Abstract
The palladium-catalyzed (3 + 2) cycloaddition reaction between vinylcyclopropanes (VCPs) bearing geminal EWG’s and imines represents a straightforward and flexible entry to polysubstituted pyrrolidine derivatives. In this paper, we demonstrate that using a synergistic catalysis approach, based on the combination of phosphoric acid and palladium catalysts, it is possible to engage for the first time N-aryl and N-benzyl imines in this cycloaddition reaction. A range of polysubstituted pyrrolidines is obtained with moderate to good yields and diastereoselectivities, using a simple palladium species (Pd(PPh3)4) and an archetypical phosphoric acid as catalyst combination. A two-step scheme which exploits the same palladium catalyst for two consecutive and mechanistically distinct reactions (the cycloaddition and a Suzuki–Miyaura cross-coupling) is also presented. This synergistic catalysis approach is well posited for the development of the enantioselective version of this reaction. A screening of common BINOL-derived chiral phosphoric acids as catalyst component identified a species giving the product with moderate, yet promising, enantioselectivity (64% ee).
1. Introduction
Vinylcyclopropanes (VCPs) bearing two germinal electron-withdrawing groups can be activated by Pd(0) complexes, resulting in zwitterionic η3-allyl Pd(II) species (Scheme 1). These species undergo formal (3 + 2) cycloaddition reactions with electron poor π-systems (i.e., Michael acceptors, carbonyl compounds, etc.), affording five-membered ring compounds upon Pd(0) release from their vinyl group [1,2,3,4,5]. The study of this VCPs reactivity, disclosed by Tsuji and co-workers in 1985 using Michael acceptors as electron poor π-partners [6], has considerably intensified in recent times, paralleling other processes based on strain-releasing formation of zwitterions by palladium chemistry (e.g., from vinylepoxides and 2-vinylaziridines). Overall, this Pd-catalyzed activation of VCPs is a versatile catalytic platform for the synthesis of five membered ring compounds. The cycloaddition has been extended to a large variety of π-acceptors, and several catalytic enantioselective transformations, mostly relying on chiral palladium complexes, have been developed.
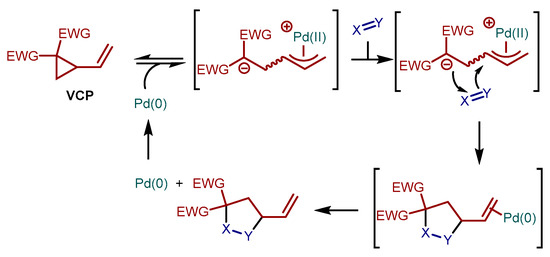
Scheme 1.
Activation of VCPs by Pd(0) and formal (3 + 2) cycloaddition reaction with electron-poor π-systems X = Y.
Somewhat surprisingly, palladium catalyzed (3 + 2) cycloadditions of VCPs with imines have been reported only very recently (2018–2019), despite the unquestionable synthetic value of the resulting pyrrolidines. Four examples have been published on this reaction. A work coordinated by Xiao and Guo described the cycloaddition between (mainly) gem-dicyano VCPs and sulfamate-derived, cyclic, imines [7] (Scheme 2, top). Vitale and co-workers reported closely related results [8], but extended the reaction to other VCPs, and to N-tosyl aldimines derived from aromatic and aliphatic aldehydes (Scheme 2, middle). Sulfamate-derived cyclic imines were also employed by de Figueiredo, Campagne and co-workers in palladium catalyzed cycloadditions with more elaborated 2-alkyl-2-alkylidene cyclopropane 1,1-dicarboxylates VCPs [9]. More recently, Chu, He, Liu and co-workers published the first examples of highly enantioselective (3 + 2) cycloadditions between VCPs and imines [10] (Scheme 2, bottom). The reactions, catalyzed by chiral palladium complexes bearing phosphoramidite ligands, give very good results with both N-sulfonyl aldimines derived from aromatic aldehydes, and N-Boc isatin ketimines. Less recently, Plietker, Kurahashi, Matsubara and their co-workers demonstrated that low valent iron and nickel complexes catalyze the (3 + 2) cycloaddition reaction between VCPs and imines [11,12,13]. In all cases, which included a moderately enantioselective example, electron-poor N-sulfonyl aldimines were used as reaction partners.
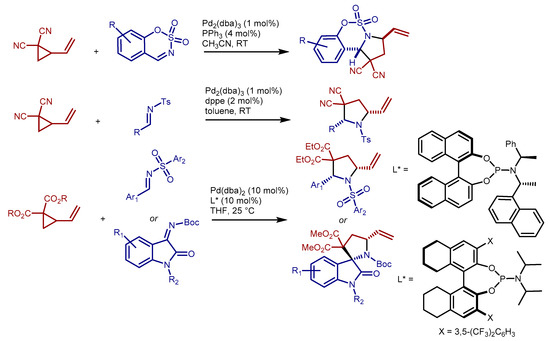
Scheme 2.
Palladium catalyzed (3 + 2) cycloadditions of VCPs with imines: selected literature examples. dba: dibenzylidene acetone; dppe: 1,3-di(triphenylphosphino)propane.
Herein, we present a conceptually new approach to the catalytic (3 + 2) cycloaddition between VCPs and imines, based on the synergistic combination of a palladium catalyst (A), for zwitterion formation from the VCP, and a phosphoric acid catalyst (B), for imine activation, along the lines of the simplified working model sketched in Scheme 3. Weaker acids (e.g., AcOH) have been used as sub-stoichiometric additives in palladium catalyzed cycloadditions [14]. However, the role of these acids is to buffer anionic intermediates, ultimately resulting in improved stereoselectivities in the reactions. In contrast, in our work, the acid serves to activate the electrophilic reaction partner. In fact, with this synergistic catalysis approach [15,16], we are able to engage less electrophilic N-aryl and N-benzyl imines (vs. N-sulfonyl imines, see Scheme 2) in reactions with VCPs, thus augmenting the structural diversity achievable. Implementation to a three-component version by forming the imine in situ is also possible, while the acid catalyst component (B) introduces a new option [17] for enantioinduction in VCPs reactions, beyond the common strategy based on chiral ligands at palladium. A similar approach has been briefly explored, but then abandoned in favor of chiral ligands, by de Figueredo, Campagne and co-workers during their studies on palladium catalyzed enantioselective 2-vinylaziridines cycloadditions [9]. On the other hand, a distinct synergistic catalysis combination was used in enantioselective cycloadditions between VCPs and enals [18,19,20,21,22], wherein chiral secondary amine and achiral palladium catalysis were successfully merged [23].
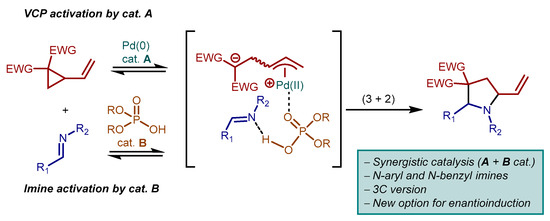
Scheme 3.
This work: synergistically (palladium A + phosphoric acid B catalysts) catalyzed (3 + 2) cycloaddition between N-aryl and N-benzyl imines and VCPs: simplified working model.
