Transfer Hydrogenation from 2-propanol to Acetophenone Catalyzed by [RuCl2(η6-arene)P] (P = monophosphine) and [Rh(PP)2]X (PP = diphosphine, X = Cl−, BF4−) Complexes




Abstract
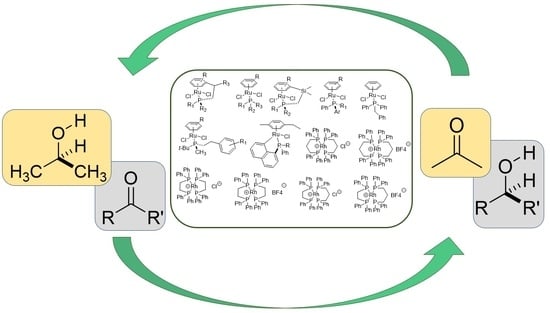
1. Introduction
2. Results
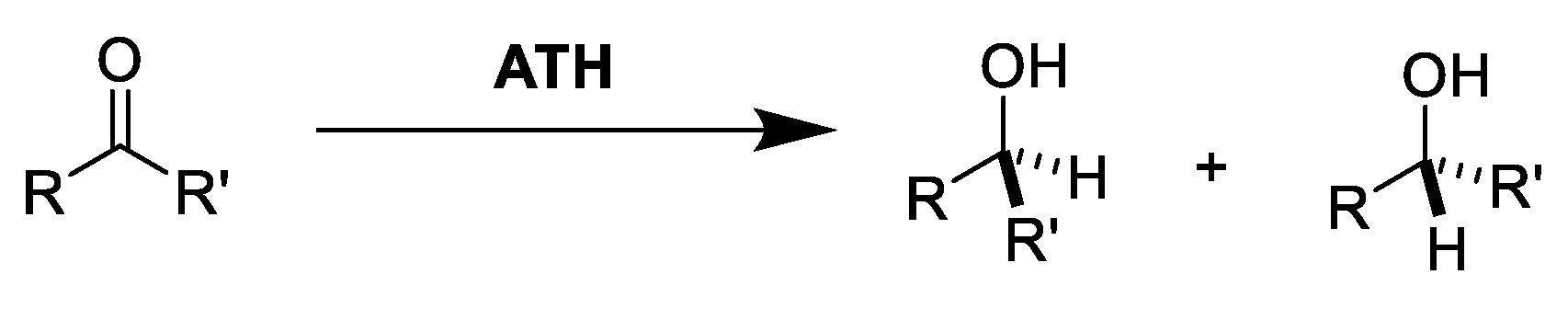
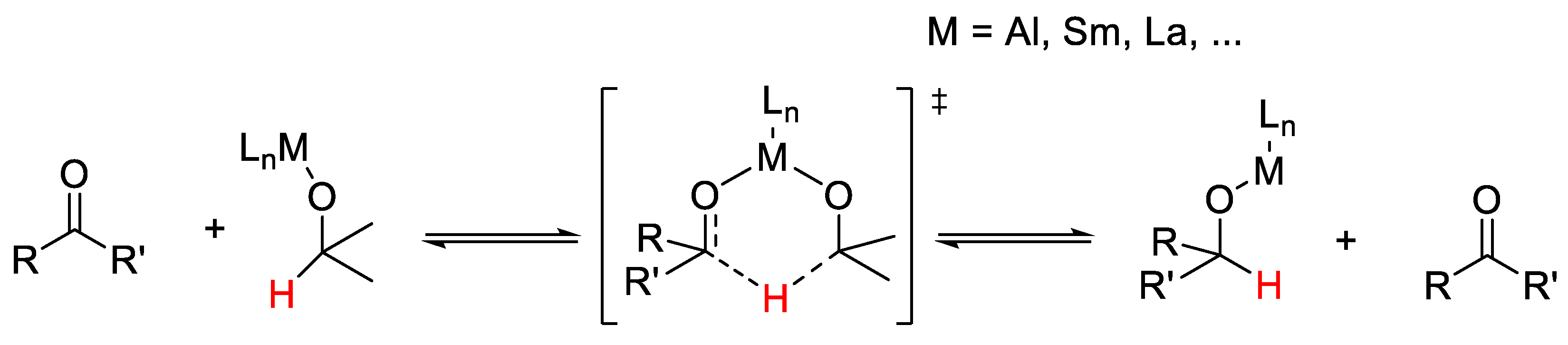
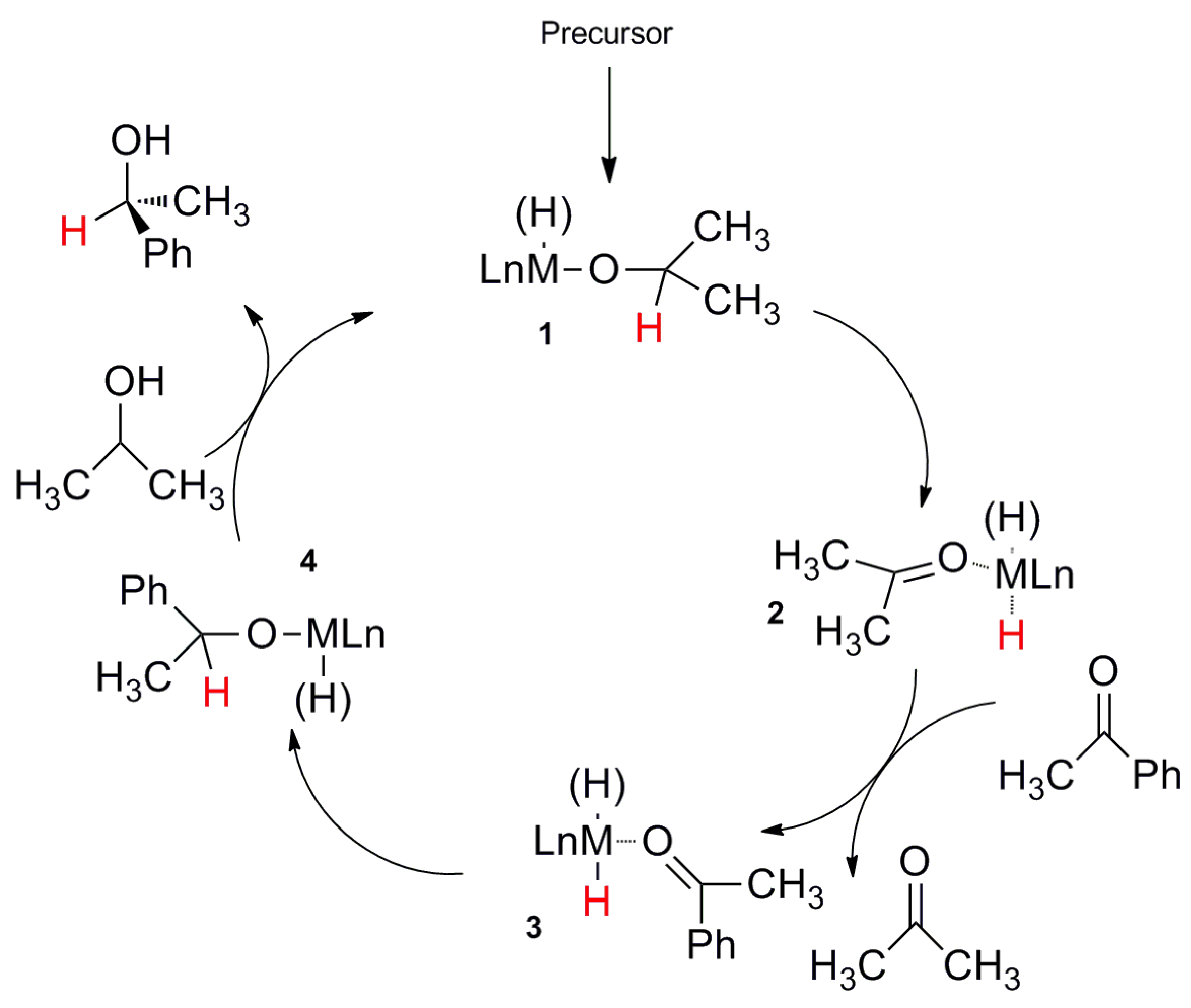
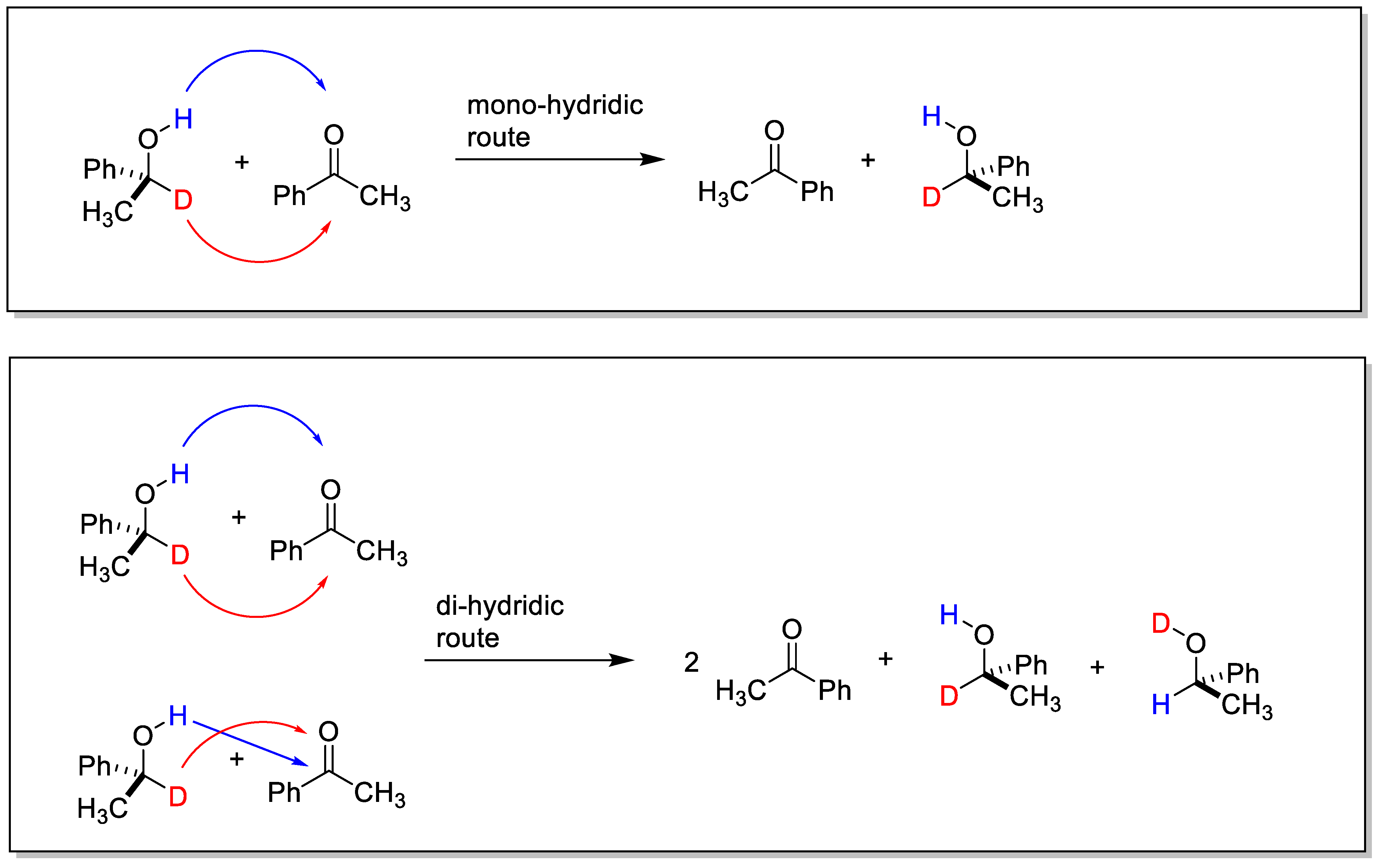
