Theoretical and Applied Aspects of Hydrodechlorination Processes—Catalysts and Technologies

Abstract
:1. Introduction
- Electrochemical reduction.
- Radiolysis of organochlorine compounds.
- Hydrogenation with hydrogen—thermal and catalytic.
- Reduction with metal hydrides and complex hydrides of elements.
- Metal reduction in the presence of hydrogen donors.
- Reduction with metal salts of lower valency.
2. Electrochemical Reduction
3. Radiolytic Reduction and Dechlorination
4. Hydrodechlorination Using Hydrogen
- thermal hydrodechlorination resulting in formation of a mixture of hydrocarbons and hydrogen chloride
- exhaustive catalytic hydrodechlorination also resulting in formation of hydrocarbons and hydrogen chloride
- selective catalytic hydrodechlorination resulting in formation of target organic products (hydrocarbons and chlorohydrocarbons) and hydrogen chloride

4.1. Thermal Hydrodechlorination
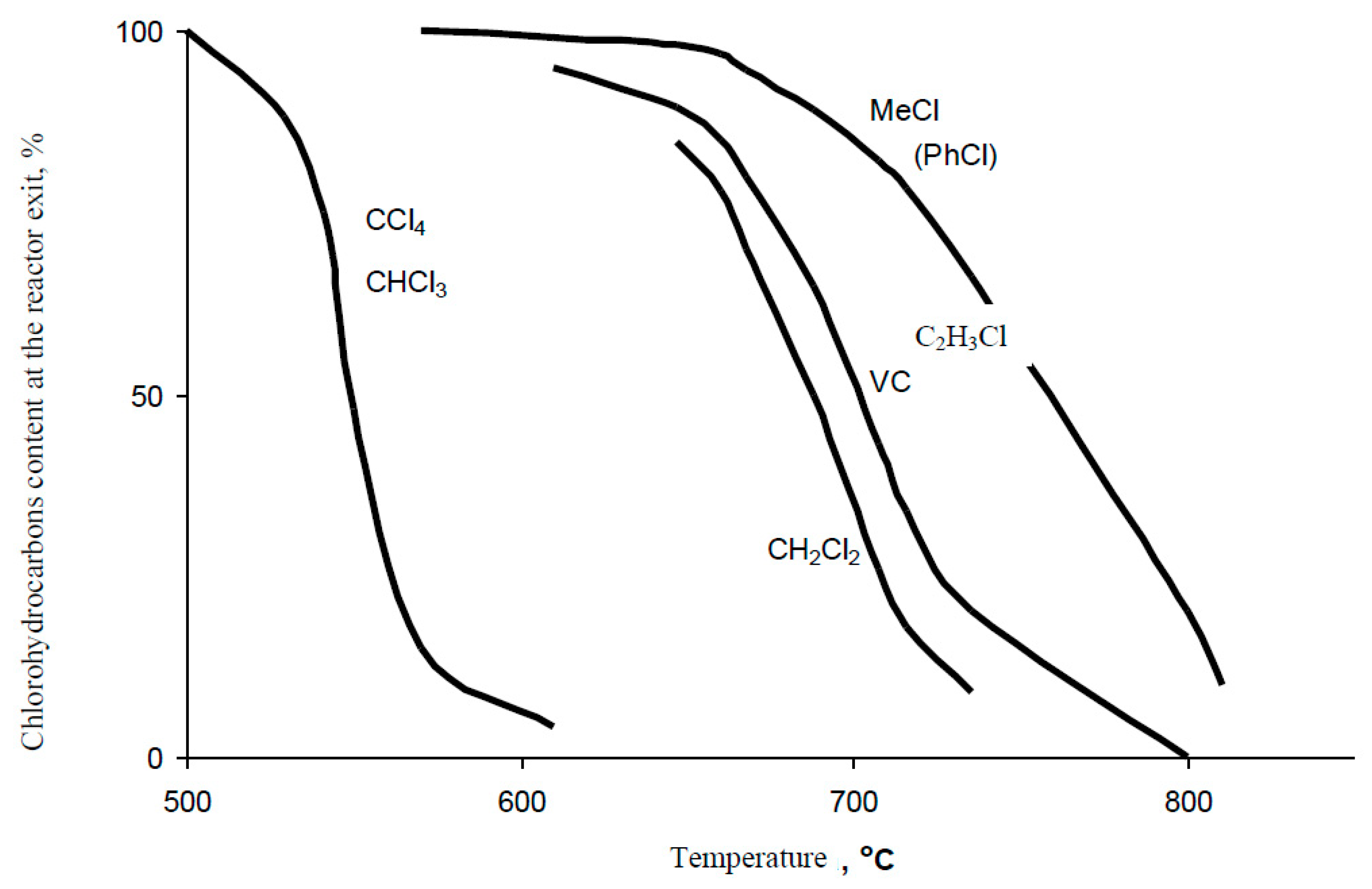
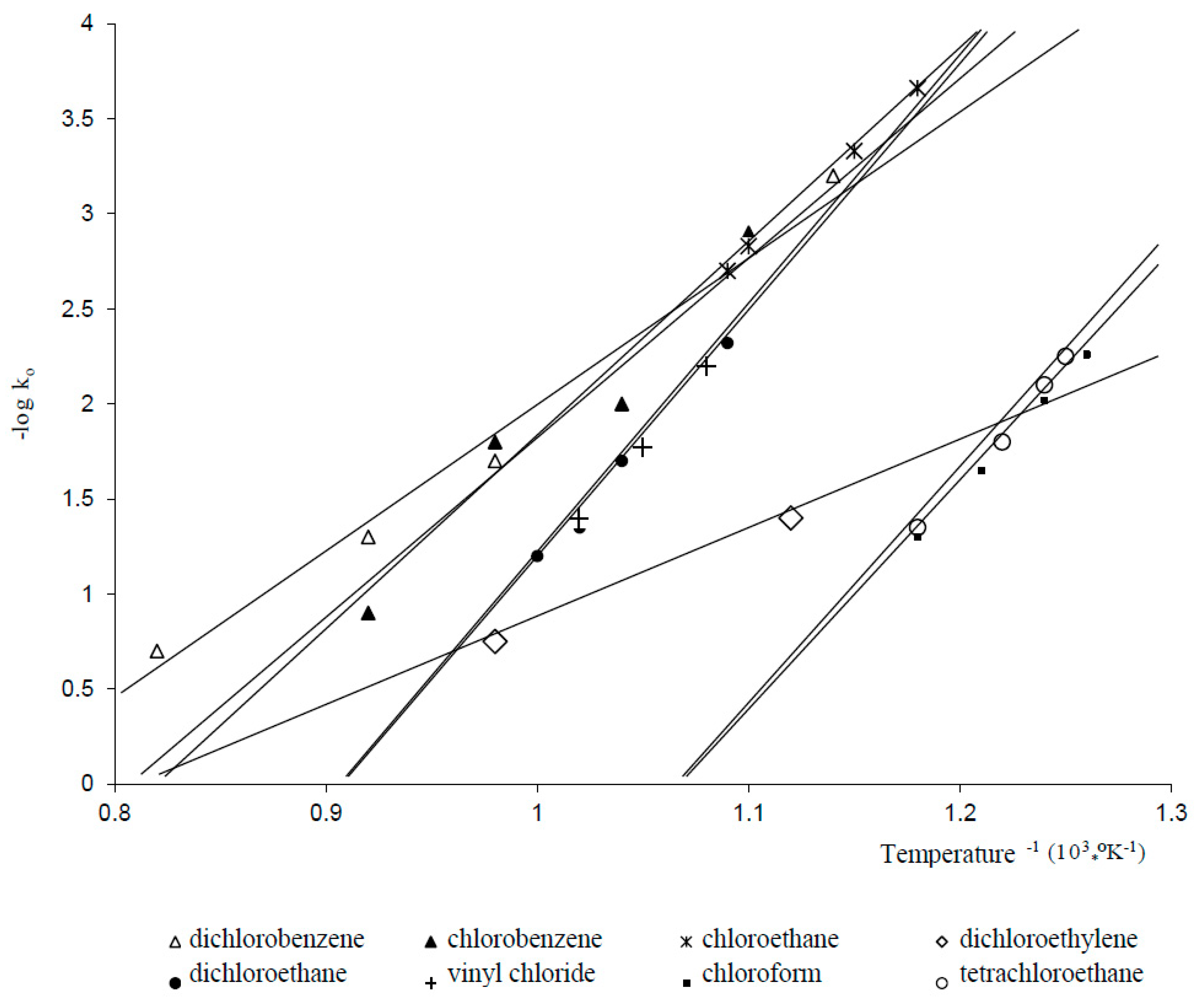
Thermal Hydrodechlorination Process
4.2. Catalytic Hydrodechlorination
4.2.1. Catalytic Systems
4.2.2. Comparative Data of Hydrodechlorination of Chlorohydrocarbons
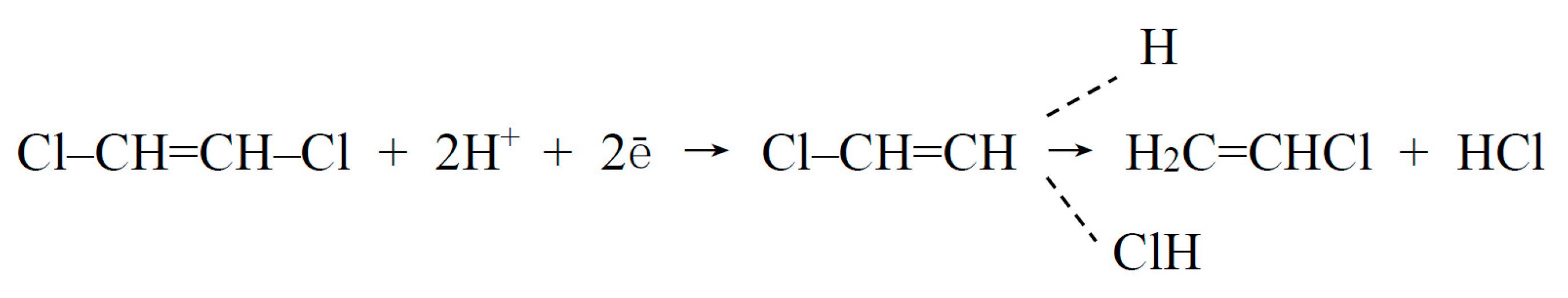
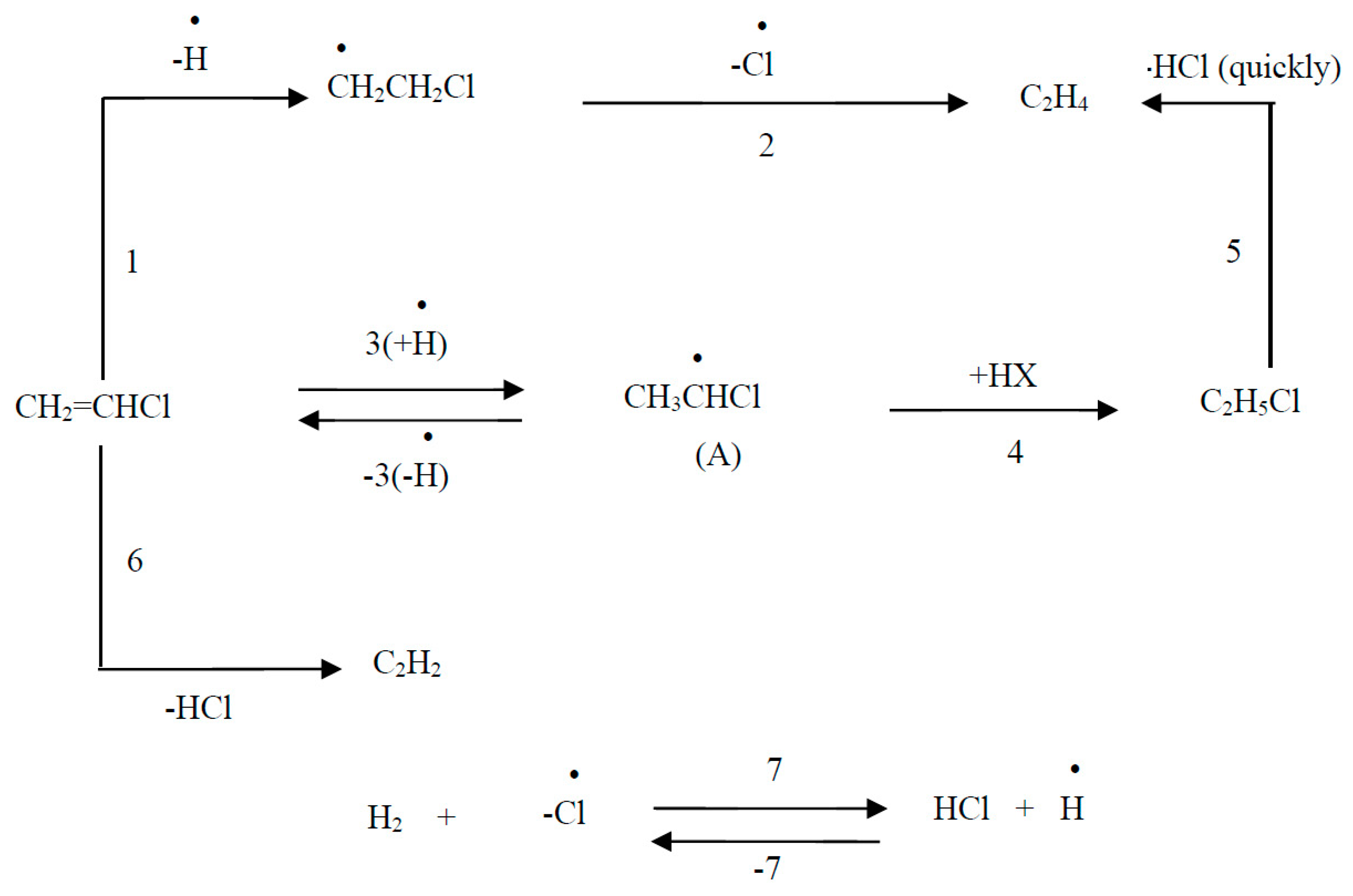
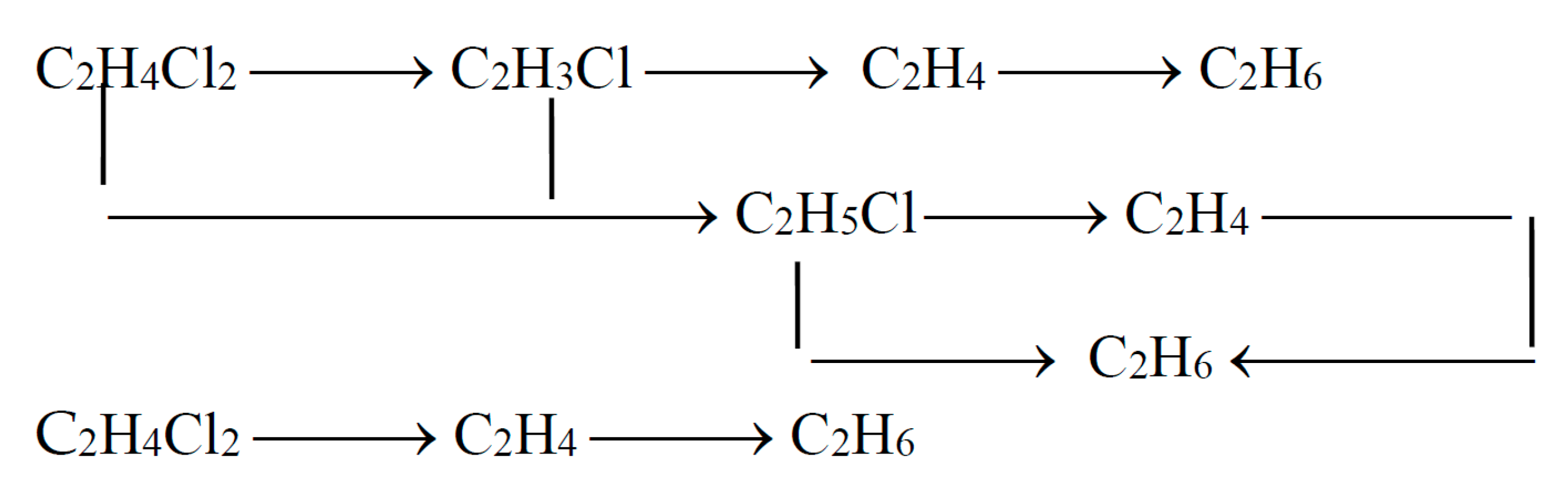
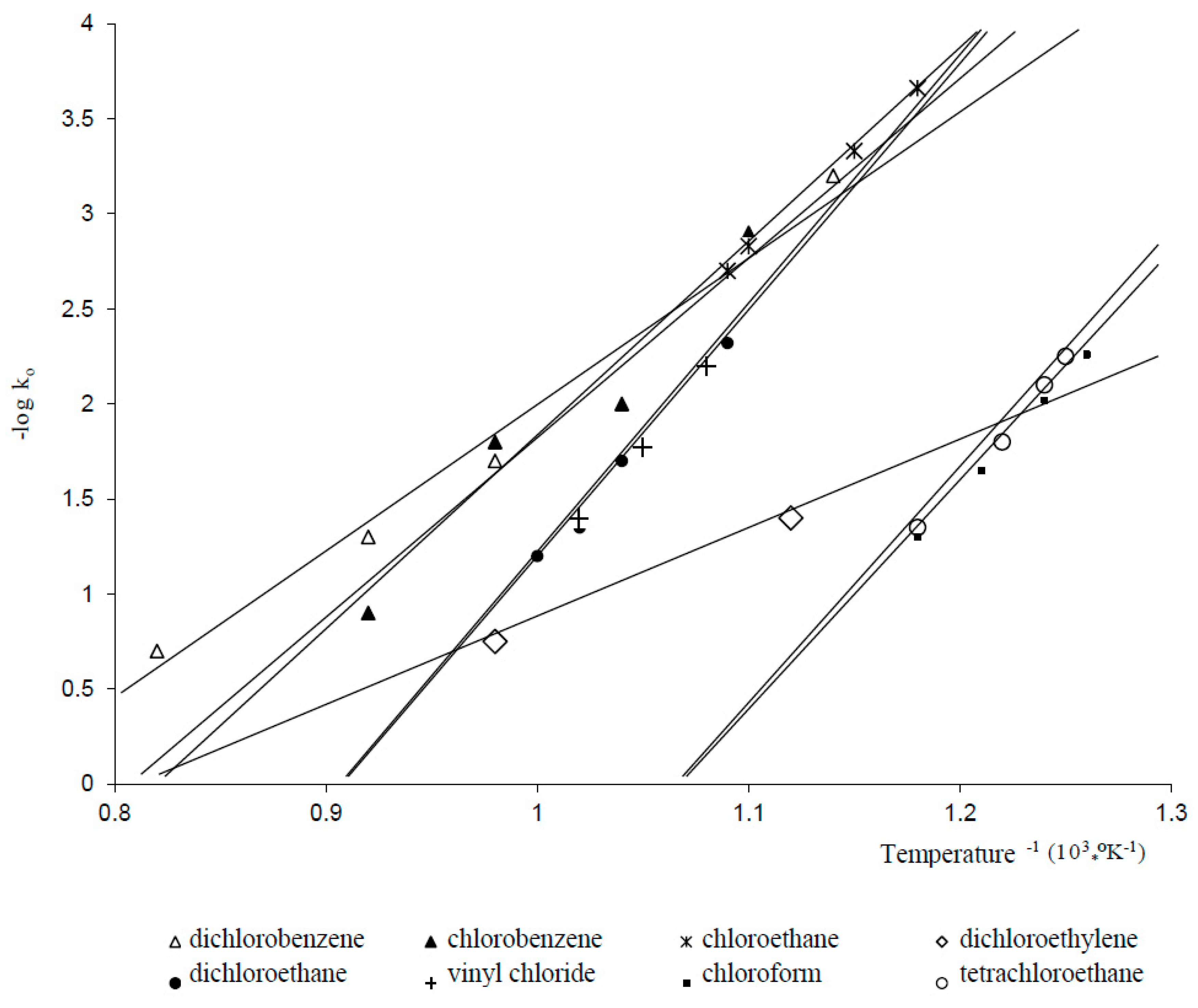
4.2.3. Mechanism and Kinetics of the Process
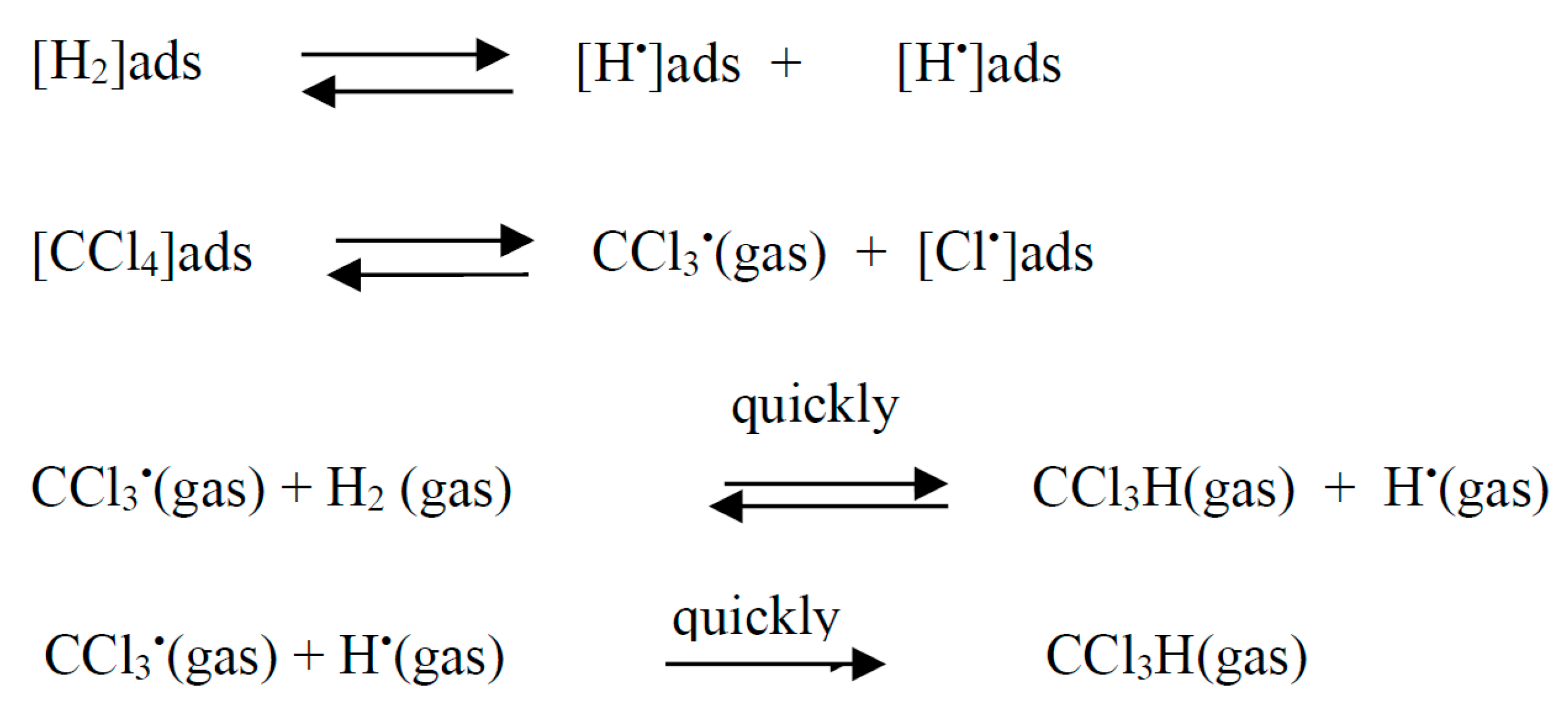 This scheme assumes predominant adsorption of chlorohydrocarbons on the surface of the catalyst (metal).