2. Results and Discussion
We performed exploratory experiments with different VCPs and N-aryl imines, under a variety of reaction conditions. The first convincing proof of the feasibility of our plans was obtained by employing indan-1,3-dione derived VCP 1, N-phenyl imine 2a, and a combination of Pd(PPh3)4 and diphenylphosphoric acid 3a as catalysts (10 mol% each) in toluene (Table 1, entry 1). The reaction performed with such combination resulted complete in less than one hour, affording the corresponding pyrrolidine 4a in very good yield and moderate diastereoselectivity, in favor of the trans-isomer. The next two entries (2,3) of Table 1 report two crucial experiments, wherein the presence of both catalyst components (Pd(PPh3)4 and 3a) was found to be essential to achieve an efficient transformation. While, unsurprisingly, omitting the palladium catalyst led to no reactivity, the reaction performed without 3a gave full consumption of 1 after prolonged time. However, the poor yield of 4a (49% yield after 24 h, vs. 92% yield after 1 h in the presence of 3a) indicates that the dipolar intermediate was not being channeled efficiently towards product 4a formation. Variation in the acid component 3 showed that a weaker acid catalyst, such as benzoic acid 3b (pKa (3b) = 11.1 in DMSO [24], vs. pKa (3a) = 3.88 in DMSO [25]) was much less effective (entry 4), substantiating our assumption on the requirement of imine activation for reactivity. More acidic (+)-camphor-10-sulphonic acid 3c as catalyst (pKa of methanesulfonic acid = 1.6 in DMSO [26]) afforded results (entry 5) comparable to the phosphoric acid 3a. With 3a, we proceeded to evaluate the influence of the loadings of the two catalytic species. The experiments reported in entries 6–8 show that decreasing the amount of palladium improves the diastereoselectivity of the reaction, reaching up to ca. 5:1 when 2.5 mol% of Pd(PPh3)4 was applied. On the contrary, the amount of 3a does not seem to influence strongly the reaction outcome. Pd(dba)2 as palladium component could not promote efficiently the reaction (entry 9), in line with the literature [8]. Thus, keeping a 2.5 mol% Pd(PPh3)4 and 10 mol% 3a loadings, a short solvent screening was performed. The reaction proceeded smoothly in all solvents testes, with diastereoselectivities comparable or lower than in toluene (compare entries 10–12 with entry 6). Halide salts are often used as additives in reactions involving zwitterionic allyl palladium intermediates [9,27]. By coordinating palladium, these additives increase the rate of π-σ-π isomerization of the allyl palladium complex [28], ultimately influencing the stereochemical outcome of the reactions (vide infra). In our case, application of one equivalent of tetra-n-butyl ammonium bromide and iodide did improve the stereoselectivity of the reaction. However, an unacceptable rate lowering was also observed (entries 14, 15). On the contrary, the corresponding chloride salt gave a less noticeable effect (entry 13).

Table 1.
Variations of catalysts and reaction conditions in the (3 + 2) cycloaddition between VCP 1 and imine 2a: selected results.
We then moved to study the scope of the reaction, by applying conditions related to entry 6 of Table 1 to different imines 2 (Scheme 4, Method A). However, first, we developed a related three-component protocol (Scheme 4, Method B): the imine 2 is formed in situ, taking advantage of the activity of catalyst 3a in promoting the condensation reaction, and then VCP 1 and Pd(PPh3)4 are added. As shown in Scheme 4, product 4a was obtained in comparable yields and diastereomeric ratios with the two methods, demonstrating the viability of the three-component protocol as well as the tolerance of the process to traces of water. Then, we applied imines 2b–f derived from different aldehydes and aniline. The resulting pyrrolidines 4b–f were obtained in good yields, including 4f from an aliphatic imine. The poor stability of this imine was overcome by applying the three-component protocol (Method B). Diastereomeric ratios of products 4b–f, measured on the crude mixtures, ranged from 4:1 for 4b to 7.4:1 for 4e. Imines 2g–j derived by combining benzaldehyde with different amines were also employed, affording the pyrrolidines 4g–j in good yields and variable diastereoselectivities. The success of the reaction of benzylamine-derived 2j shows the applicability of these protocols to a different class of imines, and its potential in delivering N-benzyl pyrrolidines. Finally, three VCPs bearing other gem-EWGs, derived from diethylmalonate, malononitrile, and Meldrum’s acid, respectively, were put to test with imine 2a. Unfortunately, none of these substrates afforded the desired pyrrolidines with good results. Even by attempting to fine-tune the reaction conditions, extensive degradation of these substrates (and of imine 2a) occurred. However, traces (<10%) of products could be observed at least for the malononitrile and Meldrum’s acid derived VCPs, leaving hope for future developments.
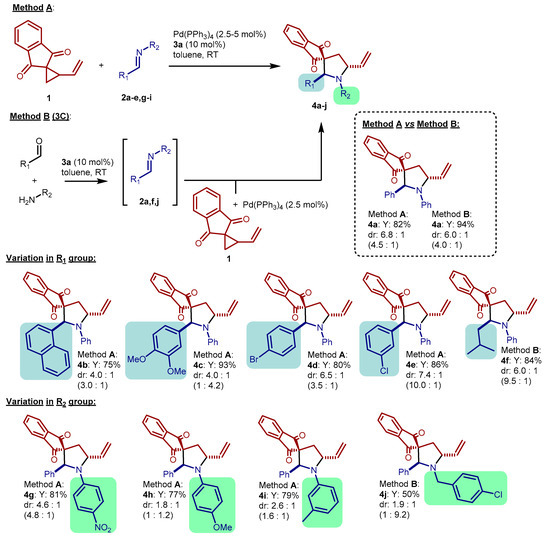
Scheme 4.
Scope of the reaction. Method A: VCP 1 (39.6 mg, 0.20 mmol), imine 2 (0.30 mmol), 3a (5.0 mg, 0.020 mmol, 10 mol%), Pd(PPh3)4 (5.8–11.6 mg, 0.0025–0.005 mmol, 2.5–5 mol%), toluene (2.4 mL), RT, 0.5–24 h. Method B: aldehyde (0.30 mmol), amine (0.30 mmol), 3a (5.0 mg, 0.020 mmol, 10 mol%), toluene (1.2 mL), RT, 30 min; then, VCP 1 (40 mg, 0.20 mmol), Pd(PPh3)4 (5.8 mg, 0.0025 mmol, 2.5 mol%), toluene (1.2 mL), RT-40 °C, 1–42 h. Yields of compounds 4 determined after purification by chromatography on silica gel. Dr determined on the crude mixtures by 1H NMR; dr in brackets refer to dr after chromatography. For further details, see Experimental Section.
In most cases, variations in the diastereomeric ratios measured before and after (dr in brackets in Scheme 4) chromatographic purification on silica gel were observed. These discrepancies were attributed to two reasons. First, the diastereosiomers of compounds 4 feature slightly different Rfs. Partial losses during chromatographic purification, due to uncollected fractions, led to slightly different diastereomeric ratios between crude and purified products 4a,b,d–g,i. Nevertheless, the different Rfs allowed to obtain the two diastereoisomers of 4a in diastereoenriched form, and to assign the relative configuration of the major one as trans, and of the minor one as cis, by 1H NOESY NMR experiments (see Supplementary Materials). Similarities between the 1H NMR spectra led us to conclude that all catalytic reactions reported in Scheme 4 furnished the trans-diastereoisomeric products 4a–j as the major isomers. Additionally, silica gel induced epimerization of the trans-isomers to their more stable [8] cis-counterparts is the second reason for the differences in dr before and after purification. Such process is particularly relevant in the case of the products 4c, 4h, and 4j, derived from the most electron rich imines 2c, 2h, and 2j. In these cases, we observed an inversion of the relative stereochemistry between the crude and the purified products 4, which resulted enriched in the cis-isomers.
We can thus assume that the catalytic reactions, which lead to less stable trans-4 as major isomers, are under kinetic control. A rationalization of such kinetic trans-diastereoselectivity can be built upon the typical Curtin–Hammett control scenario proposed in the literature for palladium catalyzed VCPs cycloaddition reactions [29,30]. The combination of such model with our working hypothesis-simplified in Scheme 3—results in the tentative mechanistic picture sketched in Scheme 5. Activation of the VCP 1 and imine 2a by the two catalytic species triggers a Mannich-type addition, giving two diastereomeric π-allyl palladium intermediates I and II. These intermediates evolve to the two allylic amination transition states III and IV, heading to the trans-4a and the cis-4a product, respectively. Under Curtin-Hammett control, intermediates I and II are in fast equilibrium, and trans-selectivity is given by the lower energy of transition state III compared to transition state IV. This energy difference is due to severe 1,3-diaxial strain in the latter one. Equilibration between I and II can occur via σ-π-σ allyl palladium isomerization, or even retro-Mannich reaction. The results obtained by using halide additives, in which diastereoselectivities increased (Table 1, entries 13–15), reinforce the plausibility of this proposed pathway. On the other hand, the exact role of the acid component in the different steps of the catalytic cycle, as well as its potential coordination to palladium species/intermediates, are far from being fully understood. The potential reversibility of many of the steps of the catalytic cycle brings additional complexity. Nevertheless, the results obtained with a chiral acid (vide infra) constitute a convincing demonstration that the acid is indeed involved in the stereo-determining step(s) of the reaction. Therefore, alternative roles, such as simple activation of the palladium complex by ligand displacement, can be excluded. Last, silica gel equilibration between trans-4a and cis-4a can occur via a retro-Mannich–Mannich sequence. The intermediate V of this process bears a positive charge on the imine. Accordingly, as observed experimentally, compounds carrying electron-donating groups able to stabilize such positive charge (e.g., 4c, 4h, and 4j) are more prone to epimerize compared to 4a.
