Theoretical Study on Electronic Structural Properties of Catalytically Reactive Metalloporphyrin Intermediates

 , , ,
, , ,
Abstract
1. Introduction
2. Computational Methods
3. Results and Discussion
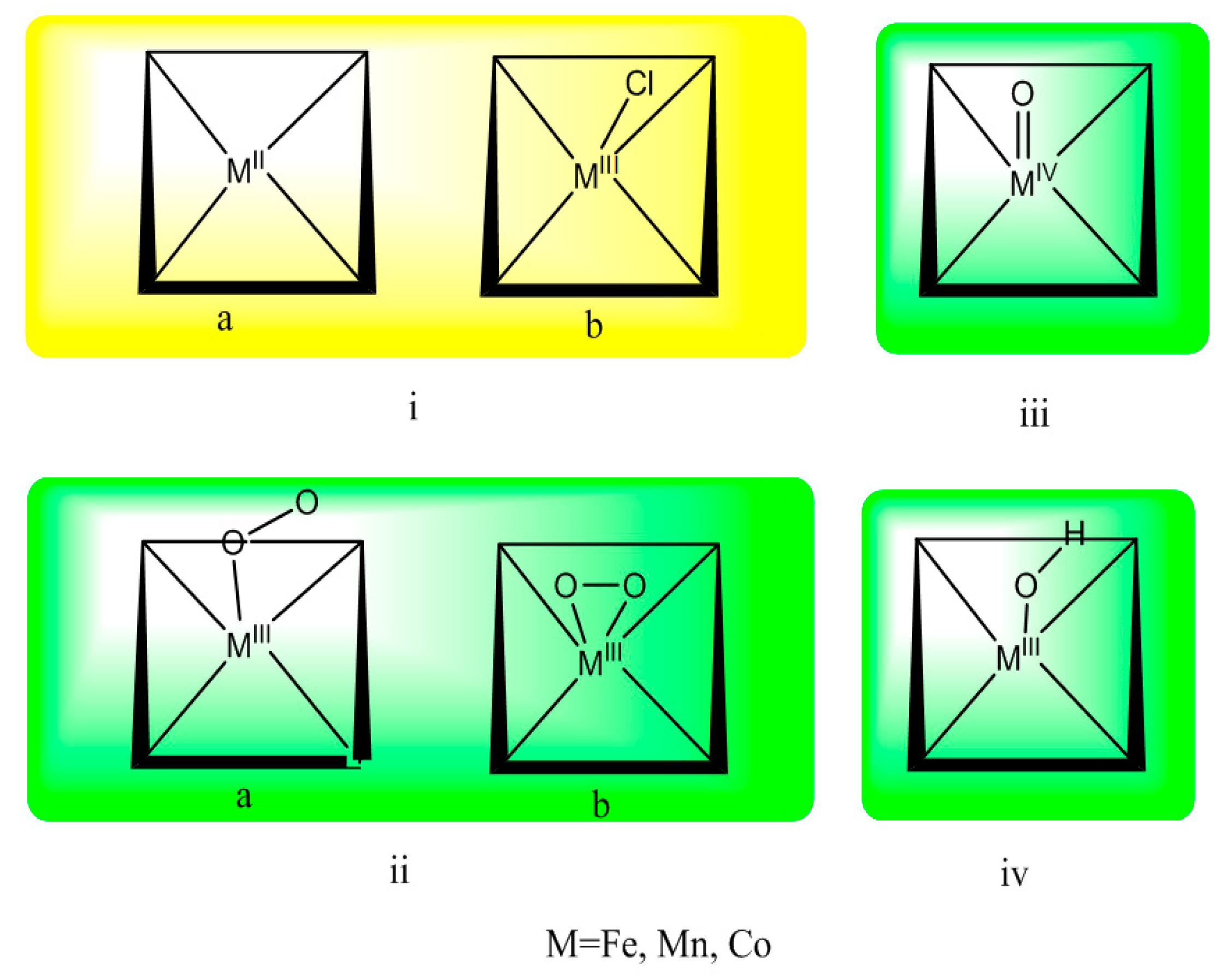
3.1. The Ground State
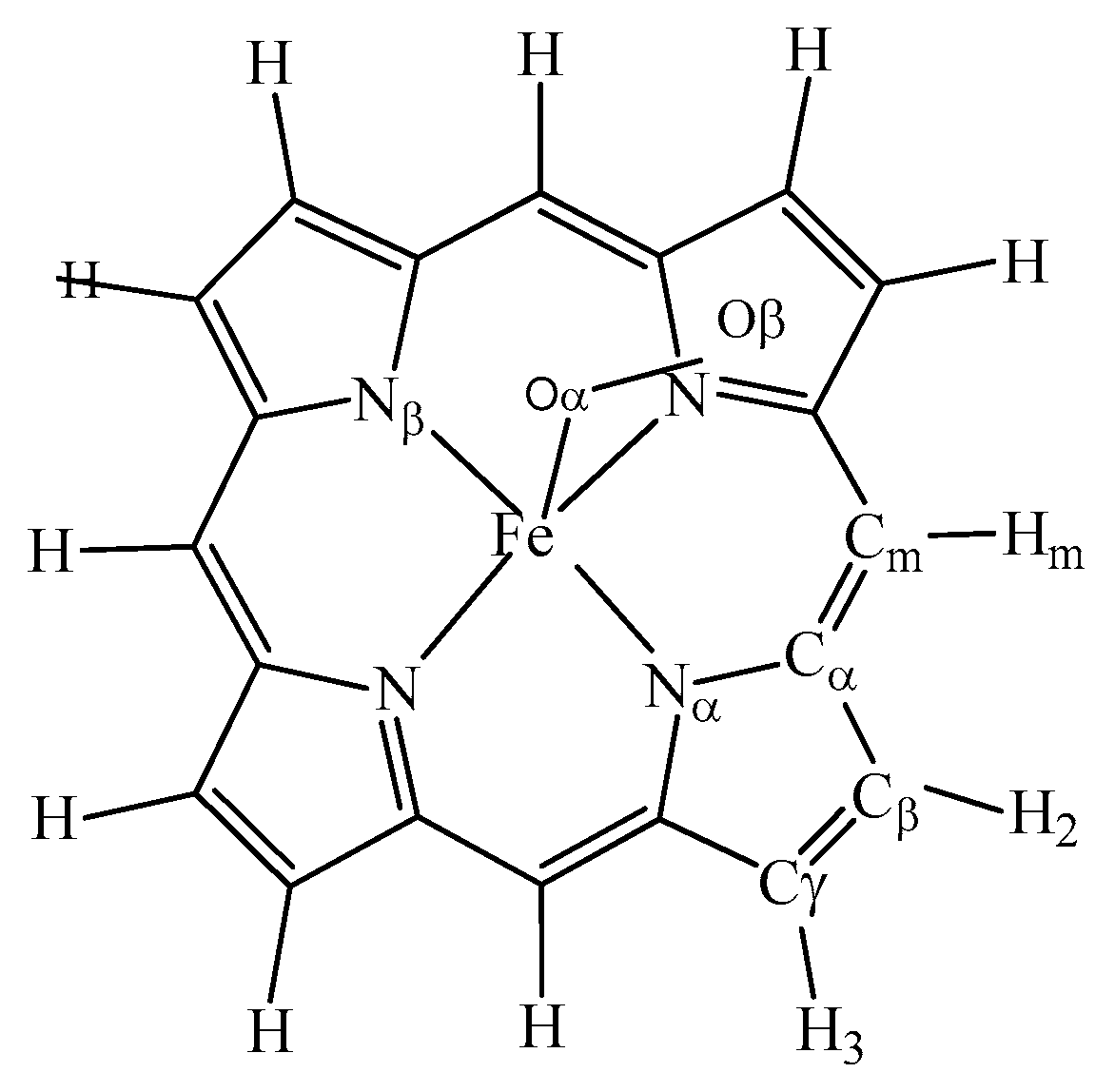
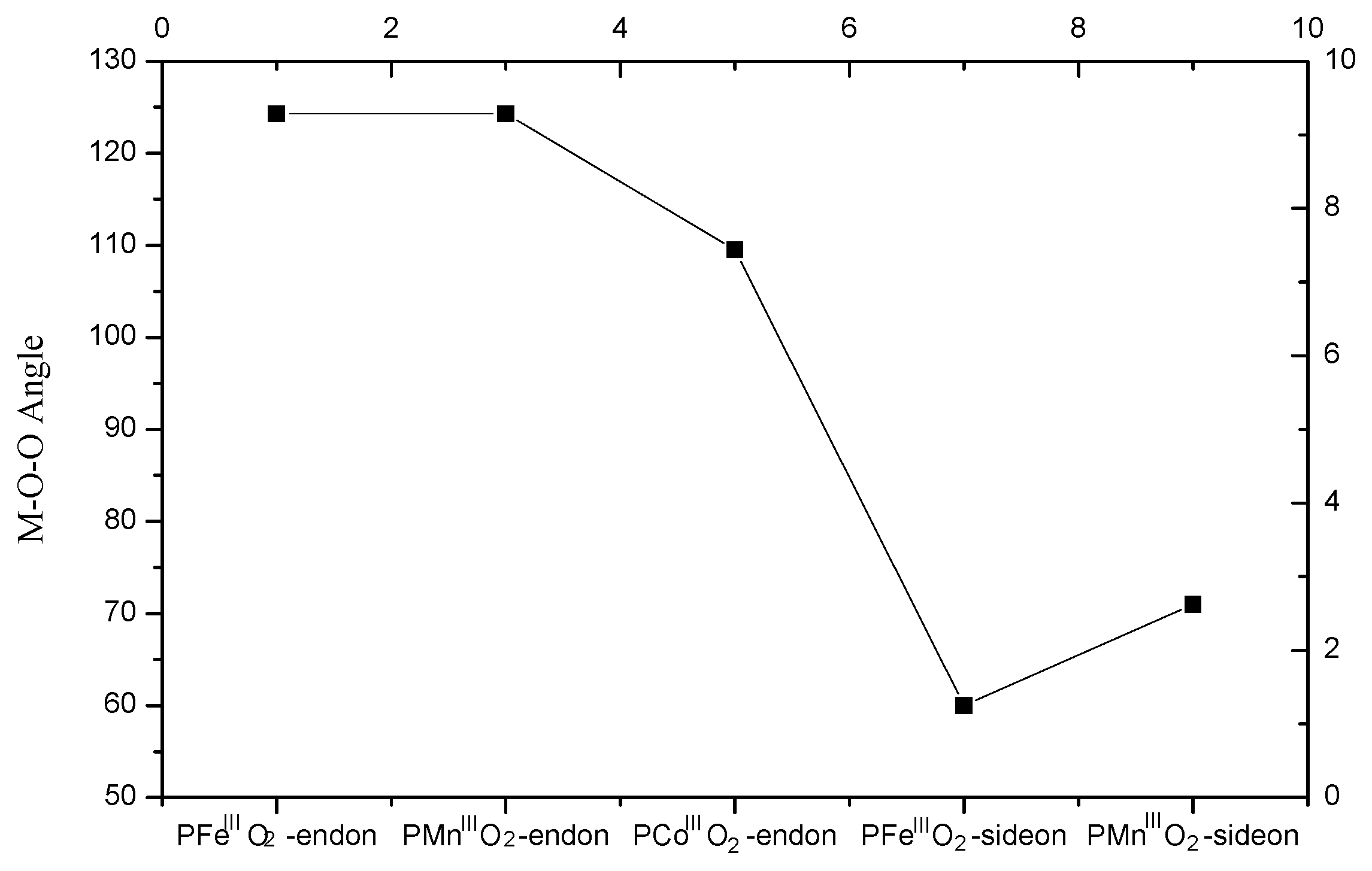
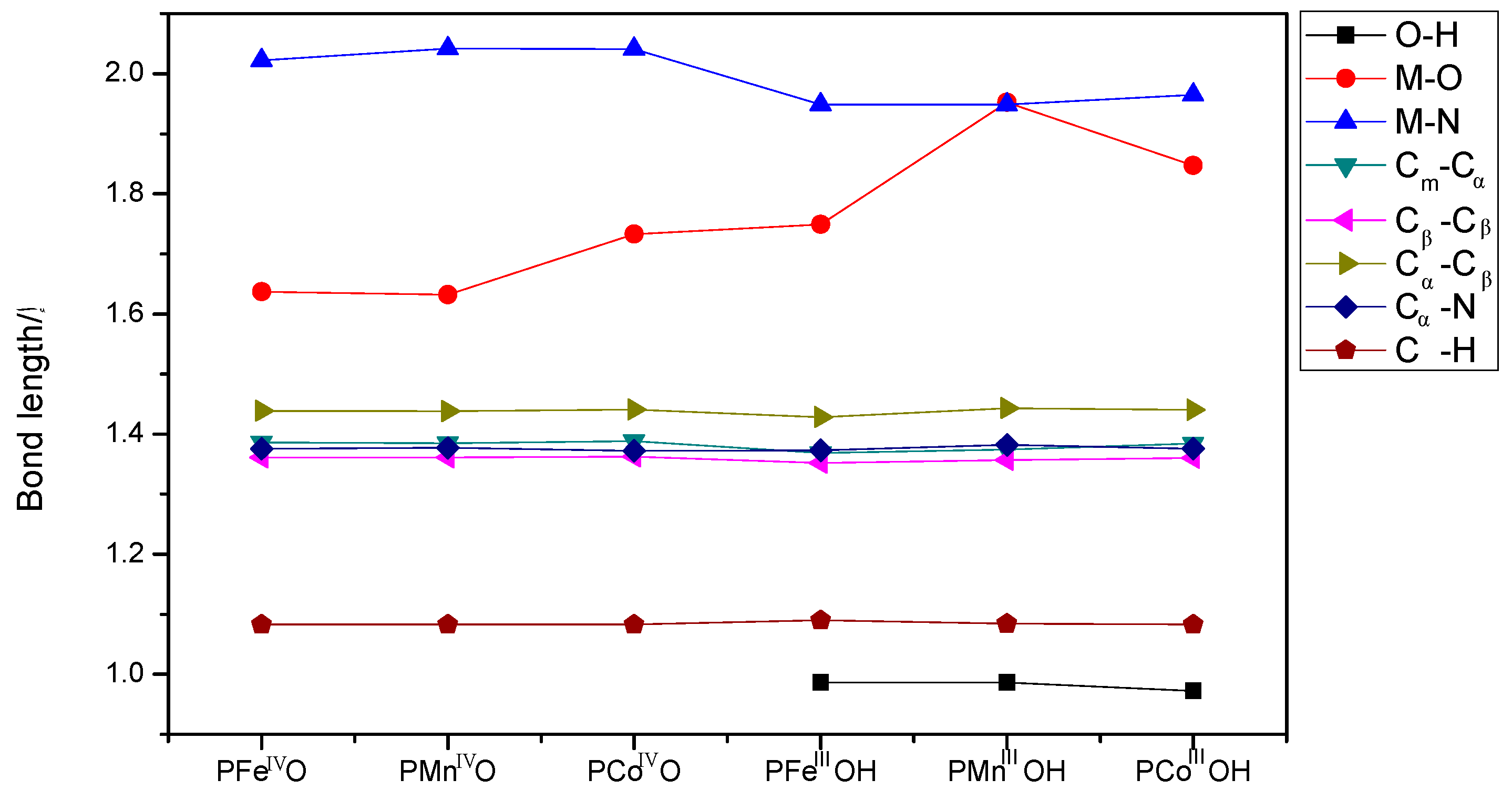
3.2. The Geometric Structure
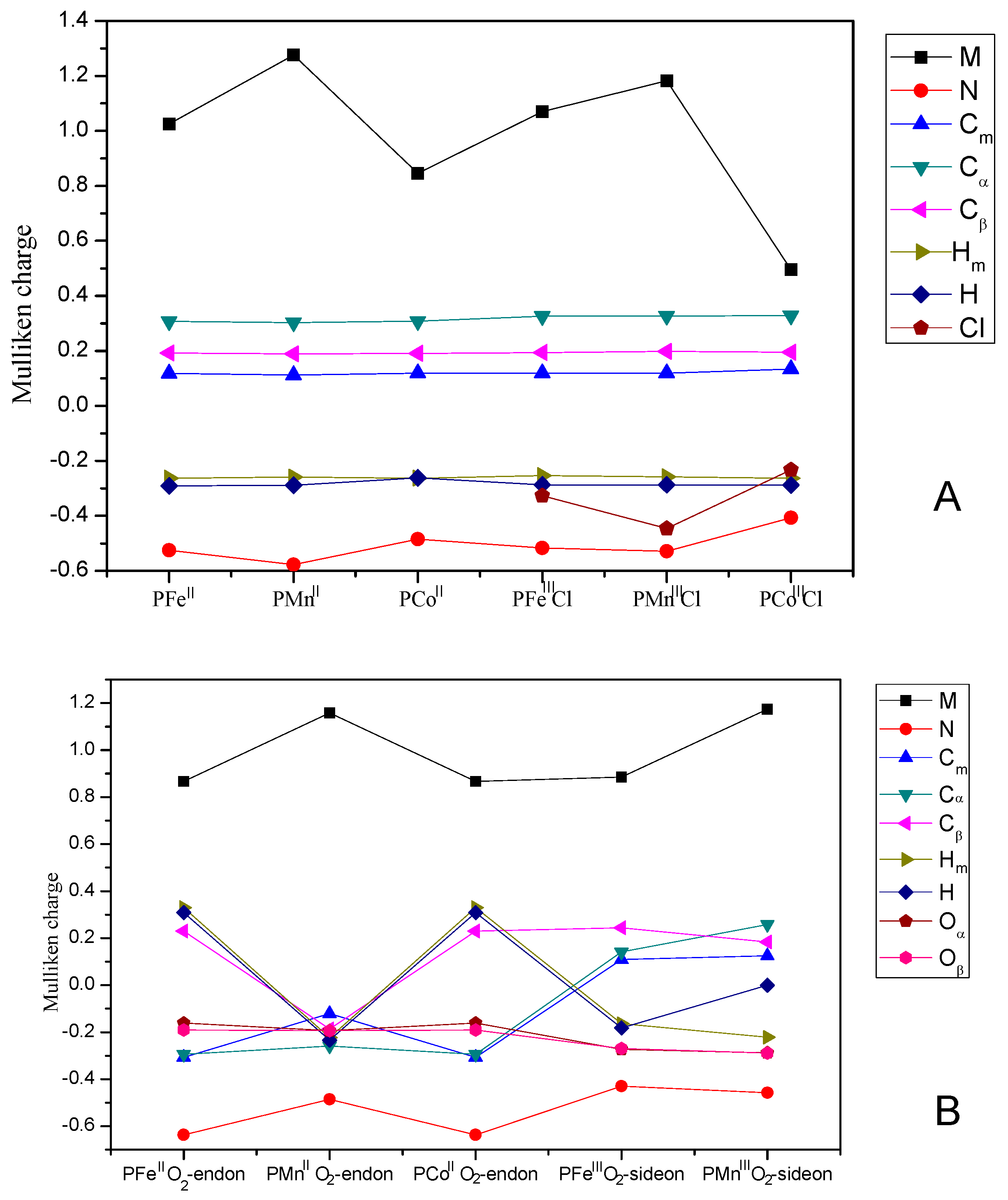
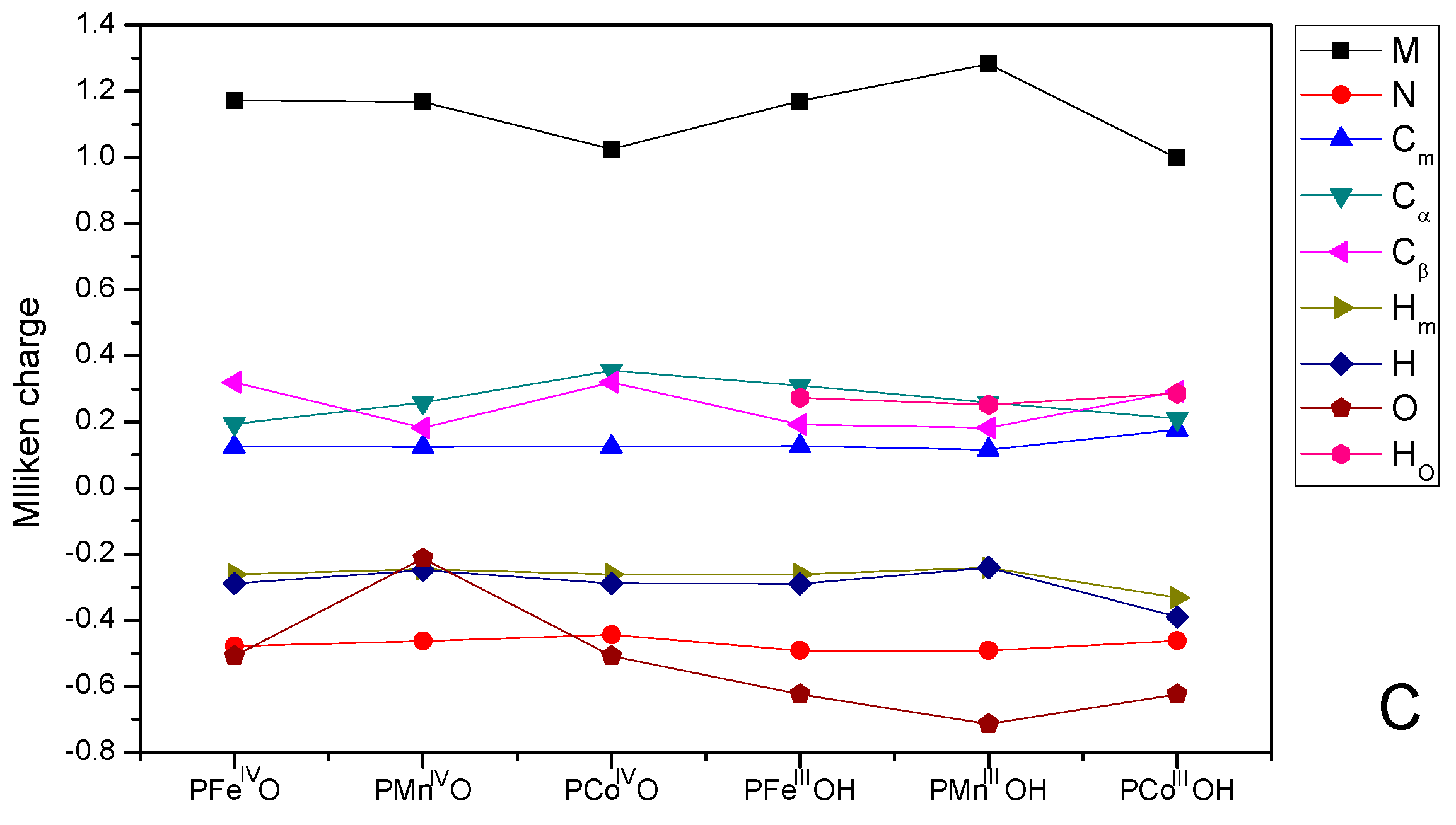
3.3. Mulliken Charge
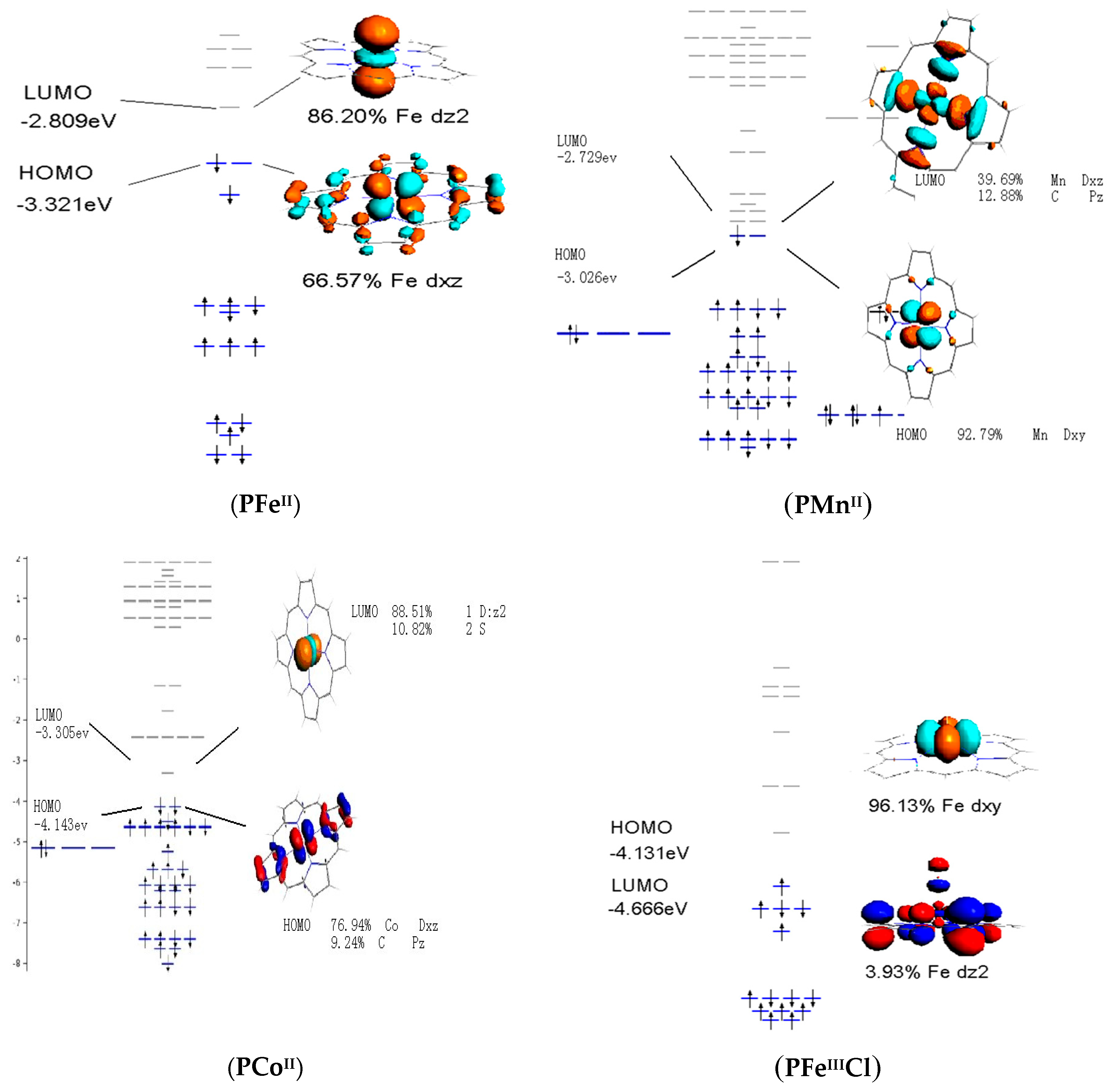
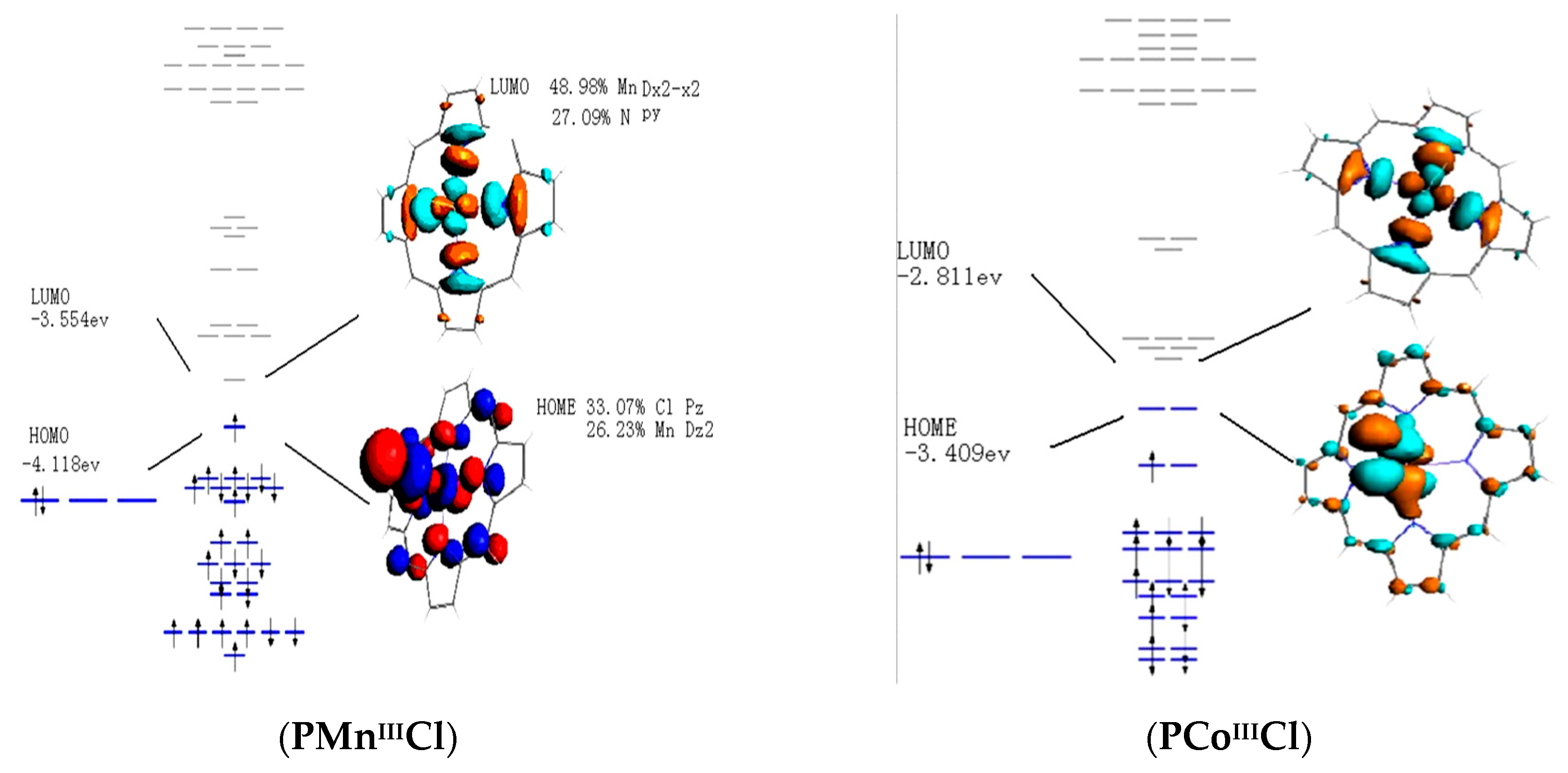
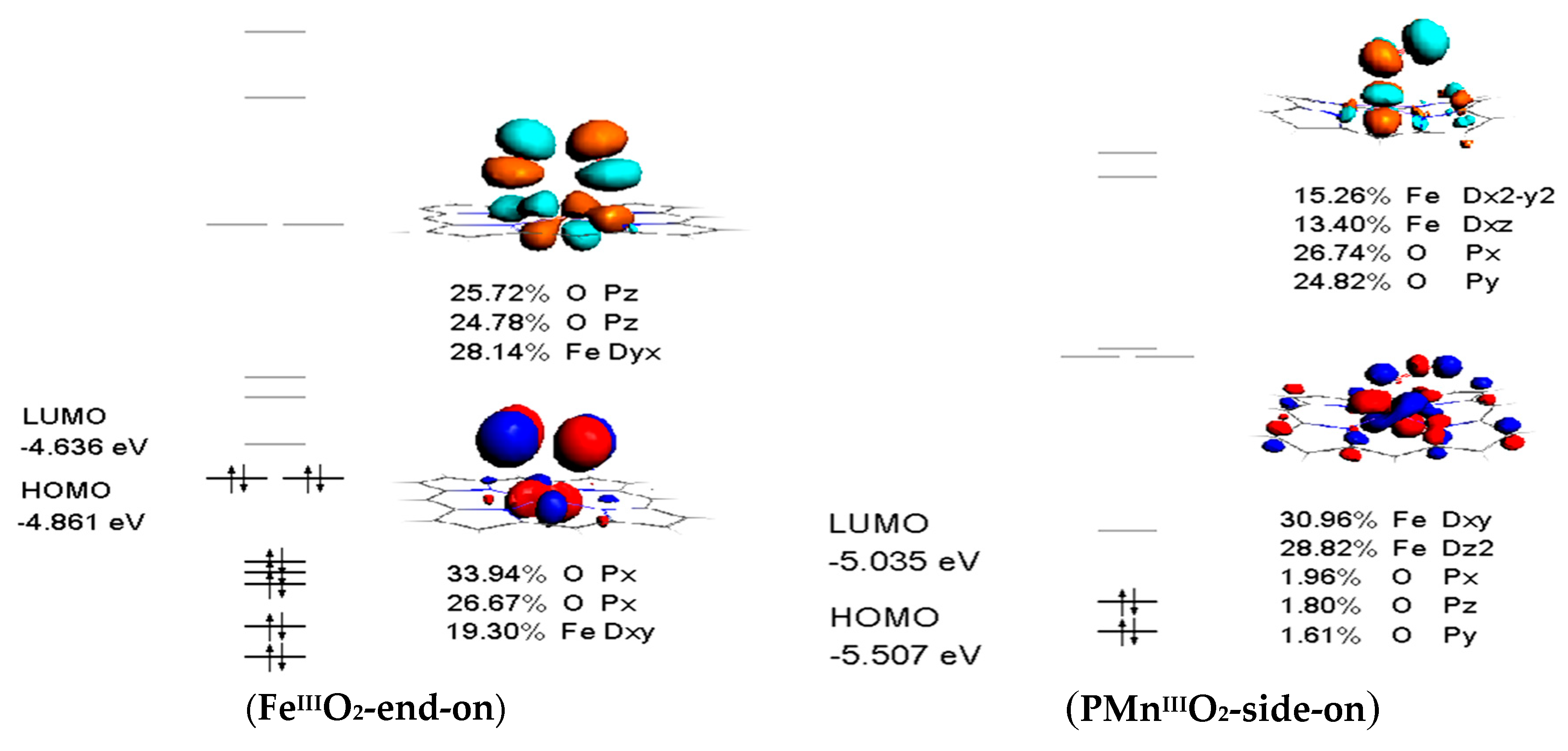
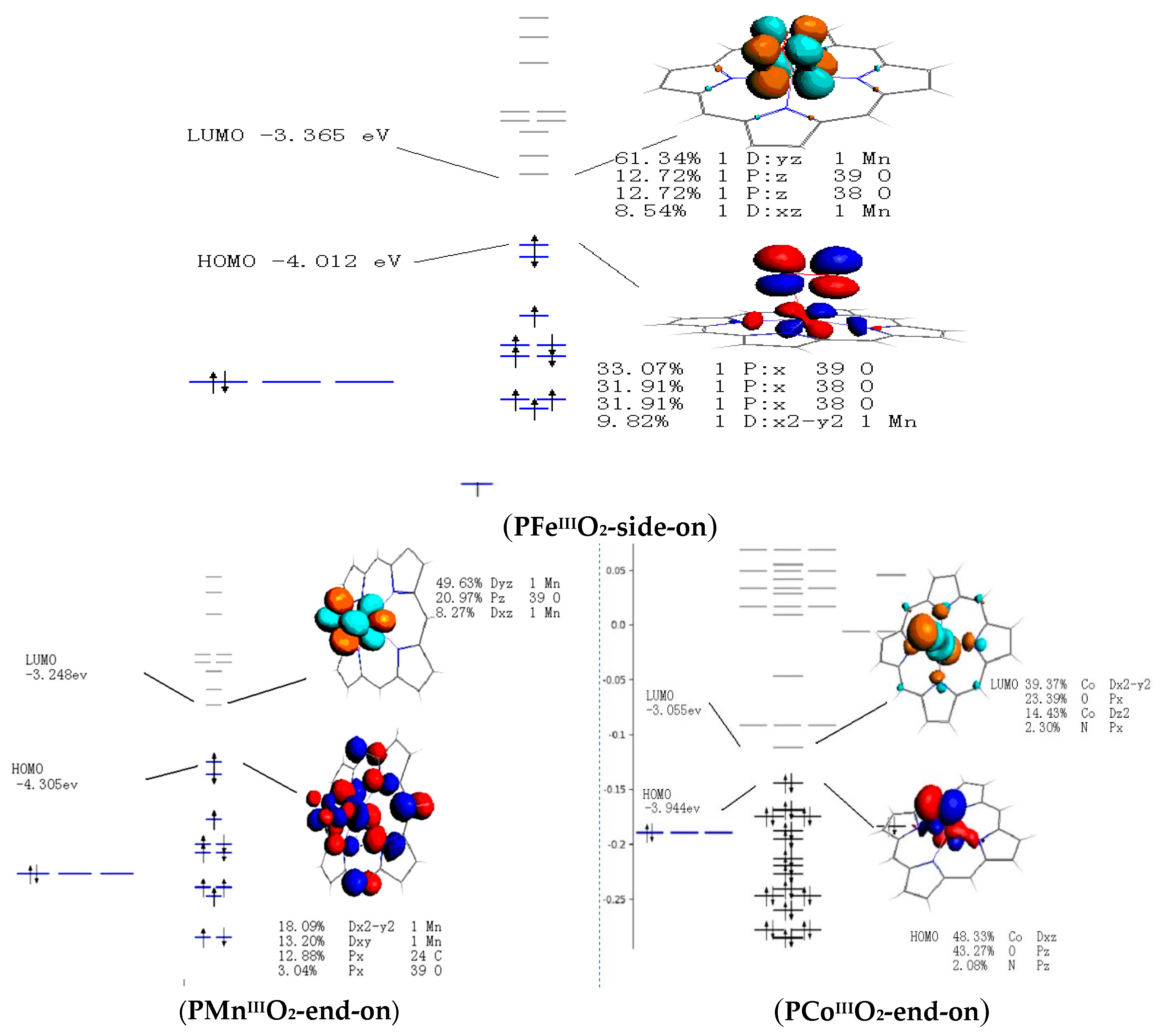
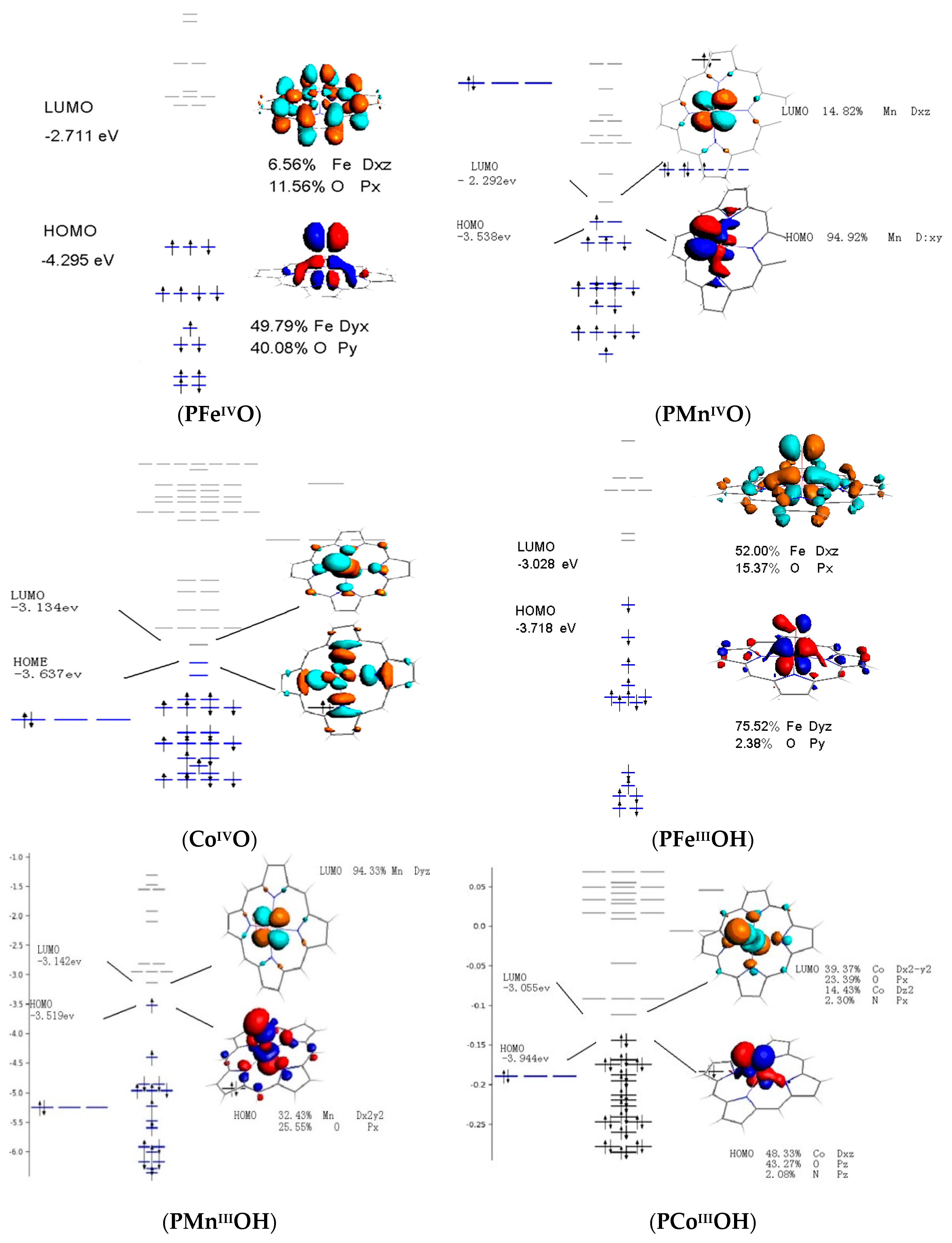
3.4. Frontier Molecular Orbitals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seo, M.S.; Kim, N.H.; Cho, K.-B.; So, J.E.; Park, S.K.; Clémancey, M.; Garcia-Serres, R.; Latour, J.-M.; Shaik, S.; Nam, W. A mononuclear nonheme iron (iv)-oxo complex which is more reactive than cytochrome P450 model compound I. Chem. Sci. 2011, 2, 1039–1045. [Google Scholar] [CrossRef]
- Shin, J.W.; Rowthu, S.R.; Hyun, M.Y.; Song, Y.J.; Kim, C.; Kim, B.G.; Min, K.S. Monomeric, trimeric, and tetrameric transition metal complexes (Mn, Fe, Co) containing N, N-bis (2-pyridylmethyl)-2-aminoethanol/-ate: Preparation, crystal structure, molecular magnetism and oxidation catalysis. Dalton Trans. 2011, 40, 5762–5773. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Groves, J.T. Manganese porphyrins catalyze selective C−H bond halogenations. J. Am. Chem. Soc. 2010, 132, 12847–12849. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.E.; Ellis, P.E., Jr.; Durante, V.A. Active iron oxo centers for the selective catalytic oxidation of Alkanes. In Studies in Surface Science and Catalysis; Elsevier: Boston, MA, USA, 1991; Volume 67, pp. 99–116. [Google Scholar]
- Pereira, M.M.; Dias, L.D.; Calvete, M.J. Metalloporphyrins: Bioinspired oxidation catalysts. ACS Catal. 2018, 8, 10784–10808. [Google Scholar] [CrossRef]
- Calvete, M.J.; Piñeiro, M.; Dias, L.D.; Pereira, M.M. Hydrogen peroxide and metalloporphyrins in oxidation catalysis: Old dogs with some new tricks. ChemCatChem 2018, 10, 3615–3635. [Google Scholar] [CrossRef]
- Yamazaki, S.-I. Metalloporphyrins and related metallomacrocycles as electrocatalysts for use in polymer electrolyte fuel cells and water electrolyzers. Coord. Chem. Rev. 2018, 373, 148–166. [Google Scholar] [CrossRef]