This scheme assumes predominant adsorption of chlorohydrocarbons on the surface of the catalyst (metal).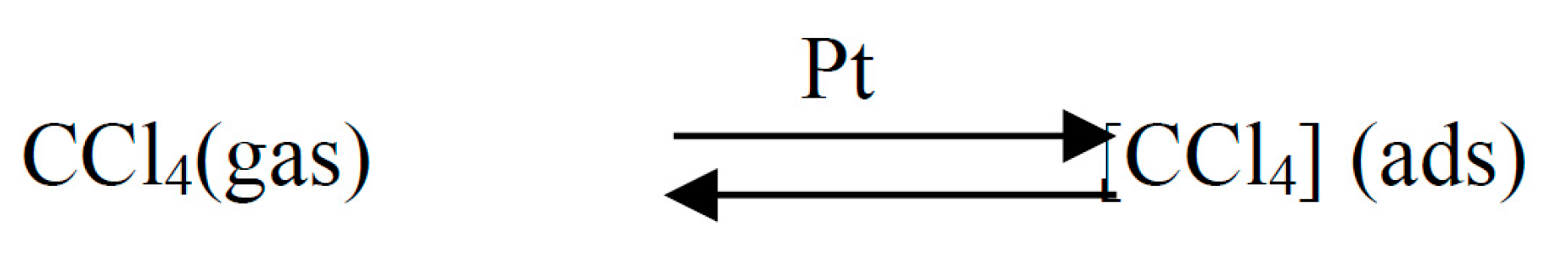
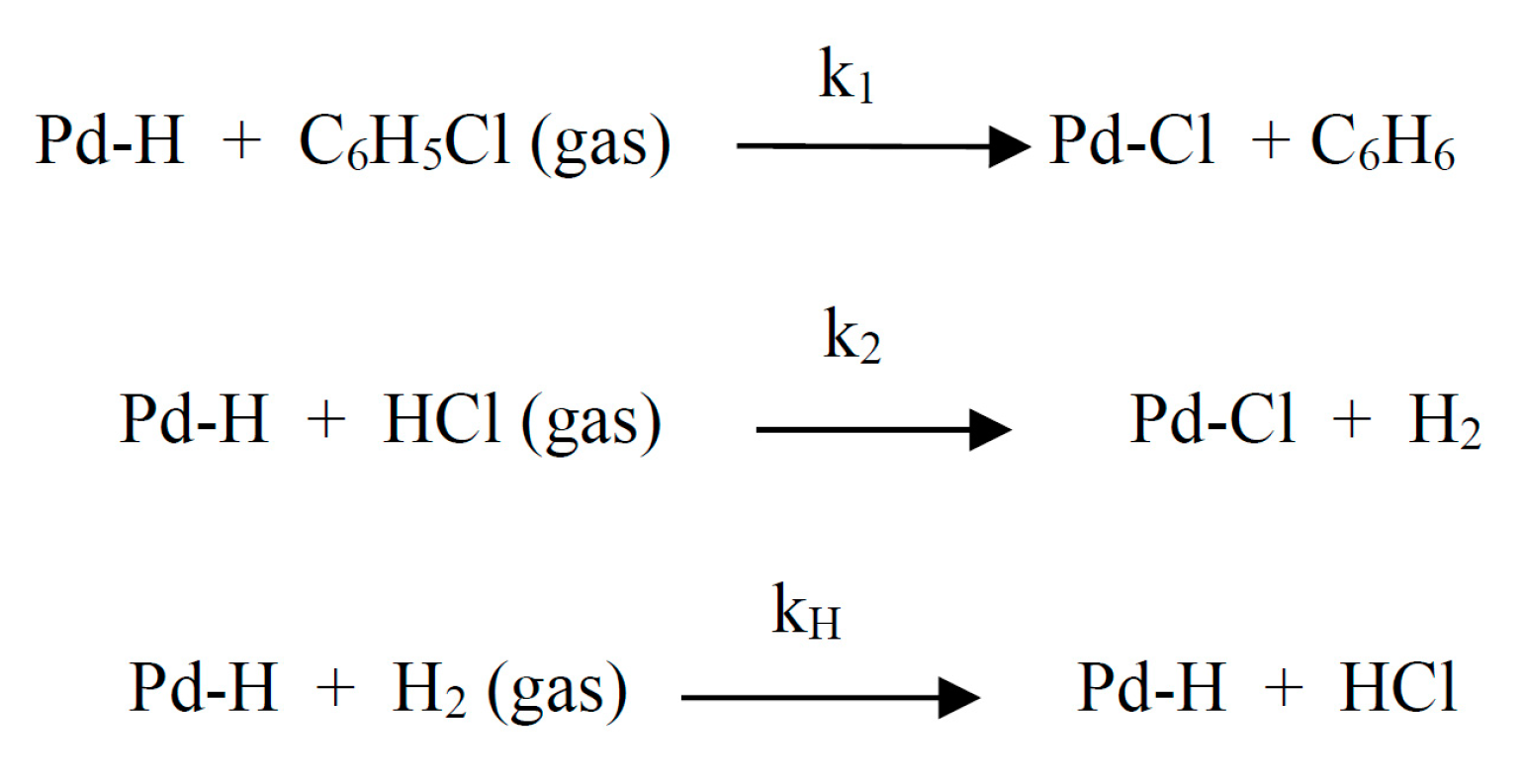
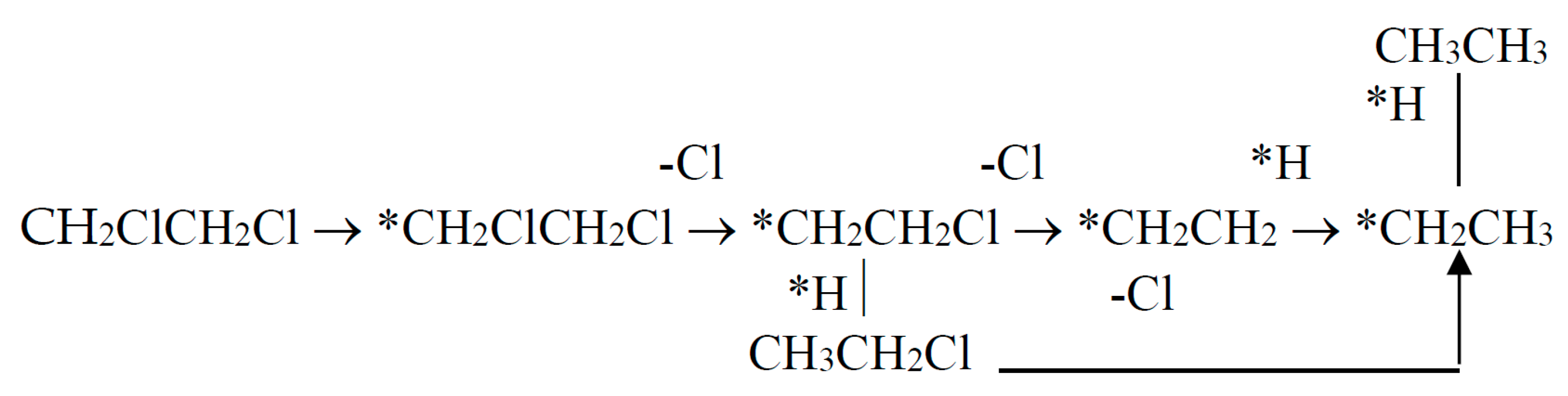 where *H is adsorbed hydrogen; *CH2CH3 – adsorbed ethyl.
where *H is adsorbed hydrogen; *CH2CH3 – adsorbed ethyl.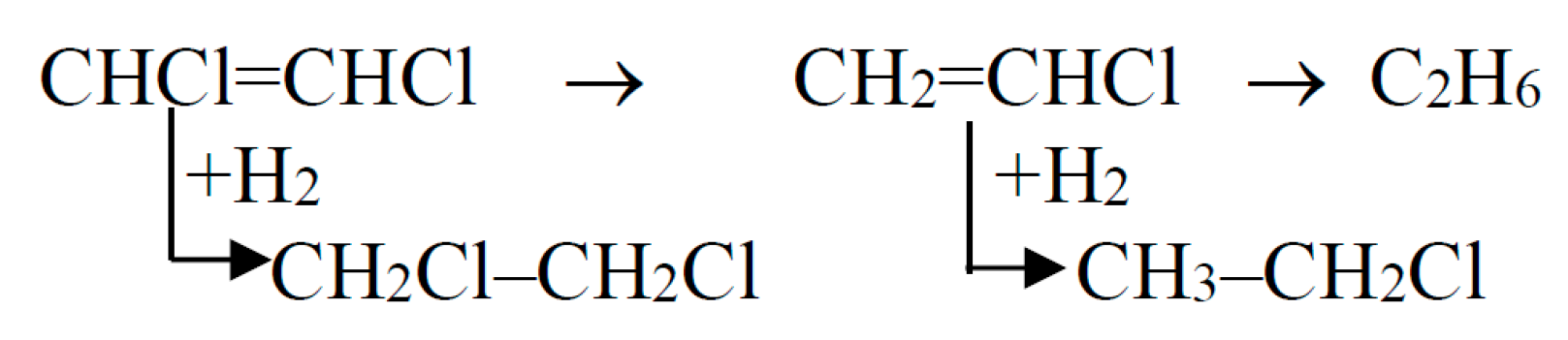
- (a)
- addition of hydrogen across a double bond in the molecule of 1,3-dichloropropene;
- (b)
- sequential removal of chlorine atoms;
- (c)
- multiplet removal of chlorine atoms.

4.2.4. Catalyst Deactivation and Stability
- drying of the catalyst in an atmosphere of diluent gas (nitrogen) for 2–6 h at an elevated temperature (100–500 °C);
- catalyst treatment with hydrochloric acid for 2–4 h at an elevated temperature (150–300 °C);
- catalyst recovery with a reducing agent (hydrogen, hydrazine, formaldehyde) for 2–24 h; at elevated temperature (150–500 °C);
- catalyst reprocessing with hydrochloric acid from 15 min to 2 h at a temperature of 80–150 °C.
4.2.5. Selective Catalytic Hydrodechlorination
Hydrodechlorination on Noble Metal Based Catalysts
Catalysts Based on Nickel, Chromium, and other Metals of Variable Valency, Binary, and Complex Catalytic Systems
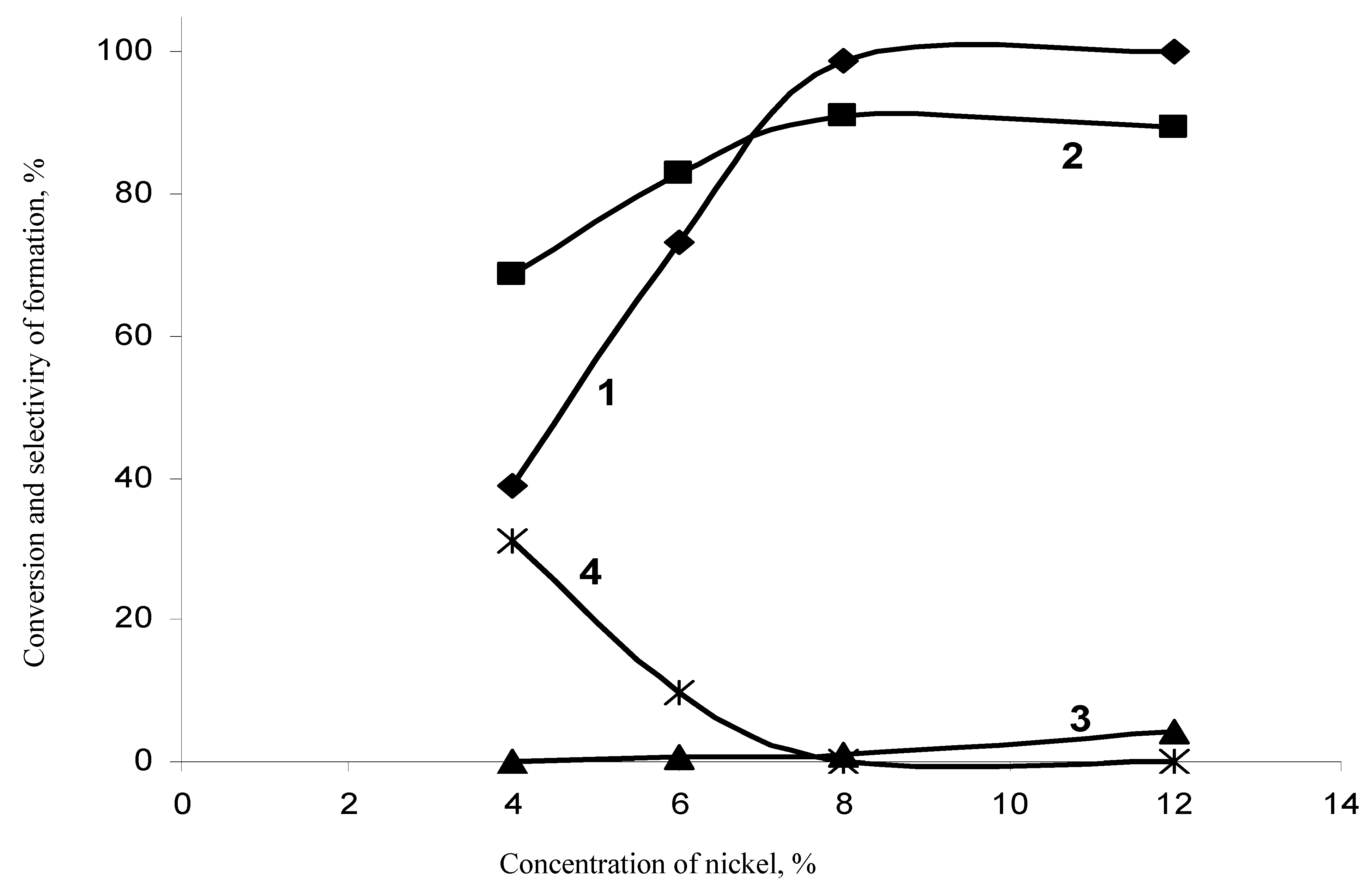
Hydrodechlorination in the Presence of a Nickel-Chromium Catalyst
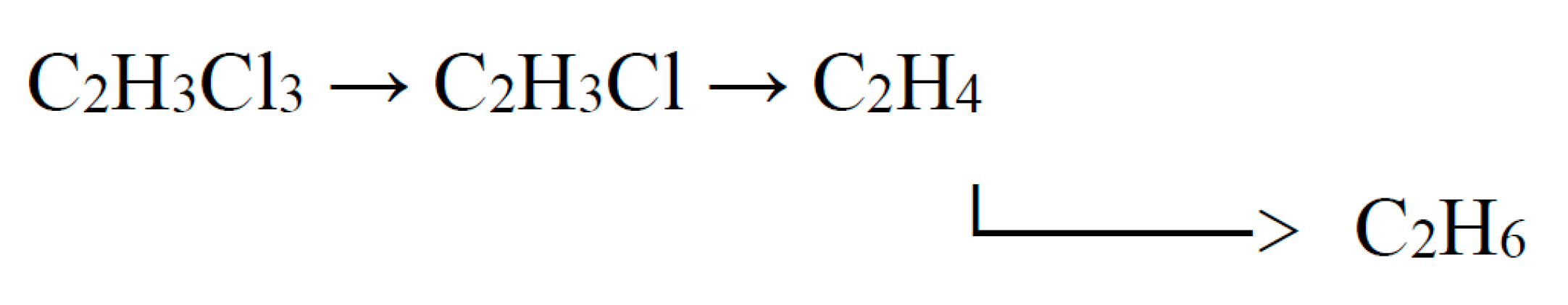
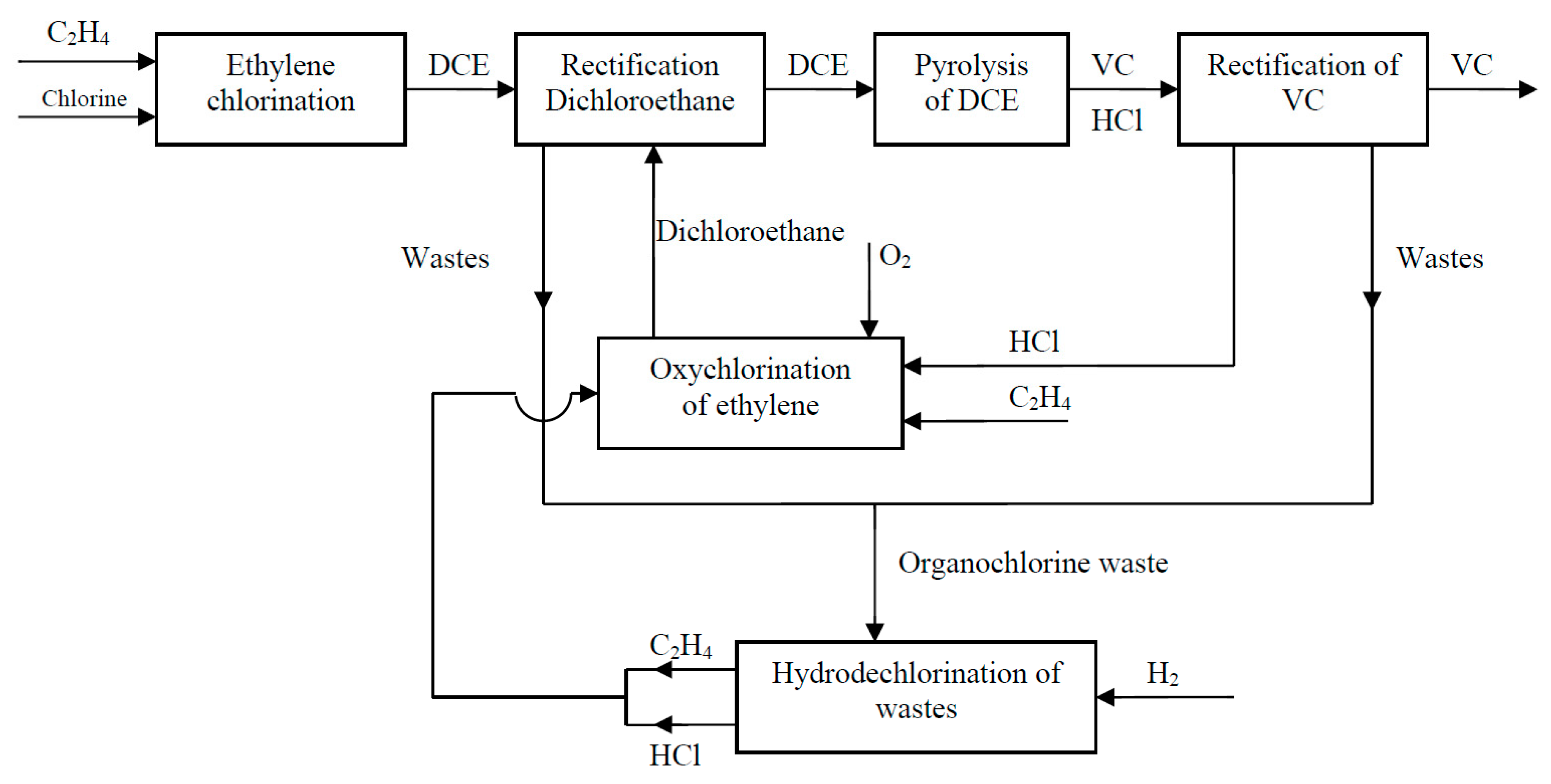
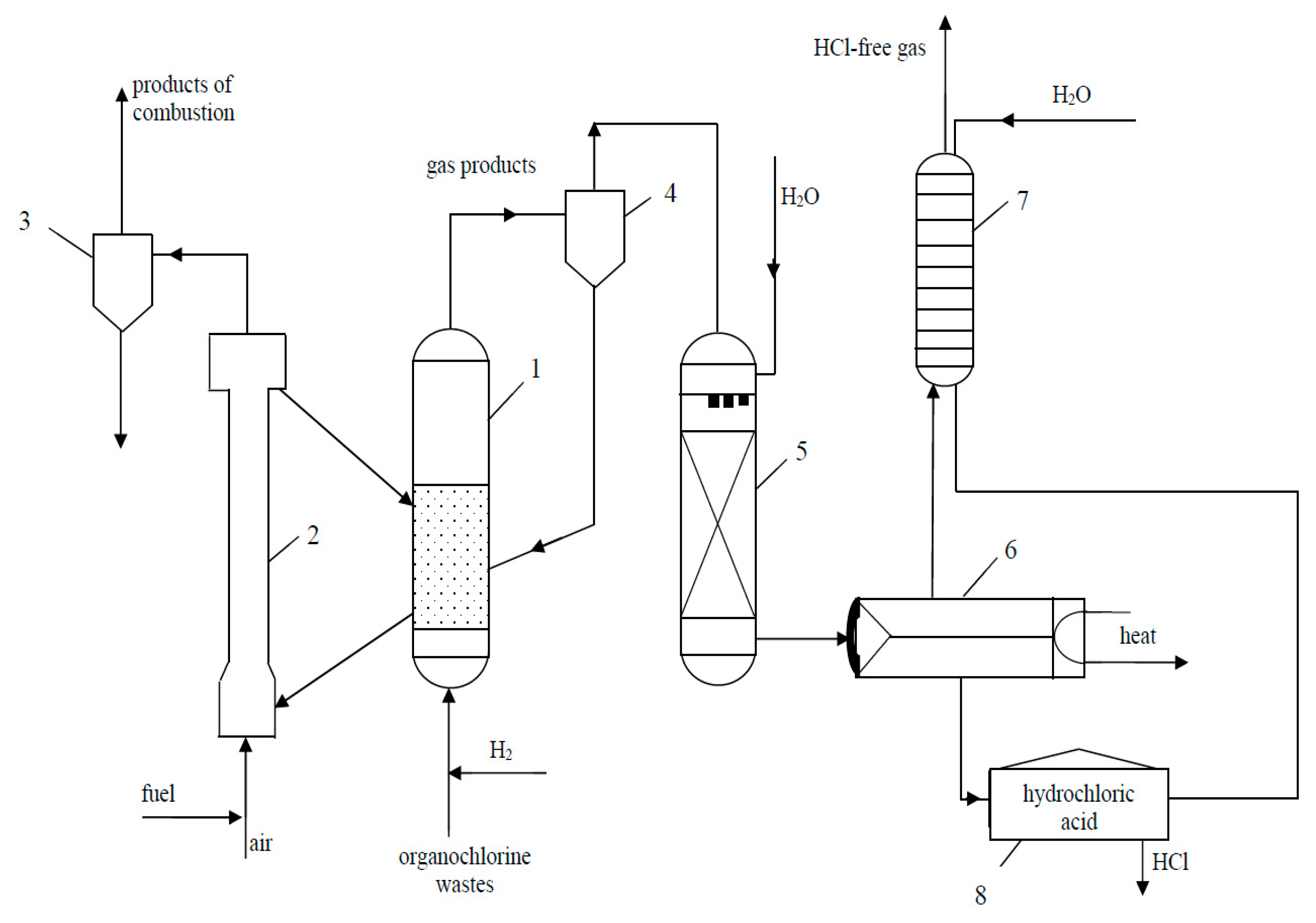
Implementation of Catalytic Hydrodechlorination Process
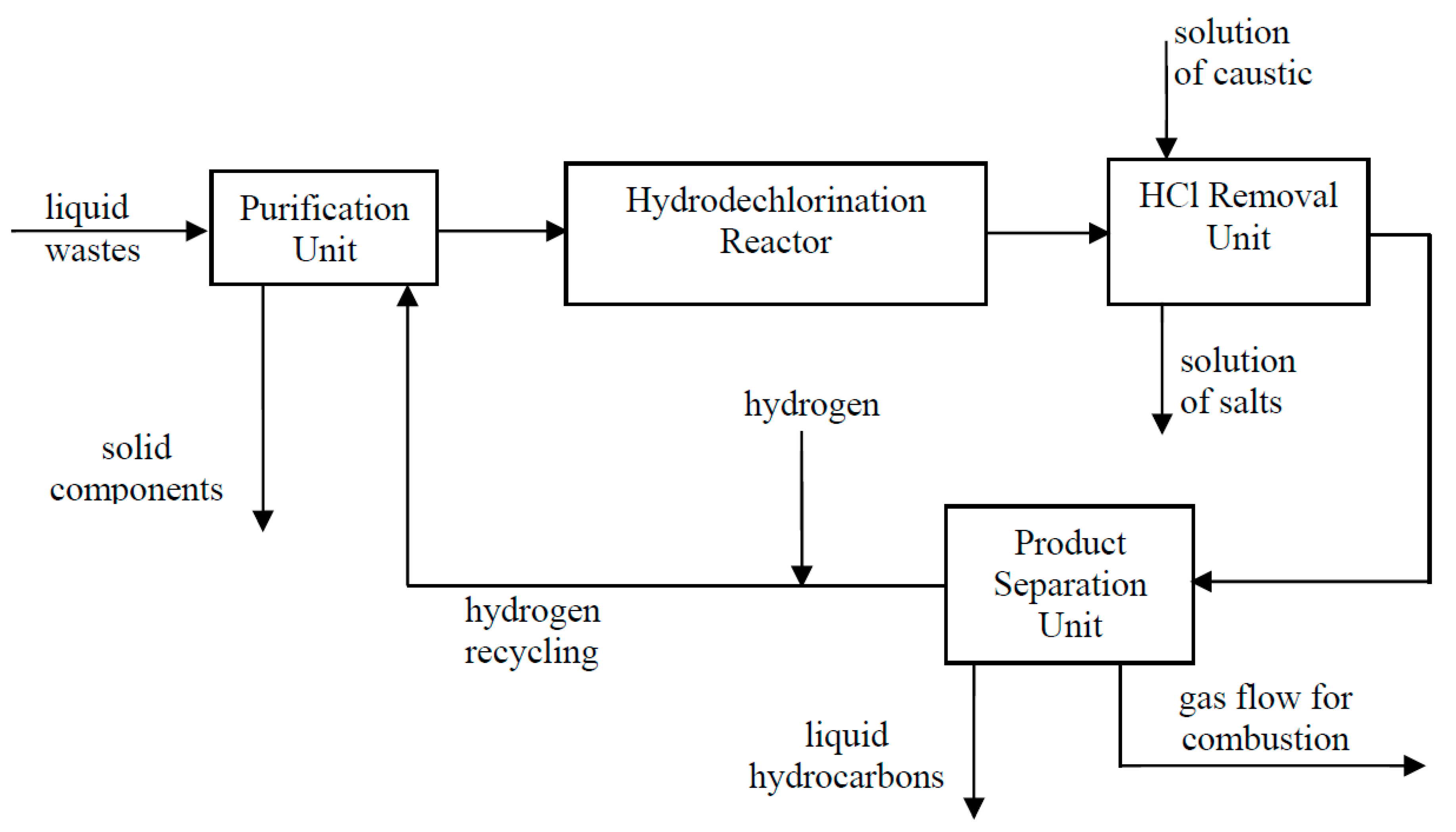
4.2.6. Catalytic Methods for Organochlorine Compounds Reduction in an Aqueous Medium
5. Reduction of Organochlorine Compounds with Metal Hydrides and Complex Hydrides of Elements
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nesmeyanov, A.N.; Kocheshkov, K.A. (Eds.) Methods of Elementoorganic Chemistry; Chloroaliphatic Compounds; Nauka: Moscow, Russia, 1973; pp. 491–548. (In Russian) [Google Scholar]
- Pinder, A.R. The Hydrogenolysis of Organic Halides. Synthesis 1980, 1980, 425–452. [Google Scholar] [CrossRef]
- Johnstone, R.A.; Wilby, A.H. Heterogeneous catalytic transfer hydrogenation and its relation to other methods for reduction of organic compounds. Chem Rev. 1985, 85, 129–170. [Google Scholar] [CrossRef]
- Zanaveskin, L.N.; A Aver’Yanov, V.; A Treger, Y. Prospects for the development of methods for the processing of organohalogen waste. Characteristic features of the catalytic hydrogenolysis of halogen-containing compounds. Russ. Chem. Rev. 1996, 65, 617–624. [Google Scholar]
- Flid, M.R.; Treger, Y.A. Vinyl Chloride: Chemistry and Technology; Kalvis: Moscow, Russia, 2008; Volume 2, pp. 163–231. (In Russian) [Google Scholar]
- Otromke, H.; Theerkorn, U.; Hedemann, G. Verfahren und vorrichtung zur gezielten zeriegung (Cracken) borganisch Stoffe in der Produktion und zur umweltfeundlichen Aufbereitung der gecrackten Stosse. EU Patent 0295454, 21 December 1988. [Google Scholar]
- Zanaveskin, L.N.; Averyanov, V.A.; Popov, S.A. Method for producing chlorobenzene. RF Patent No. 2039731, 18 May 1993. [Google Scholar]
- Rylander, P.N. Catalytic Hydrogenation in Organic Syntheses; Aldrichimica Acta: St. Louis, MI, USA, 1979; Volume 12, pp. 53–58. [Google Scholar]
- Lokteva, E.; Golubina, E.; Likholobov, V.; Lunin, V. Disposal of Chlorine-Containing Wastes. In Chemistry Beyond Chlorine; Tundo, P., Lokteva, E., Liang-Nian, H., Mota, C., Eds.; Springer International Publishing: Basel, Switherland, 2016; pp. 559–584. [Google Scholar]
- Lunin, V.V.; Lokteva, E.S. Catalytic hydrodehalogenation of organic compounds. Russ. Chem. Bull. 1996, 45, 1519–1534. [Google Scholar] [CrossRef]