2.1. TH: Generalities and Applications
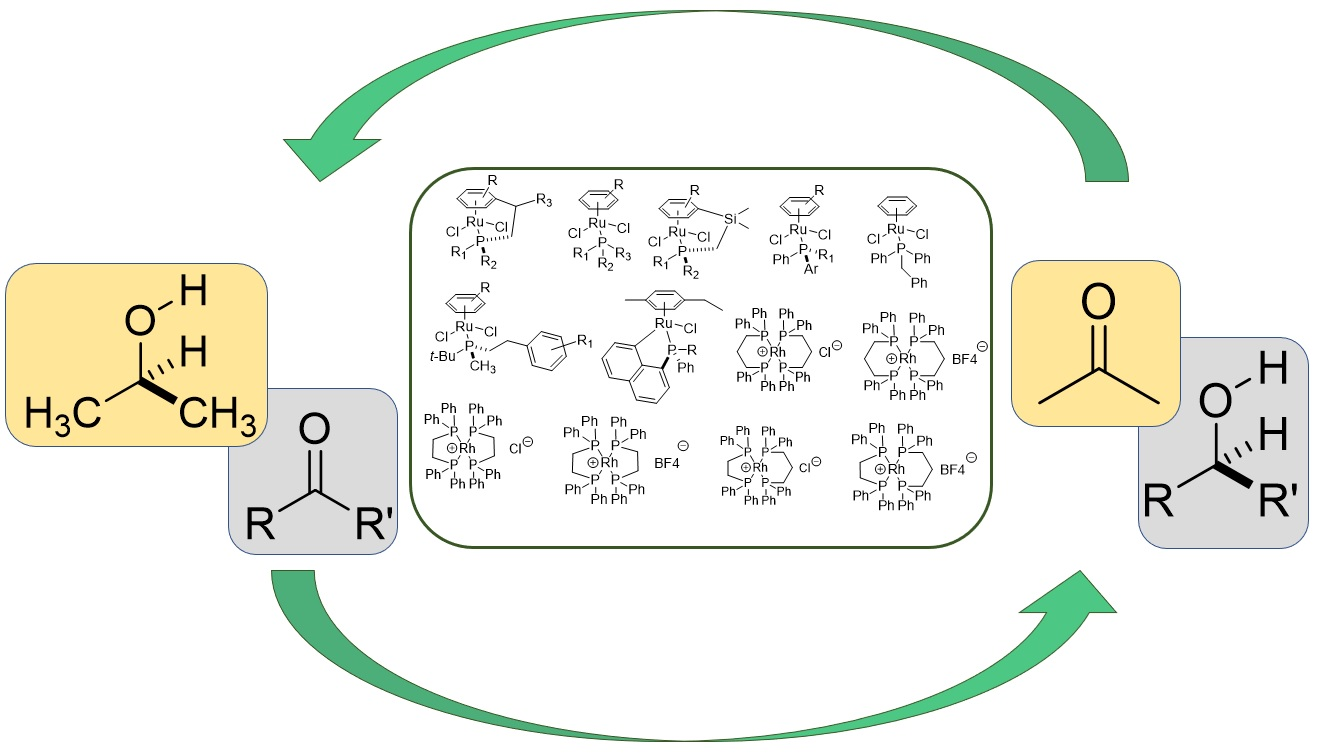
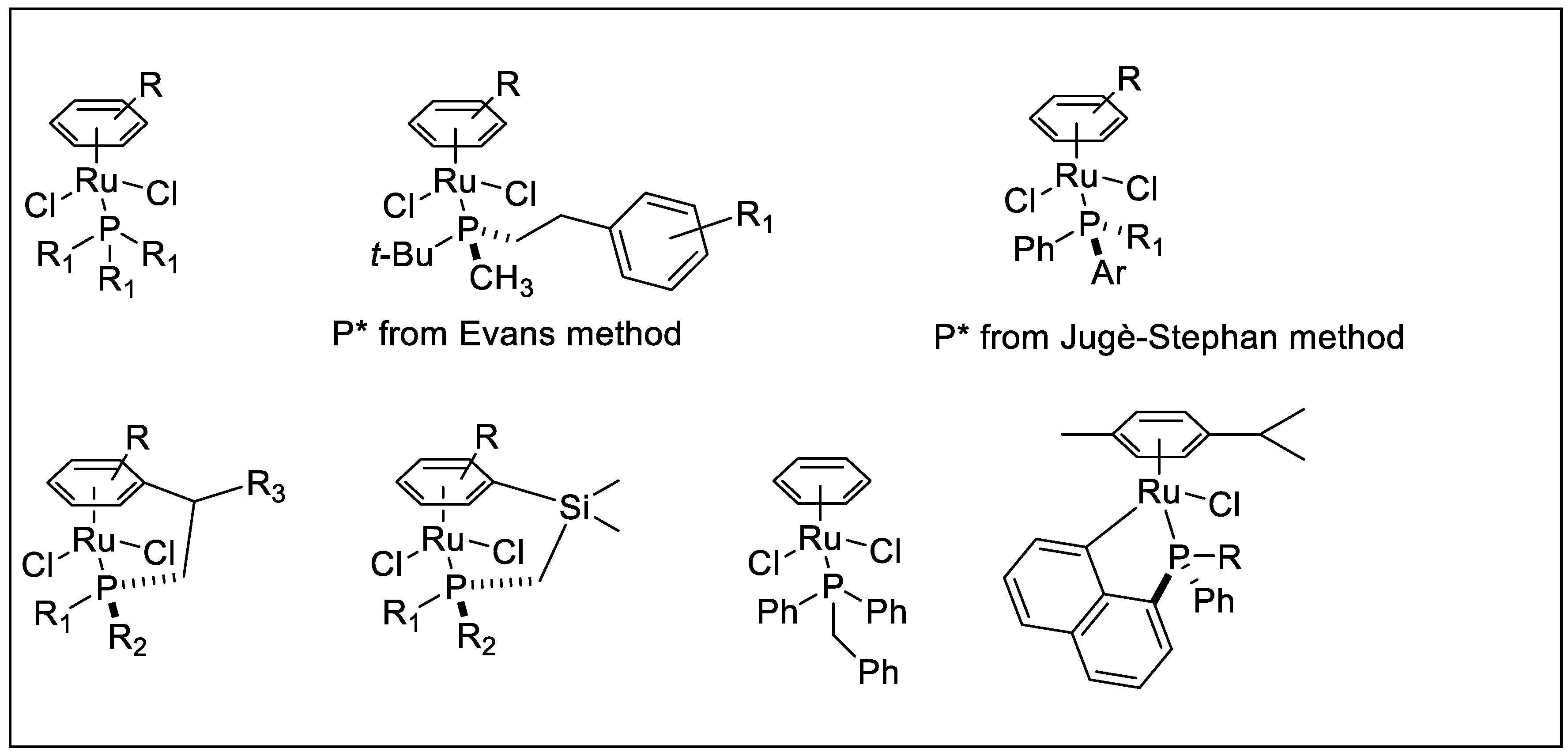
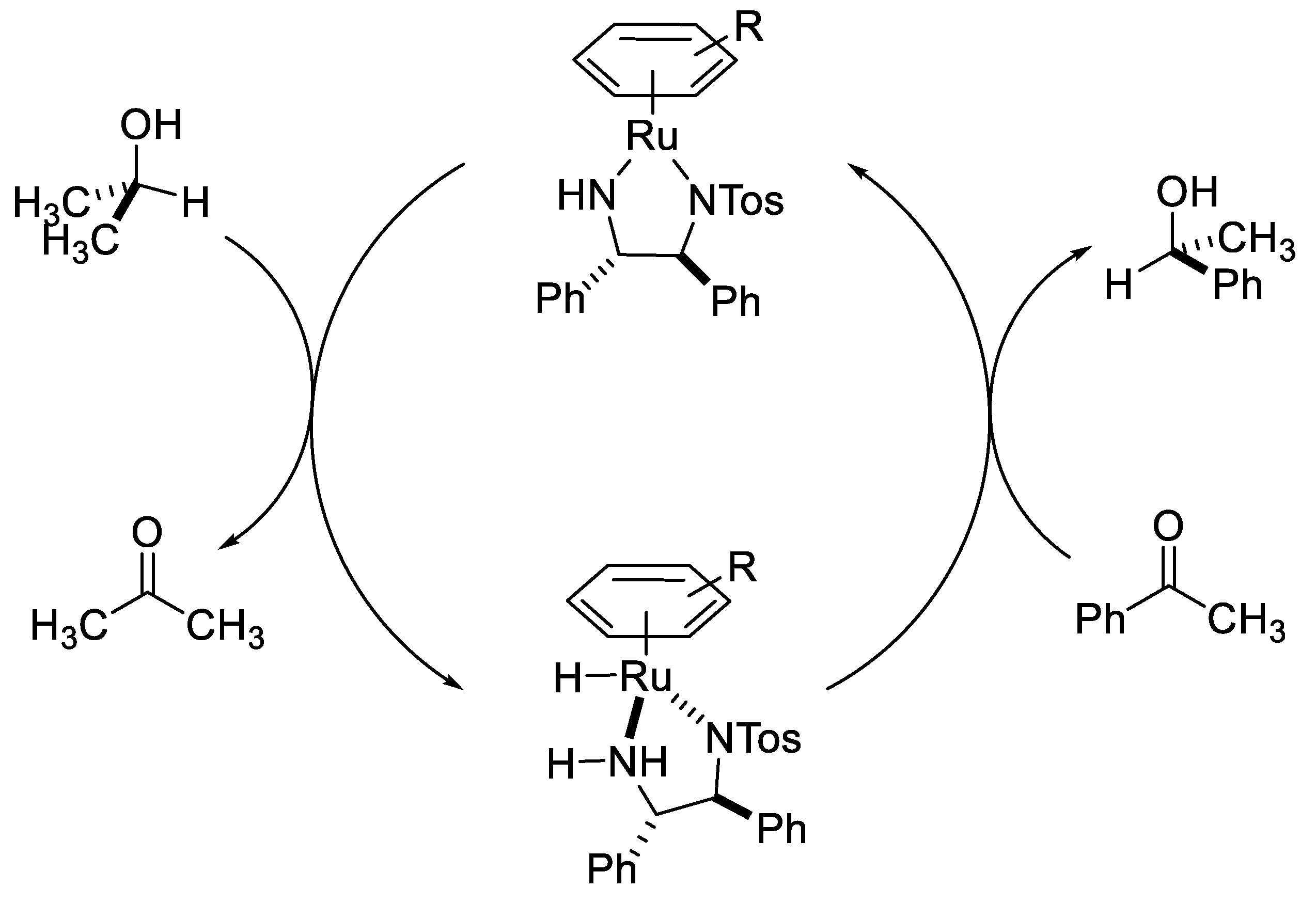
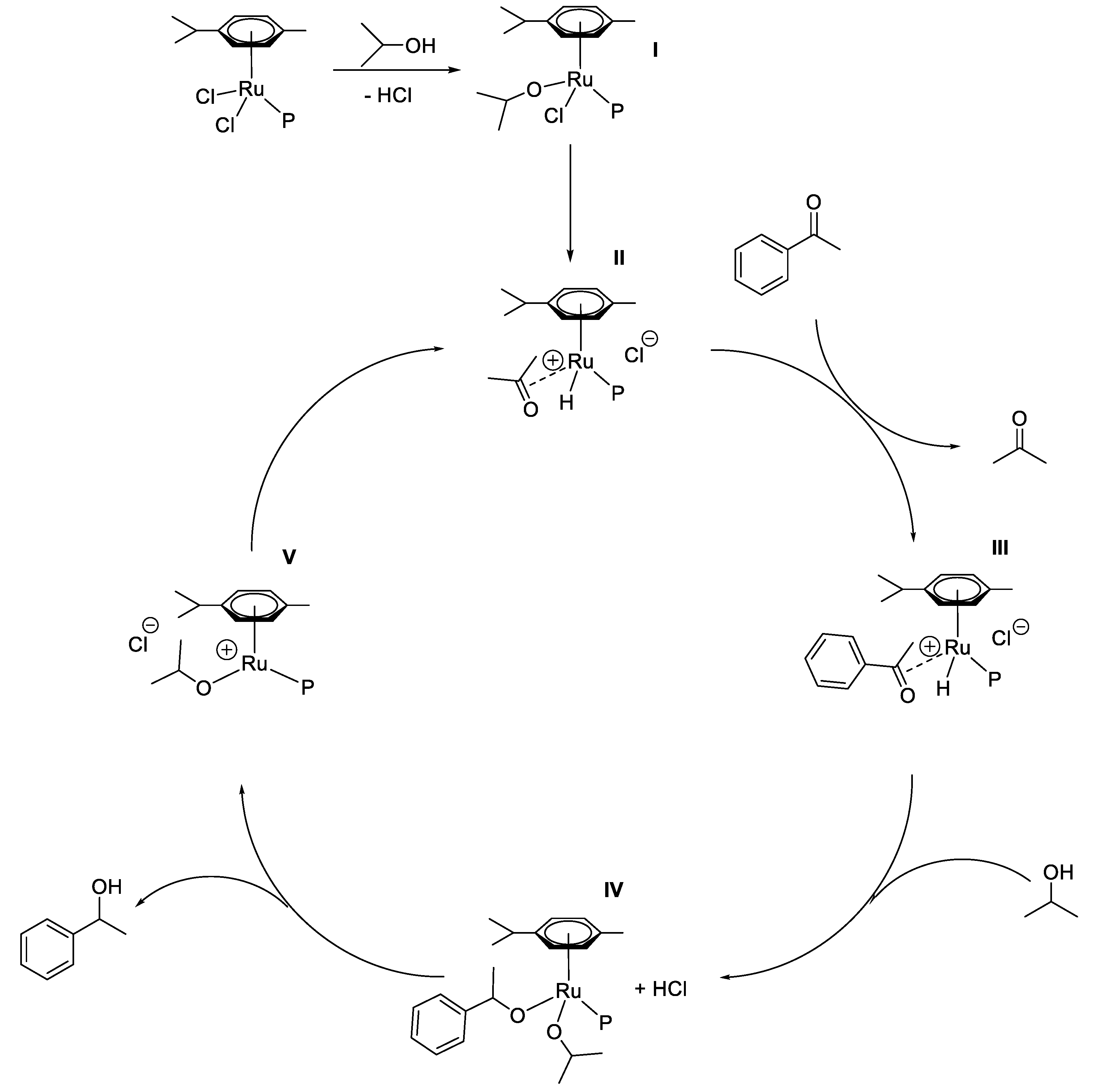
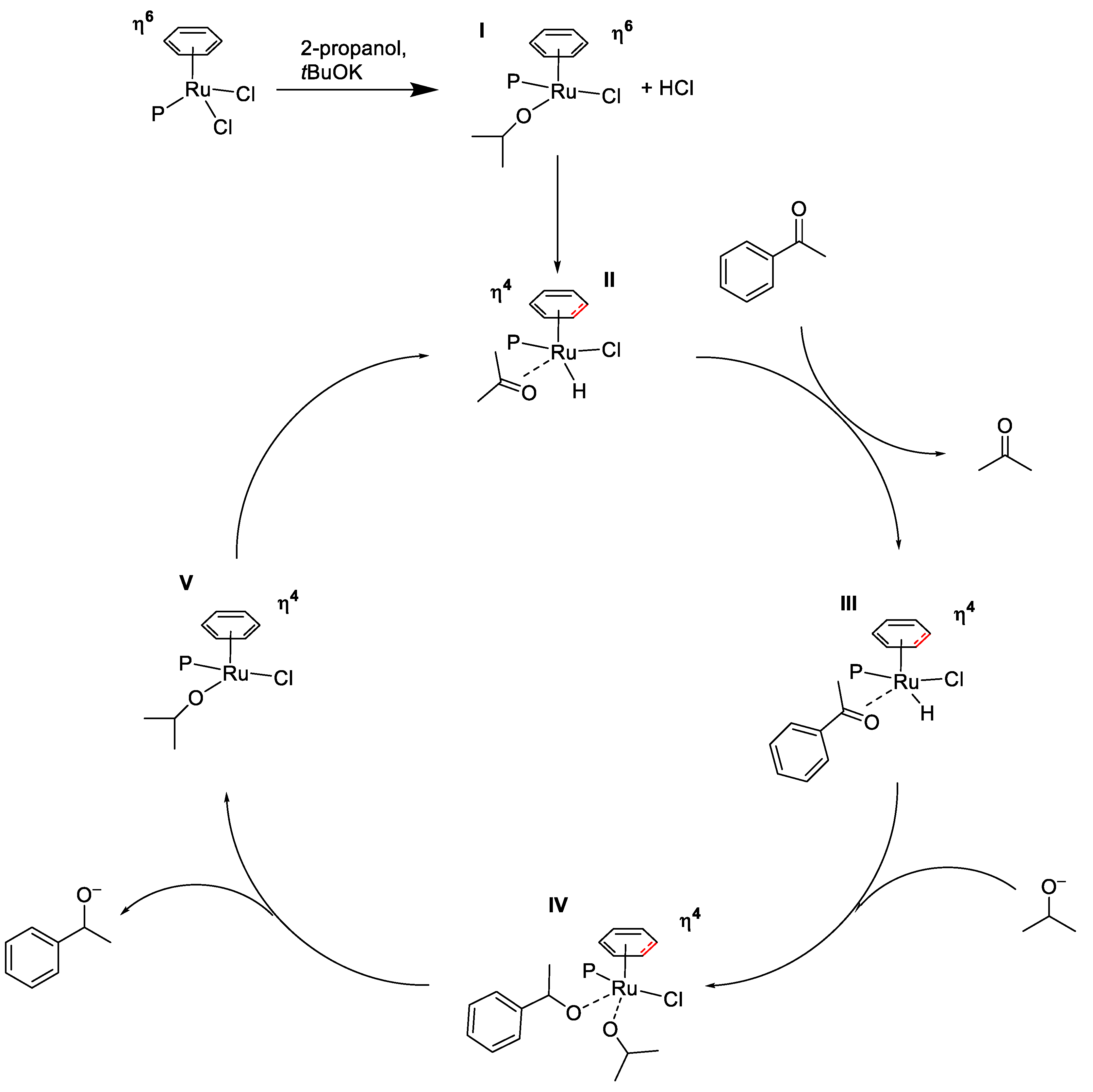
2.2. TH Catalyzed by Ru-arene Complexes Stabilized by Phosphines
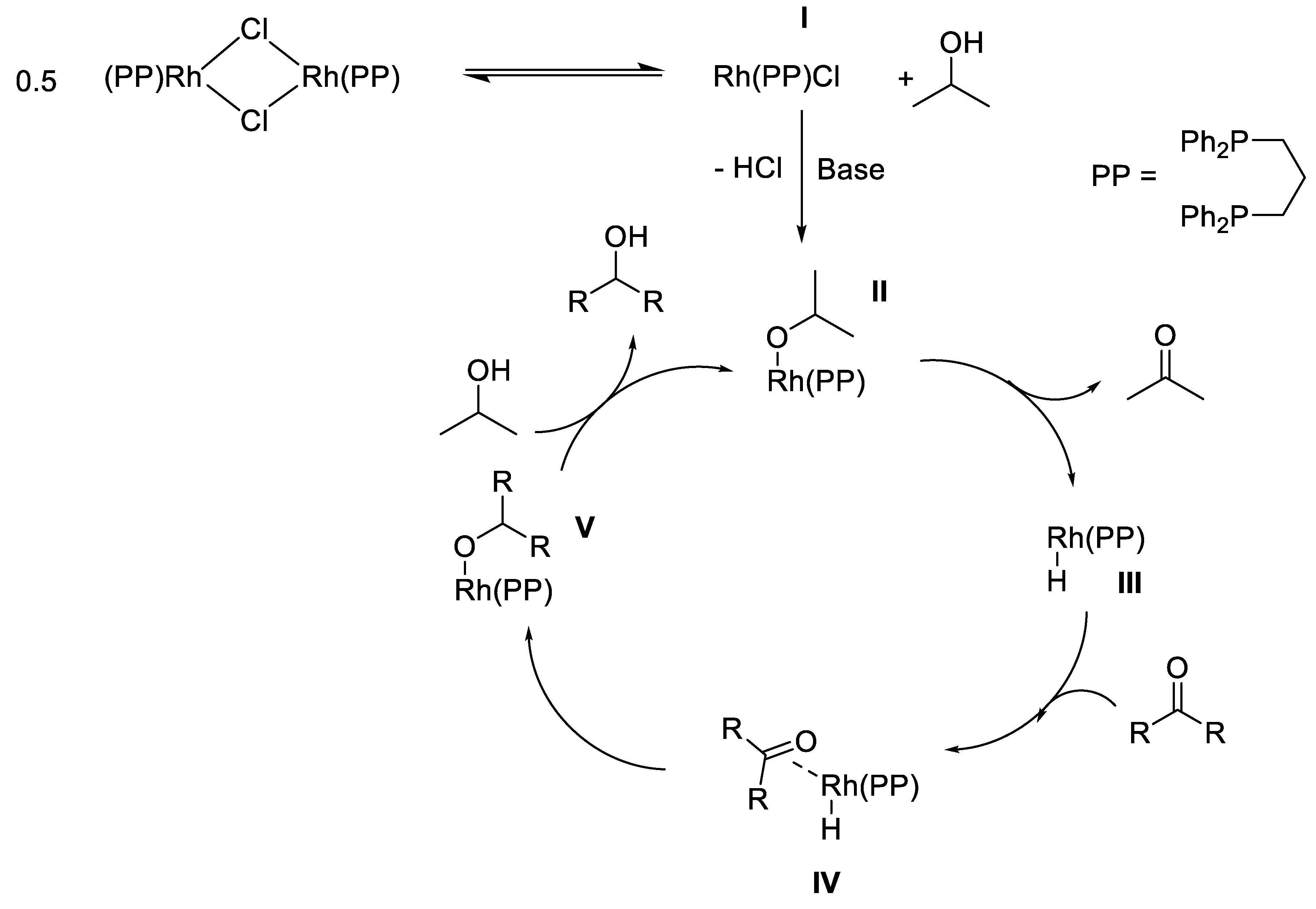
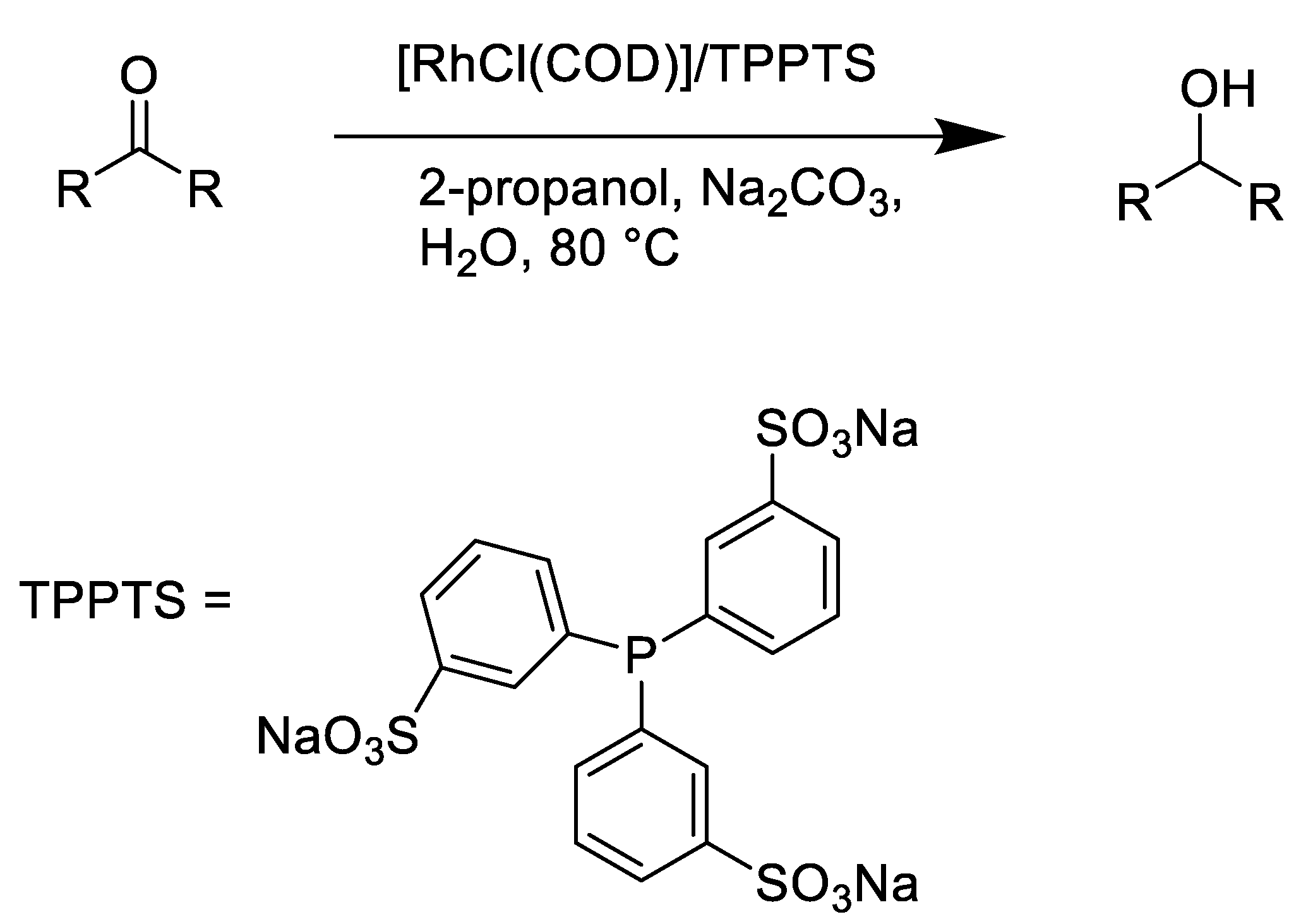