- Guimarães, A.S.; Schmitberger, B.; Meireles, A.M.; da Silva Martins, D.C.; DeFreitas-Silva, G. An eco-friendly approach to the cyclohexane oxidation catalyzed by manganese porphyrins: Green and solvent-free systems. Polyhedron 2019, 163, 144–152. [Google Scholar] [CrossRef]
- Dong, H.; Xu, S.; Wang, J.; Chen, Y.; Bi, L.; Zhao, Z. Selective aerobic allylic oxidation of α-pinene catalyzed by metalloporphyrins in the absence of solvents and additives. J. Chem. Res. 2019, 43, 419–425. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, X.; Chen, S.; Luo, R.; Jiang, J.; Liang, Z.; Ji, H. Direct aerobic liquid phase epoxidation of propylene catalyzed by Mn (iii) porphyrin under mild conditions: Evidence for the existence of both peroxide and Mn (iv)-oxo species from in situ characterizations. RSC Adv. 2015, 5, 30014–30020. [Google Scholar] [CrossRef]
- Zhou, X.; Ji, H. Manganese porphyrin immobilized on montmorillonite: A highly efficient and reusable catalyst for the aerobic epoxidation of olefins under ambient conditions. J. Porphyr. Phthalocyanines 2012, 16, 1032–1039. [Google Scholar] [CrossRef]
- Hunt, A.P.; Lehnert, N. The Thiolate Trans Effect in Heme {FeNO} 6 Complexes and Beyond: Insight into the Nature of the Push Effect. Inorg. Chem. 2019, 58, 11317–11332. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Luo, R.; Zhou, X.; Chen, Y.; Ji, H. Photocatalytic Properties and Mechanistic Insights into Visible Light-Promoted Aerobic Oxidation of Sulfides to Sulfoxides via Tin Porphyrin-Based Porous Aromatic Frameworks. Adv. Synth. Catal. 2018, 360, 4402–4411. [Google Scholar] [CrossRef]
- Li, C.; Lang, K.; Lu, H.; Hu, Y.; Cui, X.; Wojtas, L.; Zhang, X.P. Catalytic Radical Process for Enantioselective Amination of C (sp3)−H Bonds. Angew. Chem. Int. Ed. 2018, 130, 17079–17083. [Google Scholar] [CrossRef]
- Lang, K.; Torker, S.; Wojtas, L.; Zhang, X.P. Asymmetric Induction and Enantiodivergence in Catalytic Radical C–H Amination via Enantiodifferentiative H-Atom Abstraction and Stereoretentive Radical Substitution. J. Am. Chem. Soc. 2019, 141, 12388–12396. [Google Scholar] [CrossRef]
- Lv, X.-L.; Wang, K.; Wang, B.; Su, J.; Zou, X.; Xie, Y.; Li, J.-R.; Zhou, H.-C. A Base-Resistant Metalloporphyrin Metal–Organic Framework for C–H Bond Halogenation. J. Am. Chem. Soc. 2017, 139, 211–217. [Google Scholar] [CrossRef]
- Gan, T.; Zhang, H.; Liu, Y.; He, Q.; Zhang, Y.; He, X.; Ji, H. Self-Assembled Metalloporphyrins–Magnesium Phosphate Hybrid Spheres as Efficient Catalysts for Cycloaddition of Carbon Dioxide. ChemistrySelect 2019, 4, 8233–8236. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, R.; Yang, Z.; Zhou, X.; Ji, H. Imidazolium-based ionic liquid decorated zinc porphyrin catalyst for converting CO2 into five-membered heterocyclic molecules. Sustain. Energy Fuels 2018, 2, 125–132. [Google Scholar] [CrossRef]
