Influence of Remaining Acid Sites of an Amorphous Aluminosilicate on the Oligomerization of n-Butenes after Impregnation with Nickel Ions

 ,
,  ,
, 
Abstract
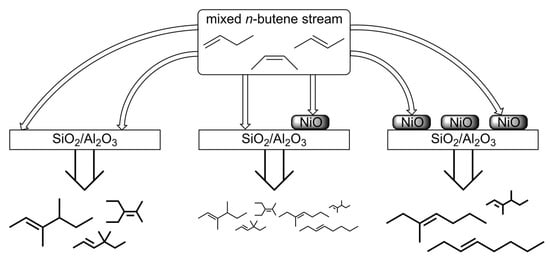
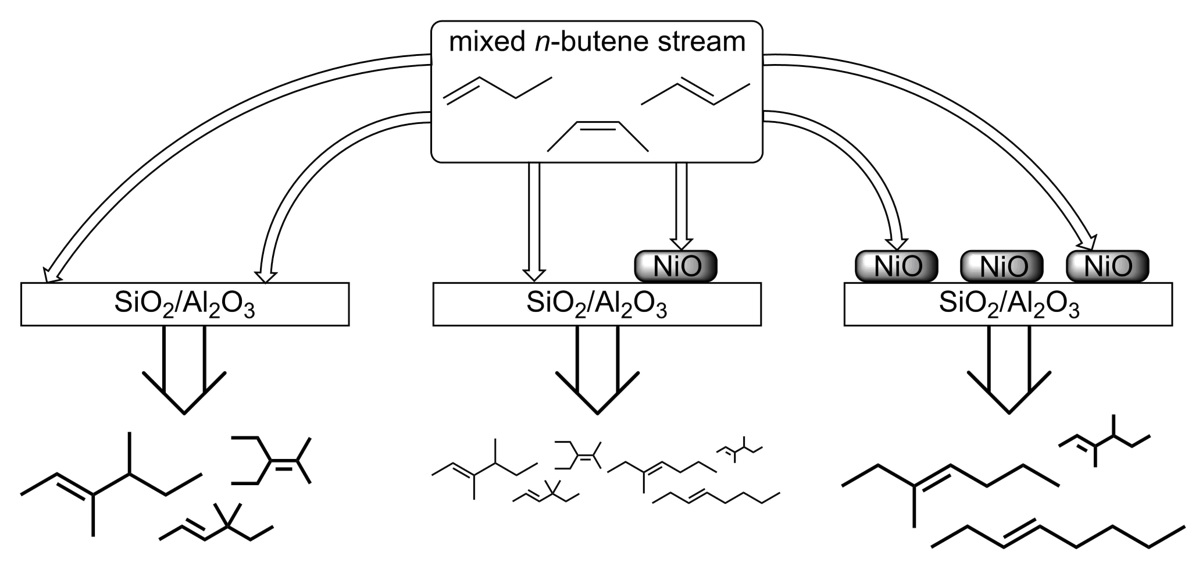
1. Introduction
2. Results and Discussion
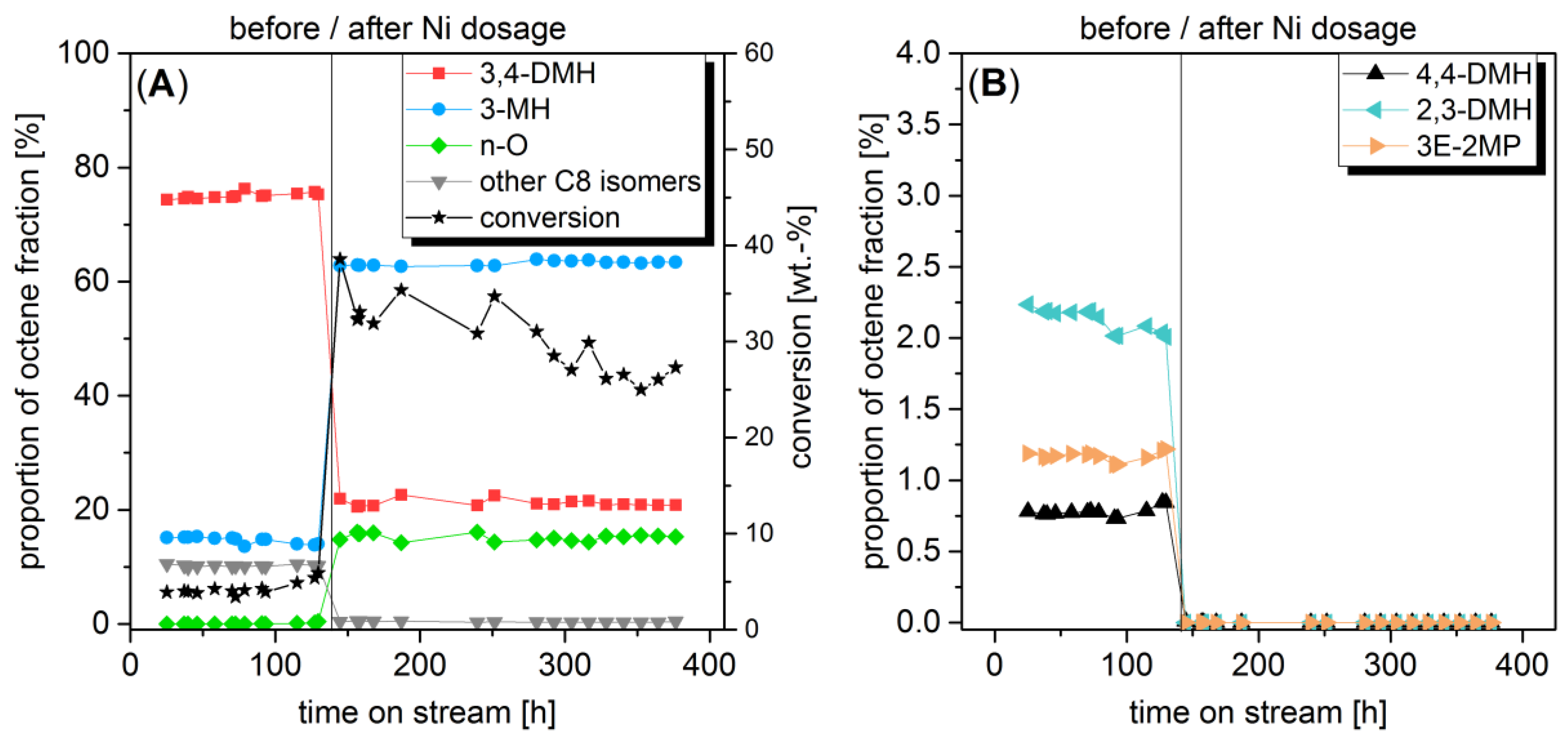
2.1. Change of Octene Isomer Distribution by In Situ Impregnation in a Differential Loop-Type Reactor
2.2. Correlation between Yield of Side Products and Feed Rate in Fixed Bed Reactor
2.3. Determination of Acid-Catalyzed Oligomerization on Nickel-Containing Catalysts
3. Materials and Methods
3.1. Catalysts
3.2. Characterization
3.3. Analysis of Products and Feed
3.4. Testing and In Situ Impregnation in a Loop-Type Reactor
3.5. Testing in a Fixed Bed Reactor
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Geilen, F.M.A.; Stochniol, G.; Peitz, S.; Schulte-Körne, E. Butenes. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2013; pp. 1–13. [Google Scholar]
- Hommeltoft, S.I. Isobutane alkylation: Recent developments and future perspectives. Appl. Catal. A 2001, 221, 421–428. [Google Scholar] [CrossRef]
- Broellos, K.; Volkamer, K.; Lindner, A. Process for Obtaining a Conjugated Diolefine from a C4-C5 Hydrocarbon Mixture. Patent DE 3119540 A1, 5 May 1982. [Google Scholar]
- Collignon, F.; Mariani, M.; Moreno, S.; Remy, M.; Poncelet, G.J. Gas Phase Synthesis of MTBE from Methanol and Isobutene over Dealuminated Zeolites. J. Catal. 1997, 166, 53–66. [Google Scholar] [CrossRef]
- Catani, R.; Mandreoli, M.; Rossini, S.; Vaccari, A. Mesoporous catalysts for the synthesis of clean diesel fuels by oligomerisation of olefins. Catal. Tod. 2002, 75, 125–131. [Google Scholar] [CrossRef]
- Ipatieff, V.N.; Corson, B.B.; Egloff, G. Polymerization, a New Source of Gasoline. Ind. Eng. Chem. 1935, 27, 1077–1081. [Google Scholar] [CrossRef]
- Nicholas, C.P. Applications of light olefin oligomerization to the production of fuels and chemicals. Appl. Catal. A Gen. 2017, 543, 82–97. [Google Scholar] [CrossRef]
- Busca, G. Acid Catalysts in Industrial Hydrocarbon Chemistry. Chem. Rev. 2007, 107, 5366–5410. [Google Scholar] [CrossRef]
- Silva, A.F.; Fernandes, A.; Antunes, M.M.; Neves, P.; Rocha, S.M.; Ribeiro, M.F.; Pillinger, M.; Ribeiro, J.; Silva, C.M.; Valente, A.A. Kinetics and Thermochemistry of C4-C6 Olefin Cracking on H-ZSM-5. Fuel 2017, 209, 371–382. [Google Scholar] [CrossRef]
