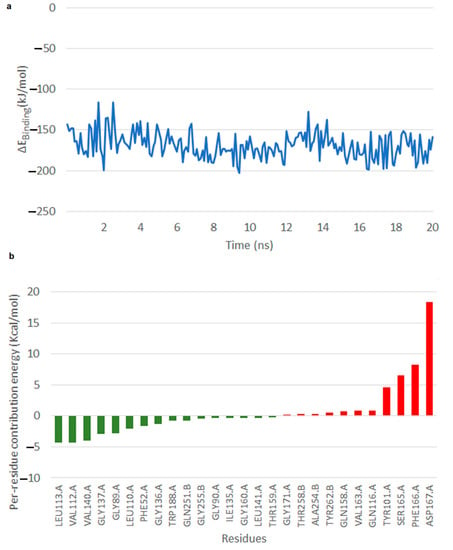
Figure 6.
Interaction of NS with OMB. (a) Free energy of binding of the NS–OMB interaction. (b) Per residue energy graph.
2.5.2. Simulations Reveal That Intermediate Structure 2 is More Stable in Its Interaction with Thioesterase
In the case of IS 1, it is seen that within 2 ns of the simulation the CoA shifted from the N-terminal of the beta strand of residues 215–219 to the C-terminal (Video S3). The substrate in this conformation forms a hydrogen bond between its ortho-hydroxyl group of the ring and oxygen of the Glu 198. The head group of the substrate is found to be stable in this state. The initial conformation of CoA had an interaction with amino acids Asp 278, Asp 279 and Ala 280. Later we saw that the CoA group swept over residues 215–219 and showed a flipped conformation away from the initial binding site, at the end of simulation. The binding free energy after 2 ns was observed to be between −200 and −100 kJ/mol until the end of simulation (Figure 7a). The per residue energy contribution revealed that Glu 198 contributed to stability with −80 kcal/mol, other residues included Leu 219, Leu 270 and Gly 216 with −10 to −5 kcal/mol (Figure 7b).
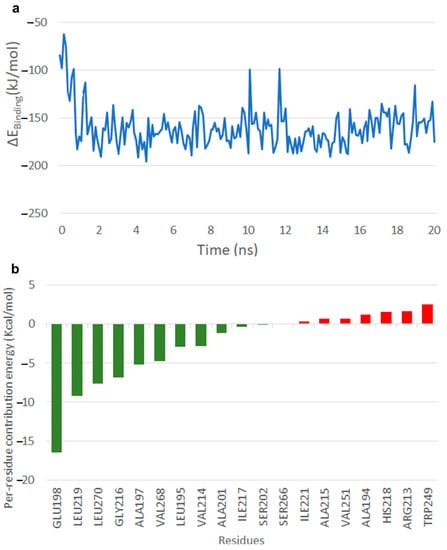
Figure 7.
Interaction of TE with IS 1. (a) Free energy of binding of the TE–IS 1 interaction. (b) Per residue energy contribution.
In the case of IS 2, throughout the simulation the OMB part remained close to the alpha-helix residues 192–207, and the CoA seemed to be stable in close association with Gly 216, Ala 215 and Val 214. The head group of IS 2 was stabilized by Leu 219 interacting with the aromatic group of the head group forming aliphatic-pi interaction across the simulation. The Enzyme–Substrate (E-S) complex seemed to be stable (Video S4) with binding free energy between −220 and −150 kJ/mol after 8 ns (Figure 8a). The per residue energy contribution revealed that residues Arg 213, Arg 267, Leu 219, Ala 215, Val 268 and Val 140 of the A chain among those that contributed to binding free energy, of which Arg 213 and Arg 267 seemed to very much stabilize the reaction with binding energy of approximately −60 kcal/mol (Figure 8b).
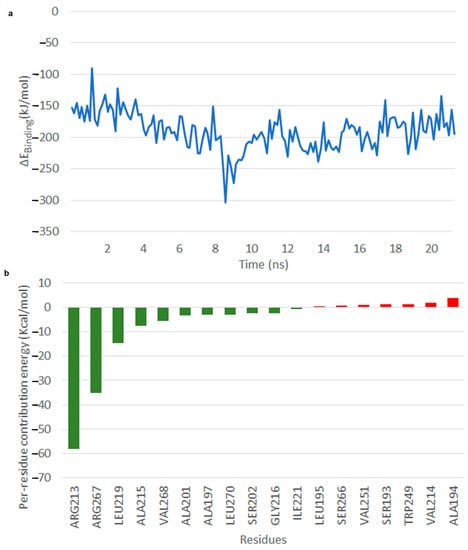
Figure 8.