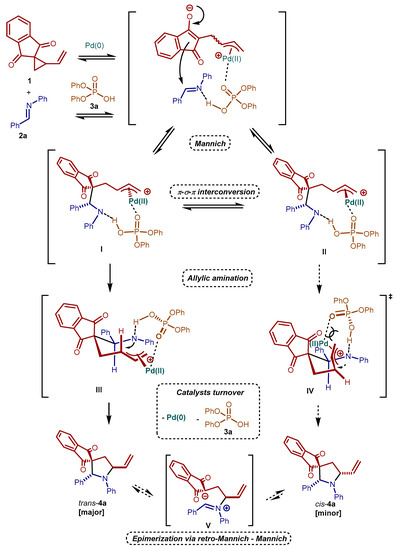
Scheme 5.
Proposed reaction pathway for the synergistically catalyzed (3 + 2) formal cycloaddition reaction between VCP 1 and imine 2a.
Inspired by a recent work [31], we decided to include our synergistically catalyzed reaction into a more complex, sequential process. We aimed to use the palladium catalyst for two sequential, mechanistically distinct, catalytic reactions: the synergistically catalyzed cycloaddition, and a Suzuki–Miyaura cross-coupling (Scheme 6). To our delight, addition of (2,3-dimethoxyphenyl)boronic acid, an inorganic base, and cyclopentyl methyl ether to the mixture of the reaction between 1 and 4-bromobenzaldehyde derived imine 2d, once complete, followed by heating to reflux for 18 h, afforded product 5 derived from this sequential transformation in good yield. Thus, no significant deactivation of the palladium catalyst occurred during the cycloaddition, allowing its involvement in subsequent transformations. Careful inspection of the 1H NMR data of compound 5, obtained as diastereomeric mixture, suggested cis-5 as the major diastereoisomer. In contrast, the cycloaddition provided trans-4d as major product. The cross-coupling conditions—heating at reflux temperature for prolonged time—are likely the cause of such epimerization of 4d and/or of 5.
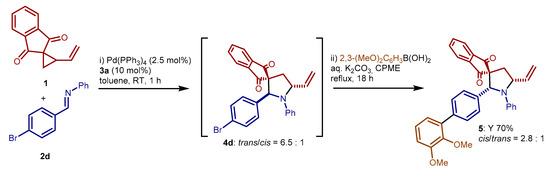
Scheme 6.
One-pot, sequential cycloaddition—Suzuki–Miyaura reaction.
A reasonable implementation of this synergistic methodology is the utilization of chiral phosphoric acid catalysts [32], conveying an enantioselective version of this reaction. To this end, we screened several common phosphoric acids derived from BINOL and at the same explored variations in reaction conditions (solvent, temperature, stoichiometry, concentration, additives, phosphine ligands, etc.; see Supplementary Materials). After a multitude of experiments, we reached the result reported in Scheme 7. Using (R)-H8-BINOL derived catalyst 3d [33], in combination with Pd(PPh3)4 and performing the reaction at low temperature, the pyrrolidine 4a is produced with high trans-selectivity, and moderate enantiomeric excess. Curiously, as shown in Scheme 7, better results in terms of diastereo- and enantioselectivities were obtained by using a higher catalyst loading of the palladium component, compared to the phosphoric acid. This contrasts the reactions with achiral 3a (Table 1). The different reaction temperature might be the main reason for such divergent behavior.
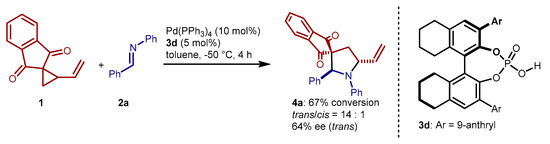
Scheme 7.
Formal (3 + 2) cycloaddition catalyzed by Pd(PPh3)4 and chiral phosphoric acid 3d.
3. Experimental Section
3.1. Materials and Methods
1H and 13C NMR spectra were recorded on a Varian Mercury 400 spectrometer. Chemical shifts (δ) are reported in ppm relative to solvent signals for 1H (CHCl3, 7.26 ppm) and 13C NMR (CDCl3, 77.0 ppm) [34]. 13C NMR were acquired under 1H broadband decoupling. Mass spectra were recorded on a micromass LCT spectrometer using electronspray ionization techniques. Chromatographic purifications were performed using 70–230 mesh silica gel. The relative configuration of the major and minor diastereoisomers of pyrrolidine 4a was assigned as 2,5-trans and 2,5-cis, respectively, by 1 D NOESY NMR experiments (see Supplementary Materials). The relative configuration of the remaining pyrrolidines 4 was assigned by analogy. Analytical grade solvents and commercially available reagents were used as received, unless otherwise stated. Imines 2 were prepared by refluxing equimolar amounts of the corresponding benzaldehydes and anilines in EtOH, and collected by filtration, or in toluene, using a Dean–Stark apparatus. VCP 1 was prepared following a literature protocol [35]. Catalyst 3d was synthesized as previously reported [33].
3.2. General Procedures for the Synthesis of Pyrrolidines 4
Method A: VCP 1 (39.6 mg, 0.20 mmol), imine 2 (0.30 mmol) and diphenylphosphoric acid 3a (5.0 mg, 0.020 mmol, 10 mol%) were sequentially added to a vial equipped with a magnetic stirring bar. Toluene (2.40 mL) was then added, followed by Pd(PPh3)4 (5.8 or 11.6 mg, 0.0025 or 0.005 mmol, 2.5 or 5.0 mol%). The resulting solution was vigorously stirred for 0.5–24 h, then passed through a short plug of silica, and the plug flushed with Et2 O. After evaporation of the solvents, the residue was analyzed by 1H NMR to determine the diastereomeric ratios of the pyrrolidines 4 produced in the reaction. Chromatographic purification (n-hexane/EtOAc mixtures) afforded pure pyrrolidines 4 as diastereomeric mixtures.
Method B (three-component reaction): Aldehyde (0.30 mmol), amine (0.30 mmol) and diphenylphosphoric acid 3a (5.0 mg, 0.020 mmol, 10 mol%) were added to a vial equipped with a magnetic stirring bar, and dissolved in toluene (1.20 mL). The resulting solution was stirred at RT for 30 min. Then, VCP 1 (39.6 mg, 0.20 mmol) was added, followed by additional toluene (1.20 mL) and Pd(PPh3)4 (5.8 mg, 0.0025 mmol, 2.5 mol%). The mixture was stirred at RT or 40 °C for 1–42 h, then passed through a short plug of silica, and the plug flushed with Et2O. After evaporation of the solvents, the residue was analyzed by 1H NMR to determine the diastereomeric ratios of the pyrrolidines 4 produced in the reaction. Chromatographic purification (n-hexane/EtOAc mixtures) afforded pure pyrrolidines 4 as diastereomeric mixtures.