- Keim, W.; Kowaldt, F.W.; Goddard, R.; Krüger, C. Novel coordination of (benzoylmethylene)-triphenylphosphorane in a nickel olgomerization catalyst. Angew. Chem. Int. Ed. Engl. 1978, 17, 466–467. [Google Scholar] [CrossRef]
- Keim, W.; Hoffmann, B.; Lodewick, R.; Peuckert, M.; Schmitt, G.; Fleischhauer, J.; Meier, U. Linear Oligomerization of olefins via nickel chelate complexes and mechanistic considerations based on semi-empirical calculations. J. Mol. Catal. 1979, 6, 79–97. [Google Scholar] [CrossRef]
- Behr, A.; Rentmeister, N.; Seidensticker, T.; Faßbach, T.A.; Peitz, S.; Maschmeyer, D. Nickel catalyzed dimerization reactions of vinylidene compounds: Head-to-head couplings and catalyst stabilization. J. Mol. Catal. A 2014, 395, 355–363. [Google Scholar] [CrossRef]
- Keim, W.; Behr, A.; Kraus, G. Influences of olefinic and diketonate ligands in the nickel-catalyzed linear oligomerization of 1-butene. J. Organomet. Chem. 1983, 251, 377–391. [Google Scholar] [CrossRef]
- Forestière, A.; Olivier-Bourbigou, H.; Saussine, L. Oligomerization of Monoolefins by Homogeneous Catalysts. Oil Gas. Sci. Technol. 2009, 64, 649–667. [Google Scholar] [CrossRef]
- Nierlich, F.; Neumeister, J.; Wildt, T.; Droste, W.; Obenaus, F. Process for Oligomerising Olefins. Patent EP 0395857, 4 May 1990. [Google Scholar]
- Forget, S.; Olivier-Bourbigou, H.; Delcroix, D. Homogeneous and Heterogeneous Nickel-Catalyzed Olefin Oligomerization: Experimental Investigation for a Common Mechanistic Proposition and Catalyst Optimization. ChemCatChem 2017, 9, 2408–2417. [Google Scholar] [CrossRef]
- Franke, R.; Selent, D.; Börner, A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef]
- Börner, A.; Franke, R. Hydroformylation—Fundamentals, Processes and Applications in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2016; Volume 2. [Google Scholar]
- Wendt, G.; Finster, J.; Schöllner, R. Untersuchungen an Nickeloxid-Mischkatalysatoren. VIII. Katalytische Eigenschaften von NiO-Al2O3/SiO2-Katalysatoren. Z. Anorg. Allg. Chem. 1982, 488, 197–206. [Google Scholar] [CrossRef]
- Wendt, G.; Jusek, M.; May, M. Untersuchungen an Nickeloxid-Mischkatalysatoren. XVI. Reduktionsverhalten amorpher NiO-Al2O3/SiO2-Katalysatoren. Z. Anorg. Allg. Chem. 1987, 550, 177–183. [Google Scholar] [CrossRef]
- Wendt, G.; Jusek, M.; Schöllner, R. Untersuchungen an Nickeloxid-Mischkatalysatoren. VII. Strukturelle Eigenschaften von NiO-Al2O3/SiO2-Katalysatoren. Z. Anorg. Allg. Chem. 1982, 488, 187–196. [Google Scholar] [CrossRef]
- Kießling, D.; Wendt, G.; Jusek, M.; Schöllner, R. Reduction of an Amorphous NiO-Al2O3/SiO2 Catalysts by Different Olefins and Hydrogen. React. Kinet. Catal. Lett. 1991, 43, 255–259. [Google Scholar] [CrossRef]