- Evans, M.G.; Hush, N.S. Ionogenic reactions involving bond breaking at electrodes. J. de Chim. Phys. 1952, 49, C159–C171. [Google Scholar] [CrossRef]
- Elving, P.J.; Pullman, B. Mechanisms of Organic Electrode Reactions. Adv. Chem. Phys. 1961, 3, 1–31. [Google Scholar]
- Tomilov, A.P.; Mairanovsky, S.G.; Fioshin, M.Y.; Smirnov, V.A. Electrochemistry of Organic Compounds; Chimiya: Leningrad, Russia, 1968. (In Russian) [Google Scholar]
- Popp, F.D.; Schultz, H.P. Electrolytic Reduction of Organic Compounds. Chem. Rev. 1962, 62, 19–40. [Google Scholar] [CrossRef]
- Stackelberg, M.; Stracke, W.Z. Das polarographische Verhalten ungesättigter und halogenierter Kohlenwasserstoffe. ElectrochemI 1949, 53, 118–125. [Google Scholar]
- Jura, W.H.; Gaul, R.J. Polarographic Behavior of Unsymmetrical Polyhalogenated Compounds. Polychlorinated Propionitrile and Derivatives1. J. Am. Chem. Soc. 1958, 80, 5402–5409. [Google Scholar] [CrossRef]
- Pikaev, A.K.; Ershov, B.G. Primary products of water radiolysis and their reactivity. Russ. Chem. Rev. 1967, 36, 1427–1459. (In Russian) [Google Scholar] [CrossRef]
- Anbar, M.; Neta, P. Reactions of halogenoaliphatic acids with free radicals in aqueous solution. Part I. Reactions with hydrogen atoms. J. Chem. Soc. 1967, 5, 834–837. [Google Scholar] [CrossRef]
- Anbar, M.; Meyerstein, D.; Neta, P. Reactivity of aliphatic compounds towards hydroxyl radicals. J. Chem. Soc. B 1966, 742. [Google Scholar] [CrossRef]
- Abramson, F.P.; Buckhold, B.M.; Firestone, R.F. The Effects of Temperature and Various Solutes on the Radiolysis of CCl4. J. Am. Chem. Soc. 1962, 84, 2285–2288. [Google Scholar] [CrossRef]
- Louw, R.; Dijks, H.; Mulder, P. Thermal hydro-dechlorination of (poly)chlorinated organic compounds. Chem. Ind. 1983, 10, 759–760. [Google Scholar]
- Louw, R.; Manion, J.A.; Mulder, P. Gas-phase thermal hydrogenolysis of organic chlorine compounds: An alternative to incineration. Resour. Conserv. 1987, 14, 365–368. [Google Scholar] [CrossRef]
- Flid, M.R. Sustainable chlorine-balanced technologies for the production of vinyl chloride from ethane-ethylene raw materials. Dis. Doc. Chem. Sci. 2002, 333. (In Russian) [Google Scholar]
- Treger, J.A.; Kartashov, L.M. The aim — waste-free production. Ind. Russ. 1997, 8, 51–54. (In Russian) [Google Scholar]
- Treger, J.A.; Kartashov, L.M. The problem of organochlorine production wastes processing and methods for its solving. creation of waste-free technologies. Russ. J. Gen. Chem. 1998, 42, 58–66. (In Russian) [Google Scholar]
- Zanaveskin, L.N.; Averyanov, V.A. Thermal hydrogenolysis of organochlorine compounds. Basic patterns and development prospects. Russ. Chem. Ind. 1999, 2, 3–13. (In Russian) [Google Scholar]
- Terekhov, A.V.; Zanaveskin, L.N.; Zanaveskin, K.L.; Konorev, O.A. Catalytic hydrodechlorination of chlorohydrocarbons in sodium hydroxide solutions. Catal. Ind. 2012, 6, 39–47. (In Russian) [Google Scholar]
- Zanaveskin, L.N.; Averyanov, V.A. Carbon tetrachloride. Methods of processing into environmentally friendly products and prospects for their development (review). Russ. Chem. Ind. 2002, 9, 4–21. (In Russian) [Google Scholar]
- Converti, A.; Zilli, M.; De Faveri, D.M.; Ferraiolo, G. Hydrogenolysis of organochlorinated pollutants: Kinetics and thermodynamics. J. Hazard. Mater. 1991, 27, 127–135. [Google Scholar] [CrossRef]
- Louw, R.; Senden, M.M.G.; Mulden, P.; Tels, M. Thermal hydro-dechlorination of chlorinated waste, an alternative to incineration. In Recycling International Industrial and Hazardous Waste; Thome-Kozmiensky, K.J., Ed.; E. Freitag: Berlin, Germany, 1984; pp. 999–1004. [Google Scholar]
- Louw, R.; Manion, J.A.; Mulder, P. Presented at the 3rd International Symposium on Materials and Energy from Refuses, Antwerp, Belgium, 18–20 March 1986.