3.3. Experimental Results and Characterization Data of Pyrrolidines 4
1′,2′-Diphenyl-5′-vinylspiro[indene-2,3′-pyrrolidine]-1,3-dione (4a). Following the general procedure, Method A (5 mol% Pd(PPh3)4, 0.5 h), the title compound was obtained as a pale yellow solid in 82% yield (trans/cis ratio: in the crude mixture, 6.8:1; after chromatographic purification, 4.5:1). Following the general procedure, Method B (2.5 mol% Pd(PPh3)4, 40 °C, 24 h), the title compound was obtained as a pale yellow solid in 94% yield (trans/cis ratio: in the crude mixture, 6.0:1; after chromatographic purification, 4.0:1). Extensive chromatography enabled the isolation of small amounts of diastereoenriched trans and cis isomers. 1H NMR [trans-4a]: 7.94 (dt, J = 7.6, 1.1 Hz, 1 H), 7.73 (td, J = 7.4, 1.3 Hz, 1 H), 7.66 (td, J = 7.4, 1.2 Hz, 1 H), 7.58 (ddd, J = 7.5, 1.3, 0.8 Hz, 1 H), 7.02 (dd, J = 8.7, 7.3 Hz, 2 H), 6.97–6.85 (m, 5 H), 6.62 (tt, J = 7.3, 1.1 Hz, 1 H), 6.56–6.49 (m, 2 H), 5.80 (ddd, J = 17.1, 9.8, 8.7 Hz, 1 H), 5.42–5.33 (m, 2 H), 5.29–5.17 (m, 2 H), 2.54 (dd, J = 13.0, 7.1 Hz, 1 H), 2.36 (dd, J = 13.0, 7.8 Hz, 1 H); 1H NMR [cis-4a]: 8.03–7.96 (m, 1 H), 7.86–7.70 (m, 3 H), 7.24–7.07 (m, 6 H), 6.78–6.65 (m, 4 H), 6.41 (ddd, J = 17.5, 10.3, 7.4 Hz, 1 H), 5.48 (d, J = 17.3 Hz, 1 H), 5.34 (d, J = 10.2 Hz, 1 H), 4.95 (s, 1 H), 4.82 (q, J = 7.5 Hz, 1 H), 2.55 (dd, J = 13.1, 7.8 Hz, 1 H), 2.44 (dd, J = 13.2, 7.3 Hz, 1 H); 13C NMR [signals of both diastereoisomers]: 201.4, 200.8, 200.2, 198.1, 147.5, 144.3, 142.5, 142.3, 141.7, 141.2, 141.0, 140.7, 139.2, 136.4, 136.0, 135.8, 135.7, 135.4, 129.3, 128.6, 128.5, 128.0, 127.9, 127.5, 127.3, 126.7, 123.5, 123.3, 123.0, 122.9, 118.3, 118.2, 117.6, 117.4, 116.1, 114.6, 72.9, 70.4, 64.4, 64.2, 64.0, 63.4, 38.1, 37.4; ESI-MS: 380 m/z [M + H]+.
2′-(Naphthalen-1-yl)-1′-phenyl-5′-vinylspiro[indene-2,3′-pyrrolidine]-1,3-dione (4b) Following the general procedure, Method A (5 mol% Pd(PPh3)4, 0.5 h), the title compound was obtained as a white solid in 75% yield (trans/cis ratio: in the crude mixture, 4.0:1; after chromatographic purification, 3.0:1). 1H NMR: 8.00 (d, J = 7.7 Hz, 1 Hcis), 7.93 (d, J = 7.2 Hz, 1 Hcis), 7.88–7.72 (m, 3 Hcis, 2 Htrans), 7.70–7.62 (m, 1 Htrans, 1 Hcis), 7.57–7.50 (m, 2 Htrans, 2 Hcis), 7.51–7.41 (m, 1 Htrans, 1 Hcis), 7.40–7.20 (m, 4 Htrans, 3 Hcis), 7.12–6.94 (m, 3 Htrans, 1 Hcis), 6.76–6.70 (m, 1 Hcis), 6.68–6.64 (m, 2 Hcis), 6.65–6.56 (m, 1 Htrans), 6.56–6.50 (m, 2 Htrans), 6.39 (ddd, J = 17.4, 10.2, 7.3 Hz, 1 Hcis), 6.24 (s, 1 Htrans), 6.11 (ddd, J = 17.1, 10.1, 8.7 Hz, 1 Htrans), 5.76 (s, 1 Hcis), 5.59 (dd, J = 17.3, 1.2 Hz, 1 Hcis), 5.48–5.40 (m, 1 Htrans, 1 Hcis), 5.35–5.26 (m, 2 Htrans), 4.99 (q, J = 7.8 Hz, 1 Hcis), 2.79 (dd, J = 13.2, 7.7 Hz, 1 Htrans), 2.59 (dd, J = 13.1, 9.0 Hz, 1 Hcis), 2.51–2.40 (m, 1 Htrans, 1 Hcis); 13C NMR [signals of both diastereoisomers]: 202.1, 201.5, 199.6, 197.4, 147.5, 144.5, 142.6, 141.9, 141.4, 141.1, 140.6, 140.4, 135.9, 135.9, 135.6, 135.6, 135.1, 134.4, 133.7, 133.5, 131.8, 130.9, 130.2, 129.4, 129.1, 128.7, 128.5, 128.1, 128.0, 127.9, 126.8, 125.8, 125.7, 125.6, 125.4, 125.3, 125.1, 123.4, 123.1, 122.9, 122.5, 122.1, 121.8, 118.2, 117.6, 117.1, 116.8, 114.5, 112.7, 68.9, 64.9, 64.2, 64.1, 62.7, 62.3, 38.5, 38.4; ESI-MS: 430 m/z [M + H]+.
2′-(3,4-Dimethoxyphenyl)-1′-phenyl-5′-vinylspiro[indene-2,3′-pyrrolidine]-1,3-dione (4c) Following the general procedure, Method A (5 mol% Pd(PPh3)4, 2 h), the title compound was obtained as a pale yellow solid in 93% yield (trans/cis ratio: in the crude mixture, 4.0:1; after chromatographic purification, 1:4.2). 1H NMR: 7.98 (d, J = 6.7 Hz, 1 Hcis), 7.93 (d, J = 7.5 Hz, 1 Htrans), 7.83–7.70 (m, 3 Hcis, 1 Htrans), 7.67 (td, J = 7.4, 1.2 Hz, 1 Htrans), 7.61 (d, J = 7.4 Hz, 1 Htrans), 7.18–7.06 (m, 2 Hcis), 7.02 (dd, J = 8.6, 7.3 Hz, 2 Htrans), 6.76–6.65 (m, 6 Hcis), 6.65–6.58 (m, 2 Htrans), 6.56–6.52 (m, 2 Htrans), 6.49–6.37 (m, 2 Hcis, 2 Htrans), 5.76 (ddd, J = 17.1, 10.0, 8.7 Hz, 1 Htrans), 5.47 (dt, J = 17.3, 1.2 Hz, 1 Hcis), 5.40–5.29 (m, 1 Hcis, 2 Htrans), 5.27–5.19 (m, 2 Htrans), 4.90 (s, 1 Hcis), 4.83–4.71 (m, 1 Hcis), 3.80 (s, 3 Hcis), 3.67 (s, 3 Hcis), 3.65 (s, 3 Htrans), 3.53 (s, 3 Htrans), 2.59 (dd, J = 13.1, 8.1 Hz, 1 Hcis), 2.50 (dd, J = 12.9, 7.0 Hz, 1 Htrans), 2.44–2.30 (m, 1 Hcis, 1 Htrans); 13C NMR [signals of both diastereoisomers]: 201.4, 201.0, 200.4, 198.2, 148.8, 148.4, 148.3, 148.0, 147.6, 144.4, 142.7, 142.4, 141.8, 141.2, 141.2, 140.5, 136.0, 135.9, 135.6, 135.3, 131.4, 128.6, 127.9, 123.4, 123.2, 123.1, 122.8, 118.9, 118.5, 118.2, 117.8, 117.4, 115.8, 114.8, 110.9, 109.7, 73.0, 70.3, 64.5, 64.2, 64.1, 63.7, 55.8, 55.7, 55.6, 55.6, 37.9, 37.0; ESI-MS: 440 m/z [M + H]+.