- Dong, Y.; Nie, R.; Wang, J.; Yu, X.; Tu, P.; Chen, J.; Jing, H. Photoelectrocatalytic CO2 reduction based on metalloporphyrin-modified TiO2 photocathode. Chin. J. Catal. 2019, 40, 1222–1230. [Google Scholar] [CrossRef]
- Leung, K.; Nielsen, I.M.; Sai, N.; Medforth, C.; Shelnutt, J.A. Cobalt−porphyrin catalyzed electrochemical reduction of carbon dioxide in water. 2. mechanism from first principles. J. Phys. Chem. A 2010, 114, 10174–10184. [Google Scholar] [CrossRef]
- Ali, M.E.; Sanyal, B.; Oppeneer, P.M. Electronic structure, spin-states, and spin-crossover reaction of heme-related Fe-porphyrins: A theoretical perspective. J. Phys. Chem. B 2012, 116, 5849–5859. [Google Scholar] [CrossRef]
- Kaizer, J.; Klinker, E.J.; Oh, N.Y.; Rohde, J.-U.; Song, W.J.; Stubna, A.; Kim, J.; Münck, E.; Nam, W.; Que, L. Nonheme FeIVO complexes that can oxidize the C−H bonds of cyclohexane at room temperature. J. Am. Chem. Soc. 2004, 126, 472–473. [Google Scholar] [CrossRef] [PubMed]
- Nam, W. High-valent iron (IV)–oxo complexes of heme and non-heme ligands in oxygenation reactions. Acc. Chem. Res. 2007, 40, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Altun, A.; Shaik, S.; Thiel, W. What is the active species of cytochrome P450 during camphor hydroxylation? QM/MM studies of different electronic states of compound I and of reduced and oxidized iron−oxo intermediates. J. Am. Chem. Soc. 2007, 129, 8978–8987. [Google Scholar] [CrossRef] [PubMed]
- Song, W.J.; Seo, M.S.; DeBeer George, S.; Ohta, T.; Song, R.; Kang, M.-J.; Tosha, T.; Kitagawa, T.; Solomon, E.I.; Nam, W. Synthesis, Characterization, and Reactivities of Manganese(V)−Oxo Porphyrin Complexes. J. Am. Chem. Soc. 2007, 129, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Charnock, J.M.; Garner, C.D.; Trautwein, A.X.; Bill, E.; Winkler, H.; Ayougou, K.; Mandon, D.; Weiss, R. Characterization of an Oxo (porphyrinato) manganese (iv) Complex by X-ray Absorption Spectroscopy. Angew. Chem. Int. Ed. 1995, 34, 343–346. [Google Scholar] [CrossRef]
- Arasasingham, R.D.; He, G.X.; Bruice, T.C. Mechanism of manganese porphyrin-catalyzed oxidation of alkenes. Role of manganese (IV)-oxo species. J. Am. Chem. Soc. 1993, 115, 7985–7991. [Google Scholar] [CrossRef]
- Chen, J.; Lee, Y.-M.; Davis, K.M.; Wu, X.; Seo, M.S.; Cho, K.-B.; Yoon, H.; Park, Y.J.; Fukuzumi, S.; Pushkar, Y.N. A Mononuclear Non-Heme Manganese(IV)–Oxo Complex Binding Redox-Inactive Metal Ions. J. Am. Chem. Soc. 2013, 135, 6388–6391. [Google Scholar] [CrossRef]