- Mackie, R.; Smith, D. Guidebook for Organic Synthesis (Russian translation); Mir: Moscow, Russia, 1985. [Google Scholar]
- Manion, J.A.; Louw, R. Gas-phase hydrogenolysis of chloroethene: rates, products, and computer modelling. J. Chem. Soc. Perkin Trans. 2 1988, 2, 1547. [Google Scholar] [CrossRef]
- Manion, J.A.; Louw, R. The gas-phase thermolyses of di-, tri- and tetrachloroethene in hydrogen between 828 and 1050 K. Rec. Trav. Chim. Pays-Bas. 1989, 108, 235–241. [Google Scholar] [CrossRef]
- Kondratiev, V.N. Rate Constants of Gas Phase Reactions; Nauka: Moscow, Russia, 1970. (In Russian) [Google Scholar]
- Weiss, A.H.; Krieger, K.A. Hydrodechlorination kinetics and reaction mechanisms. J. Catal. 1969, 6, 167–185. [Google Scholar] [CrossRef]
- Louw, R.; Mulder, P. Man and His Ecosystem. In Proceedings of the 8th World Clean Air Congress, The Hague, The Netherlands, 11–15 September 1989. [Google Scholar]
- Johnson, R.W.; Hilfman, L. Non-catalytic process for the conversion of a hydrocarbonaceous stream containing halogenated organic compounds. US Patent No. 4840722, 1 April 1990. [Google Scholar]
- Moreau, C.; Durand, R.; Geneste, P. Chemical evidence for the existence of two types of catalytic sites for hydroprocessing of substituted benzenes over NiW(Mo)/γ-A12O3. Am. Chem. Soc. Div. Pet. Chem. Preprint. Amer. 1987, 32, 298–307. [Google Scholar]
- Coq, B.; Tijani, A.; Figureueras, F. Pt/γ-Al2O3 catalytic membranes vs. Pt on γ-Al3 powders in the selective hydrogenation of p-chloronitrobenzene. Mol. Catal. 1991, 2, 331–338. [Google Scholar] [CrossRef]
- Coq, B.; Hub, S.; Figueras, F.; Tournigant, D. Conversion under hydrogen of dichlorodifluoromethane over bimetallic palladium catalysts. Appl. Catal. A Gen. 1993, 101, 41–50. [Google Scholar] [CrossRef]
- Mishchenko, A.P.; Senina, E.V. Investigation of carbon tetrachloride transformations on membrane catalysts. Russ. Chem. Bull. 1987, 7, 1664–1666. (In Russian) [Google Scholar]
- Ohnishi, R.; Wang, W.-L.; Ichikawa, M. Selective hydrodechlorination of CFC-113 on Bi- and Tl-modified palladium catalysts. Appl. Catal. A: Gen. 1994, 113, 29–41. [Google Scholar] [CrossRef]
- Heisler, M.; Gabler, W.; Pichl, E.; Strasser, R. Process for the preparation of trichloroethylene. Pat. DE No. 3804265, 11 February 1988. [Google Scholar]
- Weiss, A.; Krieger, K. Hydrodechlorination kinetics and reaction mechanisms. J. Catal. 1966, 6, 167–185. [Google Scholar] [CrossRef]
- Zanaveskin, L.N. Catalytic Hydrogenolysis of Chloroethanes and Chloroethylenes; GOSNIIHLORPROEKT: Moscow, Russia, 1987. (In Russian) [Google Scholar]
- Zaidman, O.A.; Verkhutova, E.I.; Zanaveskin, L.N.; Treger, Y.A. Substitution catalytic hydrogenation of C2 chlorine derivatives. Russ. Chem. Ind. 1991, 9, 5–10. (In Russian) [Google Scholar]
- Dasaeva, G.S.; Flid, M.R.; Dmitriev, Y.K.; Kartashov, L.M.; Treger, Y.A. Catalytic hydrodechlorination of 1,2-dichloroethane. Russ. Chem. Ind. 2000, 3, 49–54. (In Russian) [Google Scholar]
- Dasaeva, G.S.; Flid, M.R.; Dmitriev, Y.K.; Kartashov, L.M.; Treger, Y.A. Catalytic hydrodechlorination of 1,1,2-trichloroethane. Russ. Chem. Ind. 2000, 4, 44–46. (In Russian) [Google Scholar]
- Ordonez, S.; Diez, F.V.; Sastre, H. Catalytic Hydrodechlorination of Chlorinated Olefins over a Pd/Al2O3 Catalyst: Kinetics and Inhibition Phenomena. Ind. Ing. Chem. Res. 2002, 41, 505–511. [Google Scholar] [CrossRef]
- Gambhir, B.S.; Weiss, A.H. Depletion of reactants on a catalytic surface during reaction. J. Catal. 1972, 26, 82–91. [Google Scholar] [CrossRef]
- Ito, L.N.; Murchion, C.B. Processes for converting chlorinated alkenes to useful, less chlorinated alkenes. US Patent No. 5476979, 14 April 1994. [Google Scholar]
- Heinrichs, B.; Delhez, P. Palladium–silver sol–gel catalysts for selective hydrodechlorination of 1,2-dichloroethane into ethylene: 1 synthesis and characterization. J. Catal. 1997, 172, 322–335. [Google Scholar] [CrossRef]
- James, R.B.; Kalnes, T.N. Process for the simultaneous hydroconversion of a first feedstock comprising unsaturated, halogenated organic compounds and a second feedstock comprising saturated, halogenated organic compounds. US Patent No. 4895995, 2 December 1988. [Google Scholar]
- Bozzelli, J.W.; Chen, Y.M.; Chuang, S.S.C. Catalytic hydrodechlorination of 1,2-dichloroethane and trichloroethylene over Rh/SiO2 catalysts. Chem. Eng. Commun. 1992, 115, 1–11. [Google Scholar] [CrossRef]
- Scharfe, G.; Wilhelms, R.-E. Catalytic conversion of hydrocarbon chlorides to hydrogen chloride and hydrocarbons. US Patent No. 3892818, 7 December 1972. [Google Scholar]
- Shapovalov, V.V.; Ivanov, A.Y. Catalytic hydrogenation of dichloroethane. J. Appl. Chem. 1996, 69, 513–514. (In Russian) [Google Scholar]
- Mishakov, I.V.; Chesnokov, V.V.; Buyanov, R.A.; Pakhomov, N.A. Regularities of chlorine organic car bons decomposition flatiron group metals. Kinet. Katal. 2001, 42, 598–603. (In Russian) [Google Scholar] [CrossRef]
- Shapovalov, V.V.; Ivanov, A.Y. Features of the catalytic dechlorination of 1,2-dichloroethane on nickel. Russ. J. Phys. Chem. 1998, 72, 1326–1327. (In Russian) [Google Scholar]
- Timmons, R.B.; Jang, W.L.; He, Y.; Houpt, D.J. Catalytic hydrodehalogenation of polyhalogenated hydrocarbons. US Patent No. 5276240, 16 October 1992. [Google Scholar]
- Ordóñez, S.; Sastre, H.; DíezF, V. Hydrodechlorination of aliphatic organochlorinated compounds over commercial hydrogenation catalysts. Appl. Catal. B: Environ. 2000, 25, 49–58. [Google Scholar] [CrossRef]
- Martino, M.; Rosal, R.; Sastre, H.; DíezF, V. Hydrodechlorination of dichloromethane, trichloroethane, trichloroethylene and tetrachloroethylene over a sulfided Ni/Mo–γ-alumina catalyst. Appl. Catal. B Environ. 1999, 20, 301–307. [Google Scholar] [CrossRef]
- Dasaeva, G.S.; Flid, M.R.; Kartashov, L.M.; Treger, Y.A. Hydrodechlorination of trichlorosubstituted C2 hydrocarbons on nickel catalyst. Catal. Ind. 2002, 5, 24–29. (In Russian) [Google Scholar]
- Kim, D.I.; Allen, D.T. Catalytic Hydroprocessing of Chlorinated Olefins. Ind. Eng. Chem. Res. 1997, 36, 3019–3026. [Google Scholar] [CrossRef]
- Weiss, A.H.; Valinski, S.; Antoshin, G.V. Hydrodechlorination and oligomerization of carbon tetrachloride over nickel Y zeolites. J. Catal. 1982, 74, 136–143. [Google Scholar] [CrossRef]
- Meyer, R.J.; Kim, D.I.; Allen, D.T.; Jo, J. Catalytic hydrodechlorination of 1,3-dichloropropene. Chem. Eng. Sci. 1999, 54, 3627–3634. [Google Scholar] [CrossRef]
- Campbell, J.S.; Kemball, C. Catalytic fission of the carbon-halogen bond. Part 1.—Reactions of ethyl chloride and ethyl bromide with hydrogen on evaporated metal films. Trans. Faraday Soc. 1961, 57, 809. [Google Scholar] [CrossRef]
- Gomez-Sainero, L.M.; Cortés, A.; Seoane, X.L.; Arcoya, A. Hydrodechlorination of Carbon Tetrachloride to Chloroform in the Liquid Phase with Metal-Supported Catalysts. Effect of the Catalyst Components. Ind. Eng. Chem. Res. 2000, 39, 2849–2854. [Google Scholar] [CrossRef]
- Gómez-Sainero, L.M.; Seoane, X.L.; Fierro, J.L.; Arcoya, A. Liquid-Phase Hydrodechlorination of CCl4 to CHCl3 on Pd/Carbon Catalysts: Nature and Role of Pd Active Species. J. Catal. 2002, 209, 279–288. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, F.; Liao, S.; Yu, D.; Sun, N. Active catalyst for the hydrodechlorination of perchlorobenzene. React. Funct. Polym. 2000, 44, 65–69. [Google Scholar] [CrossRef]
- Kulkarni, P.P.; Kovalchuk, V.I.; D’Itri, J.L. Oligomerization pathways of dichlorodifluoromethane hydrodechlorination catalyzed by activated carbon supported Pt-Cu, Pt-Ag, Pt-Fe, and Pt-Co. Appl. Catal. B: Environ. 2002, 36, 299–309. [Google Scholar] [CrossRef]
- Golubina, E.V.; Lokteva, E.S.; Lunin, V.V.; Telegina, N.S.; Stakheev, A.Y.; Tundo, P. The role of Fe addition on the activity of Pd-containing catalysts in multiphase hydrodechorination. Appl. Catal. A. Gen. 2006, 302, 32–41. [Google Scholar] [CrossRef]
- Wang, C.-B.; Lin, H.-K.; Ho, C.-M. Effects of the addition of titania on the thermal characterization of alumina-supported palladium. J. Mol. Catal. A: Chem. 2002, 180, 285–291. [Google Scholar] [CrossRef]
- Gregori, M.; Benito, P.; Fornasari, G.; Migani, M.; Millefanti, S.; Ospitali, F.; Albonetti, S. Preparation of Pd/Cu MCM-41 catalysts for hydrodechlorination: Influence of the synthesis procedure. Microporous Mesoporous Mater. 2014, 190, 1–9. [Google Scholar] [CrossRef]
- Turakulova, A.O.; Golubina, E.V.; Lokteva, E.S.; Korotkov, A.V.; Lunin, V.V. ZrO2-Al2O3 binary oxides as promising supports for palladium catalysts of hydrodechorination. Russ. J. Phys. Chem. A. 2011, 85, 402–407. (In Russian) [Google Scholar] [CrossRef]