2.3. TH Mediated by Rh Complexes Stabilized by Phosphine Ligands
3. Conclusions
Funding
Conflicts of Interest
References and Note
- Gladiali, S.; Alberico, E. Asymmetric transfer hydrogenation: chiral ligands and applications. Chem. Soc. Rev. 2006, 35, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Wills, M. Imino Transfer Hydrogenation Reductions. Top Curr Chem (Z). 2016, 14, 374. [Google Scholar]
- Ferrini, P.; Rinaldi, R. Catalytic Biorefining of Plant Biomass to Non-Pyrolytic Lignin Bio-Oil and Carbohydrates through Hydrogen Transfer Reactions. Angew. Chem. Int. Ed. 2014, 53, 8634–8639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gyngazova, M.S.; Lolli, A.; Grazia, L.; Tabanelli, T.; Cavani, F.; Albonetti, A. Chapter 10 - Hydrogen Transfer Reaction as an Alternative Reductive Process for the Valorization of Biomass-Derived Building Blocks. Stud. Surf. Sci. Catal. 2019, 178, 195–214. [Google Scholar]
- Noyori, R. Asymmetric catalysis: science and opportunities (Nobel lecture). Angew. Chem. Int. Ed. 2002, 41, 2008–2022. [Google Scholar] [CrossRef]
- The Nobel Prize in Chemistry 2001. Available online: https://www.nobelprize.org/prizes/chemistry/2001/summary/ (accessed on 27 December 2019).
