Spray-Dried Ni Catalysts with Tailored Properties for CO2 Methanation

 , , and
, , and 
Abstract
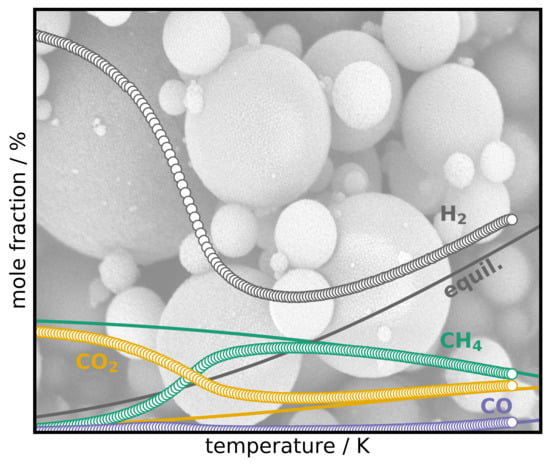
1. Introduction
2. Results and Discussion
2.1. Morphological Examination
2.2. Elemental Analysis
2.3. Physisorption
2.4. XRD
2.5. Adsorption and Degree of Reduction
2.6. Temperature-Programmed Reduction
2.7. Temperature-Programmed Desorption
2.8. Activity of the Catalysts
2.9. Activation Energies and Turnover Frequencies
3. Materials and Methods
3.1. Materials
3.2. Experimental Setup and Synthesis of the NiO/SiO2 Nanoparticles
3.3. Physical Characterization
3.4. Chemical Characterization
3.5. Methanation Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TPR | Temperature-programmed reduction |
| TPD | Temperature-programmed desorption |
| XRD | X-ray diffraction |
| TEM | Transmission Electron Microscopy |
| SEM | Scanning Electron Microscopy |
| TCD | Thermal conductivity detector |
| MS | Mass spectrometer |
| BB | Building-Blocks |
| SAXS | Small-angle X-ray scattering |
| BET | Brunauer-Emmett-Teller method |
| BJH | Barrett-Joyner-Halenda model |
List of Symbols
| Latin symbols | ||
| a | area | m2 |
| D | dispersion | - |
| DOR | degree of reduction | % |
| d | diameter | m |
| f | calibration factor | mol m−2 |
| K | Scherrer form factor | - |
| m | mass | g |
| p | pressure | bar |
| Q | adsorbed amount | mol g−1 |
| R | ideal gas constant | J mol−1 K−1 |
| r | reaction rate | mol s−1 g−1 |
| S | selectivity | - |
| T | temperature | K |
| volume flow | mL min−1 | |
| W | molar mass | g mol−1 |
| w | metal loading | - |
| WHSV | weight hourly space velocity | LN h−1 g−1 |
| X | conversion | - |
| x | molar fraction | - |
| Y | yield | - |
| z | adsorption stoichiometry | - |
| Greek symbols | ||
| α | volume reduction | - |
| β | temperature ramp | K min−1 |
| Θ | Bragg angle | ∘ |
| wavelength | m | |
| density | kg m−3 | |
| surface area | m2 | |
| Subscripts | ||
| calibration | calibration | |
| cat | catalyst | |
| f | form factor | |
| N | normal conditions | |
| Superscripts | ||
| in | inlet | |
| out | outlet | |
References
- Tomiyama, S.; Takahashi, R.; Sato, S.; Toshiaki, S.; Satoshi, Y. Preparation of Ni/SiO2 catalyst with high thermal stability for CO2-reforming of CH4. Appl. Catal. A Gen. 2003, 241, 349–361. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W.; Wang, Z.; Zhou, X.; Wang, Z.; Liu, C.J. Steam reforming of methane over Ni/SiO2 catalyst with enhanced coke resistance at low steam to methane ratio. Catal. Today 2015, 256, 130–136. [Google Scholar] [CrossRef]
- Huang, X.; Reimert, R. Kinetics of steam reforming of ethane on Ni/YSZ (yttria-stabilised zirconia) catalyst. Fuel 2013, 106, 380–387. [Google Scholar] [CrossRef]
- Xie, Z.; Yan, B.; Lee, J.H.; Wu, Q.; Li, X.; Zhao, B.; Su, D.; Zhang, L.; Chen, J.G. Effects of oxide supports on the CO2 reforming of ethane over Pt-Ni bimetallic catalysts. Appl. Catal. B Environ. 2019, 245, 376–388. [Google Scholar] [CrossRef]