- Dafinger, W.; Schmidhammer, L. Process for the hydrogenation of chloromethanes. EP. Patent No. 0523553, 15 July 1991. [Google Scholar]
- Ohnishi, R.; Suzuki, H.; Wang, W.-L.; Ichikawa, M. Promoting role of metal additives in modified Pd catalysts for selective hydrodechlorination of CFC-113. Prep. Catal. V - Sci. Bases Prep. Heterog. Catal. Proc. Fifth Int. Symp. 1993, 77, 429–432. [Google Scholar]
- Lingaiah, N.; Uddin, A.; Muto, A.; Sakata, Y. Hydrodechlorination of chlorinated hydrocarbons over metal–carbon composite catalysts prepared by a modified carbothermal reduction method. Chem. Commun. 1999, 1657–1658. [Google Scholar] [CrossRef]
- Bonarowska, M.; Pielaszek, J.; Semikolenov, V.A. Pd-Au/Sibunit carbon catalysts: characterization and catalytic activity in hydrodechorination of dichlorodifluoromethane (CFC-12). J Catal. 2002, 209, 528–538. [Google Scholar] [CrossRef]
- Yuan, G.; Louis, C.; Delannoy, L.; Keane, M.A. Silica- and titania-supported Ni–Au: Application in catalytic hydrodechlorination. J. Catal. 2007, 247, 256–268. [Google Scholar] [CrossRef]
- Bonarowska, M.; Machynskyy, O.; Łomot, D.; Kemnitz, E.; Karpinski, Z. Supported palladium–copper catalysts: Preparation and catalytic behavior in hydrogen-related reactions. Catal. Today 2014, 235, 144–151. [Google Scholar] [CrossRef]
- Pirard, S.L.; Mahy, J.G.; Pirard, J.-P.; Heinrichs, B.; Raskinet, L.; Lambert, S.D. Development by the sol–gel process of highly dispersed Ni–Cu/SiO2 xerogel catalysts for selective 1,2-dichloroethane hydrodechlorination into ethylene. Microporous Mesoporous Mater. 2015, 209, 197–207. [Google Scholar] [CrossRef]
- Baran, R.; Śrębowata, A.; Casale, S.; Łomot, D.; Dzwigaj, S. Hydrodechlorination of 1,2-dichloroethane on nickel loaded Beta zeolite modified by copper: Influence of nickel and copper state on product selectivity. Catal. Today 2014, 226, 134–140. [Google Scholar] [CrossRef]
- Baddeley, C.; Bloxham, L.; Laroze, S.; Raval, R.; Noakes, T.; Bailey, P. The dynamic catalytic surface: probing bimetallic active sites with medium energy ion scattering. Surf. Sci. 1999, 433, 827–832. [Google Scholar] [CrossRef]
- Setterfild, C. The Practical Course of Heterogeneous Catalysis (Russian Translation); Mir: Moscow, Russia, 1984. [Google Scholar]
- Śrębowata, A.; Juszczyk, W.; Kaszkur, Z.; Sobczak, J.W.; Kępiński, L.; Karpiński, Z. Hydrodechlorination of 1,2-dichloroethane and dichlorodifluoromethane over Ni/C catalysts: The effect of catalyst carbiding. Appl. Catal. A Gen. 2007, 319, 181–192. [Google Scholar] [CrossRef]
- Díaz, E.; Faba, L.; Ordóñez, S. Effect of carbonaceous supports on the Pd-catalyzed aqueous-phase trichloroethylene hydrodechlorination. Appl. Catal. B Environ. 2011, 104, 415–417. [Google Scholar] [CrossRef]
- Bae, J.W.; Kim, I.G.; Lee, J.S.; Lee, K.H.; Jang, E.J. Hydrodechlorination of CCl4 over Pt/Al2O3: Effects of platinum particle size on product distribution. Appl. Catal. A Gen. 2003, 240, 129–142. [Google Scholar] [CrossRef]
- Amorim, C.; Yuan, G.; Patterson, P.; Keane, M. Catalytic hydrodechlorination over Pd supported on amorphous and structured carbon. J. Catal. 2005, 234, 268–281. [Google Scholar] [CrossRef]
- Cuenya, B.R. Synthesis and catalytic properties of metal nanoparticles: size, shape support, composition, and oxidation state effects. Thin Solid Film. 2010, 518, 3127–3150. [Google Scholar] [CrossRef]
- Telas, E.; Margitfalvi, J.L.; Nedelus, M.; Godolos, S.; Ruckenbauer, A. Presented at the 6-th International Symposium on Relations between Homogeneous and Heterogeneous Catalysis, Pisa, Italy, 25–29 September 1989; p. 6959.
- Kavalerskaya, N.E.; Lokteva, E.S.; Rostovshchikova, T.N. Hydrodechlorination of chlorobenzene in the presence of Ni/Al2O3 prepared by laser electrodispersion and from a colloidal dispersion. Kinet. Catal. 2013, 54, 597–606. (In Russian) [Google Scholar] [CrossRef]
- Kulkarni, P.P.; Deshmukh, S.S.; Kovalchuk, V.I.; D’Itri, J.L. Hydrodechlorination of dichlorodifluoromethane on carbon-supported Group VIII noble metal catalysts. Catal. Lett. 1999, 61, 161–166. [Google Scholar] [CrossRef]
- Ordonez, S.; Diez, F.V.; Sastre, H. Characterization of the deactivation of platinum and palladium supported on activated carbon used as hydrodechlorination catalysts. Appl. Catal. B Environ. 2001, 31, 113–122. [Google Scholar] [CrossRef]
- Yoneda, T.; Takido, T.; Konuma, K. Hydrodechlorination reactivity of para-substituted chlorobenzenes over Pt/C catalyst. J. Mol. Catal. A Chem. 2006, 256, 80–89. [Google Scholar]
- Keane, M.A. Hydrodehalogenation of haloarenes over Silica supported Pd and Ni: a consideration of catalytic activity/selectivity and haloarene reactivity. Appl. Catal. A Gen. 2004, 271, 109–118. [Google Scholar] [CrossRef]
- Shin, E.-J.; Keane, M.A. Catalytic Hydrogen Treatment of Aromatic Alcohols. J. Catal. 1998, 173, 450–459. [Google Scholar] [CrossRef]
- Bae, J.W.; Park, E.D.; Lee, J.S. Hydrodechorination of CCl4 over Pt/Al2O3: Effects of reaction pressure and diluetnt gases on distribution of productsand catalyst stability. Appl. Catal. A Gen. 2001, 217, 79–89. [Google Scholar] [CrossRef]
- Diaz, E.; Mohedano, A.; Casas, J.A.; Calvo, L.; Gilarranz, M.; Rodriguez, J. Deactivation of a Pd/AC catalyst in the hydrodechlorination of chlorinated herbicides. Catal. Today 2015, 241, 86–91. [Google Scholar] [CrossRef]
- Campbell, J.S.; Kemball, C. Catalytic fission of the carbon-halogen bond. Part 2. Reactions of tertiary butyl chloride with hydrogen and deuterium on evaporated platinum and palladium films. Trans. Faraday Soc. 1963, 59, 2583. [Google Scholar] [CrossRef]
- Treger, Y.A.; Rozanov, V.N. Obtaining of organochlorine compounds based on monocarbon molecules. Russ. Chem. Rev. 1989, 58, 138–160. (In Russian) [Google Scholar] [CrossRef]
- Weiss, A.H.; Gambhir, B.S.; Leon, R.B. Hydrodechlorination of carbon tetrachloride. J. Catal. 1971, 22, 245–254. [Google Scholar] [CrossRef]
- Noelke, C.J.; Rase, H.F. Improved Hydrodechlorination Catalysis: Chloroform over Platinum-Alumina with Special Treatments. Ind. Eng. Chem. Prod. Res. Dev. 1979, 18, 325–328. [Google Scholar] [CrossRef]
- Golubina, E.V.; Lokteva, E.S.; Lunin, V.V.; O Turakulova, A.; I Simagina, V.; Stoyanova, I.V. Modification of the supported palladium catalysts surface during hydrodechlorination of carbon tetrachloride. Appl. Catal. A Gen. 2003, 241, 123–132. [Google Scholar] [CrossRef]
- Bonarowska, M.; Lin, K.-N.; Legawiec-Jarzyna, M. Multi-Wall Carbon Nanotubes as a Support for Platinum Catalysts for the Hydrodechlorination of Carbon Tetrachloride and Dichlorodifluoromethane. Solid State Phenom. 2007, 128, 261–271. [Google Scholar] [CrossRef]
- Santo, V.D.; Dossi, C.; Recchia, S.; Colavita, P.; Vlaic, G.; Psaro, R. Carbon tetrachloride hydrodechlorination with organometallics-based platinum and palladium catalysts on MgO. J. Mol. Catal. A Chem. 2002, 182, 157–166. [Google Scholar] [CrossRef]
- Rossi, M.; Rubini, C.; Pasquale, A.; Cavalli, L. Catalyst for hydrodechlorination of carbon tetrachloride to chloroform. RF Patent No. 2268773, 30 October 2001. [Google Scholar]
- Avetisov, A.K.; Tarasova, D.V.; Gelperin, E.I. Method for vapor phase hydrodechlorination of carbon tetrachloride. RF Patent No. 2125034, 27 November 1995. [Google Scholar]
- Kartashov, L.M.; Rozanov, V.N.; Treger, Y.A.; Flid, M.R.; Kalyuzhnaya, T.L.; Tkach, D.V. Processing the wastes from the production of methyl chloride in the synthesis of olefins from natural gas. Catal. Ind. 2010, 2, 230–238. [Google Scholar] [CrossRef]
- Treger, Y.A.; Rozanov, V.N.; Kartashov, L.M.; Flid, M.R. An integrated method for catalytic processing of natural gas to produce lower olefins. RF Patent No. 2451005, 30 May 2011. [Google Scholar]
- Hagh, B.F.; Allen, D.T. Catalytic hydroprocessing of chlorobenzene and 1,2-dichlorobenzene. AIChE J. 1990, 36, 773–778. [Google Scholar] [CrossRef]
- Tijani, A.; Coq, B.; Figueras, F. Hydrogenation ofpara-chloronitrobenzene over supported ruthenium-based catalysts. Appl. Catal. 1991, 76, 255–266. [Google Scholar] [CrossRef]
- Anderson, J.; McConkey, B. Reactions of methyl chloride and of methylene chloride at metal surfaces II. Reactions over evaporated films of titanium and other metals. J. Catal. 1968, 11, 54–70. [Google Scholar] [CrossRef]
- Coq, B.; Figureueras, F. Presented at the VIII Soviet-French Seminar on Catalysis, Novosibirsk, Russia, 18–21 June 1990; p. 218.