2′-(4-Bromophenyl)-1′-phenyl-5′-vinylspiro[indene-2,3′-pyrrolidine]-1,3-dione (4d) Following the general procedure, Method A (5 mol% Pd(PPh3)4, 0.5 h), the title compound was obtained as a yellow solid in 80% yield (trans/cis ratio: in the crude mixture, 6.5:1; after chromatographic purification, 3.5:1). 1H NMR: 7.99 (dd, J = 6.4, 1.2 Hz, 1 Hcis), 7.95 (dd, J = 7.5, 1.1 Hz, 1 Htrans), 7.86–7.67 (m, 2 Htrans, 3 Hcis), 7.68–7.61 (m, 1 Htrans), 7.39 (d, J = 7.5 Hz, 1 Hcis), 7.33 (d, J = 8.6 Hz, 1 Hcis), 7.15–7.07 (m, 2 Htrans, 2 Hcis), 7.03 (dd, J = 8.6, 7.3 Hz, 2 Htrans, 2 Hcis), 6.82 (d, J = 8.0 Hz, 2 Htrans), 6.75 (t, J = 7.3 Hz, 1 Hcis), 6.65 (t, J = 7.3 Hz, 1 Htrans, 2 Hcis), 6.50 (d, J = 7.6 Hz, 2 Htrans), 6.39 (ddd, J = 17.5, 10.3, 7.4 Hz, 1 Hcis), 5.76 (ddd, J = 17.1, 10.0, 8.7 Hz, 1 Htrans), 5.47 (d, J = 17.3 Hz, 1 Hcis), 5.42–5.16 (m, 4 Htrans, 1 Hcis), 4.92 (s, 1 Hcis), 4.79 (q, J = 7.4 Hz, 1 Hcis), 2.70–2.50 (m, 1 Htrans, 1 Hcis), 2.42–2.30 (m, 1 Htrans, 1 Hcis); 13C NMR [signals of both diastereoisomers]: 201.1, 200.6, 199.9, 198.1, 147.3, 143.9, 142.3, 142.1, 141.6, 141.0, 140.9, 140.3, 138.4, 136.2, 136.1, 135.9, 135.73, 135.71, 131.7, 131.2, 128.7, 128.4, 128.0, 123.6, 123.4, 123.3, 123.2, 123.1, 121.8, 121.4, 118.5, 118.4, 118.0, 117.6, 116.2, 114.7, 72.1, 69.5, 64.2, 64.2, 64.0, 63.1, 38.5, 37.8; ESI-MS: 458, 460 m/z [M + H]+.
2′-(3-Chlorophenyl)-1′-phenyl-5′-vinylspiro[indene-2,3′-pyrrolidine]-1,3-dione (4e) Following the general procedure, Method A (2.5 mol% Pd(PPh3)4, 24 h), the title compound was obtained as a pale yellow solid in 86% yield (trans/cis ratio: in the crude mixture, 7.4:1; after chromatographic purification, 10.0:1). 1H NMR [signals of trans-4e]: 7.96 (dt, J = 7.8, 0.9 Hz, 1 H), 7.76 (td, J = 7.4, 1.3 Hz, 1 H), 7.69 (td, J = 7.6, 1.2 Hz, 1 H), 7.62 (dt, J = 7.5, 0.9 Hz, 1 H), 7.07–7.00 (m, 2 H), 6.95–6.87 (m, 3 H), 6.83 (br d, J = 7.6 Hz, 1 H), 6.64 (td, J = 7.4, 1.0 Hz, 1 H), 6.54–6.49 (m, 2 H), 5.78 (ddd, J = 18.8, 10.0, 8.8 Hz, 1 H), 5.37 (d, J = 17.1 Hz, 1 H), 5.32 (s, 1 H), 5.25–5.17 (m, 2 H), 2.54 (dd, J = 13.2, 7.2 Hz, 1 H), 2.34 (dd, J = 12.9, 7.4 Hz, 1 H); 13C NMR [signals of trans-4e]: 200.5, 199.8, 143.9, 142.1, 141.6, 140.4, 138.9, 136.1, 135.6, 134.0, 129.4, 128.1, 127.9, 127.4, 125.7, 123.2, 123.1, 118.3, 118.0, 117.5, 69.6, 64.21, 64.16, 38.2; ESI-MS: 414, 416 m/z [M + H]+.
2′-Isobutyl-1′-phenyl-5′-vinylspiro[indene-2,3′-pyrrolidine]-1,3-dione (4f) Following the general procedure, Method B (2.5 mol% Pd(PPh3)4, RT, 1 h), the title compound was obtained as a pale orange solid in 84% yield (trans/cis ratio: in the crude mixture, 6.0:1; after chromatographic purification, 9.5:1). 1H NMR [signals of trans-4f]: 8.00–7.96 (m, 1 H), 7.94–7.91 (m, 1 H), 7.86–7.82 (m, 2 H), 7.24–7.17 (m, 2 H), 6.77–6.71 (m, 3 H), 5.94 (ddd, J = 17.1, 10.3, 6.8 Hz, 1 H), 5.30 (dt, J = 17.2, 1.2 Hz, 1 H), 5.17 (dt, J = 10.4, 1.2 Hz, 1 H), 4.59 (q, J = 8.0 Hz, 1 H), 4.01 (dd, J = 9.6, 4.0 Hz, 1 H), 2.47 (dd, J = 13.2, 8.6 Hz, 1 H), 2.37 (dd, J = 13.1, 7.7 Hz, 1 H), 1.94 (ddd, J = 14.1, 9.4, 4.2 Hz, 1 H), 1.56 (ddd, J = 14.3, 9.1, 4.1 Hz, 1 H), 1.23–1.13 (m, 1 H), 0.91 (d, J = 6.5 Hz, 3 H), 0.83 (d, J = 6.5 Hz, 3 H); 13C NMR [signals of trans-4e]: 201.3, 199.2, 147.2, 142.2, 140.7, 140.3, 135.9, 135.8, 128.9, 123.7, 123.1, 117.3, 115.3, 113.0, 65.1, 62.3, 60.5, 41.3, 37.8, 25.8, 23.4, 22.1; ESI-MS: 360 m/z [M + H]+.
1′-(4-Nitrophenyl)-2′-phenyl-5′-vinylspiro[indene-2,3′-pyrrolidine]-1,3-dione (4g) Following the general procedure, Method A (5 mol% Pd(PPh3)4, 18 h), the title compound was obtained as a yellow solid in 81% yield (trans/cis ratio: in the crude mixture, 4.6:1; after chromatographic purification, 4.8:1). 1H NMR: 8.01 (ddd, J = 7.3, 1.4, 0.8 Hz, 1 Hcis), 8.00–7.96 (m, 2 Hcis), 7.95 (dt, J = 7.7, 1.1 Hz, 1 Htrans), 7.92–7.86 (m, 2 Htrans), 7.86–7.79 (m, 3 Hcis), 7.76 (td, J = 7.4, 1.1 Hz, 1 Htrans), 7.69 (td, J = 7.4, 1.1 Hz, 1 Htrans), 7.60 (dt, J = 7.6, 1.0 Hz, 1 Htrans), 7.28–7.24 (m, 3 Hcis), 7.10–7.03 (m, 2 Hcis), 7.03–6.97 (m, 3 Htrans), 6.87–6.81 (m, 2 Htrans), 6.64–6.58 (m, 2 Hcis), 6.54–6.44 (m, 2 Htrans), 6.38 (ddd, J = 17.6, 10.3, 7.6 Hz, 1 Hcis), 5.77 (ddd, J = 17.1, 10.1, 8.5 Hz, 1 Htrans), 5.56–5.45 (m, 1 Htrans, 2 Hcis), 5.43 (s, 1 Htrans), 5.37–5.26 (m, 2 Htrans), 5.03 (s, 1 Hcis), 4.93 (q, J = 7.6 Hz, 1 Hcis), 2.64–2.43 (m, 1 Htrans, 2 Hcis), 2.40 (dd, J = 13.1, 8.6 Hz, 1 Htrans); 13C NMR [signals of both diastereoisomers]: 200.9, 199.8, 199.5, 197.2, 151.8, 149.6, 142.4, 141.9, 141.5, 140.6, 139.2, 139.0, 138.7, 138.1, 137.3, 136.4, 136.2, 136.1, 136.0, 134.8, 128.9, 128.5, 128.2, 127.2, 126.5, 126.3, 125.3, 124.6, 123.8, 123.5, 123.3, 123.2, 118.8, 117.7, 116.4, 113.2, 72.1, 70.5, 64.4, 63.9, 63.6, 62.6, 38.6, 37.4; ESI-MS: 447 m/z [M + Na]+.