- Sehested, J.; Dahl, S.; Jacobsen, J.; Rostrup-Nielsen, J.R. Methanation of CO over nickel: Mechanism and kinetics at high H2/CO ratios. J. Phys. Chem. B 2005, 109, 2432–2438. [Google Scholar] [CrossRef]
- Nematollahi, B.; Rezaei, M.; Lay, E.N. Preparation of highly active and stable NiO-CeO2 nanocatalysts for CO selective methanation. Int. J. Hydrogen Energy 2015, 40, 8539–8547. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Gao, J.; Liu, Q.; Gu, F.; Liu, B.; Zhong, Z.; Su, F. Recent advances in methanation catalysts for the production of synthetic natural gas. RSC Adv. 2015, 5, 22759–22776. [Google Scholar] [CrossRef]
- Kreitz, B.; Wehinger, G.D.; Turek, T. Dynamic simulation of the CO2 methanation in a micro-structured fixed-bed reactor. Chem. Eng. Sci. 2019, 195, 541–552. [Google Scholar] [CrossRef]
- Kreitz, B.; Friedland, J.; Güttel, R.; Wehinger, G.D.; Turek, T. Dynamic Methanation of CO2—Effect of Concentration Forcing. Chem. Ing. Tech. 2019, 91, 576–582. [Google Scholar] [CrossRef]
- Kreitz, B.; Brauns, J.; Wehinger, G.D.; Turek, T. Modeling the Dynamic Power–to–Gas Process: Coupling Electrolysis with CO2 Methanation. Chem. Ing. Tech. 2020, 43, 20332. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported Catalysts for CO2 Methanation: A Review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Ashok, J.; Pati, S.; Hongmanorom, P.; Tianxi, Z.; Junmei, C.; Kawi, S. A review of recent catalyst advances in CO2 methanation processes. Catal. Today 2020. [Google Scholar] [CrossRef]
- Wei, W.; Jinlong, G. Methanation of carbon dioxide: An overview. Front. Chem. Sci. Eng. 2011, 5, 2–10. [Google Scholar] [CrossRef]
- Pan, Q.; Peng, J.; Sun, T.; Wang, S.; Wang, S. Insight into the reaction route of CO2 methanation: Promotion effect of medium basic sites. Catal. Commun. 2014, 45, 74–78. [Google Scholar] [CrossRef]
- Aziz, M.; Jalil, A.A.; Triwahyono, S.; Mukti, R.R.; Taufiq-Yap, Y.H.; Sazegar, M.R. Highly active Ni-promoted mesostructured silica nanoparticles for CO2 methanation. Appl. Catal. B Environ. 2014, 147, 359–368. [Google Scholar] [CrossRef]
- Chen, C.S.; Budi, C.S.; Wu, H.C.; Saikia, D.; Kao, H.M. Size-Tunable Ni Nanoparticles Supported on Surface-Modified, Cage-Type Mesoporous Silica as Highly Active Catalysts for CO2 Hydrogenation. ACS Catal. 2017, 7, 8367–8381. [Google Scholar] [CrossRef]
- Ye, R.P.; Gong, W.; Sun, Z.; Sheng, Q.; Shi, X.; Wang, T.; Yao, Y.; Razink, J.J.; Lin, L.; Zhou, Z.; et al. Enhanced stability of Ni/SiO2 catalyst for CO2 methanation: Derived from nickel phyllosilicate with strong metal-support interactions. Energy 2019, 188, 116059. [Google Scholar] [CrossRef]
- Zhu, P.; Chen, Q.; Yoneyama, Y.; Tsubaki, N. Nanoparticle modified Ni-based bimodal pore catalysts for enhanced CO2 methanation. RSC Adv. 2014, 4, 64617–64624. [Google Scholar] [CrossRef]
- Huang, Y.J.; Schwarz, J.A. The effect of catalyst preparation on catalytic activity: I. The catalytic activity of Ni/Al2O3 catalysts prepared by wet impregnation. Appl. Catal. 1987, 30, 239–253. [Google Scholar] [CrossRef]