- Gurvich, L.V.; Karachevtsev, G.V.; Kondratyev, V.N. Energies of Chemical Bond Breaking. Ionization Potentials and Electron Affinity; Nauka: Moscow, Russia, 1974. (In Russian) [Google Scholar]
- Lokteva, E.S.; Simagina, V.I.; Golubina, E.V.; Stoyanova, I.V.; Lunin, V.V. Formation of C1-C5 Hydrocarbons from CCl4 in the Presence of Carbon-Supported Palladium Catalysts. Kinet. Catal. 2000, 41, 855–860. (In Russian) [Google Scholar] [CrossRef]
- Van Barneveld, W.; Ponec, V. On the apparent controversy regarding the effect of alloying on the selectivity of the Fischer-Tropsch synthesis. J. Catal. 1984, 89, 542–544. [Google Scholar] [CrossRef]
- Hagh, B.F.; Allen, D.T. Catalytic hydroprocessing of chlorinated benzenes. Chem. Eng. Sci. 1990, 45, 2695–2701. [Google Scholar] [CrossRef]
- Mishakov, I.V. Development of catalytic methods for the disposal of chlorohydrocarbons and their mixtures. Dis. Cand. Chem. 2002, 180. (In Russian) [Google Scholar]
- Kim, S.Y.; Choi, H.C.; Yanga, O.B.; Lee, K.H.; Lee, J.S.; Kim, Y.G. Hydrodechlorination of tetrachloromethane over supported Pt catalysts. J. Chem. Soc. Chem. Commun. 1995, 21, 2169. [Google Scholar] [CrossRef] [Green Version]
- Wiersma, A.; Van De Sandt, E.; Makkee, M.; Luteijn, C.; Van Bekkum, H.; Moulijn, J. Process for the selective hydrogenolysis of CCl2F2 (CFC-12) into CH2F2 (HFC-32). Catal. Today 1996, 27, 257–264. [Google Scholar] [CrossRef]
- Choi, H.C.; Choi, S.H.; Yang, O.B.; Lee, J.S.; Lee, K.H.; Kim, Y.G. Hydrodechlorination of Carbon Tetrachloride over Pt/MgO. J. Catal. 1996, 161, 790–797. [Google Scholar] [CrossRef]
- Bucsi, I.; Beard, B.C. Selective monochlorination of methane over solid acid and zeolite catalysts. Catal. Lett. 1992, 16, 27–38. [Google Scholar] [CrossRef]
- Dasaeva, G.S.; Velichko, S.M.; Treger, Y.A.; Moiseev, I.I. Hydrodechlorination of CCl4 in the presence of Pd(OAc)2. Kinet. Catal. 1990, 31, 858–862. (In Russian) [Google Scholar]
- Dasaeva, G.S.; Treger, Y.A.; Moiseev, I.I.; Zanaveskin, L.N. Liquid phase catalytic hydrodechlorination of carbon tetrachloride. Russ. Chem. Ind. 1996, 6, 346–350. (In Russian) [Google Scholar]
- Zhang, Z. Treatment to improve the durability of a hydrodechlorination catalyst and catalyst. World Patent WO1999017876B, 10 February 1997. [Google Scholar]
- Prati, L. Reductive catalytic dehalogenation of light chlorocarbons. Appl. Catal. B Environ. 1999, 23, 135–142. [Google Scholar] [CrossRef]
- Kellner, C.S.; Lerou, J.J.; Rao, V.N.M.; Wuttke, K.G. Regeneration or activation of noble metal catalysts using fluorohalocarbons or fluorohalohydrocarbons. US Patent No. 4980324, 25 December 1990. [Google Scholar]
- Kellner, C.S. Regeneration of noble metal catalysts used in hydrodehalogenation of halogen-substituted hydrocarbons containing fluorine and at least one other halogenus. US Patent No. 5057470, 22 May 1990. [Google Scholar]
- Holbrook, M.T.; Harley, A.D. Vapor phase hydrogenation of carbon tetrachlorideus. US Patent No. 5105032, 4 October 1990. [Google Scholar]
- Bozon, A.; Lakatos, E.; Koberstein, E.; Pletka, H.-D.; Voelker, H. Process for application of a catalytically active coating containing platinum, palladium or rhodium or their mixtures to a carrier and product made by said process. US Patent No. 4374047, 15 February 1983. [Google Scholar]
- Novak, M. and Zdrazil, M. Effects of Sulfidation and Synergism in Hydrodechlorination of o-Dichlorobenzene Over NiMo/Alumina Catalyst. Bull. Soc. Chim. Belg. 1993, 102, 271–279. [Google Scholar] [CrossRef]
- Mullin, C.R.; Wymore, C.E. Hydrogenolysis of carbon tetrachloride and chloroform. US Patent No. 3579596, 29 March 1968. [Google Scholar]
- Kincey, P.M.B. 2-Aminopurine preparation process. EP Patent No. 0355986, 18 July 1990. [Google Scholar]
- Hoke, J.B.; Gramiccioni, G.A.; Balko, E.N. Catalytic hydrodechlorination of chlorophenols. Appl. Catal. B: Environ. 1992, 1, 285–296. [Google Scholar] [CrossRef]
- Simakova, I.L.; Semikolenov, V.A. Study of the principles of liquid-phase hydrodechlorination of organochlorine compounds on a Pd/C catalyst. Kinet. Catal. 1991, 32, 989–993. (In Russian) [Google Scholar]
- Sharf, V.Z.; Gurovets, A.S.; Slinyakova, I.B. Investigation of the catalytic activity of metal complexes fixed on a solid support. Message I. Hydrogenolysis of chlorobenzenes in the presence of palladium chloride complex deposited on modified silica gel. Russ. Chem. Bull. 1980, 1, 114–117. (In Russian) [Google Scholar]
- Kartashov, L.M.; Flid, M.R.; Treger, Y.A. Stability of catalytic systems in hydrodechlorination of chlorohydrocarbons and chlorocarbons C1–C6. In Proceedings of the 10th International Symposium on Catalyst Deactivation, Berlin, Germany, 5–8 February 2006. [Google Scholar]
- Heinrichs, B.; Schoebrechts, J.-P.; Pirard, J.-P. Palladium–Silver Sol–Gel Catalysts for Selective Hydrodechlorination of 1,2-Dichloroethane into Ethylene. J. Catal. 2001, 200, 309–320. [Google Scholar] [CrossRef]
- Job, N.; Heinrichs, B.; Ferauche, F.; Noville, F.; Marien, J.; Pirard, J.-P. Hydrodechlorination of 1,2-dichloroethane on Pd–Ag catalysts supported on tailored texture carbon xerogels. Catal. Today 2005, 102, 234–241. [Google Scholar] [CrossRef]
- Kiperman, S.L. Introduction to Kinetics of Heterogeneous Catalytic Reactions; Nauka: Moscow, Ruissia, 1964. (In Russian) [Google Scholar]
- Śrębowata, A.; Juszczyk, W.; Kaszkur, Z.; Karpiński, Z. Hydrodechlorination of 1,2-dichloroethane on active carbon supported palladium–nickel catalysts. Catal. Today 2007, 124, 28–35. [Google Scholar] [CrossRef]
- Baran, R.; Kamińska, I.; Śrębowata, A.; Dzwigaj, S. Selective hydrodechlorination of 1,2-dichloroethane on NiSiBEA zeolite catalyst: Influence of the preparation procedure on a high dispersion of Ni centers. Microporous Mesoporous Mater. 2013, 169, 120–127. [Google Scholar] [CrossRef]
- Śrębowata, A.; Baran, R.; Łomot, D.; Lisovytskiy, D.; Onfroy, T.; Dzwigaj, S. Remarkable effect of postsynthesis preparation procedures on catalytic properties of Ni-loaded BEA zeolites in hydrodechlorination of 1,2-dichloroethane. Appl. Catal. B Environ. 2014, 147, 208–220. [Google Scholar] [CrossRef]
- Kaminska, I.I.; Srebowata, A. Active carbon-supported nickel-palladium catalysts for hydrodechlorination of 1,2-dichloroethane and 1,1,2-trichloroethane. Rec. Chem. Intermed. 2015, 41, 2007. [Google Scholar]
- Kalnes, T.N.; James, R.B. Hydrogenation and recycle of organic waste streams. Environ. Prog. 1988, 7, 185–191. [Google Scholar] [CrossRef]
- Huang, B.; Lei, C.; Wei, C.; Zeng, G. Chlorinated volatile organic compounds (Cl-VOCs) in environment — sources, potential human health impacts, and current remediation technologies. Environ. Int. 2014, 71, 118–138. [Google Scholar] [CrossRef]
- Wang, X.; Chen, C.; Chang, Y.; Liu, H. Dechlorination of chlorinated methanes by Pd/Fe bimetallic nanoparticles. J. Hazard. Mater. 2009, 161, 815–823. [Google Scholar] [CrossRef]
- Zhang, W.-H.; Quan, X.; Zhang, Z.-Y. Catalytic reductive dechlorination of p-chlorophenol in water using Ni/Fe nanoscale particles. J. Environ. Sci. 2007, 19, 362–366. [Google Scholar] [CrossRef]
- Zhou, T.; Li, Y.; Lim, T.-T. Catalytic hydrodechlorination of chlorophenols by Pd/Fe nanoparticles: Comparisons with other bimetallic systems, kinetics and mechanism. Sep. Purif. Technol. 2010, 76, 206–214. [Google Scholar] [CrossRef]
- Trujillo-Reyes, J.; Peralta-Videa, J.; Gardea-Torresdey, J. Supported and unsupported nanomaterials for water and soil remediation: Are they a useful solution for worldwide pollution? J. Hazard. Mater. 2014, 280, 487–503. [Google Scholar] [CrossRef]
- Huang, C.-C.; Lo, S.-L.; Tsai, S.-M.; Lien, H.-L. Catalytic hydrodechlorination of 1,2-dichloroethane using copper nanoparticles under reduction conditions of sodium borohydride. J. Environ. Monit. 2011, 13, 2406. [Google Scholar] [CrossRef]
- Roginskaya, E.V. Lithium aluminum hydride and its use in organic chemistry. Russ. Chem. Rev. 1952, 21, 3–39. (In Russian) [Google Scholar]
- Brown, H.S. Organic Reactions; IL: Moscow, Russia, 1952. [Google Scholar]
- Gaylord, N.C. Reduction with complex metal hydrides. J. Chem. Educ. 1956, 34, 367. [Google Scholar] [CrossRef]
- Haszeldine, R.N.; Osborne, J.E. 13. Addition of free radicals to unsaturated systems. Part XII. Free-radical and electrophilic attack on fluoro-olefins. J. Chem. Soc. 1956, 61. [Google Scholar] [CrossRef]
- Brown, H.C.; Bell, H.M. Alkali-Catalyzed Dealkilation of Two Diastereometric Benzyl Boranes. J. Org. Chem. 1962, 27, 1928. [Google Scholar]
- Eliel, E.L.; Herrmann, C.; Traxler, J.T. The Mechanism of Halide Reductions with Lithium Aluminum Hydride. III. Reduction of α-Chloro Acids and Esters. J. Am. Chem. Soc. 1956, 78, 1193–1198. [Google Scholar] [CrossRef]
- Jacobs, T.L.; Wilcox, R.D. Dehalogenation of Propargyl and Allenyl Halides. II. J. Am. Chem. Soc. 1964, 86, 2240–2247. [Google Scholar] [CrossRef]
- Johnson, J.E.; Blizzard, R.H.; Carhart, H.W. Hydrogenolysis of Alkyl Halides by Lithium Aluminum Hydride1. J. Am. Chem. Soc. 1948, 70, 3664–3665. [Google Scholar] [CrossRef]
- Bell, H.M.; Brown, H.C. Selective Reductions. XI. The Reaction of Sodium Borohydride with Alkyl Halides under Solvolytic Conditions. Borohydride as a Convenient Trap for Carbonium Ions1,2. J. Am. Chem. Soc. 1966, 88, 1473–1477. [Google Scholar] [CrossRef]
- Bell, H.M.; Vanderslice, C.W.; Spehar, A. Reduction of organic halogen compounds by sodium borohydride. J. Org. Chem. 1969, 34, 3923–3926. [Google Scholar] [CrossRef]
- Hutchins, R.O.; Hoke, D.; Keogh, J.; Koharski, D. Sodium borohydride in dimethyl sulfoxide or sulfolane. Convenient systems for selective reductions of primary, secondary and certain tertiary halides and tosylates. Tetrahedron Lett. 1969, 10, 3495–3498. [Google Scholar] [CrossRef]
- Jacobus, J. The mechanism of the reduction of alkyl halides with sodium borohydride in dimethyl sulphoxide. Chem. Comm. 1970, 6, 338. [Google Scholar]
- Simagina, V.I.; Stojanova, I.V.; Gentsler, A.G.; Tajban, E.S. Catalyst and method for hydrodechlorination of chloroaromatic compounds. RF Patent No. 2214864, 22 July 2002. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Heat of Formation ∆H298, kJ/mol | Thermal Effect of the Reaction, kJ/mol | ||
|---|---|---|---|---|
| to Ethylene | to Ethane | to Methane and Benzene | ||
| Chloroform | −100.3 | - | - | +251.6 (to methane) |
| Tetrachloromethane | −105.3 | - | - | +339.0 (to methane) |