- Rabeah, J.; Radnik, J.; Briois, V.; Maschmeyer, D.; Stochniol, G.; Peitz, S.; Reeker, H.; La Fontaine, C.; Brückner, A. Tracing Active Sites in Supported Ni Catalysts during Butene Oligomerization by Operando Spectroscopy under Pressure. ACS Catal. 2016, 6, 8224–8228. [Google Scholar] [CrossRef]
- Brückner, A.; Bentrup, U.; Zanthoff, H.; Maschmeyer, D. The role of different Ni sites in supported nickel catalysts for butene dimerization under industry-like conditions. J. Catal. 2009, 266, 120–128. [Google Scholar] [CrossRef]
- Albrecht, S.; Kießling, D.; Maschmeyer, D.; Nierlich, F.; Wendt, G. Oligomerisierung von n-Butenen. Chem. Ing. Tech. 2005, 77, 695–709. [Google Scholar] [CrossRef]
- Nierlich, F.; Neumeister, J.; Wildt, T. The oligomerization of lower olefins by heterogeneous catalysis. Prepr. Am. Chem. Soc. Div Pet. Chem. 1991, 36, 585–595. [Google Scholar]
- Ehrmaier, A.; Liu, Y.; Peitz, S.; Jentys, A.; Chin, Y.-H.C.; Sanchez-Sanchez, M.; Bermejo-Deval, R.; Lercher, J. Dimerization of Linear Butenes on Zeolite-Supported Ni2+. ACS Catalysis 2019, 9, 315–324. [Google Scholar] [CrossRef]
- Ehrmaier, A.; Peitz, S.; Sanchez-Sanchez, M.; Bermejo-Deval, R.; Lercher, J. On the role of co-cations in nickel exchanged LTA zeolite for butene dimerization. Microporous Mesoporous Mater. 2019, 284, 241–246. [Google Scholar] [CrossRef]
- Ehrmaier, A.; Löbbert, L.; Sanchez-Sanchez, M.; Bermejo-Deval, R.; Lercher, J. Impact of Alkali and Alkali-Earth Cations on Ni-Catalyzed Dimerization of Butene. ChemCatChem 2020, 12, 3705–3711. [Google Scholar] [CrossRef]
- Alscher, F.; Nadolny, F.; Frenzel, H.; Knossalla, J.; Peitz, S.; Borovinskaya, E.; Breitkopf, C.; Franke, R.; Reschetilowski, W. Determination of the prevailing n-butenes dimerization mechanism over nickel containing Al-MCM-41/ZSM-5 mixed-phase catalysts. Catal. Sci. Technol. 2019, 9, 2456–2468. [Google Scholar] [CrossRef]
- Zhao, D.; Xu, Z.; Chada, J.P.; Carrero, C.A.; Rosenfeld, D.C.; Rogers, J.L.; Hermans, I.; Huber, G.W. Cobalt Oxide on N-Doped Carbon for 1-Butene Oligomerization to Produce Linear Octenes. ACS Catalysis 2017, 7, 7479–7489. [Google Scholar] [CrossRef]
- Franken, J.; Kirschhock, C.E.A.; Mathys, G.M.; Martens, J.A.; Franken, J.; Kirschhock, C.E.A.; Mathys, G.M.; Martens, J.A. Design of a Cobalt–Zeolite Catalyst for Semi-Linear Higher-Olefin Synthesis. ChemCatChem 2012, 4, 1245–1248. [Google Scholar] [CrossRef]