- Li, G.; Hu, L.; Hill, J.M. Comparison of reducibility and stability of alumina-supported Ni catalysts prepared by impregnation and co-precipitation. Appl. Catal. A Gen. 2006, 301, 16–24. [Google Scholar] [CrossRef]
- Ashok, J.; Ang, M.L.; Kawi, S. Enhanced activity of CO2 methanation over Ni/CeO2-ZrO2 catalysts: Influence of preparation methods. Catal. Today 2017, 281, 304–311. [Google Scholar] [CrossRef]
- Koschany, F.; Schlereth, D.; Hinrichsen, O. On the kinetics of the methanation of carbon dioxide on coprecipitated NiAl(O)x. Appl. Catal. B Environ. 2016, 181, 504–516. [Google Scholar] [CrossRef]
- Chiarello, G.; Rossetti, I.; Forni, L. Flame-spray pyrolysis preparation of perovskites for methane catalytic combustion. J. Catal. 2005, 236, 251–261. [Google Scholar] [CrossRef]
- Compagnoni, M.; Tripodi, A.; Di Michele, A.; Sassi, P.; Signoretto, M.; Rossetti, I. Low temperature ethanol steam reforming for process intensification: New Ni/MxO-ZrO2 active and stable catalysts prepared by flame spray pyrolysis. Int. J. Hydrogen Energy 2017, 42, 28193–28213. [Google Scholar] [CrossRef]
- Saib, A.M.; Claeys, M.; van Steen, E. Silica supported cobalt Fischer–Tropsch catalysts: Effect of pore diameter of support. Catal. Today 2002, 71, 395–402. [Google Scholar] [CrossRef]
- Borg, Ø.; Eri, S.; Blekkan, E.A.; Storsæter, S.; Wigum, H.; Rytter, E.; Holmen, A. Fischer–Tropsch synthesis over γ-alumina-supported cobalt catalysts: Effect of support variables. J. Catal. 2007, 248, 89–100. [Google Scholar] [CrossRef]
- Song, D.; Li, J. Effect of catalyst pore size on the catalytic performance of silica supported cobalt Fischer–Tropsch catalysts. J. Mol. Catal. A Chem. 2006, 247, 206–212. [Google Scholar] [CrossRef]
- Koirala, R.; Pratsinis, S.E.; Baiker, A. Synthesis of catalytic materials in flames: Opportunities and challenges. Chem. Soc. Rev. 2016, 45, 3053–3068. [Google Scholar] [CrossRef]
- Gradon, L.; Balgis, R.; Hirano, T.; Rahmatika, A.M.; Ogi, T.; Okuyama, K. Advanced aerosol technologies towards structure and morphologically controlled next-generation catalytic materials. J. Aerosol Sci. 2020, 149, 105608. [Google Scholar] [CrossRef]
- Martínez Arias, A.; Weber, A.P. Aerosol synthesis of porous SiO2-cobalt-catalyst with tailored pores and tunable metal particle size for Fischer-Tropsch synthesis (FTS). J. Aerosol Sci. 2019, 131, 1–12. [Google Scholar] [CrossRef]
- Wojciechowski, B. The temperature scanning reactor I: Reactor types and modes of operation. Catal. Today 1997, 36, 167–190. [Google Scholar] [CrossRef]
- Asprey, S. The temperature scanning reactor III: Experimental procedures and data processing. Catal. Today 1997, 36, 209–226. [Google Scholar] [CrossRef]
- Wojciechowski, B.; Asprey, S.P. Kinetic studies using temperature-scanning: The oxidation of carbon monoxide. Appl. Catal. A Gen. 2000, 190, 1–24. [Google Scholar] [CrossRef]
- Liebner, C.; Wolf, D.; Baerns, M.; Kolkowski, M.; Keil, F.J. A high-speed method for obtaining kinetic data for exothermic or endothermic catalytic reactions under non-isothermal conditions illustrated for the ammonia synthesis. Appl. Catal. A Gen. 2003, 240, 95–110. [Google Scholar] [CrossRef]