- Taguchi, T.; Gupta, R.; Lassalle-Kaiser, B.; Boyce, D.W.; Yachandra, V.K.; Tolman, W.B.; Yano, J.; Hendrich, M.P.; Borovik, A. Preparation and properties of a monomeric high-spin MnV–oxo complex. J. Am. Chem. Soc. 2012, 134, 1996–1999. [Google Scholar] [CrossRef]
- Liu, J.G.; Ohta, T.; Yamaguchi, S.; Ogura, T.; Sakamoto, S.; Maeda, Y.; Naruta, Y. Spectroscopic Characterization of a Hydroperoxo–Heme Intermediate: Conversion of a Side-On Peroxo to an End-On Hydroperoxo Complex. Angew. Chem. Int. Ed. 2009, 48, 9262–9267. [Google Scholar] [CrossRef]
- Liu, J.-G.; Shimizu, Y.; Ohta, T.; Naruta, Y. Formation of an end-on ferric peroxo intermediate upon one-electron reduction of a ferric superoxo heme. J. Am. Chem. Soc. 2010, 132, 3672–3673. [Google Scholar] [CrossRef]
- Liao, M.-S.; Watts, J.D.; Huang, M.-J. Electronic structure of some substituted iron (II) porphyrins. Are they intermediate or high spin? J. Phys. Chem. A 2007, 111, 5927–5935. [Google Scholar] [CrossRef] [PubMed]
- Rutkowska-Zbik, D.; Tokarz-Sobieraj, R.; Witko, M. Quantum chemical description of oxygen activation process on Co, Mn, and Mo porphyrins. J. Chem. Theory Comput. 2007, 3, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cho, K.-B.; Lai, W.; Nam, W.; Shaik, S. Dioxygen activation by a non-heme iron (II) complex: Theoretical study toward understanding ferric–superoxo complexes. J. Chem. Theory Comput. 2012, 8, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Silaghi-Dumitrescu, R. A density functional investigation of hydrogen peroxide activation by high-valent heme centers: Implications for the catalase catalytic cycle. J. Porphyr. Phthalocyanines 2010, 14, 371–374. [Google Scholar] [CrossRef]
- Hill, C.L.; Schardt, B.C. Alkane activation and functionalization under mild conditions by a homogeneous manganese (III) porphyrin-iodosylbenzene oxidizing system. J. Am. Chem. Soc. 1980, 102, 6374–6375. [Google Scholar] [CrossRef]
- Kameyama, H.; Narumi, F.; Hattori, T.; Kameyama, H. Oxidation of cyclohexene with molecular oxygen catalyzed by cobalt porphyrin complexes immobilized on montmorillonite. J. Mol. Catal. A Chem. 2006, 258, 172–177. [Google Scholar] [CrossRef]
- Conradie, M.M.; Conradie, J.; Ghosh, A. A DFT overview of high-valent iron, cobalt and nickel tetraamidomacrocyclic ligand (TAML) complexes: The end of innocence? J. Inorg. Biochem. 2006, 100, 620–626. [Google Scholar] [CrossRef]