- Martens, J.A.; Ravishankar, R.; Mishin, I.E.; Jacobs, P.A. Tailored alkene oligomerization with H-ZSM-57 zeolite. Angew. Chem. Int. Ed. 2000, 39, 4376–4379. [Google Scholar] [CrossRef]
- Kärger, J.; Freude, D. Mass Transfer in Micro- and Mesoporous Materials. Chem. Eng. Technol. 2002, 25, 769–778. [Google Scholar] [CrossRef]
- Andrei, R.D.; Popa, M.I.; Fajula, F.; Hulea, V. Heterogeneous oligomerization of ethylene over highly active and stable NiAlSBA-15 mesoporous catalysts. J. Catal. 2015, 323, 76–84. [Google Scholar] [CrossRef]
- Kießling, D.; Wendt, G.; Hagenau, K.; Schöllner, R. Dimerization of n-butenes on amorphous NiO-Al2O3/SiO2 catalysts. Appl. Catal. 1991, 71, 69–78. [Google Scholar] [CrossRef]
- Brouwer, D.M.; Hogeveen, H. Electrophilic Substitutions at Alkanes and in Alkylcarbonium Ions. Progr. Phys. Org. Chem. 1972, 9, 179–240. [Google Scholar]
- Olah, G.A.; Jeuell, C.L.; Kelly, D.P.; Porter, R.D. Stable carbocations. CXIV Structure of cyclopropylcarbinyl and cyclobutyl cations. J. Am. Chem. Soc. 1972, 94, 146–156. [Google Scholar] [CrossRef]
- Liu, H.; Lei, G.D.; Sachtler, W.M.H. Alkane isomerization over solid acid catalysts—Effects of one-dimensional micropores. Appl. Catal. A Gen. 1996, 137, 167–177. [Google Scholar] [CrossRef]
- Hu, H.; Ke, M.; Zhang, K.; Liu, Q.; Yu, P.; Liu, Y.; Li, C.; Liu, W. Designing ferrierite-based catalysts with improved properties for skeletal isomerization of of n-butene to isobutene. RSC Adv. 2017, 7, 31535–31543. [Google Scholar] [CrossRef]
- Kim, Y.T.; Chada, J.P.; Xu, Z.; Pagan-Torres, Y.J.; Rosenfeld, D.C.; Winniford, W.L.; Schmidt, E.; Huber, G. Low-temperature oligomerization of 1-butene with H-ferrierite. J. Catal. 2015, 323, 33–44. [Google Scholar] [CrossRef]
- Lallemand, M.; Rusu, O.A.; Dumitriu, E.; Finiels, A.; Fajula, F.; Hulea, V. NiMCM-36 and NiMCM-22 catalysts for the ethylene oligomerization: Effect of zeolite texture and nickel cations/acid sites ratio. Appl. Catal. A Gen. 2008, 338, 37–43. [Google Scholar] [CrossRef]
- Bekker, R.; Prinsloo, N.M. Butene Oligomerization over Phosphoric Acid: Structural Characterization of Products. Ind. Eng. Chem. Res. 2009, 48, 10156–10162. [Google Scholar] [CrossRef]
- Reschetilowski, W. Einführung in Die Heterogene Katalyse; Springer Spektrum: Berlin/Heidelberg, Germany, 2014; pp. 91–107. [Google Scholar]
- Hulea, V.; Fajula, F. Ni-exchanged AlMCM-41—An efficient bifunctional catalyst for ethylene oligomerization. J. Catal. 2004, 225, 213–222. [Google Scholar] [CrossRef]