Interaction of TE with IS 2. (a) Free energy of binding of the TE–IS 2 interaction. (b) Per residue energy contribution.
3. Materials and Methods
3.1. Molecular Modeling and Docking
In the JBP, OSB CoA is cyclized by the enzyme NS to di-hydroxynaphthoic acid CoA (DHNA CoA), which is hydrolyzed to DHNA by thioesterase (TE). As NS is not yet sequenced in the family Plumbaginaceae, the NS sequence from the predominantly studied Arabidopsis thaliana Accession ID, Q8GYN9.2 [6] was chosen for modeling. A BLAST search of the same was performed against sequences specifically from order Caryophyllales under which falls the family Plumbaginaceae. This resulted in XP_010682844 of Beta vulgaris to be the closest homolog, which was modeled using the Swiss model. The same methodology was followed for TE also using Arabidopsis thaliana NP_563705 TE superfamily protein as the reference sequence, which returned TE XP_021853154.1 from Spinacia oleracea as the closest homolog. TE was modeled using the same.
Docking experiments were conducted to derive the reported binding conformation of OSB-CoA in NS in the JBP and this conformation was used as the reference for comparing the docking results of OMB-CoA with NS in the PBP. Docking of the predicted IS-1 and IS-2 with the modeled TE were performed using AutoDock 4.2 [7], the results of which correlate well with the active site and mechanism of action of TE in previously reported studies [8]. The 3-D grid box has been generated with a grid centre co-ordinates comprising of grid spacing 0.375 Å and 42, 58, 50 point size considering. For each compound, the most stable docking model was selected based upon conformation of best score predicted by the AutoDock scoring function.
3.2. Molecular Dynamics Simulations and Quantum Mechanics Optimization
The ES complexes were simulated for 50 nanoseconds (ns) each using GROMACS 4.5.4 package [9] with Amber ff12SB force field. The charges for the ligands were assigned using an antechamber [10] and the topologies file for GROMACS were prepared using the program Topolbuild, developed by Bruce D. Ray (personal communication). Amber ff12SB and GAFF force fields [11] were consistently used for defining charges and topologies for the protein and ligand respectively [12]. Molecular Dynamics (MD) simulations were performed in isothermal–isobaric conditions. Simulations were conducted by placing the proteins with respective substrates in a box containing water molecules using the explicit solvent SPC/E model (a water model used to simulate and thermodynamically calculate water clusters). The total charge of the system having was neutralized using K+ ions. During the MD simulations, we initially performed 50,000 steps of steepest descent minimization and 200 picoseconds (ps) position restrained dynamics (PSD) to equilibrate the water molecules throughout the system. During PSD, the protein was restrained, and the water atoms were allowed to move. The electrostatics were calculated using the PME method (Particle mesh Ewald, an algorithm used in calculating electrostatic forces in molecular dynamics simulations) with a real space cut-off of 10 Å, using an order of 4 and a relative tolerance between long and short range energies of 10−5. Short range interactions were evaluated using a neighbor list of 10 Å and the Lennard–Jones (LJ) interactions and the real space electrostatic interactions were truncated at 9 Å. The V-rescale thermostat was used to maintain the temperature; hydrogen bonds were constrained using LINCS algorithm [13]. Visualization studies were done using VMD 1.9 [14] and Chimera 1.7. [15] Affinity between the enzyme and substrate were assessed using empirical scoring function and the molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) [16] method described below. Quantum mechanics (QM) optimization of the structures was done using GAMESS [17].
3.3. Calculation of Free Energy of Binding Using the MM/PBSA Method
Free energy of binding of the ligands (Gbinding) was calculated using the components such as potential energy in vacuum (EMM), polar solvation energy (Gpolar) and nonpolar solvation energy (Gnonpolar). The change in these energy components across time, i.e., ΔEMM, ΔGpolar, ΔGnonpolar and ΔGbinding were plotted separately and analyzed. All these calculations were carried out using g_mmpbsa package [18]. Default parameters provided by the tool were used for calculations.
3.4. Prediction of Intermediates by Quantum Mechanics Studies
The reaction mechanism of NS and TS for the synthesis of plumbagin in PBP from OMB was proposed in this QM study. The initial geometry of OMB and the final expected naphthoquinone intermediate was provided to the hybrid density function B3LYP method [19] with 6-311G** bases set using GAMESS [20]. QM optimization was done without the Co-A part.