- Gladiali, S.; Taras, R. Reduction of Carbonyl Compounds by Hydrogen Transfer. In Modern Reduction Methods; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 135–157. [Google Scholar]
- Štefane, B.; Požgan, F. Asymmetric Hydrogenation and Transfer Hydrogenation of Ketones. In Hydrogenation; Karamé, L., Ed.; Intech Open: London, UK, 2012. [Google Scholar]
- Ponndorf, W. Der reversible Austausch der Oxydationsstufen zwischen Aldehyden oder Ketonen einerseits und primären oder sekundären Alkoholen anderseits. Angew. Chem. 1926, 39, 138. [Google Scholar] [CrossRef]
- De Graauw, C.F.; Peters, J.A.; Van Berkkum, H.; Huskens, J. Meerwein-Ponndorf-Verley Reductions and Oppenauer Oxidations: An Integrated Approach. Synthesis 1994, 1107–1117. [Google Scholar] [CrossRef]
- Djerassi, C. The Oppenauer Oxidation in Organic Reactions; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar] [CrossRef]
- Ooi, T.; Miura, T.; Maruoka, K. Highly Efficient, Catalytic Meerwein-Ponndorf-Verley Reduction with a Novel Bidentate Aluminum Catalyst. Angew. Chem. Int. Ed. 1998, 37, 2347–2349. [Google Scholar] [CrossRef]
- Campbell, E.J.; Zhou, H.; Nguyen, S.T. The Asymmetric Meerwein–Schmidt–Ponndorf–Verley Reduction of Prochiral Ketones with iPrOH Catalyzed by Al Catalysts. Angew. Chem. Int. Ed. 2002, 41, 1020–1022. [Google Scholar] [CrossRef]
- Campbell, E.J.; Zhou, H.; Nguyen, S.T. Catalytic Meerwein-Pondorf-Verley reduction by simple aluminum complexes. Org. Lett. 2001, 3, 2391–2393. [Google Scholar] [CrossRef] [PubMed]
- Doering, W.W.; Young, R.W.J. Partially asymmetric Meerwein-Ponndorf-Verley reductions. J. Am. Chem. Soc. 1950, 72, 631. [Google Scholar] [CrossRef]
- Meerwein, H.; Schmidt, R. Ein neues Verfahren zur Reduktion von Aldehyden und Ketonen. Liebig Ann. Chem. 1925, 444, 221–238. [Google Scholar] [CrossRef]
- Bailar, J.C.; Hitatani, H.J. Homogeneous Catalysis in the Reactions of Olefinic Substances. VI. Selective Hydrogenation of Methyl Linoleate and Isomerization of Methyl Oleate by Homogeneous Catalysis with Platinum Complexes Containing Triphenylphosphine, -arsine, or -stibine. J. Am. Chem. Soc. 1967, 89, 1592–1599. [Google Scholar] [CrossRef]
- Trocha-Grimshaw, J.; Henbest, H.B. Catalysis of the transfer of hydrogen from propan-2-ol to αβ-unsaturated ketones by organoiridium compounds. A carbon–iridium compound containing a chelate keto-group. Chem. Commun. 1967, 544. [Google Scholar] [CrossRef]
- Sasson, Y.; Blum, J. Dichlorotris(triphenylphosphine)ruthenium-catalyzed hydrogen transfer from alcohols to saturated and alpha, beta-unsaturated ketones. J. Org. Chem. 1975, 40, 1887–1896. [Google Scholar] [CrossRef]
- Matteoli, U.; Frediani, P.; Bianchi, M.; Botteghi, C.; Gladiali, S. Asymmetric homogeneous catalysis by ruthenium complexes. J. Mol. Catal. 1981, 12, 265–319. [Google Scholar] [CrossRef]
- Wang, C.; Wu, X.; Xiao, J. Broader, Greener, and More Efficient: Recent Advances in Asymmetric Transfer Hydrogenation. Chem. Asian J. 2008, 3, 1750–1770. [Google Scholar] [CrossRef]
- Farrar-Tobar, R.A.; Wei, Z.; Jiao, H.; Hinze, S.; de Vries, J.G. Selective Base-free Transfer Hydrogenation of α,β-Unsaturated Carbonyl Compounds using iPrOH or EtOH as Hydrogen Source. Chem. Eur. J. 2018, 24, 2725–2734. [Google Scholar] [CrossRef]
- Ruff, A.; Kirby, C.; Chan, B.C.; O’Connor, A.R. Base-Free Transfer Hydrogenation of Ketones Using Cp*Ir(pyridinesulfonamide)Cl Precatalysts. Organometallics 2016, 35, 327–335. [Google Scholar] [CrossRef]
- Carrión, M.C.; Sepúlveda, F.; Jalón, F.A.; Manzano, B.R.; Rodríguez, A.M. Base-Free Transfer Hydrogenation of Ketones Using Arene Ruthenium(II) Complexes. Organometallics 2009, 28, 3822–3833. [Google Scholar] [CrossRef]
- Lagaditis, P.O.; Lough, A.J.; Morris, R.H. Mechanism of the Hydrogenation of Ketones Catalyzed by trans-Dihydrido(diamine)ruthenium(II) Complexes. J. Am. Chem. Soc. 2011, 133, 9662–9665. [Google Scholar] [CrossRef] [PubMed]
- Laxmi, Y.R.S.; Bäckvall, J.-E. Mechanistic studies on ruthenium-catalyzed hydrogen transfer reactions. Chem. Commun. 2000, 611–612. [Google Scholar] [CrossRef]
- Yang, J.W.; Hechevarria Fonseca, M.T.; Vignola, N.; List, B. Metal-Free, Organocatalytic Asymmetric Transfer Hydrogenation of α,β-Unsaturated Aldehydes. Angew. Chem. Int. Ed. 2005, 44, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Bagnali, R.; Strauss, C.R. Uncatalysed hydrogen-transfer reductions of aldehydes and ketones. Chem. Commun. 1999, 287–288. [Google Scholar] [CrossRef]
- Clapham, S.E.; Hadzovic, A.; Morris, R.H. Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes. Coord. Chem. Rev. 2004, 248, 2201–2237. [Google Scholar] [CrossRef]
- Zassinovich, G.; Bettella, R.; Mestroni, G.; Bresciani-Pahor, N.; Geremia, S.; Randaccio, L. Enantioselective hydrogen transfer reactions from propan-2-ol to ketones catalyzed by pentacoordinate iridium(I) complexes with chiral Schiff bases. J. Organomet. Chem. 1989, 370, 187–202. [Google Scholar] [CrossRef]
- Petra, D.G.I.; Kamer, P.C.J.; Speck, A.L.; Schoemaker, H.E.; Van Leeuwen, P.W.N.M. Aminosulf(ox)ides as Ligands for Iridium(I)-Catalyzed Asymmetric Transfer Hydrogenation. J. Org. Chem. 2000, 65, 3010–3017. [Google Scholar] [CrossRef]
- Handgraaf, J.W.; Reek, J.H.N.; Meijer, E.J. Iridium (I) versus ruthenium (II). A computational study of the transition metal catalyzed transfer hydrogenation of ketones. Organometallics 2003, 22, 3150–3157. [Google Scholar] [CrossRef]
- Pàmies, O.; Bäckvall, J.-E. Studies on the Mechanism of Metal-Catalyzed Hydrogen Transfer from Alcohols to Ketones. Chem. Eur. J. 2001, 7, 5052–5058. [Google Scholar] [CrossRef]
- Samec, J.S.M.; Bäckvall, J.-E.; Andersson, P.G.; Brandt, P. Mechanistic aspects of transition metal-catalyzed hydrogen transfer reactions. Chem. Soc. Rev. 2006, 35, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Haack, K.J.; Hashiguchi, S.; Fujii, A.; Ikariya, T.; Noyori, R. The Catalyst Precursor, Catalyst, and Intermediate in the RuII-Promoted Asymmetric Hydrogen Transfer between Alcohols and Ketones. Angew. Chem. Int. Ed. Engl. 1997, 36, 285. [Google Scholar] [CrossRef]
- Ikariya, T.; Murata, K.; Noyori, R. Bifunctional transition metal-based molecular catalysts for asymmetric syntheses. Org. Biomol. Chem. 2006, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Hounjet, L.J.; Ferguson, M.J.; Cowie, M. Phosphine–Amido Complexes of Ruthenium and Mechanistic Implications for Ketone Transfer Hydrogenation Catalysis. Organometallics 2011, 30, 4108–4114. [Google Scholar] [CrossRef]
- Hashiguchi, S.; Fujii, A.; Takehara, J.; Ikariya, T.; Noyori, R. Asymmetric transfer hydrogenation of aromatic ketones catalyzed by chiral ruthenium (II) complexes. J. Am. Chem. Soc. 1995, 117, 7562–7563. [Google Scholar] [CrossRef]
- Noyori, R.; Hashiguchi, S. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Noyori, R.; Yamakawa, M.; Hashiguchi, S. Metal− ligand bifunctional catalysis: a nonclassical mechanism for asymmetric hydrogen transfer between alcohols and carbonyl compounds. J. Org. Chem. 2001, 66, 7931–7944. [Google Scholar] [CrossRef]
- Giboulot, S.; Baldino, S.; Ballico, M.; Nedden, H.G.; Zuccaccia, D.; Baratta, W. Cyclometalated dicarbonyl ruthenium catalysts for transfer hydrogenation and hydrogenation of carbonyl compounds. Organometallics 2018, 37, 2136–2146. [Google Scholar] [CrossRef]
- Sun, R.; Chu, X.; Zhang, S.; Li, T.; Wang, Z.; Zhu, B. Synthesis, Structure, Reactivity, and Catalytic Activity of Cyclometalated (Phosphine)-and (Phosphinite) ruthenium Complexes. Eur. J. Inorg. Chem. 2017, 3174–3183. [Google Scholar] [CrossRef]
- Hey, D.A.; Fischer, P.J.; Baratta, W.; Kühn, F.E. Ru(O2CCF3)2(PPh3)2 and ruthenium phosphine complexes bearing fluoroacetate ligands: synthesis, characterization and catalytic activity. Dalton Trans. 2019, 48, 4625–4635. [Google Scholar] [CrossRef]
- Giboulot, S.; Comuzzi, C.; Del Zotto, A.; Figliolia, R.; Lippe, G.; Lovison, D.; Strazzolini, P.; Susmel, S.; Zangrando, E.; Zuccaccia, D.; et al. Preparation of monocarbonyl ruthenium complexes bearing bidentate nitrogen and phosphine ligands and their catalytic activity in carbonyl compound reduction. Dalton Trans. 2019, 48, 12560–12576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Baratta, W. Synthesis of Pincer Ruthenium RuCl(CNN)(PP) Catalysts from [RuCl(μ-Cl)(η6-p-cymene)]2. Organometallics 2013, 32, 3339–3342. [Google Scholar] [CrossRef]
- Zhang, S.; Baldino, S.; Baratta, W. Synthesis of [RuX(CO)(dppp)(NN)] Cl (X= H, Cl; NN= en, ampy) Complexes and Their Use as Catalysts for Transfer Hydrogenation. Organometallics 2013, 32, 5299–5304. [Google Scholar] [CrossRef]
- For the TH with 2-propanol see: Baldino, S.; Facchetti, S.; Zanotti-Gerosa, A.; Nedden, H.G.; Baratta, W. ChemCatChem 2016, 8, 2279–2288.