- Vogt, C.; Wijten, J.; Madeira, C.L.; Kerkenaar, O.; Xu, K.; Holzinger, R.; Monai, M.; Weckhuysen, B.M. Alkali Promotion in the Formation of CH4 from CO2 and Renewably Produced H2 over Supported Ni Catalysts. ChemCatChem 2020, 12, 2792–2800. [Google Scholar] [CrossRef]
- Le, T.A.; Kim, T.W.; Lee, S.H.; Park, E.D. Effects of Na content in Na/Ni/SiO2 and Na/Ni/CeO2 catalysts for CO and CO2 methanation. Catal. Today 2018, 303, 159–167. [Google Scholar] [CrossRef]
- Beierlein, D.; Häussermann, D.; Pfeifer, M.; Schwarz, T.; Stöwe, K.; Traa, Y.; Klemm, E. Is the CO2 methanation on highly loaded Ni-Al2O3 catalysts really structure-sensitive? Appl. Catal. B Environ. 2019, 247, 200–219. [Google Scholar] [CrossRef]
- Zeng, L.; Weber, A.P. Aerosol synthesis of nanoporous silica particles with controlled pore size distribution. J. Aerosol Sci. 2014, 76, 1–12. [Google Scholar] [CrossRef]
- Okuyama, K.; Wuled Lenggoro, I. Preparation of nanoparticles via spray route. Chem. Eng. Sci. 2003, 58, 537–547. [Google Scholar] [CrossRef]
- Röhrbein, J.; Arias, A.M.; Weber, A.P. Aerosol-Synthese von porösen Katalysatorpartikeln mit einstellbaren Porengrößen und Katalysatordurchmessern. Chem. Ing. Tech. 2017, 89, 1739–1751. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Griboval-Constant, A.; Bechara, R.; Zholobenko, V.L. Pore Size Effects in Fischer Tropsch Synthesis over Cobalt-Supported Mesoporous Silicas. J. Catal. 2002, 206, 230–241. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- St. O’Neill, H.C.; Dollase, W.A.; Ross, C.R. Temperature dependence of the cation distribution in nickel aluminate (NiAl2O4) spinel: A powder XRD study. Phys. Chem. Miner. 1991, 18, 302–319. [Google Scholar] [CrossRef]
- Zhou, R.S.; Snyder, R.L. Structures and transformation mechanisms of the η, γ and θ transition aluminas. Acta Crystallogr. Sect. B Struct. Sci. 1991, 47, 617–630. [Google Scholar] [CrossRef]
- Cao, A.; Lu, R.; Veser, G. Stabilizing metal nanoparticles for heterogeneous catalysis. Phys. Chem. Chem. Phys. PCCP 2010, 12, 13499–13510. [Google Scholar] [CrossRef]
- Hansen, T.W.; Delariva, A.T.; Challa, S.R.; Datye, A.K. Sintering of catalytic nanoparticles: Particle migration or Ostwald ripening? Accounts Chem. Res. 2013, 46, 1720–1730. [Google Scholar] [CrossRef]
- Cao, A.; Veser, G. Exceptional high-temperature stability through distillation-like self-stabilization in bimetallic nanoparticles. Nat. Mater. 2010, 9, 75–81. [Google Scholar] [CrossRef]
- Petroski, J.M.; Wang, Z.L.; Green, T.C.; El-Sayed, M.A. Kinetically Controlled Growth and Shape Formation Mechanism of Platinum Nanoparticles. J. Phys. Chem. B 1998, 102, 3316–3320. [Google Scholar] [CrossRef]
- Van de Loosdrecht, J.; van der Kraan, A.M.; van Dillen, A.J.; Geus, J.W. Metal-Support Interaction: Titania-Supported and Silica-Supported Nickel Catalysts. J. Catal. 1997, 170, 217–226. [Google Scholar] [CrossRef]