4. Discussion
The presence of the same amino acids Tyr, Phe, Leu, Val, Thr, Gln, Gly, Ser and Ala in NS–OMB CoA in our docking and simulation studies reveal that OMB CoA was well suited to be the substrate of NS based on comparable results [21]. The stability observed in the simulation of this E-S complex supported this in a stronger manner.
Molecular docking of both IS 1 and IS 2 with TE revealed common amino acids in the substrate binding region, also showing common acids that make contacts with the substrates. However the simulation results of IS 1 and IS 2 with TE varied much from each other. In the case of IS 2, CoA was stable across the simulation making the binding conformation of the head group of the substrate stable. The head group of CoA showed better stability in IS 1 than IS 2. In IS 2, the PO3 group showed interactions with a backbone of Arg 244 and Leu 246, whereas in IS 1, the initial interaction of the PO3 group with backbone hydrogen of Arg 244, and Leu 246 changed from the side chains of Arg 244 after 1 ns of simulation. After 6 ns the flexible C-terminal of the protein caused the Arg 288 side chain to form a hydrogen bond interaction with PO3 and within 10 ns Lys 285 also formed hydrogen bonds with the same group. These interactions made the head group of the CoA highly flexible across the simulation of IS 1. The pantothenate group of IS 1 seemed to flip as the simulation proceeded, making the head group more loosely bind to the active site, hence making it unstable for binding. From these observations, we proposed that IS 2 was the more appropriate structure to be the substrate of TE, which remained stable through 20 ns in the E-S complex.
5. Conclusions
The QM studies conducted here showed two possible structures (IS 1 and IS 2) that could be the substrate for enzyme TE, however the MD simulations of TE conducted with IS 1 and IS 2 revealed IS 2 to be the most probable because of stable interactions with the enzyme throughout 20 ns with binding free energy between −220 and −150 kJ/mol. Though the structure and possible interactions of IS 2 that led to the formation of plumbagin was unveiled in this study, there remained further more enzymes and intermediates in the biosynthetic pathway of plumbagin that are to be identified.
Plumbagin being a natural compound with proven therapeutic properties can be exploited to treat various disease conditions. Identification of crucial enzymes as NS and TE with specific intermediates shall open opportunities for biocatalytic routes for the production of this chemical with such great medicinal value.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/10/3/280/s1, Figure S1: Building block and the end products of Shikimate and Acetate Polymalonate Pathway, Figure S2: Similar intermediates in Juglone and Plumbagin Biosynthesis, Figure S3: Hypothetical Enzyme Reaction for Biosynthesis of Plumbagin, Figure S4: Acetate Polymalonate Pathway for synthesis of Plumbagin [22,23], Figure S5: Plumbagin synthesis [4,24], Figure S6: Docked conformations of NS with OSBCoA as reported previously [5] and NS with OMBCoA as per our studies. Video S1. 20 ns MD Simulation of OMB with Naphthoate Synthase. The average binding energy is −190 +/−0.19 kJ/mol. Across 20ns OMB seems to stay very stable within the active site. Hydroxyl group of the OMB head rearranges to form hydrogen bonding with the backbone oxygen of Phe 166 which in turn stabilizes the E-S complex. Video S2. Movie of the QM optimised intermediate formation from OMB. The starter molecule shows the existence of two transition states (TSs) and two intermediate states (ISs). In TS 1 the reactive positively charged carbon interacts with polar carbonyl carbon with a distance of 1.56 Å and ΔG of 62.7 Kcal/mol. This initiates the reaction and proceeds to show two intermediate states IS 1 and IS 2 that corresponds to structures b and c in Figure 5. The energy of the starting and final conformations are −502.01 kcal/mol and −690.26 kcal/mol respectively. Video S3. 20 ns MD Simulation of intermediate structure 1 with Thioesterase. Within 2ns of the simulation the CoA shifts from N-terminal of the beta strand of residues 215–219 to the C-terminal. The head group of the substrate is found to be stable in this state. The initial conformation of CoA had interaction with amino acids Asp 278, Asp 279 and Ala 280. Later we see that the CoA group sweeps over residues 215–219 and shows a flipped conformation away from the initial binding site, at the end of simulation. The binding free energy after 2ns is observed to be between −200 to −100 kJ/mol till the end of simulation. Video S4. 20 ns MD Simulation of intermediate structure 2 with Thioesterase. The OMB part remains close to the alpha-helix residues 192–207, and the CoA seems to be stable in close association with Gly 216, Ala 215 and Val 214. The head group of IS 2 is stabilized by Leu219 interacting with the aromatic group of the head group forming aliphatic-pi interaction across the simulation. The E-S complex seems to be stable with binding free energy between −220 to −150 kJ/mol after 8ns.