- Misono, M.; Yoneda, Y. The Acid Strength and the Catalytic Activity of Solid Catalysts in the Oligomerization of Propylene. Bull. Chem. Soc. Jap. 1967, 40, 42–49. [Google Scholar] [CrossRef]
- Villegas, J.I.; Kumar, N.; Heikkilä, T.; Lehto, V.-P.; Salmi, T.; Murzin, D.Y. A study on the dimerization of 1-butene over Beta zeolite. Top. Catal. 2007, 45, 187–190. [Google Scholar] [CrossRef]
- Kresnawahjuesa, O.; Kühl, G.H.; Gorte, R.J.; Quierini, C.A. An Examination of Bronsted Acid Sites in H-[Fe]-ZSM-5 for Oligomerization and Adsorption. J. Catal. 2002, 210, 106–115. [Google Scholar] [CrossRef]
- Wendt, G.; Kießling, D. Dimerization of n-butenes on NiO-Al2O3/SiO2 catalysts. Chem. Tech. 1995, 47, 136–143. [Google Scholar]
- Katada, N.; Niwa, N. Analysis of Acidic Properties of Zeolitic and Non-Zeolitic Solid Acid Catalysts Using Temperature-Programmed-Desorption of Ammonia. Catal. Surv. Asia 2004, 8, 161–170. [Google Scholar] [CrossRef]
- Arena, F.; Dario, R.; Parmaliana, A. A characterization study of the surface acidity of solid catalysts by temperature programmed methods. Appl. Catal. A Gen. 1998, 170, 127–137. [Google Scholar] [CrossRef]
- Wu, M.-C.; Truong, C.M.; Goodman, D.W. Interactions of ammonia with a nickel oxide (100) surface studied by high-resolution electron energy loss spectroscopy and temperature programmed spectroscopy. J. Phys. Chem. 1993, 97, 4182–4186. [Google Scholar] [CrossRef]
- Vaughan, J.S.; O´Connor, C.T.; Fletcher, J.C.Q. High-Pressure Oligomerization of Propene over Heteropoly Acids. J. Catal. 1994, 147, 441–454. [Google Scholar] [CrossRef]
- Li, X.; Han, D.; Wang, H.; Liu, G.; Wang, B.; Li, Z.; Wu, J. Propene oligomerization to high-quality liquid fuels over Ni/HZSM-5. Fuel 2015, 144, 9–14. [Google Scholar] [CrossRef]
- Reschetilowski, W. Metal-support interaction in bifunctional metal zeolite catalysts and the HSAB principle. Erdöl Erdgas Kohle 1996, 112, 467–470. [Google Scholar]
- Hoang, D.L.; Berndt, H.; Miessner, H.; Schreier, E.; Völter, J.; Lieske, H. Nickel modified H-ZSM-5 catalysts. Appl. Catal. A Gen. 1994, 114, 295–311. [Google Scholar] [CrossRef]
- Moussa, S.; Concepión, P.; Arribas, M.A.; Martínez, A. Nature of Active Nickel Sites and Initiation Mechanism for Ethylene Oligomerization on Heterogeneous Ni-Beta Catalysts. ACS Catal. 2018, 8, 3903–3912. [Google Scholar] [CrossRef]
- Barret, E.P.; Joyner, L.G.; Halenda, P.H. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Rouquérol, J.; Avnir, D.; Fairbridge, C.W.; Everett, D.H.; Haynes, J.H.; Pernicone, N.; Ramsay, J.D.F.; Sing, K.S.W.; Unger, K.K. Recommendations for the characterization of porous solids. Pure Appl. Chem. 1994, 66, 1739–1758. [Google Scholar] [CrossRef]