- Zoz, W.; Gonzalez, R. Stabilization and sintering of porous Pt/SiO2: A new approach. Appl. Catal. A Gen. 1993, 102, 181–200. [Google Scholar] [CrossRef]
- Khodakov, A.Y. Enhancing cobalt dispersion in supported Fischer-Tropsch catalysts via controlled decomposition of cobalt precursors. Braz. J. Phys. 2009, 39, 171–175. [Google Scholar] [CrossRef]
- Friedland, J.; Kreitz, B.; Grimm, H.; Turek, T.; Güttel, R. Measuring Adsorption Capacity of Supported Catalysts with a Novel Quasi-Continuous Pulse Chemisorption Method. ChemCatChem 2020, 12, 4373–4386. [Google Scholar] [CrossRef]
- Mile, B.; Stirling, D.; Zammitt, M.A.; Lovell, A.; Webb, M. The location of nickel oxide and nickel in silica-supported catalysts: Two forms of “NiO” and the assignment of temperature-programmed reduction profiles. J. Catal. 1988, 114, 217–229. [Google Scholar] [CrossRef]
- Burattin, P.; Che, M.; Louis, C. Metal Particle Size in Ni/SiO2 Materials Prepared by Deposition-Precipitation: Influence of the Nature of the Ni(II) Phase and of Its Interaction with the Support. J. Phys. Chem. B 1999, 103, 6171–6178. [Google Scholar] [CrossRef]
- Zieliński, J. Reductibility of silica supported nickel oxide. Catal. Lett. 1995, 31, 47–56. [Google Scholar] [CrossRef]
- Ho, S.C.; Chou, T.C. The Role of Anion in the Preparation of Nickel Catalyst Detected by TPR and FTIR Spectra. Ind. Eng. Chem. Res. 1995, 34, 2279–2284. [Google Scholar] [CrossRef]
- Louis, C.; Cheng, Z.X.; Che, M. Characterization of nickel/silica catalysts during impregnation and further thermal activation treatment leading to metal particles. J. Phys. Chem. 1993, 97, 5703–5712. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, J.; Chen, Y.W. Studies of Surface NiO Species in NiO/SiO2 Catalysts using Temperature-programmed Reduction and X-Ray Diffraction. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1992, 88, 2075–2078. [Google Scholar] [CrossRef]
- Burattin, P.; Che, M.; Louis, C. Ni/SiO2 Materials Prepared by Deposition-Precipitation: Influence of the Reduction Conditions and Mechanism of Formation of Metal Particles. J. Phys. Chem. B 2000, 104, 10482–10489. [Google Scholar] [CrossRef]
- Coenen, J.W. Characterization of the standard nickel/silica catalyst EuroNi-1. Appl. Catal. 1989, 54, 65–78. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Chemistry of Nickel-Alumina Catalysts. J. Catal. 1976, 45, 41–53. [Google Scholar] [CrossRef]
- Ewald, S.; Hinrichsen, O. On the interaction of CO2 with Ni-Al catalysts. Appl. Catal. A Gen. 2019, 580, 71–80. [Google Scholar] [CrossRef]
- Goodwin, D.G.; Speth, R.L.; Moffat, H.K.; Weber, B.W. Cantera: An Object-oriented Software Toolkit for Chemical Kinetics, Thermodynamics, and Transport Processes. Version 2.4.0. 2018. Available online: https://www.cantera.org (accessed on 24 August 2020).
- Rusic, B.; Bross, D.H. Active Thermochemical Tables (ATcT) Values Based on ver. 1.122 g of the Thermochemical Network. 2019. Available online: https://atct.anl.gov/ (accessed on 7 August 2020).