- Chen, Y.; She, Y.; Xu, J.; Li, Y. Studies on QSAR of metalloporphyrin catalysts in the oxidation of cyclohexane to adipic acid. Front. Chem. Eng. China 2007, 1, 155–161. [Google Scholar] [CrossRef]
- Conradie, J.; Ghosh, A. Iron (III)−Nitro Porphyrins: Theoretical Exploration of a Unique Class of Reactive Molecules. Inorg. Chem. 2006, 45, 4902–4909. [Google Scholar] [CrossRef]
- Conradie, J.; Ghosh, A. Electronic Structure of an Iron-Porphyrin−Nitrene Complex. Inorg. Chem. 2009, 49, 243–248. [Google Scholar] [CrossRef]
- Que, L.; Watanabe, Y. Oxygenase pathways: Oxo, peroxo, and superoxo. Science 2001, 292, 651–653. [Google Scholar] [PubMed]
- Groves, J.T.; Han, Y.-Z. Models and mechanisms of cytochrome P450 action. In Cytochrome P450; Springer: Boston, MA, USA, 1995; pp. 3–48. [Google Scholar]
- Haber, J.; Matachowski, L.; Pamin, K.; Połtowicz, J. Manganese porphyrins as catalysts for oxidation of cyclooctane in Lyons system. J. Mol. Catal. A Chem. 2000, 162, 105–109. [Google Scholar] [CrossRef]
- Chin, D.-H.; La Mar, G.N.; Balch, A.L. Mechanism of autoxidation of iron (II) porphyrins. Detection of a peroxo-bridged iron (III) porphyrin dimer and the mechanism of its thermal decomposition to the oxo-bridged iron (III) porphyrin dimer. J. Am. Chem. Soc. 1980, 102, 4344–4350. [Google Scholar] [CrossRef]
- Ghiladi, R.A.; Kretzer, R.M.; Guzei, I.; Rheingold, A.L.; Neuhold, Y.-M.; Hatwell, K.R.; Zuberbühler, A.D.; Karlin, K.D. (F8TPP)FeII/O2 Reactivity Studies {F8TPP = Tetrakis(2,6-difluorophenyl)porphyrinate(2−)}: Spectroscopic (UV−Visible and NMR) and Kinetic Study of Solvent-Dependent (Fe/O2 = 1:1 or 2:1) Reversible O2-Reduction and Ferryl Formation. Inorg. Chem. 2001, 40, 5754–5767. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, M.R.; Johansson, A.J.; Siegbahn, P.E. O−O Bond Cleavage in Dinuclear Peroxo Complexes of Iron Porphyrins: A Quantum Chemical Study. Inorg. Chem. 2007, 46, 7992–7997. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.P.; Lehnert, N. Heme-nitrosyls: Electronic structure implications for function in biology. Acc. Chem. Res. 2015, 48, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.F.; Snijders, J.; te Velde, G.; Baerends, E.J. Towards an order-N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar]
- Te Velde, G.t.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Baerends, E.J. Kohn-Sham density functional theory: Predicting and understanding chemistry. Rev. Com. Chem. 2000, 15, 1–86. [Google Scholar]