Author Contributions
Conceptualization, M.K.S. and S.G.; Data curation, M.N.S. and M.J.; Investigation, M.K.S., R.L., P.K.R. and A.P.S.; Methodology, R.L. and P.K.R.; Project administration, P.K.R.; Resources, P.K.R.; Supervision, S.G.and N.M.; Validation, M.K.S., R.L., S.G. and N.M.; Visualization, R.L.; Writing—Original draft, M.K.S. and R.L.; Writing—Review & editing, M.K.S. All authors have read and agree to the published version of the manuscript.
Funding
This research received no external funding
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aziz, M.H.; Dreckschmidt, N.E.; Verma, A.K. Plumbagin, a medicinal plant-derived naphthoquinone, is a novel inhibitor of the growth and invasion of hormone-refractory prostate cancer. Cancer Res. 2008, 68, 9024–9032. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, A.; Ubukata, M.; Sakamura, S. Synthesis of Plumbagin by the Retro-Diels-Alder Reaction. Agric. Biol. Chem. 1980, 44, 211–213. [Google Scholar] [CrossRef]
- Mbaveng, A.T.; Kuete, V. Review of the Chemistry and Pharmacology of 7-Methyljugulone. Afr. Health Sci. 2014, 14, 201–205. [Google Scholar] [CrossRef] [PubMed]
- McCoy, R.M.; Utturkar, S.M.; Crook, J.W.; Thimmapuram, J.; Widhalm, J.R. The origin and biosynthesis of the naphthalenoid moiety of juglone in black walnut. Hortic. Res. 2008, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Li, X.K.; Liu, N.N.; Zhang, H.N.; Truglio, J.J.; Mishra, S.; Kisker, C.; Garcia-Diaz, M.; Tonge, P.J. Mechanism of the Intramolecular Claisen Condensation Reaction Catalyzed by MenB, a Crotonase Superfamily Member. Biochemistry 2011, 50, 9532–9544. [Google Scholar] [CrossRef]
- Tzin, V.; Galili, G. The Biosynthetic Pathways for Shikimate and Aromatic Amino Acids in Arabidopsis thaliana. Arabidopsis Book/Am. Soc. Plant Biol. 2010, 8, e0132. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef]
- Truglio, J.J.; Theis, K.; Feng, Y.; Gajda, R.; Machutta, C.; Tonge, P.J.; Kisker, C. Crystal structure of Mycobacterium tuberculosis MenB, a key enzyme in vitamin K2 biosynthesis. J. Biol. Chem. 2003, 278, 42352–42360. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindah, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.; Couch, G.; Greenblatt, D.M.; Meng, E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Alexeev, Y.; Mazanetz, M.P.; Ichihara, O.; Fedorov, D.G. GAMESS as a Free Quantum-Mechanical Platform for Drug Research. Curr. Top. Med. Chem. 2012, 12, 2013–2033. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for highthroughput MM-PBSA calculations open source drug discovery consortium. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.N.; Su, S.J.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Sun, Y.; Song, H.; Li, J.; Li, Y.; Jiang, M.; Zhou, J.; Guo, Z. Structural basis of the induced-fit mechanism of 1,4-dihydroxy-2-naphthoyl coenzyme A synthase from the crotonase fold superfamily. PLoS ONE 2013, 8, e63095. [Google Scholar] [CrossRef] [PubMed]
- Dewick, P.M. Medicinal Natural Products: A Biosynthetic Approach, 2nd ed.; John Wiley and Sons, Ltd.: West Sussex, UK, 2002. [Google Scholar] [CrossRef]
- Aphacha, J.; Karin, S.; Jürgen, S.; Wanchai, D.-E.; Toni, M.K. Pyrone polyketides synthesized by a type III polyketide synthase from Drosophyllum lusitanicum. Phytochemistry 2008, 69, 3043–3053. [Google Scholar] [CrossRef]
- Widhalm, J.R.; Rhodes, D. Biosynthesis and molecular actions of specialized 1,4-naphthoquinone natural products produced by horticultural plants. Hortic. Res. 2016, 3, 16046. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).