- Burcat, A.; Ruscic, B. Third Millenium Ideal Gas and Condensed Phase Thermochemical Database for Combustion (with Update from Active Thermochemical Tables); Technical Report; Argonne National Lab. (ANL): Argonne, IL, USA, 2005. [Google Scholar]
- Bjørgum, E.; Chen, D.; Bakken, M.G.; Christensen, K.O.; Holmen, A.; Lytken, O.; Chorkendorff, I. Energetic mapping of Ni catalysts by detailed kinetic modeling. J. Phys. Chem. B 2005, 109, 2360–2370. [Google Scholar] [CrossRef]
- Weatherbee, G.D.; Bartholomew, C.H. Hydrogenation of CO2 on Group VIII Metal: I. Specific Activity of Ni/SiO2. J. Catal. 1981, 68, 67–76. [Google Scholar] [CrossRef]
- Karam, L.; Reboul, J.; El Hassan, N.; Nelayah, J.; Massiani, P. Nanostructured Nickel Aluminate as a Key Intermediate for the Production of Highly Dispersed and Stable Nickel Nanoparticles Supported within Mesoporous Alumina for Dry Reforming of Methane. Molecules 2019, 24, 4170. [Google Scholar] [CrossRef]
- Aziz, M.; Jalil, A.A.; Triwahyono, S.; Saad, M. CO2 methanation over Ni-promoted mesostructured silica nanoparticles: Influence of Ni loading and water vapor on activity and response surface methodology studies. Chem. Eng. J. 2015, 260, 757–764. [Google Scholar] [CrossRef]
- Vrijburg, W.L.; van Helden, J.W.A.; van Hoof, A.J.F.; Friedrich, H.; Groeneveld, E.; Pidko, E.A.; Hensen, E.J.M. Tunable colloidal Ni nanoparticles confined and redistributed in mesoporous silica for CO2 methanation. Catal. Sci. Technol. 2019, 9, 2578–2591. [Google Scholar] [CrossRef]
- Ren, J.; Guo, H.; Yang, J.; Qin, Z.; Lin, J.; Li, Z. Insights into the mechanisms of CO2 methanation on Ni(111) surfaces by density functional theory. Appl. Surf. Sci. 2015, 351, 504–516. [Google Scholar] [CrossRef]
- Vogt, C.; Groeneveld, E.; Kamsma, G.; Nachtegaal, M.; Lu, L.; Kiely, C.J.; Berben, P.H.; Meirer, F.; Weckhuysen, B.M. Unravelling structure sensitivity in CO2 hydrogenation over nickel. Nat. Catal. 2018, 1, 127–134. [Google Scholar] [CrossRef]
- Blaylock, D.W.; Zhu, Y.A.; Green, W.H. Computational Investigation of the Thermochemistry and Kinetics of Steam Methane Reforming Over a Multi-Faceted Nickel Catalyst. Top. Catal. 2011, 54, 828–844. [Google Scholar] [CrossRef]
- Weber, A.P.; Seipenbusch, M.; Kasper, G. Size Effects in the Catalytic Activity of Unsupported Metallic Nanoparticles. J. Nanoparticle Res. 2003, 5, 293–298. [Google Scholar] [CrossRef]
- Ertl, G.; Knözinger, H.; Schüth, F.; Weitkamp, J. Handbook of Heterogeneous Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Unit | 1a | 1b | 1c | 2a | 2b | 2c | 3a | 3b | 3c | 4a | 4b | 4c | Method |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Surface area | m2 g−1 | - | 110 | 96 | - | 111 | 103 | - | 184 | 153 | - | 199 | 181 | BET |
| Pore volume | mm3 g−1 | - | 281 | 269 | - | 290 | 272 | - | 357 | 334 | - | 398 | 375 | BJH |
| Average pore size | - | 9.0 | 9.3 | - | 9.1 | 8.6 | - | 6.4 | 6.6 | - | 6.7 | 6.6 | BJH | |