- Hoe, W.-M.; Cohen, A.J.; Handy, N.C. Assessment of a new local exchange functional OPTX. Chem. Phys. Lett. 2001, 341, 319–328. [Google Scholar] [CrossRef]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Conradie, M.M.; Conradie, J.; Ghosh, A. Capturing the spin state diversity of iron (III)-aryl porphyrins: OLYP is better than TPSSh. J. Inorg. Biochem. 2011, 105, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Conradie, J.; Ghosh, A. Electronic Structure of Trigonal-Planar Transition-Metal−Imido Complexes: Spin-State Energetics, Spin-Density Profiles, and the Remarkable Performance of the OLYP Functional. J. Chem. Theory Comput. 2007, 3, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Radoń, M.; Pierloot, K. Binding of CO, NO, and O2 to Heme by Density Functional and Multireference ab Initio Calculations. J. Phys. Chem. A 2008, 112, 11824–11832. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; Brothers, P.J.; Ghosh, A. Density Functional Theory Calculations on Ruthenium(IV) Bis(amido) Porphyrins: Search for a Broader Perspective of Heme Protein Compound II Intermediates. J. Phys. Chem. B 2010, 114, 15380–15388. [Google Scholar] [CrossRef]
- Jaworska, M.; Lodowski, P. Electronic structure and spectra of nitrosyl complexes with cobalt and manganese porphyrins. Struct. Chem. 2012, 23, 1333–1348. [Google Scholar] [CrossRef]
- Liao, M.-S.; Scheiner, S. Electronic structure and bonding in unligated and ligated Fe II porphyrins. J. Chem. Phys. 2002, 116, 3635–3645. [Google Scholar] [CrossRef]
- Mispelter, J.; Momenteau, M.; Lhoste, J. Proton magnetic resonance characterization of the intermediate (S= 1) spin state of ferrous porphyrins. J. Chem. Phys. 1980, 72, 1003–1012. [Google Scholar] [CrossRef]
- Liao, M.S.; Scheiner, S. Comparative study of metal-porphyrins,-porphyrazines, and-phthalocyanines. J. Comput. Chem. 2002, 23, 1391–1403. [Google Scholar] [CrossRef]
- Lin, W. d-Orbital energies and low-lying excited states of cobalt porphyrins. Inorg. Chem. 1976, 15, 1114–1118. [Google Scholar] [CrossRef]
- Nakamura, K.; Kitaoka, Y.; Akiyama, T.; Ito, T.; Weinert, M.; Freeman, A.J. Constraint density functional calculations for multiplets in a ligand-field applied to Fe-phthalocyanine. Phys. Rev. B 2012, 85, 235129. [Google Scholar] [CrossRef]
- Panchmatia, P.M.; Ali, M.E.; Sanyal, B.; Oppeneer, P.M. Halide ligated iron porphines: A DFT+ U and UB3LYP study. J. Phys. Chem. A 2010, 114, 13381–13387. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.P.; Sundholm, D. Spin and charge distribution in iron porphyrin models: A coupled cluster and density-functional study. J. Chem. Phys. 2004, 120, 3229–3236. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.J.; Murray, K.S.; Zwack, P.R.; Homborg, H.; Kalz, W. Spin states in iron (III) phthalocyanines studied by Moessbauer, magnetic susceptibility, and ESR measurements. Inorg. Chem. 1986, 25, 2539–2545. [Google Scholar] [CrossRef]