1′-(4-Methoxyphenyl)-2′-phenyl-5′-vinylspiro[indene-2,3′-pyrrolidine]-1,3-dione (4h) Following the general procedure, Method A (5 mol% Pd(PPh3)4, 0.5 h), the title compound was obtained as a dark yellow solid in 77% yield (trans/cis ratio: in the crude mixture, 1.8:1; after chromatographic purification, 1:1.2). 1H NMR: 7.99–7.96 (m, 1 Hcis), 7.96–7.90 (m, 1 Htrans), 7.83–7.67 (m, 2 Hcis, 2 Htrans), 7.63 (td, J = 7.4, 1.1 Hz, 1 Hcis), 7.56 (dt, J = 7.5, 1.0 Hz, 1 Htrans), 7.21–7.10 (m, 3 Hcis, 3 Htrans), 6.93 (br s, 4 Hcis), 6.69 (br s, 4 Htrans), 6.65–6.59 (m, 2 Htrans), 6.54–6.47 (m, 2 Hcis), 6.37 (ddd, J = 17.6, 10.2, 7.5 Hz, 1 Hcis), 5.79 (ddd, J = 17.1, 10.0, 8.9 Hz, 1 Htrans), 5.43 (dt, J = 17.3, 1.2 Hz, 1 Hcis), 5.38–5.26 (m, 1 Hcis, 2 Htrans), 5.22–5.11 (m, 2 Htrans), 4.88 (s, 1 Hcis), 4.73–4.62 (m, 1 Hcis), 3.68 (s, 3 Hcis), 3.64 (s, 3 Htrans), 2.59–2.50 (m, 1 Hcis, 1 Htrans), 2.46–2.31 (m, 1 Hcis, 1 Htrans); 13C NMR [signals of both diastereoisomers]: 201.7, 201.1, 200.4, 198.5, 152.9, 152.0, 142.5, 142.4, 141.8, 141.7, 141.4, 141.1, 140.6, 139.1, 138.4, 136.4, 135.9, 135.8, 135.6, 135.3, 128.4, 127.9, 127.8, 127.5, 127.4, 126.9, 123.4, 123.2, 122.9, 122.9, 119.8, 117.4, 117.0, 116.1, 114.2, 113.5, 73.8, 70.7, 65.1, 64.7, 63.5, 55.5, 55.3, 37.7, 37.4; ESI-MS: 410 m/z [M + H]+.
2′-Phenyl-1′-(m-tolyl)-5′-vinylspiro[indene-2,3′-pyrrolidine]-1,3-dione (4i) Following the general procedure, Method A (5 mol% Pd(PPh3)4, 18 h), the title compound was obtained as an orange waxy solid in 79% yield (trans/cis ratio: in the crude mixture, 2.6:1; after chromatographic purification, 1.6:1). 1H NMR: 8.02–7.97 (m, 1 Hcis), 7.95 (dd, J = 7.6, 1.1 Hz, 1 Htrans), 7.85–7.69 (m, 1 Htrans, 3 Hcis), 7.65 (td, J = 7.4, 1.2 Hz, 1 Htrans), 7.58 (dt, J = 7.6, 1.1 Hz, 1 Htrans), 7.24–7.19 (m, 1 Htrans, 1 Hcis), 7.01–6.87 (m, 3 Htrans, 5 Hcis), 6.60–6.35 (m, 4 Htrans, 4 Hcis), 6.30 (dd, J = 8.1, 2.4 Hz, 1 Htrans), 5.82 (ddd, J = 17.1, 10.0, 8.8 Hz, 1 Htrans), 5.50 (dt, J = 17.3, 1.2 Hz, 1 Hcis), 5.42–5.32 (m, 2 Htrans, 1 Hcis), 5.27–5.20 (m, 1 Htrans), 4.95 (s, 1 Htrans), 4.83 (q, J = 7.5 Hz, 1 Hcis), 2.59–2.51 (m, 1 Htrans, 1 Hcis), 2.43 (dd, J = 13.1, 7.3 Hz, 1 Hcis), 2.36 (dd, J = 13.0, 7.7 Hz, 1 Htrans), 2.20 (s, 3 Hcis), 2.15 (s, 3 Htrans); 13C NMR [signals of both diastereoisomers]: 201.4, 200.9, 200.2, 198.1, 147.6, 144.3, 142.5, 142.3, 141.7, 141.2, 141.0, 140.8, 139.4, 138.2, 137.4, 136.1, 135.9, 135.8, 135.7, 135.3, 128.5, 128.5, 128.0, 127.9, 127.7, 127.5, 127.3, 126.7, 123.5, 123.3, 123.3, 123.0, 119.2, 119.1, 118.6, 117.2, 116.0, 115.5, 115.3, 111.9, 73.0, 70.5, 64.4, 64.2, 64.0, 63.4, 38.0, 37.3, 21.8, 21.7; ESI-MS: 394 m/z [M + H]+.
1′-(4-Chlorobenzyl)-2′-phenyl-5′-vinylspiro[indene-2,3′-pyrrolidine]-1,3-dione (4j) Following the general procedure, Method B (2.5 mol% Pd(PPh3)4, 40 °C, 42 h), the title compound was obtained as a white solid in 50% yield (trans/cis ratio: in the crude mixture, 1.9:1; after chromatographic purification, 1:9.2). 1H NMR [signals of cis-4j]: 7.85 (dt, J = 7.5, 0.9 Hz, 1 H), 7.66 (td, J = 7.3, 1.0 Hz, 1 H), 7.59 (td, J = 7.5, 1.2 Hz, 1 H), 7.49 (br d, J = 7.7 Hz, 1 H), 7.19–7.15 (m, 2 H), 7.09–7.01 (m, 5 H), 6.98–6.93 (m, 2 H), 5.97 (ddd, J = 18.6, 10.0, 8.6 Hz, 1 H), 5.27 (dd, J = 17.2, 1.3 Hz, 1 H), 5.15 (dd, J = 10.1, 1.6 Hz, 1 H), 3.97 (s, 1 H), 3.80 (d, J = 14.6 Hz, 1 H), 3.59 (d, J = 14.6 Hz, 1 H), 3.54 (q, J = 8.5 Hz, 1 H), 2.27 (dd, J = 13.3, 8.2 Hz, 1 H), 2.13 (dd, J = 13.0, 8.5 Hz, 1 H); 13C NMR [signals of cis-4j]: 202.6, 200.6, 142.7, 141.5, 140.0, 135.8, 135.5, 135.0, 134.2, 132.7, 131.2, 128.5, 128.03, 127.96, 127.94, 127.8, 122.7, 117.3, 75.6, 65.9, 64.0, 52.2, 36.4; ESI-MS: 428, 430 m/z [M + H]+.