| Chlorethyl | −105.3 | −64.8 | +71.9 | - |
| Vinyl chloride | +155.5 | +77.3 | +214.0 | - |
| Vinylidene chloride | +1.3 | +133.8 | +270.4 | - |
| 1,1-dichloroethane | −130.0 | +2.9 | +139.6 | - |
| 1,2-dichloroethane | −130.0 | +2.9 | +139.6 | - |
| 1,1,2-trichloroethane | −138.4 | +86.5 | +223.2 | - |
| Perchloroethene | −15.0 | +302.2 | +438.9 | - |
| 1,1,2,2-tetrachloroethane | −152.6 | +164.7 | +301.4 | - |
| Chlorobutanes and Chlorobutenes | −191.4 | +26.8 | +27.2 | - |
| Chlorobenzene | −111.6 | - | - | +102.0 (to benzene) |
| Methane | −74.8 | - | - | - |
| Ethylene | +52.3 | - | - | - |
| Ethane | −84.4 | - | - | - |
| Butene | −125.4 | - | - | |
| Butane | −126.2 | - | - | - |
| Benzene | +82.8 | - | - | - |
| HCl | −92.4 | - | - | - |
| Component | Temperature, °C | Residence Time, s | Conversion, % | Reference |
|---|---|---|---|---|
| CH3Cl | 604–840 | 11 | 19.6–98.4 | [31] |
| CH2Cl2 | 650–720 | 11 | 12.0–88.0 | [31] |
| CHCl3 | 526–550 | 11 | 44.5–78.5 | [31] |
| CCl4 | 532–573 | 11 | 16.4–74.7 | [31] |
| CH2=CH–Cl | 660–800 | 11 | 43.2–80.0 | [31] |
| Cl–CH=CH–Cl | 610–750 | 6.2–8.0 | 50.0–96.0 | [21,30] |
| CCl2=CCl2 | 750 | 6 | 96 | [32] |
| C2H4Cl2 | 580 | 10 | 8 | [32] |
| C6H5Cl | 604–840 | 7.5–11.0 | 19.6–98.4 | [21,30,31] |
| C6H4Cl2 | 600–950 | 4.6–8.8 | 0.7–93.0 | [21,30] |
| Component | Eact (kJ/mol) | ∆Sact (kJ/mol K) | ∆Gact (kJ/mol) | ko (c−1•103) | logA |
|---|---|---|---|---|---|
| CH3Cl | 184.8 | –103.9 | 285.8 | 9 | 7.88 |
| CH2Cl2 | 237.9 | –37.7 | 274.4 | 40 | 11.35 |
| CHCl3 | 243.9 | 8.4 | 235.5 | 4790 | 13.74 |
| CCl4 | 256.8 | 23 | 234.6 | 5517 | 15.52 |
| CH2=CH–Cl | 244.7 | –30.6 | 274 | 41 | 11.72 |
| Cl–CH=CH–Cl | 90.9 | –181 | 267.3 | 96 | 3.86 |
| C6H5Cl | 200.7 | –113.1 | 285.8 | 10 | 7.40 |
| C6H4Cl2 | 146.7 | –146.2 | 288.7 | 7 | 5.68 |
| Noble Metal Systems | ||||
| Catalyst | Raw materials | Process conditions | Products | Source |
| Pd/Al2O3, Pd/SiO2 palladium content 0.5–4 wt % | 1,2-dichloroethane | 200–350 °C | ethane, ethylene | [46,47,48] |
| 1,1,2-trichloroethane | 200–350 °C | ethane, ethylene, vinyl chloride | [46,47,49] | |
| tetrachloroethane | 200-350 | ethane, ethylene, dichloroethenes | [46,49] | |
| Pt/Al2O3, Pd/Al2O3 | dichloroethenes | 90 °C 170–350 °C, 0,5 MPa | ethane, chloroethenes ethane | [45,50,51] |
| Pd-Cu-Sn/C | Chloralkenes, (perchloroethene) | 160–250 °C 0.2–2 MPa | less chlorinated alkenes | [52] |
| Pd-Ag | 1,2-dichloroethane | 250–350 °C | ethylene | [53] |
| Pd/Al2O3 | Trichloroethene, perchloroethene | 170–350 °C 0.5 MPa | ethane | [50] |
| Pd/Al2O3 | Unsaturated halogenated hydrocarbons, saturated halogenated hydrocarbons | 80 °C, 5 MPa 320 °C, 5 MPa | saturated hydrocarbons | [54] |
| Rh/SiO2 Rh/carrier with promotor: Pd, Pt, Ru, Fe, Co, etc. | 1,2-dichloroethane Trichloroethene Hydrocarbons C1–C30 (dichloropropane) | 90–280 °C 120 °C 180–250 °C | ethane, ethyl chloride ethane (propane, propene) | [55,56] [56] |
| Pd, Pt, Rh/carrier | Dichloropropane | 200–300 °C | propene, propane | [25] |
| Nickel Based Systems | ||||
| Catalyst | Raw materials | Process conditions | Products | Source |
| 9.6% Ni on pumice | 1,2-dichloroethane | 350–500 °C | ethane | [57] |
| Nickel aluminum | 1,2-dichloroethane | 400–550 °C | methane, ethane | [58] |
| Ni on fireclay | 1,2-dichloroethane | 350–400 °C | ethylene | [59] |
| Ni-Cr on C | 1,2-dichloroethane | 280-370 °C | ethylene, ethane, monochlorinated C2 | [48] |
| Ni-Cr on C | 1,1,2-trichloroethane | 280–370 °C | ethylene, ethane, vinyl chloride, dichloroethenes | [49] |
| Ni on ZSM-5/Al2O3 | Trichloroethene Trichloroethane | 250–450 °C | non-chlorinated olefins, paraffins, aromatic compounds | [60] |
| Ni-Mo/Al2O3 | 1,1,1-trichloroethane | 300 °C | ethane, vinyl chloride, dichloroethenes | [61] |
| Ni-Mo/γ-Al2O3 sulphurized | 1,1,1-trichloroethane, per- and trichloroethene | 250–350 °C | non-chlorinated hydrocarbons | [62] |
| Ni-Mo/Al2O3 | Trichloroethene | 300 °C | ethane, dichloroethenes | [61] |
| Ni-Cr on C | Trichloroethene | 300–350 °C | ethylene, vinyl chloride | [63] |
| Ni-Mo/Al2O3 | Perchloroethene Trichloroethene | 175–275 °C | dichloroethenes, non-chlorinated aliph. compounds | [64] |
| Ni-Mo/Al2O3 | Perchloroethene | 300 °C | ethane, trichloroethene, dichloroethenes, ethyl chloride | [61] |
| Ni on Y zeolites | tetrachloromethane | 370 | chlorinated and non-chlorinated alkanes | [65] |
| Ni-Mo/γ-Al2O3 | 1,3-dichloropropene | 325 °C | propene, propane, allyl chloride | [66] |
| Ni/Al2O3/SiO2 | 1,3-dichloropropene | 325 °C | propene, chloropropenes | [66] |
| Ni on H-ZSM-5/Al2O3 | 1,3-dichloropropene | 325 °C | propene, chloropropenes | [66] |
| Reaction | Catalyst | E, kJ/mol | Source |
|---|---|---|---|
| CH3Cl + H2 → CH4 + HCl | Ti film | 67.3 | [113] |
| CH2Cl2 + H2 → CH3Cl + HCl | Ti film | 56.0 | [113] |
| CH2Cl2 + 2H2 → CH4 + 2HCl | Ti film | 55.6 | [113] |
| (CH3)3CCl + H2 → (CH3)3CH + HCl | Pt film | 79.4 | [100] |
| (CH3)3CCl + H2 → (CH3)3CH + HCl | Pd film | 75.2 | [100] |
| C6H5Cl + H2 → C6H6 + HCl | Pd/Al2O3 | 104.5 | [114] |
| ClHC = CHCl + H2 → H2C = CHCl + HCl | Pt/Al2O3 | 115.0 | [45] |
| Temperature, °C | Molar Ratio, H2:DCE | Composition of the Reaction Products, % | DCE Conversion Degree, % | |||
|---|---|---|---|---|---|---|
| CH2CHCl | CH3CH2Cl | C2H6 | C2H4 | |||
| 200 | 1:1 | - | - | 100 | - | 11 |
| 250 | 1:1 | 1.4 | - | 90.9 | 7.7 | 40 |
| 300 | 1:1 | 5.5 | - | 79.3 | 15.2 | 47 |
| 350 | 1:1 | 1.8 | 12.0 | 71.0 | 15.2 | 55 |
| 350 | 2:1 | 0.75 | 8.4 | 83.9 | 7.0 | 45 |
| 350 | 1:1 | 1.8 | 12.0 | 71.0 | 15.2 | 24 |
| 350 | 1:2 | 8.0 | 8.0 | 48.2 | 35.9 | 10 |
| 350 | 1:4 | 10.0 | 5.3 | 37.1 | 47.6 | 5 |
| Temperature, °C | Molar Ratio, H2:DCE | Composition of the Reaction Products, % | 1,1,2-TCE Conversion Degree, % | ||||
|---|---|---|---|---|---|---|---|
| CH2=CHCl | CH3CH2Cl | CH2CLCH2Cl | C2H6 | C2H4 | |||
| 200 | 3:1 | - | 7.7 | 1.1 | 91.0 | 0.2 | 46 |
| 250 | 3:1 | - | 15.7 | 2.4 | 81.5 | 0.4 | 58 |
| 300 | 3:1 | 2.1 | 18.0 | 0.7 | 71.8 | 9.5 | 96 |
| 350 | 3:1 | 3.2 | 20.0 | - | 53.7 | 23.1 | 99 |
| 300 | 4:1 | 1.3 | 13.3 | - | 78.5 | 6.9 | 97 |
| 300 | 3:1 | 2.1 | 18.0 | 0.7 | 71.8 | 9.5 | 96 |
| 300 | 1:2 | 9.8 | 14.3 | 0.7 | 48.8 | 26.1 | 62 |
| 300 | 1:1 | 21.3 | 5.6 | 1.0 | 32.5 | 38.7 | 21 |
| Catalyst | Temperature, | Conversion, | Selectivity, mol% | |||
|---|---|---|---|---|---|---|
| °C | 1,2-DCE, % of Feed | C2H4 | C2H6 | C2H3Cl | C2H5Cl | |
| 0.25% Pd on Al2O3 | 280 | 58.7 | 41.9 | 52.4 | 2.0 | 3.7 |
| 350 | 83.0 | 62.9 | 28.6 | 7.6 | 0.9 | |
| 0.5% Pd on Al2O3 | 280 | 68.7 | 28.1 | 65.8 | 1.1 | 4.9 |
| 350 | 90.3 | 55.1 | 33.5 | 9.0 | 2.3 | |
| Catalyst | Temperature, | Conversion, | Selectivity, mol% | |||
|---|---|---|---|---|---|---|
| °C | 1,1,2-TCE, % of Feed | C2H4 | C2H6 | C2H3Cl | i-C2H2Cl2 | |
| 0.25% Pd on Al2O3 | 280 | 78.7 | 7.8 | 70.2 | 19.8 | 2.2 |
| 350 | 98.9 | 23.9 | 62.1 | 11.4 | 2.6 | |
| 0.5 % Pd on Al2O3 | 280 | 89.2 | 6.2 | 75.2 | 15.8 | 2.8 |
| 350 | 99.2 | 25.6 | 67.7 | 5.6 | 1.1 | |
| Metal Component of Catalyst | Temperature, °C | DCE Conversion, % | Selectivity, mol% | Yield of Ethylene, % | |||
|---|---|---|---|---|---|---|---|
| Ethane | Ethylene | Propylene | Vinyl Chloride | ||||
| Ni | 250 | 3.4 | - | 63.0 | - | 100.0 | 0.0 |
| 350 | 39.0 | - | 68.7 | - | 31.2 | 26.8 | |
| Cr | 250 | 3.3 | - | 39.7 | - | 60.3 | 1.3 |
| 350 | 83.7 | - | 24.0 | 3.1 | 72.9 | 20.0 | |
| Cu | 250 | 3.9 | - | – | - | 100.0 | 0.0 |
| 350 | 21.3 | - | 35.9 | - | 64.1 | 7.6 | |
| Mn | 250 | 2.1 | - | 33.3 | - | 66.7 | 0.7 |
| 350 | 50.8 | - | 2.3 | - | 97.7 | 1.2 | |
| Cd | 250 | 2.0 | - | 39.4 | - | 60.6 | 0.8 |
| 350 | 84.3 | - | 11.3 | 1.7 | 87.0 | 9.5 | |
| Mo | 250 | 1.6 | - | 17.1 | - | 82.9 | 0.3 |
| 350 | 69.3 | - | 45.5 | 4.3 | 50.2 | 31.5 | |
| Bi | 250 | 5.4 | - | 36.5 | - | 63.5 | 2.0 |
| 350 | 82.8 | - | 5.6 | 1.3 | 93.1 | 4.6 | |
| V | 250 | 1.4 | - | 45.4 | - | 54.6 | 0.6 |
| 350 | 68.0 | - | 30.7 | 5.3 | 64.0 | 20.9 | |
| The Content in the Catalyst, wt% | DCE Conversion, % | Selectivity, mol% | Yield of C2H4, mol% | ||||
|---|---|---|---|---|---|---|---|
| Ni | Mo | Cr | Cu | C2H4 | VC | ||
| 4.0 | - | - | - | 39.0 | 68.7 | 31.2 | 26.8 |
| 2.0 | 2.0 | - | - | 54.7 | 63.2 | 36.0 | 34.6 |
| - | - | 4.0 | - | 83.7 | 24.0 | 72.9 | 20.0 |
| - | 2.0 | 2.0 | - | 53.9 | 60.2 | 37.8 | 32.3 |
| 2.0 | - | 2.0 | - | 58.1 | 78.1 | 20.3 | 45.3 |
| - | - | 2.0 | 2.0 | 99.0 | 99.5 | 0.5 | 98.1 |
| Temp., °C | Fed H2, L/h | H2: DCE, mol | Residence Time, s | DCE Conversion, % | Selectivity, mol% | Yield of Ethylene, % | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ethane | Ethylene | Propane | Propylene | Vinyl Chloride | ||||||
| 250 | 0.7 | 2:1 | 8.9 | 2.1 | - | 72.2 | - | - | 27.8 | 1.5 |
| 350 | 0.7 | 2:1 | 7.5 | 54.7 | - | 63.2 | - | 0.8 | 36.0 | 34.6 |
| 350 | 1.4 | 4:1 | 6.5 | 77.2 | - | 90.2 | - | 0.9 | 8.9 | 69.6 |
| Temp, °C | Fed H2, L/h | H2: DCE, mol | Residence Time, s | DCE Conversion, % | Selectivity, mol% | Yield of Ethylene, % | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ethane | Ethylene | Propane | Propylene | Vinyl Chloride | ||||||
| 250 | 0.7 | 2:1 | 8.7 | 2.3 | - | 73.9 | - | 0.1 | 23.1 | 2.7 |
| 350 | 0.7 | 2:1 | 7.6 | 53.9 | - | 60.2 | - | 0.9 | 37.8 | 30.9 |
| 350 | 1.4 | 4:1 | 6.6 | 77.8 | - | 91.6 | - | 1.1 | 9.6 | 72.4 |
| A, L/Lcat sec | E/R | |
|---|---|---|
| 1,2-DCE-an | 9.4●104 | 8.51 |
| 1,2-DCE-en | 2.67●105 | 8.91 |
| A, L/Lcat sec | E/R | |
|---|---|---|
| 1,1,2-trichloroethane | 1.0●106 | 8.51 |
| Trichlorethylene | 4.1●106 | 10.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flid, M.R.; Kartashov, L.M.; Treger, Y.A. Theoretical and Applied Aspects of Hydrodechlorination Processes—Catalysts and Technologies. Catalysts 2020, 10, 216. https://doi.org/10.3390/catal10020216
Flid MR, Kartashov LM, Treger YA. Theoretical and Applied Aspects of Hydrodechlorination Processes—Catalysts and Technologies. Catalysts. 2020; 10(2):216. https://doi.org/10.3390/catal10020216
Chicago/Turabian StyleFlid, M.R., L.M. Kartashov, and Yu.A. Treger. 2020. "Theoretical and Applied Aspects of Hydrodechlorination Processes—Catalysts and Technologies" Catalysts 10, no. 2: 216. https://doi.org/10.3390/catal10020216
APA StyleFlid, M. R., Kartashov, L. M., & Treger, Y. A. (2020). Theoretical and Applied Aspects of Hydrodechlorination Processes—Catalysts and Technologies. Catalysts, 10(2), 216. https://doi.org/10.3390/catal10020216