- Kepenekian, M.; Calborean, A.; Vetere, V.; Le Guennic, B.; Robert, V.; Maldivi, P. Toward reliable dft investigations of mn-porphyrins through CASPT2/DFT comparison. J. Chem. Theory Comput. 2011, 7, 3532–3539. [Google Scholar] [CrossRef]
- Shaik, S.; Hirao, H.; Kumar, D. Reactivity of high-valent iron–oxo species in enzymes and synthetic reagents: A tale of many states. Acc. Chem. Res. 2007, 40, 532–542. [Google Scholar] [CrossRef]
- Yin, G. Understanding the Oxidative Relationships of the Metal Oxo, Hydroxo, and Hydroperoxide Intermediates with Manganese(IV) Complexes Having Bridged Cyclams: Correlation of the Physicochemical Properties with Reactivity. Acc. Chem. Res. 2012, 46, 483–492. [Google Scholar] [CrossRef]
- Groenhof, A.R.; Swart, M.; Ehlers, A.W.; Lammertsma, K. Electronic ground states of iron porphyrin and of the first species in the catalytic reaction cycle of cytochrome P450s. J. Phys. Chem. A 2005, 109, 3411–3417. [Google Scholar] [CrossRef]
- Collman, J.P.; Hoard, J.; Kim, N.; Lang, G.; Reed, C.A. Synthesis, stereochemistry, and structure-related properties of. .alpha.,.beta.,.gamma.,.delta.-tetraphenylporphinatoiron(II). J. Am. Chem. Soc. 1975, 97, 2676–2681. [Google Scholar] [CrossRef]
- Wondimagegn, T.; Rauk, A. The Structures and Stabilities of the Complexes of Biologically Available Ligands with Fe (II) Porphine: An Ab Initio Study. J. Phys. Chem. B 2012, 116, 10301–10310. [Google Scholar] [CrossRef]
- Scheidt, W.R.; Finnegan, M.G. Structure of monoclinic chloro (meso-tetraphenylporphyrinato) iron (III). Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1989, 45, 1214–1216. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Scheidt, W.R. Chloro(5,10,15,20-tetraphenylporphyrinato)manganese(III) with 4/m Symmetry. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1996, 52, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Madura, P.; Scheidt, W.R. Stereochemistry of low-spin cobalt porphyrins. 8. .alpha.,.beta.,.gamma.,.delta.-Tetraphenylporphinatocobalt(II). Inorg. Chem. 1976, 15, 3182–3184. [Google Scholar] [CrossRef]
- Scheidt, W.R. Stereochemistry of low-spin cobalt porphyrins. III. Crystal structure and molecular stereochemistry of bis(piperidine)-.alpha.,.beta.,.gamma.,.delta.-tetraphenylporphinatocobalt(II). J. Am. Chem. Soc. 1974, 96, 84–89. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spin States(S) | Electronic Figuration | 0 | 1/2 | 1 | 3/2 | 2 | 5/2 | Ground State |