| Ni loading | wt% | 20.9 | 20.9 | 20.9 | 13.0 | 13.0 | 13.0 | 23.1 | 23.1 | 23.1 | 12.2 | 12.2 | 12.2 | ICP-OES |
| Na content | wt% | 0.01 | 0.01 | 0.01 | <0.003 | <0.003 | <0.003 | 0.12 | 0.12 | 0.12 | 0.15 | 0.15 | 0.15 | ICP-OES |
| K content | wt% | 0.018 | 0.018 | 0.018 | 0.019 | 0.019 | 0.019 | 0.014 | 0.014 | 0.014 | 0.011 | 0.011 | 0.011 | ICP-OES |
| uptake | μmol g−1 | 402 | 181 | 93 | 254 | 171 | 58 | 460 | 271 | 110 | 324 | 205 | 95 | chemisorption |
| Metal surface area | m2 g−1 | 34.3 | 16.3 | 8.9 | 28.1 | 17.6 | 8.4 | 40.5 | 23.8 | 12.0 | 34.6 | 21.1 | 11.3 | chemisorption |
| Average Ni crystal size | 4.1 (4.5) | 8.6 (6.8) | 15.9 (8.3) | 3.1 (3.2) | 5.0 (4.7) | 10.4 (5.4) | 3.9 (4.3) | 6.5 (5.1) | 12.9 (5.9) | 2.4 (2.0) | 3.9 (2.5) | 7.3 (2.8) | chemisorption (XRD) | |
| Ni dispersion | % | 24.6 | 11.7 | 6.3 | 32.4 | 20.2 | 9.7 | 26.2 | 15.4 | 7.8 | 42.4 | 25.9 | 13.9 | chemisorption |
| Degree of reduction | % | 91.9 | 87.0 | 82.7 | 70.8 | 76.6 | 53.9 | 89.2 | 89.2 | 71.4 | 73.5 | 76.3 | 66.2 | chemisorption |
| uptake | μmol g−1 | 78.5 | 26.6 | - | - | - | - | 126.3 | 53.5 | 16.9 | - | - | - | chemisorption |
| Property | Unit | 5a | 5b | 5c | 6a | 6b | 6c | Method |
|---|---|---|---|---|---|---|---|---|
| Surface area | m2 g−1 | - | 178 | 123 | - | 155 | 99 | BET |
| Pore volume | mm3 g−1 | - | 337 | 331 | - | 311 | 293 | BJH |
| Average pore size | - | 5.9 | 8.0 | - | 6.4 | 8.7 | BJH | |
| Ni loading | wt% | 9.0 | 9.0 | 9.0 | 15.1 | 15.1 | 15.1 | ICP-OES |
| Na content | wt% | 0.16 | 0.16 | 0.16 | 0.17 | 0.17 | 0.17 | ICP-OES |
| K content | wt% | 0.0055 | 0.0055 | 0.0055 | 0.0046 | 0.0046 | 0.0046 | ICP-OES |
| uptake | μmol g−1 | 7 | - | - | 143 | 69 | - | chemisorption |
| Metal surface area | m2 g−1 | 5.8 | - | - | 28.3 | 18.1 | - | chemisorption |
| Average Ni crystal size | 14.5 | - | - | 2.9 | 4.5 | - | chemisorption | |
| Ni dispersion | % | 7.1 | - | - | 34.7 | 22.3 | - | chemisorption |
| Degree of reduction | % | 9.6 | - | - | 39.6 | 29.8 | - | chemisorption |
| uptake | μmol g−1 | 140.9 | - | - | 125.6 | - | - | chemisorption |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kreitz, B.; Martínez Arias, A.; Martin, J.; Weber, A.P.; Turek, T. Spray-Dried Ni Catalysts with Tailored Properties for CO2 Methanation. Catalysts 2020, 10, 1410. https://doi.org/10.3390/catal10121410
Kreitz B, Martínez Arias A, Martin J, Weber AP, Turek T. Spray-Dried Ni Catalysts with Tailored Properties for CO2 Methanation. Catalysts. 2020; 10(12):1410. https://doi.org/10.3390/catal10121410
Chicago/Turabian StyleKreitz, Bjarne, Aurina Martínez Arias, Jan Martin, Alfred P. Weber, and Thomas Turek. 2020. "Spray-Dried Ni Catalysts with Tailored Properties for CO2 Methanation" Catalysts 10, no. 12: 1410. https://doi.org/10.3390/catal10121410
APA StyleKreitz, B., Martínez Arias, A., Martin, J., Weber, A. P., & Turek, T. (2020). Spray-Dried Ni Catalysts with Tailored Properties for CO2 Methanation. Catalysts, 10(12), 1410. https://doi.org/10.3390/catal10121410