- Beroza, M.; Sarmiento, R. Determination of the Carbon Skeleton and Other Structural Features of Organic Compounds by Gas Chromatography. Anal. Chem. 1963, 35, 1353–1357. [Google Scholar] [CrossRef]
- Soják, L.; Addová, G.; Kubinec, R.; Kraus, A.; Boháč, A. Capillary gas chromatography—Mass spectrometry of all 93 acyclic octenes and their identification in fluid catalytic cracked gasoline. J. Chrom. A 2004, 1025, 237–253. [Google Scholar] [CrossRef]
- Reisener, G.; Schreier, M.; Adler, R. Korrektur reaktionskinetischer Daten des realen Differential-Kreislaufreaktors. Chem. Ing. Techn. 2000, 72, 1192–1195. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst Sample | m3,4-DMH/gcat per h | m3-MH/gcat per h | mn-O/gcat per h |
|---|---|---|---|
| Ni-free | 2.4 | 0.1 | 0 |
| 1 wt.% Ni | 1.2 | 0.5 | 0.2 |
| 6 wt.% Ni | 0.9 | 0.6 | 0.2 |
| 14 wt.% Ni | 0.5 | 1.1 | 0.3 |
| Catalyst Sample | Measured Content of 3,4-DMH in Product Stream, wt.% | 3,4-DMH calc. via 4,4-DMH, wt.% | 3,4-DMH calc. via 3E-2MP, wt.% |
|---|---|---|---|
| 1 wt.% Ni | 13.0 | 14.8 | 14.3 |
| 6 wt.% Ni | 9.6 | 9.6 | 9.4 |
| 14 wt.% Ni | 5.1 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nadolny, F.; Alscher, F.; Peitz, S.; Borovinskaya, E.; Franke, R.; Reschetilowski, W. Influence of Remaining Acid Sites of an Amorphous Aluminosilicate on the Oligomerization of n-Butenes after Impregnation with Nickel Ions. Catalysts 2020, 10, 1487. https://doi.org/10.3390/catal10121487
Nadolny F, Alscher F, Peitz S, Borovinskaya E, Franke R, Reschetilowski W. Influence of Remaining Acid Sites of an Amorphous Aluminosilicate on the Oligomerization of n-Butenes after Impregnation with Nickel Ions. Catalysts. 2020; 10(12):1487. https://doi.org/10.3390/catal10121487
Chicago/Turabian StyleNadolny, Fabian, Felix Alscher, Stephan Peitz, Ekaterina Borovinskaya, Robert Franke, and Wladimir Reschetilowski. 2020. "Influence of Remaining Acid Sites of an Amorphous Aluminosilicate on the Oligomerization of n-Butenes after Impregnation with Nickel Ions" Catalysts 10, no. 12: 1487. https://doi.org/10.3390/catal10121487
APA StyleNadolny, F., Alscher, F., Peitz, S., Borovinskaya, E., Franke, R., & Reschetilowski, W. (2020). Influence of Remaining Acid Sites of an Amorphous Aluminosilicate on the Oligomerization of n-Butenes after Impregnation with Nickel Ions. Catalysts, 10(12), 1487. https://doi.org/10.3390/catal10121487