|---|---|---|---|---|---|---|---|---|
| PFeII | d6 | 191.66 | - | 0.00 | - | 112.90 | - | 1 [41,42] |
| PMnII | d5 | - | 1207.73 | - | 0 | - | 136.53 | 3/2 [43] |
| PCoII | d7 | -- | 0 | - | 86.64 | - | - | 1/2 [44,45] |
| PFeIIICl | d5 | - | 44.63 | - | 0.00 | - | 18.38 | 3/2 [46], 5/2 [47] |
| PMnIIICl | d4 | 388.57 | - | 68.26 | - | 0 | - | 2 [48] |
| PCoIIICl | d6 | 0 | - | 31.51 | - | 63.01 | - | - |
| Spin States(S) | Electronic Configuration | 0 | 1/2 | 1 | 3/2 | 2 | 5/2 |
|---|---|---|---|---|---|---|---|
| PFeIIIO2-side-on | - | 0.00 | - | 18.38 | - | 94.52 | - |
| PFeIIIO2-end-on | d5 | 0.00 | - | 21.00 | - | 73.51 | - |
| PMnIIIO2-side-on | d4 | - | 73.51 | - | 0 | - | 52.51 |
| PMnIIIO2-end-on | d4 | - | 10.50 | - | 2.63 | - | 0 |
| PCoIIIO2-end-on | d6 | - | 0 | - | 47.26 | - | - |
| Spin States(S) | Electronic Configuration | 0 | 1/2 | 1 | 3/2 | 2 | 5/2 | Ground State |
|---|---|---|---|---|---|---|---|---|
| PFeIVO | d4 | 84.02 | - | 0.00 | - | 68.26 | - | 1 |
| PMnIVO | d3 | - | 0 | - | 44.63 | - | - | - |
| PCoIVO | d5 | - | 433.21 | - | 0 | - | 147.29 | - |
| PFeOIIIH | d5 | - | 42.01 | - | 0.00 | - | 44.63 | 3/2 |
| PMnOIIIH | d4 | 81.39 | - | 31.51 | - | 0 | - | - |
| PCoOIIIH | d6 | 0 | - | 94.52 | - | 133.90 | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, M.; Gao, A.; Liu, Y.; Zhou, Y.; Sun, Z.; Li, Y.; He, F.; Li, L.; Mo, L.; Liu, R.; et al. Theoretical Study on Electronic Structural Properties of Catalytically Reactive Metalloporphyrin Intermediates. Catalysts 2020, 10, 224. https://doi.org/10.3390/catal10020224
Cao M, Gao A, Liu Y, Zhou Y, Sun Z, Li Y, He F, Li L, Mo L, Liu R, et al. Theoretical Study on Electronic Structural Properties of Catalytically Reactive Metalloporphyrin Intermediates. Catalysts. 2020; 10(2):224. https://doi.org/10.3390/catal10020224
Chicago/Turabian StyleCao, Meijuan, Aijing Gao, Yuanyuan Liu, Yang Zhou, Zhicheng Sun, Yaling Li, Furui He, Luhai Li, Lixin Mo, Ruping Liu, and et al. 2020. "Theoretical Study on Electronic Structural Properties of Catalytically Reactive Metalloporphyrin Intermediates" Catalysts 10, no. 2: 224. https://doi.org/10.3390/catal10020224
APA StyleCao, M., Gao, A., Liu, Y., Zhou, Y., Sun, Z., Li, Y., He, F., Li, L., Mo, L., Liu, R., Han, Y., & Yang, Y. (2020). Theoretical Study on Electronic Structural Properties of Catalytically Reactive Metalloporphyrin Intermediates. Catalysts, 10(2), 224. https://doi.org/10.3390/catal10020224