- For the TH with formate sources see: Baldino, S.; Facchetti, S.; Zanotti-Gerosa, A.; Nedden, H.G.; Baratta, W. Chemoselective Transfer Hydrogenation of Aldehydes with HCOONH4 Catalyzed by RuCl(CNNPh)(PP) Pincer Complexes. ChemCatChem 2016, 8, 3195–3198. [Google Scholar]
- Baratta, W.; Ballico, M.; Baldino, S.; Chelucci, G.; Herdtweck, E.; Siega, K.; Magnolia, S.; Rigo, P. New Benzo[h]quinoline-Based Ligands and their Pincer Ru and Os Complexes for Efficient Catalytic Transfer Hydrogenation of Carbonyl Compounds. Chem. Eur. J. 2008, 14, 9148–9160. [Google Scholar] [CrossRef]
- Carriedo, G.A.; Crochet, P.; García Alonso, F.J.; Gimeno, J.; Presa-Soto, A. Synthesis and Catalytic Activity of (η6-p-Cymene)(phosphane)ruthenium(II) Complexes Supported on Poly(biphenoxyphosphazene) or Chiral Poly(binaphthoxyphosphazene) Copolymers. Eur. J. Inorg. Chem. 2004, 3668–3674. [Google Scholar] [CrossRef]
- Grabulosa, A.; Mannu, A.; Alberico, E.; Denurra, S.; Gladiali, S.; Muller, G. Neutral η6-arene ruthenium complexes with monodentate P-donor ligands: Activation in the transfer hydrogenation reaction. J. Mol. Catal. A Chem. 2012, 49, 363–364. [Google Scholar]
- Angurell, I.; Muller, G.; Rocamora, M.; Rossell, O.; Seco, M. Single and double metallic layer-containing ruthenium dendrimers. Synthesis and catalytic properties. Dalton Trans. 2004, 2450–2457. [Google Scholar] [CrossRef]
- Wang, L.; Yang, Q.; Fu, H.-Y.; Chen, H.; Yuan, M.-L.; Li, R.-X. Ru–η6-benzene–phosphine complex-catalyzed transfer hydrogenation of ketones. Appl. Organometal. Chem. 2011, 25, 626–631. [Google Scholar] [CrossRef]
- Grabulosa, A.; Mannu, A.; Mezzetti, A.; Muller, G. Neutral p-cymene ruthenium complexes with P-stereogenic monophosphines. New catalytic precursors in enantioselective transfer hydrogenation and cyclopropanation. J. Organomet. Chem. 2012, 696, 4221–4228. [Google Scholar] [CrossRef]
- Aznar, R.; Grabulosa, A.; Mannu, A.; Muller, G.; Sainz, D.; Moreno, V.; Font-Bardia, M.; Calvet, T.; Lorenzo, J. [RuCl2(η6-p-cymene)(P*)] and [RuCl2(κ-P*-η6-arene)] Complexes Containing P-Stereogenic Phosphines. Activity in Transfer Hydrogenation and Interactions with DNA. Organometallics 2013, 32, 2344–2362. [Google Scholar] [CrossRef]
- Clavero, P.; Grabulosa, A.; Rocamora, M.; Muller, G.; Font-Bardia, M. Ruthenium complexes of P-stereogenic phosphines with a heterocyclic substituent. Dalton Trans. 2016, 45, 8513–8531. [Google Scholar] [CrossRef] [PubMed]
- Clavero, P.; Grabulosa, A.; Font-Bardia, M.; Muller, G. P-Stereogenic monophosphines with the 2-p-terphenylyl and 1-pyrenyl substituents. Application to Pd and Ru asymmetric catalysis. Journal of Mol. Catal A: Chem. 2014, 391, 183–190. [Google Scholar] [CrossRef]
- Navarro, M.; Vidal, D.; Clavero, P.; Grabulosa, A.; Muller, G. Mild Photochemical Tethering of [RuCl2(η6-arene)P*] Complexes with P-Stereogenic 2-Biphenylylphosphines. Organometallics 2015, 34, 973–994. [Google Scholar] [CrossRef]
- Grabulosa, A. Chapter 5 (Evans method), Chapter 4 (Jugé-Stephan). In P-Stereogenic Ligands in Enantioselective Catalysis; RSC Publishing: Cambridge, UK, 2010. [Google Scholar]
- Chaplin, A.B.; Dyson, P.J. Catalytic Activity of Bis-phosphine Ruthenium(II)−Arene Compounds: Chemoselective Hydrogenation and Mechanistic Insights. Organometallics 2007, 26, 4357–4360. [Google Scholar] [CrossRef]
- Chaplin, A.B.; Dyson, P.J. Catalytic Activity of Bis-phosphine Ruthenium (II)− Arene Compounds: Structure− Activity Correlations. Organometallics 2007, 26, 2447–2455. [Google Scholar] [CrossRef]
- Dinda, S.; Sebastian, K.L.; Samuelson, A.G. Mechanistic Aspects of Nucleophilic Substitution at Half-Sandwich Metal Complexes. Organometallics 2010, 29, 6209–6218. [Google Scholar] [CrossRef]
- De Pasquale, J.; Kumar, M.; Zeller, M.; Papish, E.T. Variations on an NHC theme: which features enhance catalytic transfer hydrogenation with ruthenium complexes? Organometallics 2013, 32, 966–979. [Google Scholar] [CrossRef]
- Brissos, R.F.; Clavero, P.; Gallen, A.; Grabulosa, A.; Barrios, L.A.; Caballero, A.B.; Korrodi-Gregório, L.; Pérez-Tomás, R.; Muller, G.; Soto-Cerrato, V.; et al. Highly cytotoxic ruthenium (II)-arene complexes from bulky 1-pyrenylphosphane ligands. Inorg. Chem. 2018, 57, 14786–14797. [Google Scholar] [CrossRef]
- Štefane, B.; Požgan, F. Homogenous, asymmetric hydrogenation and transfer hydrogenation. In Hydrogenation; Karamé, L., Ed.; BoD–Books on Demand: Norderstedt, Germany, 2012. [Google Scholar]
- Aydemir, M.; Ocak, Y.S.; Rafikova, K.; Kystaubayeva, N.; Kayan, C.; Zazybin, A.; Ok, F.; Baysal, A.; Temel, H. Rhodium-catalyzed transfer hydrogenation with aminophosphines and analysis of electrical characteristics of rhodium (I) complex/n-Si heterojunctions. Appl. Organomet. Chem. 2014, 28, 396–404. [Google Scholar] [CrossRef]
- Aydemir, M.; Meric, N.; Kayan, C.; Ok, F.; Baysal, A. Rhodium-catalyzed transfer hydrogenation with functionalized bis (phosphino) amine ligands. Inorg. Chim. Acta 2013, 398, 1–10. [Google Scholar] [CrossRef]
- Kayan, C.; Meric, N.; Aydemir, M.; Baysal, A.; Elma, D.; Ak, B.; Sahin, E.; Gürbüz, N.; Ozdemir, I. Ruthenium, rhodium and iridium complexes of the furfuryl-2-(N-diphenylphosphino) methylamine ligand: Molecular structure and catalytic activity. Polyhedron 2012, 42, 142–148. [Google Scholar] [CrossRef]
- Aydemir, M.; Baysal, A.; Meric, N.; Kayan, C.; Gümgüm, B.; Ozkar, S.; Sahin, E. Organometallic ruthenium, rhodium and iridium complexes containing a P-bound thiophene-2-(N-diphenylphosphino)-methylamine ligand: Synthesis, molecular structure and catalytic activity. J. Organomet. Chem. 2011, 696, 2584–2588. [Google Scholar] [CrossRef]