3.4. One Pot Cycloaddition—Suzuki–Miyaura Reaction: Preparation of Compound 5
2′-(2′,3′-Dimethoxy-[1,1′-biphenyl]-4-yl)-1′-phenyl-5′-vinylspiro[indene-2,3′-pyrrolidine]-1,3-dione (5) VCP 1 (39.6 mg, 0.20 mmol), 4-bromobenzaldehyde derived imine 2d (57.2 mg, 0.22 mmol), and diphenylphosphoric acid 3a (5.0 mg, 0.020 mmol, 10 mol%) were sequentially added to a Schlenk tube equipped with a magnetic stirring bar. Toluene (2.40 mL) was then added, followed by Pd(PPh3)4 (5.8 mg, 0.0025 mmol, 2.5 mol%). After stirring the resulting solution for 1 h to complete the cycloaddition reaction, (2,3-dimethoxyphenyl)boronic acid (72 mg, 0.40 mmol), aqueous K2 CO3 (2 M, 260 μL, 0.52 mmol) and cyclopentyl methyl ether (126 μL) were added. The mixture was degassed by three freeze-pump-thaw cycles, then heated to reflux temperature for 18 h with stirring. The mixture was passed through a short plug of silica, and the plug flushed with Et2 O. After evaporation of the solvents, the residue was analyzed by 1H NMR, showing a 2.8:1 diastereomeric ratio, likely favoring the cis-isomer. Chromatographic purification (petroleum ether/EtOAc 5:1) afforded the title compound as a waxy orange solid in 70% yield (cis/trans = 2.8:1). 1H NMR: 8.00 (dt, J = 7.5, 1.0 Hz, 1 Hcis), 7.96 (dt, J = 7.7, 1.0 Hz, 1 Htrans), 7.81 (td, J = 7.3, 1.4 Hz, 1 Hcis), 7.78–7.68 (m, 2 Hcis, 1 Htrans), 7.64–7.55 (m, 2 Htrans), 7.41–7.38 (m, 1 Hcis, 1 Htrans), 7.18–6.94 (m, 6 Hcis, 6 Htrans), 6.94–6.86 (m, 1 Hcis, 1 Htrans), 6.83 (dd, J = 8.2, 1.5 Hz, 1 Htrans), 6.79–6.70 (m, 3 Hcis, 2 Htrans), 6.65 (tt, J = 7.3, 1.1 Hz, 1 Htrans), 6.60–6.56 (m, 1 Hcis), 6.49 (ddd, J = 17.5, 10.3, 7.4 Hz, 1 Hcis), 5.80 (ddd, J = 17.1, 10.0, 8.8 Hz, 1 Htrans), 5.56–5.47 (m, 1 Hcis), 5.44–5.34 (m, 1 Hcis, 2 Htrans), 5.32–5.22 (m, 2 Htrans), 5.02 (s, 1 Hcis), 4.87–4.78 (m, 1 Hcis), 3.89 (s, 3 Hcis), 3.84 (s, 3 Htrans), 3.46 (s, 3 Hcis), 3.20 (s, 3 Htrans), 2.63 (dd, J = 13.1, 8.1 Hz, 1 Hcis), 2.55 (dd, J = 12.9, 7.0 Hz, 1 Htrans), 2.48–2.36 (m, 1 Hcis, 1 Htrans); 13C NMR [signals of both diastereoisomers]: 201.4, 200.8, 200.3, 198.2, 153.0, 152.9, 147.6, 146.6, 146.4, 144.3, 142.6, 142.4, 141.9, 141.5, 141.2, 140.6, 137.8, 137.6, 137.3, 136.0, 135.7, 135.6, 135.4, 135.3, 135.2, 135.2, 129.4, 128.9, 128.6, 127.9, 127.0, 126.4, 124.0, 124.0, 123.5, 123.2, 123.0, 122.8, 122.5, 122.3, 118.4, 118.2, 117.7, 117.4, 116.0, 114.8, 111.4, 111.3, 73.0, 70.6, 64.6, 64.5, 64.3, 63.8, 60.4, 60.1, 55.9, 55.9, 37.8, 37.2; ESI-MS: 516 m/z [M + H]+.
3.5. Enantioselective Cycloaddition with Chiral Phosphoric Acid 3d
1′,2′-Diphenyl-5′-vinylspiro[indene-2,3′-pyrrolidine]-1,3-dione (4a)
VCP 1 (9.9 mg, 0.05 mmol), imine 2a (10.9 mg, 0.055 mmol) and chiral phosphoric acid 3d (1.8 mg, 0.0025 mmol, 5 mol%) were sequentially added to a vial equipped with a magnetic stirring bar, and dissolved in toluene (0.60 mL). After cooling the resulting solution to −50 °C, Pd(PPh3)4 (5.8 mg, 0.005 mmol, 10 mol%) was added. The mixture was stirred at −50 °C for 6 h, then passed through a short plug of silica, and the plug flushed with Et2 O. After evaporation of the solvents, the residue was analyzed by 1H NMR, showing a 67% conversion with a 14:1 diastereomeric ratio favoring the trans-isomer of pyrrolidine 4a. Chromatographic purification (n-hexane/EtOAc 6:1) afforded enantioenriched pyrrolidine 4a, which spectral data were consistent with previously obtained rac-4a. HPLC analysis on chiral stationary phase (Daicel Chiralcel OD-H column, n-hexane/i-PrOH = 95:5, flow 0.75 mL/min, UV detector operating at λ = 254 nm), showed 64% ee for the major trans-isomer (tmaj = 10.1 min, tmin = 10.9 min), and 40% ee for the minor cis-isomer (tmaj = 12.3 min, tmin = 15.1 min).
4. Conclusions
We have developed a conceptually new approach to the formal (3 + 2) cycloaddition between VCPs and imines. The synergistic combination of a palladium (for VCP activation) and acidic (for imine activation) catalyst allowed to use for the first time N-aryl and N-benzyl imines, thus extending the range of products obtainable. All previous examples, based on a single palladium catalyst species, employed in fact electron-poor N-sulfonyl and N-carbamoyl imines in this reaction. By adjusting the reaction conditions and the catalysts loading, we were able to obtain a range of N-aryl and N-benzyl polysubstitued pyrrolidines in an efficient manner, applying in some cases a three-component protocol (i.e., by forming the imine in situ). The possibility to engage the palladium species in a one-pot process, including a subsequent, and mechanistically distinct, reaction (a Suzuki–Miyaura cross coupling) was demonstrated. By using common chiral phosphoric acids as catalyst component, a moderately enantioselective version of the cycloaddition reaction was developed, affording the benchmark product with a moderate, yet promising, 64% enantiomeric excess. We cannot exclude, and we are confident in, that: (i) understanding the mechanistic subtleties of the reaction, which encompasses a complex pathway characterized by many potentially reversible steps and catalysts interactions; or: (ii) the employment of different, less conventional, chiral phosphoric acid structures; will ultimately enable the development of a highly enantioselective version of this reaction.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/10/2/150/s1: copies of NMR spectra and assignment of the relative configuration of compound 4a by NOESY NMR experiments, HPLC traces for the enantioselective reaction.
Author Contributions
Conceptualization: V.C., E.M. and L.B.; methodology: V.C., E.M., M.M., and A.G.; writing—original draft preparation: L.B.; writing—review and editing: all authors; supervision: V.C., M.F., and L.B.; project administration: M.F. and L.B.; funding acquisition: M.F. and L.B. All authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge financial support by the University of Bologna (RFO program), MIUR (FFABR 2017), and F.I.S. (Fabbrica Italiana Sintetici).
Acknowledgments
We thank Luca Zuppiroli for performing mass spectrometry measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jiao, L.; Yu, Z.-X. Vinylcyclopropane derivatives in transition-metal-catalyzed cycloadditions for the synthesis of carbocyclic compounds. J. Org. Chem. 2013, 78, 6842–6848. [Google Scholar] [CrossRef]
- Venkataraman, G.; Chandrasekaran, S. Recent advances in the synthesis and reactivity of vinylcyclopropanes. Synthesis 2016, 4347–4380. [Google Scholar]
- Meazza, M.; Guo, H.; Rios, R. Synthetic applications of vinyl cyclopropane opening. Org. Biomol. Chem. 2017, 15, 2479–2490. [Google Scholar] [CrossRef]
- Allen, B.D.W.; Lakeland, C.P.; Harrity, J.P.A. Utilizing palladium-stabilized zwitterions for the construction of N-heterocycles. Chem. Eur. J. 2017, 23, 13830–13857. [Google Scholar] [CrossRef]
- Fumagalli, G.; Stanton, S.; Bower, J.F. Recent methodologies that exploit C–C single-bond cleavage of strained ring systems by transition metal complexes. Chem. Rev. 2017, 117, 9404–9432. [Google Scholar] [CrossRef]
- Shimizu, I.; Ohashi, Y.; Tsuji, J. Palladium-catalyzed [3+2] cycloaddition reaction of vinylcyclopropanes with α,β-unsaturated esters or ketones. Tetrahedron Lett. 1985, 26, 3825–3828. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, C.; Shi, W.; Xiao, Y.; Guo, H. Pd-catalyzed diastereoselective [3+2] cycloaddition of vinylcyclopropanes with sulfamate-derived cyclic imines. Org. Biomol. Chem. 2018, 16, 4881–4887. [Google Scholar] [CrossRef]
- Ling, J.; Laugeois, M.; Ratovelomanana-Vidal, V.; Vitale, M.R. Palladium(0)-catalyzed diastereroselective (3+2) cycloadditions of vinylcyclopropanes with sulfonyl-activated imines. Synlett 2018, 29, 2288–2292. [Google Scholar]
- Spielmann, K.; Tosi, E.; Lebrun, A.; Niel, G.; van der Lee, A.; de Figueiredo, R.M.; Campagne, J.-M. Vinylaziridines and cyclopropanes in Pd-catalyzed (3+2)-cycloaddition reactions with cyclic N-sulfonyl imines. Tetrahedron 2018, 74, 6497–6511. [Google Scholar] [CrossRef]
- Huang, X.-B.; Li, X.-J.; Li, T.-T.; Chen, B.; Chu, W.-D.; He, L.; Liu, Q.-Z. Palladium-catalyzed highly enantioselective cycloaddition of vinyl cyclopropanes with imines. Org. Lett. 2019, 21, 1713–1716. [Google Scholar] [CrossRef]
- Dieskau, A.P.; Holzwarth, M.S.; Plietker, B. Fe-Catalyzed allylic C−C-bond activation: Vinylcyclopropanes as versatile a1,a3,d5-synthons in traceless allylic substitutions and [3 + 2]-cycloadditions. J. Am. Chem. Soc. 2012, 134, 5048–5051. [Google Scholar] [CrossRef] [PubMed]
- Pursley, D.; Plietker, B. Insertion of imines into vinylcyclopropanes catalyzed by nucleophilic iron complexes: A formal [3+2]-cycloaddition strategy for the synthesis of substituted pyrrolidine derivatives. Synlett 2014, 25, 2316–2318. [Google Scholar] [CrossRef]
- Tombe, R.; Kurahashi, T.; Matsubara, S. Nickel-catalyzed cycloaddition of vinylcyclopropanes to imines. Org. Lett. 2013, 15, 1791–1793. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Fandrick, D.R. Dynamic kinetic asymmetric cycloadditions of isocyanates to vinylaziridines. J. Am. Chem. Soc. 2003, 125, 11836–11837. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.E.; MacMillan, D.W.C. Synergistic catalysis: A powerful synthetic strategy for new reaction development. Chem. Sci. 2012, 3, 633–658. [Google Scholar] [CrossRef]
- Chen, D.-F.; Han, Z.-Y.; Zhou, X.-L.; Gong, L.-Z. Asymmetric organocatalysis combined with metal catalysis: Concept, proof of concept, and beyond. Acc. Chem. Res. 2014, 47, 2365–2377. [Google Scholar] [CrossRef]
- Mukherjee, S.; List, B. Chiral counteranions in asymmetric transition-metal catalysis: Highly enantioselective Pd/Brønsted acid-catalyzed direct α-allylation of aldehydes. J. Am. Chem. Soc. 2007, 129, 11336–11337. [Google Scholar] [CrossRef]
- Halskov, K.S.; Næsborg, L.; Tur, F.; Jørgensen, K.A. Asymmetric [3+2] cycloaddition of vinylcyclopropanes and α,β-unsaturated aldehydes by synergistic palladium and organocatalysis. Org. Lett. 2016, 18, 2220–2223. [Google Scholar] [CrossRef]
- Meazza, M.; Rios, R. Synergistic catalysis: Enantioselective ring expansion of vinyl cyclopropanes combining four catalytic cycles for the synthesis of highly substituted spirocyclopentanes bearing up to four stereocenters. Chem. Eur. J. 2016, 22, 9923–9928. [Google Scholar] [CrossRef]
- Laugeois, M.; Ponra, S.; Ratovelomanana-Vidal, V.; Michelet, V.; Vitale, M.R. Asymmetric preparation of polysubstituted cyclopentanes by synergistic Pd(0)/amine catalyzed formal [3+2] cycloadditions of vinyl cyclopropanes with enals. Chem. Commun. 2016, 52, 5332–5335. [Google Scholar] [CrossRef]
- Zhang, K.; Meazza, M.; Izaga, A.; Contamine, C.; Gimeno, M.C.; Herrera, R.P.; Rios, R. Synergistic catalysis: Asymmetric synthesis of cyclopentanes bearing four stereogenic centers. Synthesis 2017, 49, 167–174. [Google Scholar]
- Kamlar, M.; Franc, M.; Císařová, I.; Gyepes, R.; Vesely, J. Formal [3+2] cycloaddition of vinylcyclopropane azlactones to enals using synergistic catalysis. Chem. Commun. 2019, 55, 3829–3832. [Google Scholar] [CrossRef]
- Afewerki, S.; Córdova, A. Combinations of aminocatalysts and metal catalysts: A powerful cooperative approach in selective organic synthesis. Chem. Rev. 2016, 116, 13512–13570. [Google Scholar] [CrossRef]
- Olmstead, W.N.; Bordwell, F.G. Ion-pair association constants in dimethyl sulfoxide. J. Org. Chem. 1980, 45, 3299–3305. [Google Scholar] [CrossRef]
- Christ, P.; Lindsay, A.G.; Vormittag, S.S.; Neudörfl, J.-M.; Berkessel, A.; O’Donoghue, A.C. pKa Values of chiral Brønsted acid catalysts: Phosphoric acids/amides, sulfonyl/sulfuryl imides, and perfluorinated TADDOLs (TEFDDOLs). Chem. Eur. J. 2011, 17, 8524–8528. [Google Scholar] [CrossRef] [PubMed]
- McCallum, C.; Pethybridge, A.D. Conductance of acids in dimethyl-sulphoxide—II. Conductance of some strong acids in DMSO at 25 °C. Electrochim. Acta 1975, 20, 815–818. [Google Scholar] [CrossRef]
- Spielmann, K.; van der Lee, A.; de Figueiredo, R.M.; Campagne, J.-M. Diastereoselective palladium-catalyzed (3+2)-cycloadditions from cyclic imines and vinyl aziridines. Org. Lett. 2008, 20, 1444–1447. [Google Scholar] [CrossRef]
- Trost, B.M.; Toste, F.D. Regio- and enantioselective allylic alkylation of an unsymmetrical substrate: a working model. J. Am. Chem. Soc. 1999, 121, 4545–4554. [Google Scholar] [CrossRef]
- Trost, B.M.; Morris, P.J.; Sprague, S.J. Palladium-catalyzed diastereo- and enantioselective formal [3+2]-cycloadditions of substituted vinylcyclopropanes. J. Am. Chem. Soc. 2012, 134, 17823–17831. [Google Scholar] [CrossRef]
- Laugeois, M.; Ling, J.; Férard, C.; Michelet, V.; Ratovelomanana-Vidal, V.; Vitale, M.R. Palladium(0)-catalyzed dearomative [3+2] cycloaddition of 3-nitroindoles with vinylcyclopropanes: An entry to stereodefined 2,3-fused cyclopentannulated indoline derivatives. Org. Lett. 2017, 19, 2266–2269. [Google Scholar] [CrossRef]
- Leth, L.A.; Glaus, F.; Meazza, M.; Fu, L.; Thøgersen, M.K.; Bitsch, E.A.; Jørgensen, K.A. Decarboxylative [4+2] cycloaddition by synergistic palladium and organocatalysis. Angew. Chem. Int. Ed. 2016, 55, 15272–15726. [Google Scholar] [CrossRef]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: History and classification by mode of activation; Brønsted acidity, hydrogen Bonding, ion pairing, and metal phosphates. Chem. Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef] [PubMed]
- Caruana, L.; Fochi, M.; Ranieri, S.; Mazzanti, A.; Bernardi, L. Catalytic highly enantioselective vinylogous Povarov reaction. Chem. Commun. 2013, 49, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Ren, C.-L.; Wang, D.; Liu, L. Highly enantioselective [3+2] cycloaddition of vinylcyclopropane with nitroalkenes catalyzed by palladium(0) with a chiral bis(tert-amine) ligand. Chem. Eur. J. 2015, 21, 2335–2338. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).