- Mannu, A.; Vlahopoulou, G.; Kubis, C.; Drexler, H.-J. Synthesis and characterization of [Rh (PP)(PP)] X complexes (PP = DPPE or DPPP, X= Cl− or BF4−). Phosphine exchange and reactivity in transfer hydrogenation conditions. J. Organomet. Chem. 2019, 885, 59–64. [Google Scholar] [CrossRef]
- James, B.R.; Mahajan, D. Bis(ditertiaryphosphine) complexes of rhodium(I). Synthesis, spectroscopy, and activity for catalytic hydrogenation. Can. J. Chem. 1979, 57, 180–187. [Google Scholar] [CrossRef]
- Du Bois, D.L.; Blake, D.M.; Miedaner, A.; Curtis, C.J.; Du Bois, M.R.; Franz, J.A.; Linehan, J.C. Hydride Transfer from Rhodium Complexes to Triethylborane. Organometallics 2006, 25, 4414–4419. [Google Scholar] [CrossRef]
- Mansel, S.M. Catalytic applications of small bite-angle diphosphorus ligands with single-atom linkers. Dalton Trans. 2017, 46, 15157–15174. [Google Scholar] [CrossRef]
- Bays, J.T.; Priyadershani, N.; Jeletic, M.S.; Hulley, E.B.; Miller, D.L.; Linehan, J.C.; Shaw, W.J. The Influence of the Second and Outer Coordination Spheres on Rh(diphosphine)2CO2 Hydrogenation Catalysts. ACS Catal. 2014, 4, 3663–3670. [Google Scholar] [CrossRef]
- Dorta, R.; Shimon, L.; Milstein, D. Rhodium complexes with chiral counterions: achiral catalystsin chiral matrices. J. Organomet. Chem. 2004, 689, 751–758. [Google Scholar] [CrossRef]
- van der Broeke, J.; Lutz, M.; Kooijman, H.; Spek, A.L.; Deelman, B.-J.; van Koten, G. Increasing the Lipophilic Character of Tetraphenylborate Anions through Silyl Substituents. Organometallics 2011, 20, 2114–2117. [Google Scholar] [CrossRef]
- Anderson, M.P.; Pignolet, L.H. Rhodium complexes of 1,4-bis(diphenylphosphino)butane. Crystal and molecular structures of [Rh(dppb)2]BF4.cntdot.C4H10O and [Rh(cod)(dppb)]BF4. Inorg. Chem. 1981, 20, 4101–4107. [Google Scholar] [CrossRef]
- Alberico, E.; Möller, S.; Horstmann, M.; Drexler, H.-J.; Heller, D. Activation, Deactivation and Reversibility Phenomena in Homogeneous Catalysis: A Showcase Based on the Chemistry of Rhodium/Phosphine Catalysts. Catalysts 2019, 581. [Google Scholar] [CrossRef]
- Meißner, A.; Alberico, E.; Drexler, H.-J.; Baumann, W.; Heller, D. Rhodium diphosphine complexes: a case study for catalyst activation and deactivation. Catal. Sci. Technol. 2014, 4, 3409–3425. [Google Scholar] [CrossRef]
- Mannu, A.; Drexler, H.-J.; Thede, R.; Ferro, M.; Baumann, W.; Rüger, J.; Heller, D. Oxidative addition of CH2Cl2 to neutral dimeric rhodium diphosphine complexes. J. Organomet. Chem. 2018, 871, 178–184. [Google Scholar] [CrossRef]
- Mannu, A.; Ferro, M.; Möller, S.; Heller, D. Monomerisation of [Rh2(1, 3-bis-(diphenylphosphino)-propane)2(μ2-Cl)2] detected by pulsed gradient spin echo spectroscopy and 31P NMR monitoring of metathesis experiments. J. Chem. Res. 2018, 42, 402–404. [Google Scholar] [CrossRef]
- Baráth, E. Hydrogen Transfer Reactions of Carbonyls, Alkynes, and Alkenes with Noble Metals in the Presence of Alcohols/Ethers and Amines as Hydrogen Donors. Catalysts 2018, 8, 671. [Google Scholar] [CrossRef]
- Ajjou, A.N.; Pinet, J.-L. The biphasic transfer hydrogenation of aldehydes and ketones with isopropanol catalyzed by water-soluble rhodium complexes. J. Mol. Catal. A Chem. 2004, 214, 203–206. [Google Scholar] [CrossRef]
- Kathó, A.; Szatmári, I.; Papp, G.; Joó, F. Effect of 2-propanol on the transfer hydrogenation of aldehydes by aqueous sodium formate using a rhodium(i)-sulfonated triphenylphosphine catalyst. Chimia 2015, 69, 339–344. [Google Scholar] [CrossRef]
- Joó, F.; Bényei, A. Biphasic reduction of unsaturated aldehydes to unsaturated alcohols by ruthenium complex-catalyzed hydrogen transfer. J. Organomet. Chem. 1989, 363, C19–C21. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalytic Precursor | Conversion (%) | e.e. (%) | Reference |
|---|---|---|---|---|
| 1 |  | 95% 1 | 5(S) | [53] |
| 2 |  | 93% 1 | 6(S) | [53] |
| 3 |  | 97% 1 | 4(S) | [53] |
| 4 |  | 66% 1 | - | [53] |
| 5 |  | 58% 1 | - | [53] |
| 6 |  | 73% 1 43% 2 | 20(S) 50(S) | [53] |
| 7 |  | 44% 2 | 8(S) | [53] |
| 8 |  | 98% 3 | - | [52] |
| 9 |  | 96% 3 | - | [52] |
| 10 |  | 57% 3 | - | [52] |
| 11 |  | 43% 3 | 9(S) | [52] |
| 12 |  | 46% 4 | 14(R) | [57] |
| 13 |  | 99% 4 | 70(R) | [57] |
| 14 |  | 82% 5 | 26(R) | [59] |
| 15 |  | 72% 5 | 5(R) | [59] |
| 16 |  | 94% 5 | 23(R) | [59] |
| 17 |  | 99% 1 | 12(R) | [58] |
| Entry | Catalytic Precursor | Formula 1 | Conversion |
|---|---|---|---|
| 1 |  | [Rh(DPPP)2]Cl | 92.4% |
| 2 |  | [Rh(DPPE)(DPPP)]Cl | 58.7% |
| 3 |  | [Rh(DPPE)2]Cl | 49.4% |
| 4 |  | [Rh(DPPP)2]BF4 | 77.6% |
| 5 |  | [Rh(DPPE)(DPPP)]BF4 | 36.0% |
| 6 |  | [Rh(DPPE)2]BF4 | 3.6% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mannu, A.; Grabulosa, A.; Baldino, S. Transfer Hydrogenation from 2-propanol to Acetophenone Catalyzed by [RuCl2(η6-arene)P] (P = monophosphine) and [Rh(PP)2]X (PP = diphosphine, X = Cl−, BF4−) Complexes. Catalysts 2020, 10, 162. https://doi.org/10.3390/catal10020162
Mannu A, Grabulosa A, Baldino S. Transfer Hydrogenation from 2-propanol to Acetophenone Catalyzed by [RuCl2(η6-arene)P] (P = monophosphine) and [Rh(PP)2]X (PP = diphosphine, X = Cl−, BF4−) Complexes. Catalysts. 2020; 10(2):162. https://doi.org/10.3390/catal10020162
Chicago/Turabian StyleMannu, Alberto, Arnald Grabulosa, and Salvatore Baldino. 2020. "Transfer Hydrogenation from 2-propanol to Acetophenone Catalyzed by [RuCl2(η6-arene)P] (P = monophosphine) and [Rh(PP)2]X (PP = diphosphine, X = Cl−, BF4−) Complexes" Catalysts 10, no. 2: 162. https://doi.org/10.3390/catal10020162
APA StyleMannu, A., Grabulosa, A., & Baldino, S. (2020). Transfer Hydrogenation from 2-propanol to Acetophenone Catalyzed by [RuCl2(η6-arene)P] (P = monophosphine) and [Rh(PP)2]X (PP = diphosphine, X = Cl−, BF4−) Complexes. Catalysts, 10(2), 162. https://doi.org/10.3390/catal10020162